

Abstract
Italy is the first western country suffering heavy severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) transmission and disease impact after coronavirus disease‐2019 pandemia started in China. Even though the presence of mutations on spike glycoprotein and nucleocapsid in Italian isolates has been reported, the potential impact of these mutations on viral transmission has not been evaluated. We have compared SARS‐CoV‐2 genome sequences from Italian patients with virus sequences from Chinese patients. We focussed upon three nonsynonymous mutations of genes coding for S(one) and N (two) viral proteins present in Italian isolates and absent in Chinese ones, using various bioinformatics tools. Amino acid analysis and changes in three‐dimensional protein structure suggests the mutations reduce protein stability and, particularly for S1 mutation, the enhanced torsional ability of the molecule could favor virus binding to cell receptor(s). This theoretical interpretation awaits experimental and clinical confirmation.
Keywords: bioinformatics, COVID‐19, molecular evolution, mutation, SARS coronavirus
Highlights
-
‐
two highly prevalent mutation have been developed by SARS‐CoV‐2, one on the Spike glycoprotein and one on the Nucleocapsid protein
-
‐
The enhanced torsional ability of Spike protein could favor virus binding to human cell receptors
-
‐
mutations on the Nucleocapsid region could influence the modulation of a number of virus properties, including cell signaling
-
‐
the study of SARS‐CoV‐2 mutations could be important for vaccines developmen
1. INTRODUCTION
Coronavirus disease‐2019 (COVID‐19), which is caused by a novel coronavirus termed severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), is a respiratory disease that officially started in the Chinese city of Wuhan Hubei Province during December 2019 and has since spread globally as a pandemic. 1 As of 13 May 2020, the total COVID‐19 world cases have surpassed four million with more than 250 000 deaths. Italy was swept by the virus soon after China and has long ranked second in the dreadful list of most affected countries with high rate of contagiousness and SARS‐CoV‐2‐ attributable mortality rate (https://www.worldometers.info/coronavirus/)
SARS‐CoV‐2 is a beta‐coronavirus similar to those causing severe acute respiratory syndrome (SARS‐CoV) and Middle East respiratory syndrome (MERS‐CoV). Most of the protein structures of coronaviruses, including SARS‐CoV, MERS‐CoV, HKU, MHV, and NL63, share high similarity except for their receptor binding domain (RBD). 2 SARS‐CoV‐2 genome was found to be about 80% similar to SARS‐CoV. The most variable residues among bat‐CoV, SARS‐CoV, and SARS‐CoV‐2 have been found to reside within the S1 subunit of the S protein that exposes RBD. 3 A flexible RBD was observed on S glycoproteins of MERS‐CoV and SARS‐CoV and is thought to be important for pathogenesis. 2
Searching for mutations while the virus continues to spread within the country can offer opportunities for a better understanding of virus evolution, biopathology, and transmission. The earliest nucleotide mutations of three Italian isolates of SARS‐CoV‐2 isolates were reported on 20 March 2020. Recently, Lorusso et al 4 reported on mutations in SARS‐2‐CoV isolates in the region of Abruzzo, Central Italy. In this study, we have examined all available SARS‐2‐CoV genome sequences of isolate from different Italian regions and compared them with the first original sequences isolated in Wuhan. Here, we report on identification, biochemical, and biophysical properties of three mutations one in the S1 spike component and two in the nucleocapsid proteins, shared by all Italian isolates and absent in the Chinese ones. The S1 mutation D614G is here particularly discussed in light of a potential influence on SARS‐2‐CoV transmission.
2. MATERIALS AND METHODS
All 79 COVID‐19 whole genome sequences isolated from Italian patients from 29 January 2020 to 27 April 2020 (53, 11, 6, 6, 2, and 1 isolates from regions Abruzzo, Lazio, Friuli Venezia Giulia, Lombardy, Veneto, and Marche, respectively) (Table 1) and 48 COVID‐19 sequences from 24 December 2019 to 17 January 2020 in China have been downloaded from GISAID (https://www.gisaid.org/) database. For Chinese isolates, the above time interval has been selected to rule out sequences from virus isolated from possible return infections, so allowing comparison of Italian sequences with the truly original Chinese ones. The data set has been aligned using multiple sequence alignment (MAFFT) online tool 5 and manually edited using Bioedit program v7.0.5. 6 Sequence alignments and analyses were obtained through the Jalview editor 7 and structural models have been built relying on the website I‐Tasser 8 and HHPred. 9 CUPSAT 10 and Dynamut 11 online server has been used to estimate the stability of potential mutations found using the selective pressure analysis. three‐dimensional structures have been analyzed and displayed using PyMOL. 12
Table 1.
Table reporting GISAID accession number and region of isolation of the sequences with a mutation on the spike glycoprotein
|
|
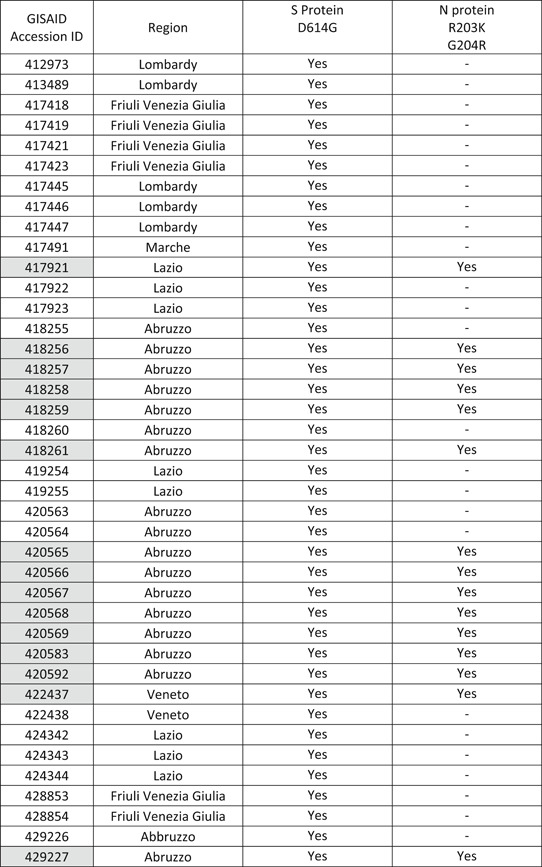
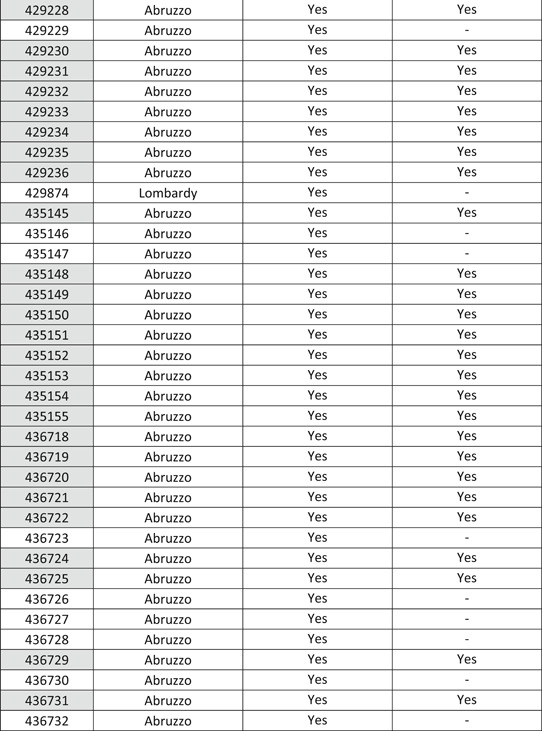
Note: The sequences showing a mutation in the nucleocapsid region have been highlighted in light gray.
3. RESULTS
The analysis of the alignment has revealed the presence of two mutations on spike and nucleocapsid (N) proteins. Regarding the spike glycoprotein, the transition from an adenosine to guanine occurring on the 1901st nucleotide has led to nonsynonymous mutation from an aspartate to a glycine residue in the 614 amino acidic position (found in all the Italian isolates). This mutation, which was first reported in italian isolates by Zehender et al, 13 is characteristic of all the Italian SARS‐CoV‐2 sequences besides those of the numerous viral isolates in Abruzzo. 4 Aspartate is a chiral and polar amino acid which frequently occurs at the N‐termini of alpha helices while glycine is nonpolar and the only achiral amino acid. This mutation is located on the SD2 region of the RBD‐containing S1 subunit. 14 Using the crystallographic three‐dimensional structure of the S protein, the implications of this mutation has been analyzed using CUPSAT and Dynamut servers. The results of these analysis have shown that the mutation from aspartate to glycine reduces the stability of the protein (ΔΔG [kcal/mol] −1.51) favouring the torsion potential. The Δ vibrational entropy energy between wild‐type and mutant spike protein has been calculated to be: ΔΔSVib ENCoM: 0.065 kcal mol−1 K−1 (Figure 1). 15
Figure 1.
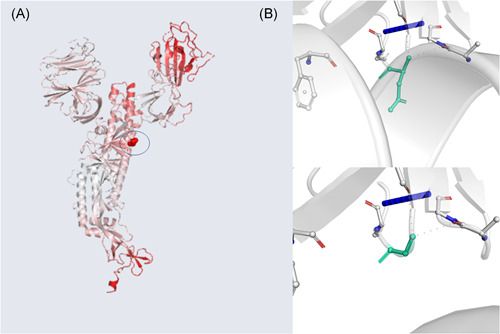
A, A model of spike glycoprotein monomer displaying the amino acids colored according to the vibrational entropy change upon mutation, red regions are those gaining in flexibility, The amino acidic mutation is blue circled; (B) the top image shows the molecular interaction between the side chain of the wild‐type amino acid and the side chains of the surrounding amino acid; the bottom image shows the molecular interaction between the side chain of the mutated amino acid and the side chains of the surrounding amino acid
Regarding the nucleocapsid protein, the two contiguous mutations (found in 56% of Italian isolates), from AGG to AAA occurring on the 649 to 651 nucleotides and GGA to CGA occurring on the 652 to 655 nucleotides both lead to nonsynonymous mutation from an arginine to a lysine and from glycine to arginine residue in the 203 and 204 amino acidic position, respectively. This latter mutation is characteristic of the SARS‐CoV‐2 sequences isolated in Abruzzo. Arginine is a chiral and polar amino acid which frequently occurs at the N‐termini of alpha helices while lysine is a chiral and polar amino acid (for glycine, see above). Using the three‐dimensional structure available on I‐tasser server, the implication of these mutations has been analyzed by CUPSAT server. The results point out both mutations reduce protein stability while favouring torsion (ΔΔG [kcal/mol] −1.92 and −2.94 for arginine to lysine and glycine to arginine, respectively). 15 The homology modeling analysis performed using HHpred server has shown structural similarity of the subdomains 22 to 184 and 261 to 378 amino acidic regions of the SARS‐CoV‐2 nucleocapsid with the RNA binding domain of nucleocapsid protein of MERS‐CoV and the nucleocapsid oligomerization domain of SARS‐CoV, respectively. For the sequence 184 to 261 amino acids no statistically significant homologous model has been found (Figure 2).
Figure 2.
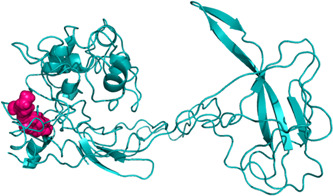
Cartoon model of the nucleocapsid of the SARS‐CoV‐2 where the mutated amino acids have been shown in purple
4. DISCUSSION
Italy is the European country that has been first and heavily hit by SARS‐CoV‐2 epidemic started from China. In fact, recent evidence 16 strongly suggests that the virus was circulating unrecognized in Lombardy since at least mid‐January, while the first official COVID‐19 Italian case was notified in Codogno (Lombardy) only the 21st of February. This happened when other European countries and United States were not yet, or minimally, affected. For these reasons, the Italian isolates of SARS‐CoV‐2 represent an interesting and useful case for investigating early virus mutations in a comparison with Chinese isolates.
In this paper, we have examined all available genome sequences of the virus isolates from different Italian regions, in comparison with 48 Chinese sequences, in an attempt to unveil changes, if any, prospecting their potential biological relevance for virus infection and transmission. While confirming previous ones, 4 , 13 , 17 we have now focussed on biostructural features on three nonsynonymous mutations, one on the 614 amino acidic position of S1 region within the spike S glycoprotein (from an aspartate to a glycine) and two consecutive mutations on the N protein (an arginine to a lysine and glycine to arginine residue in the 203 and 204 amino acidic positions, respectively. All three mutations, none of which were present in the Chinese isolates examined, affect biologically relevant, structural components of the virus and appear to confer enhanced torsional ability to the encoded proteins.
S1 spike is a major SARS‐CoV‐2 protein allowing, through a defined binding domain (RBD), virus entry into human cells expressing angiotensin‐converting enzyme 2 (ACE‐2) receptor. 18 The RBD engages ACE‐2 receptor through a conformational movement that exposes the relevant binding moieties in a “up” position. 14 For this and the additional reason that many efforts to generate a safe and efficacious COVID‐19 vaccine focus upon the spike component, changes in its composition and structure are particularly relevant. The S1 mutation in the genome of Italian isolates, first reported by Zehender et al 13 is in a position (AA 614; SD2 region) close to the S1 junction with the S2 component of the spike protein. The mutation appears to reduce protein stability through enhancement of torsional flexibility that, in theory, could favor energetically acquiring the” up” conformation of the spike glycoprotein, thus enhancing receptor binding capacity.
Regarding the mutations affecting N protein (known as LKR in SARS‐CoV) no structural information is available, 19 possibly due to its high positive charge and flexible nature. 20 However, previous studies suggested some evidence in support of the functional importance of this intrinsically disordered region, in the modulation of a number of virus properties, including cell signaling. 10
Recently, it has been shown that SARS‐CoV‐2 enters cells through endocytosis after binding of S protein to ACE‐2 receptor and phosphatidylinositol 3‐phosphate 5‐kinase (PIKfyve), two pore channel subtype 2 (TPC2), and cathepsin L are critical for this entry. 21 Nucleocapsid protein was also proposed to be important in COVID‐19 infectivity. 22
In conclusion, we have here focussed on mutations present on critical structural components of Italian SARS‐CoV‐2 isolates, that appear to be absent in early Chinese isolates of the virus. These mutations, in particular the one present in the S1 protein, are here examined for their potential to affect virus evolution and biopathological impact on the epidemic. However, our interpretation remains purely theoretical and should be taken cautiously in the absence of experimental and clinical investigations addressing the infectious and transmission capacity of SARS‐CoV‐2 bearing the above mutations. Definitely, more work is required in this area.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
DB, MC, and AC conceived and designed the study. DB, MB, and ABD collected data and prepared the data sets. DB and SP participated to bioinformatic analyses. DB, MG, RC, and AC wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank the GISAID website and all the colleagues who have immediately made available the SARS‐CoV‐2 sequences reported in Supplementary Table 1. Their laborious and important work is highly appreciated.
APPENDIX 1.
While this paper was in advanced preparation for publication, a paper was posted in BioRxiv, May 5th (see Korber et al, bioRxiv preprint https://doi.org/10.1101/2020.04.29.069054.) describing the D614G mutations in European and US SARS‐2‐CoV isolates and discussing its potential relevance for biopathological aspects of the virus. We declare that the work described here is the result of an autonomous investigation, totally independent from that reported by Korber et al above.
Benvenuto D, Demir AB, Giovanetti M, et al. Evidence for mutations in SARS‐CoV‐2 Italian isolates potentially affecting virus transmission. J Med Virol. 2020;92:2232–2237. 10.1002/jmv.26104
REFERENCES
- 1. Novel Coronavirus (2019‐nCoV) Situation Report‐1. 21 January 2020. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200121-sitrep-1-2019-ncov.pdf. Accessed April 23, 2020.
- 2. Yuan Y, Cao D, Zhang Y, et al. Cryo‐EM structures of MERS‐CoV and SARS‐CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun. 2017;8(1):15092. 10.1038/ncomms15092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade‐long structural studies of SARS coronavirus. J Virol. 2020;94:e00127‐20. 10.1128/JVI.00127-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lorusso A, Calistri P, Mercante M, T, et al. A “One‐Health” approach for diagnosis and molecular characterization of SARS‐CoV‐2 in Italy. One Health. 2020;10:100135. 10.1016/j.onehlt.2020.100135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017;20(4):1160‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hall TA. BioEdit a user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95‐98. [Google Scholar]
- 7. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview version 2‐a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189‐1191. 10.1093/bioinformatics/btp033J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I‐TASSER suite: protein structure and function prediction. Nature Methods. 2015;12:7‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimmermann L, Stephens A, Nam SZ. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430(15):2237‐2243. [DOI] [PubMed] [Google Scholar]
- 10. Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197‐208. 10.1038/nrm1589 [DOI] [PubMed] [Google Scholar]
- 11. Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation flexibility and stability. Nucleic Acids Res. 2018;46(W1):W350‐W355. 10.1093/nar/gky300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schrödinger LLC . 2015. The {PyMOL} Molecular Graphics System Version 1.8.
- 13. Zehender G, Lai A, Bergna A, et al. Genomic characterization and phylogenetic analysis of SARS‐COV‐2 in Italy [published online ahead of print, 2020 Mar 29]. J Med Virol. 2020. 10.1002/jmv.25794 [DOI] [PMC free article] [PubMed]
- 14. Wrapp D, Wang N, Corbett KS, et al. Cryo‐EM structure of the 2019‐nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260‐1263. 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parthiban V, Gromiha MM, Schomburg D. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006;34:W239‐W242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guzzetta G, Poletti P, Ajelli M, et al. Potential short‐term outcome of an uncontrolled COVID‐19 epidemic in Lombardy Italy February to March 2020. Euro Surveill. 2020;25(12), 10.2807/1560-7917.es.2020.25.12.2000293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benvenuto D, Angeletti S, Giovanetti M, et al. Evolutionary analysis of SARS‐CoV‐2: how mutation of non‐structural protein 6 (NSP6) could affect viral autophagy. J Infect. 2020. 10.1016/j.jinf.2020.03.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vinson V. How SARS‐CoV‐2 binds to human cells. Science. 2020;367(6485):1438‐1439. 10.1126/science.367.6485.1438-d [DOI] [Google Scholar]
- 19. Chang CK, Hou MH, Chang CF, Hsiao CD, Huang TH. The SARS coronavirus nucleocapsid protein–forms and functions. Antiviral Res. 2014;103:39‐50. 10.1016/j.antiviral.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu IM, Oldham ML, Zhang J, Chen J. Crystal structure of the severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein dimerization domain reveals evolutionary linkage between corona‐ and arteriviridae. J Biol Chem. 2006;281:17134‐17139. 10.1074/jbc.M602107200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS‐CoV‐2 on virus entry and its immune cross‐reactivity with SARS‐CoV. Nature Commun. 2020;11(1), 10.1038/s41467-020-15562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goh GK, Dunker AK, Foster JA, Uversky VN. Rigidity of the outer shell predicted by a protein intrinsic disorder model sheds light on the COVID‐19 (Wuhan‐2019‐nCoV) infectivity. Biomolecules. 2020;10(2):331. 10.3390/biom10020331 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information