

Abstract
Mucopolysaccharidosis type IVA (MPS IVA) is due to the deficiency of GALNS (N-acetylgalactosamine 6-sulfate sulfatase) and is characterized by systemic skeletal dysplasia. We have evaluated adeno-associated virus 8 (AAV8) vectors expressing different forms of human GALNS under a liver-specific promoter. The vectors were delivered intravenously into 4-week-old MPS IVA knockout (KO) and immune tolerant (MTOL) mice at a dose of 5 × 1013 genome copies (GC)/kg. These mice were monitored for 12 weeks post-injection. GALNS enzyme activity was elevated significantly in plasma of all treated mice at 2 weeks post-injection. The activity observed was 4- to 19-fold higher than that in wild-type mice and was maintained throughout the monitoring period. Treatment with AAV vectors resulted in a reduction of keratan sulfate (KS) levels in plasma to normal levels 2 weeks post-injection, which were maintained until necropsy. Both vectors reduced the storage in articular cartilage, ligaments, and meniscus surrounding articular cartilage and growth plate region as well as heart muscle and valves. Our results suggest that the continuous presence of high levels of circulating enzyme increases the penetration into bone and heart and reduces the KS level, thereby improving storage in these regions. The current data support a strategy for developing a novel treatment to address the bone and heart disease in MPS IVA using AAV gene therapy.
Keywords: MPS IVA, GALNS, AAV, liver targeting, keratan sulfate, skeletal dysplasia, cardiovascular disease
Graphical Abstract
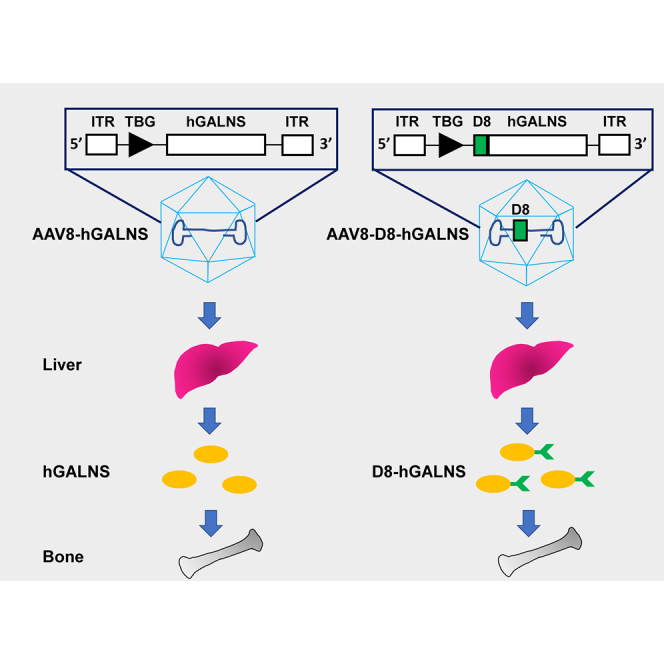
Adeno-associated virus (AAV) gene delivery is a promising approach for many diseases, but implementation in bone is hampered by the need for delivering AAV to the avascular cartilage. Unfortunately, most conventional, non-modified AAV vectors penetrate the cartilage poorly. The proposed method with marked expression of gene product may ameliorate bone lesions.
Introduction
Mucopolysaccharidosis type IVA (MPS IVA, Morquio syndrome type A) is an autosomal recessive disorder caused by the deficiency of the lysosomal enzyme N-acetylgalactosamine-6-sulfate sulfatase (GALNS).1, 2, 3, 4, 5, 6 GALNS degrades glycosaminoglycans (GAGs), both chondroitin-6-sulfate (C6S) and keratan sulfate (KS), and its deficiency causes progressive accumulation of KS and C6S in cartilage and extracellular matrix (ECM), leading to a systemic and unique skeletal dysplasia with incomplete ossification and subsequent imbalance of growth.4,7,8 Clinical manifestations include short neck and trunk, cervical spinal cord compression, tracheal obstruction, pectus carinatum, laxity of joints, kyphoscoliosis, coxa valga, and genu valgum. Patients with a severe type die of airway compromise, cervical spinal cord complications, or heart valve disease in their 20s or 30s if untreated.4,7,9, 10, 11 Enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT), and various surgical interventions are currently available for patients with MPS IVA in clinical practice. MPS IVA patients often require multiple orthopedic surgeries in the upper spine and lower extremities within the first 10 years of life. ERT may reduce fatigue and improve the quality of life in patients with MPS IVA but it has not shown any impact on bone pathology, which is the underlying cause of the most debilitating symptoms of the disease. This has been reported by several groups, either by looking at the clearance of storage in the surgical remnants of patients undergoing ERT,12,13 or by examining the bone growth improvement14,15 as well as the reduction in surgical interventions in these patients.16 Additionally, there has been no reduction in the blood KS levels after ERT.17,18 In preclinical and clinical studies, conventional ERT with intravenous (i.v.) infusion provided short elimination half-life in circulation (2 min in mouse and 35 min in human),19,20 suggesting that most infused enzyme does not reach the avascular cartilage. Moreover, ERT treatment is burdensome and requires weekly infusions for 5–6 hours.13,21 While HSCT may provide better outcomes than ERT on bone pathology, this cell-based therapy may not be applicable to all patients because of limitations in the availability of a matched donor, age limit for effective treatment, lack of well-trained staff in facilities, and the mortality risk of the procedure such as graft-versus-host disease (GVHD), infection, and other complications.22, 23, 24, 25, 26 Therefore, effective and feasible therapy for bone lesions in MPS IVA remains an unmet need.
MPS IVA mouse models recapitulate certain aspects of the human disease, specifically that they exhibit severe bone pathology but do not reveal any clinical symptoms.27,28 The knockout (KO) mouse model for MPS IVA was generated by targeted disruption of exon 2 of the murine GALNS gene.27 Another mouse model for MPS IVA was developed by introducing an inactive human GALNS (hGALNS) sequence in intron 1 along with the targeted mutation (C76S) in exon 2 of the murine GALNS gene.28 This model, referred to as MPS IVA tolerant mouse (MTOL), has inactive hGALNS ubiquitously expressed, thereby rendering it tolerant to interventions using the hGALNS enzyme.28 Both of these mouse models have no detectable enzyme activity and, as a consequence, have GAG accumulation in peripheral organs, cartilage, and bone. Additionally, they have elevated GAG levels in blood and urine. They have been widely used to test the efficacy of different treatments for MPS IVA.19,29,30
Gene therapy has the potential for long-term therapeutic efficacy in MPS diseases where the secreted enzyme from transduced cells can cross-correct the neighboring cells.31,32 Adeno-associated virus (AAV) vectors are efficient at targeting various organs or tissues for stable genetic modification and are being used in a growing number of preclinical and clinical gene therapy studies for diseases, including MPS I, II, IIIA, IIIB, and VI (ClinicalTrials.gov); however, gene therapy approaches for MPS IVA using AAV and other virus-based vectors remain under preclinical evaluation.33, 34, 35, 36 AAV-based gene therapy products have already been approved for patients suffering from debilitating genetic diseases.37,38 Certain AAV vectors, including AAV8, are preferred for liver-directed gene transfer, based on the high efficiency observed in non-human primates.39, 40, 41 Animal studies also indicate that AAV vectors in which liver-specific promoters are used for expressing therapeutic transgenes may allow for inducing immune tolerance to the transgene product and the stable, long-term expression of the transgene product.42, 43, 44, 45, 46, 47, 48 For example, treatment of murine and feline models of MPS VI with an AAV8 vector featuring a liver-specific promoter provided a significant impact on skeletal lesions.49,50
Patients with MPS IVA show the most severe skeletal abnormalities in all types of MPS,8 and a bone-targeting strategy that could supply sufficient enzyme to penetrate the cartilage region would be advantageous. We have previously demonstrated enhanced preferential translocation to bone by attaching a short acidic amino acid tag to the N or C terminus of several enzymes.29,51,52 Hydroxyapatite (HA) is the major inorganic component in bone and has a positively charged surface that contains calcium ions. Bone sialoprotein and osteopontin bind to HA, and these phosphorylated acidic glycoproteins have repeated sequences of negatively charged acidic amino acids (Asp and Glu), which are required for bone targeting.53,54 We applied the same principle to engineer a GALNS enzyme with an aspartic acid octapeptide (D8) N-terminal tag, which could potentially get translocated to bone better when compared to the native GALNS.
In this study, we have aimed to develop AAV8 vectors expressing hGALNS with or without bone-targeting signal, under the control of liver-specific thyroxin-binding globulin (TBG) promoter, and to investigate the therapeutic efficacy of these recombinant AAV8 vectors on bone and heart pathology in both MPS IVA KO and MTOL mice.
Results
AAV-hGALNS Delivery Increases GALNS Activity in Blood and Various Tissues in Mouse Models of MPS IVA
The two mouse models (KO and MTOL) of MPS IVA disease recapitulate the human disease in terms of the deficiency of hGALNS activity, increased levels of KS in blood and tissues, and storage materials (vacuoles) in various tissues, including chondrocytes, meniscus, ligaments, and heart muscle and valves. These biomarkers have been widely used to evaluate the severity of the phenotype and the therapeutic efficacy of several approaches in these mouse models.19,27, 28, 29, 30 For this study, we delivered AAV8-TBG-hGALNSco and AAV8-TBG-D8-hGALNSco (Figure 1) i.v. into 4-week-old MPS IVA KO and MTOL mice at a uniform dose of 5 × 1013 GC/kg body weight. The mice were monitored for 12 weeks post-injection, and blood samples were collected every other week to analyze the enzyme activity and KS levels. Additionally, at necropsy, tissue samples were taken from different organs for enzymatic activity and KS levels as well as knee joints and heart valves for histopathology analysis.
Figure 1.

Schematic Structure of AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS Viral Vector Genome
ITR, inverted terminal repeat; TBG, thyroxin-binding globulin; RBG pA, rabbit β-globin poly(A).
Plasma enzyme activities in KO and MTOL mice are shown in Figures 2A and 2B. No GALNS activity was detected in plasma in untreated MPS IVA mice. Two weeks post-injection, plasma hGALNS activity in KO mice treated with AAV8-TBG-hGALNS or AAV8-TBG-D8-hGALNS was significantly increased, compared to that in wild-type mice. The plasma enzyme activity from AAV8-TBG-D8-hGALNS treatment was higher than that from AAV8-TBG-hGALNS treatment 2 weeks post-injection; however, plasma hGALNS activity was not different between these two AAV vector treatments at other time points out to 12 weeks post-injection. In MTOL mice treated with both AAV vectors, plasma hGALNS activity was significantly increased compared to that in wild-type mice 2 weeks post-injection. The levels of enzyme activity from mice treated with AAV8-TBG-D8-hGALNS were higher than those from mice treated with AAV8-TBG-hGALNS throughout the entire study duration. Supraphysiological levels of circulating hGALNS activity were achieved after a single injection of AAV8-TBG-hGALNS or AAV8-TBG-D8-hGALNS in both MPS IVA mouse models, and these high levels of enzyme activity were maintained for the duration of the study.
Figure 2.

Blood and Tissue Human N-Acetylgalactosamine-6-Sulfate Sulfatase (hGALNS) Enzyme Activity in MPS IVA Mice Treated with AAV8 Vectors
(A and B) A blood sample was collected from MPS IVA mice every other week until 16 weeks of age, and plasma hGALNS enzyme activity was measured in (A) knockout (KO) and (B) tolerant (MTOL) mice. The tissue sample was collected from MPS IVA mice 12 weeks post-injection of AAV vectors with or without a bone-targeting signal. (C and D) hGALNS enzyme activity in tissues including liver, spleen, lung, kidney, heart, and bone (leg) was measured in (C) KO and (D) MTOL mice. n = 4–8. Statistics were analyzed by one-way ANOVA with a Bonferroni’s post hoc test. Data are presented as mean ± SD. ∗p < 0.05 versus AAV8-TBG-hGALNS. (A and B) Wild-type (WT), ○; AAV8-TBG-hGALNS, ▪; AAV8-TBG-D8-hGALNS, ▲. (C and D) WT, open bar; untreated, black bar; AAV8-TBG-hGALNS, gray bar; AAV8-TBG-D8-hGALNS, striped bar. N.D., not detected.
The levels of hGALNS activity in the liver and other tissues 12 weeks after i.v. delivery of AAV vectors are shown in Figures 2C and 2D. Consistent with our use of an AAV8 vector with a liver-specific TBG promoter, the hGALNS activity levels in the livers of all treated MPS IVA mice were significantly higher than those in untreated MPS IVA mice. The mean enzyme activity levels in KO mice treated with AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS were, respectively, 49- and 9-fold higher than the level observed in wild-type mice. In MTOL mice treated with AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS, hGALNS activities in the liver were, respectively, 60- and 9-fold higher than those in wild-type mice. We detected hGALNS genome copies in the livers of MPS IVA mice treated with either AAV vectors at 16 weeks of age (Figure S2). The liver genome copy numbers trended similar to the enzyme level differences detected in the two mouse models.
To evaluate the potential cross-correction of hGALNS deficiency, we evaluated hGALNS activity in tissues where we did not expect AAV vector-mediated expression of GALNS. The hGALNS activity was observed in all examined tissues, including spleen, lung, kidney, bone (leg), and heart in both KO and MTOL mice after both AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS treatments (Figures 2C and 2D). The enzyme activities were similar to or higher than wild-type levels in spleen and heart, and slightly lower levels of activities were observed in the lung and kidney. Notably, 37% and 20% of wild-type enzyme activities were observed in the bone of KO mice treated with AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS, respectively. Also, 57% and 43% of wild-type enzyme activities were observed in MTOL mice treated with these two AAV vectors. The hGALNS activity levels in bone were not statistically different between AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS.
Levels of Mono-Sulfated KS in the Blood and Tissues Decreased as a Result of AAV-GALNS Delivery
We measured mono-sulfated KS, which is the major component of KS, in plasma and tissues of MPS IVA mice. The levels of plasma mono-sulfated KS in KO and MTOL mice are shown in Figures 3A and 3B. Before the administration of AAV vectors, plasma mono-sulfated KS levels in untreated KO mice were significantly higher than those in wild-type mice (mean, 41.8 versus 16.3 ng/mL). Two weeks post-injection, mono-sulfated KS levels in plasma were completely normalized for both AAV vectors, and these levels were maintained for at least another 10 weeks (at necropsy). Mono-sulfated KS levels were similar in wild-type mice and untreated MTOL mice at 4 weeks of age. The mono-sulfated KS levels in wild-type mice were maintained at a constant level throughout the study; however, the levels of mono-sulfated KS in untreated MTOL mice gradually increased with age. MTOL mice treated with either of the AAV vectors maintained the normal levels throughout the entire study period. At 16 weeks of age, mono-sulfated KS levels in MTOL mice treated with AAV vectors were significantly lower, compared with those in the untreated MTOL mice.
Figure 3.

Blood and Tissue Glycosaminoglycan (GAG) Levels in MPS IVA Mice Treated with AAV8 Vectors
(A and B) A blood sample was collected from MPS IVA mice every other week until 16 weeks of age, and plasma mono-sulfated KS level was measured in (A) knockout (KO) and (B) tolerant (MTOL) mice. n = 4–8. The tissue sample was collected from MPS IVA mice 12 weeks post-injection of AAV vectors with or without a bone-targeting signal. (C and D) The amount of mono-sulfated KS in tissues, including (C) liver and (D) lung, was measured in KO and MTOL mice. n = 4–8. Statistics were analyzed by one-way ANOVA with a Bonferroni’s post hoc test. Data are presented as mean ± SD. (A and B) ∗p < 0.05 versus untreated. (C and D) ∗p < 0.05 versus wild-type (WT); #p < 0.05 versus untreated; $p < 0.05. (A and B) WT, ○; untreated, ●; AAV8-TBG-hGALNS, ▪; AAV8-TBG-D8-hGALNS, ▲. (C and D) WT, open bar; untreated, black bar; AAV8-TBG-hGALNS, gray bar; AAV8-TBG-D8-hGALNS, striped bar.
We also measured mono-sulfated KS levels in tissues of MPS IVA mice. At necropsy, excessive storage of GAGs was present in tissues of both KO and MTOL mice. The amount of mono-sulfated KS in livers of KO and MTOL mice and in the lungs of KO mice were significantly decreased 12 weeks post-injection with either AAV vector (Figures 3C and 3D). To assess the effect of these AAV vectors expressing hGALNS on other GAG levels, the levels of heparan sulfate (HS) were analyzed in blood and tissues of MPS IVS mice. Both KO and MTOL mice had normal levels of diHS-0S in plasma, and the levels were not affected after injection by AAV vectors (Figure S3). Tissue diHS-0S levels in the liver and lung were also not changed between all groups at 12 weeks post-injection of AAV vectors (Figure S4).
Delivery of AAV GALNS Vectors Improved Bone and Cartilage Pathology in MPS IVA Mice
Tissues including bone (femur and tibia) and heart (muscle and valve) were assessed from MPS IVA mice at 12 weeks post-injection of AAV8-TBG-hGALNS or AAV8-TBG-D8-hGALNS.
Untreated MPS IVA KO and MTOL mice at 16 weeks of age exhibited GAG storage vacuoles in the growth plate of the femur and tibia (hyaline cartilage) (Figure 4A), articular disc (Figure 4B), ligament surrounding the knee joint (Figure S5A), and meniscus (Figure S5B). The growth plate also exhibited a disorganized column structure with ballooned and vacuolated chondrocytes (Figures 4A and 4B). In KO mice treated with AAV8-TBG-hGALNS or AAV8-TBG-D8-hGALNS, the growth plate, articular cartilage, ligaments, and meniscus in the knee joint had a partial reduction of storage material, and the column structure of chondrocytes was improved but remained disorganized and distorted. In MTOL mice treated with these AAV vectors, the growth plate, articular cartilage, ligaments, and meniscus in the knee joint had a greater reduction of storage, and the column structure of the growth plate and articular cartilage showed greater recovery than in untreated MTOL mice. To objectively assess the improvement of vacuolization in cartilage cells of the growth plate, chondrocyte cell size was quantified in the growth plate lesions of KO and MTOL mice (Figure 4C). We observed a moderate reduction of chondrocyte size in these growth plate lesions, which reached statistical significance in the MTOL mice.
Figure 4.

Correction of Bone Pathology in MPS IVA Mice Treated with AAV8 Vectors
(A and B) Correction of chondrocytes vacuolization was assessed by toluidine blue staining analysis using light microscopy of (A) growth plate and (B) articular disc in the knee joint of MPS IVA mice treated with AAV8 vectors. Bone pathology in knockout (KO) and tolerant (MTOL) mice were compared with wild-type, untreated MPS IVA, and treated MPS IVA with AAV8 vectors with or without bone-targeting signal. Scale bars, 25 μm. (C and D) Chondrocyte cell size in growth plate lesions of (C) femur or (D) tibia was quantified by ImageJ software. Data expressed fold change from wild-type group. n = 4–7. Statistics were analyzed by one-way ANOVA with a Bonferroni’s post hoc test. Data are presented as mean ± SD. ∗p < 0.05 versus wild-type (WT); #p < 0.05 versus untreated. (C and D) WT, open bar; untreated, black bar; AAV8-TBG-hGALNS, gray bar; AAV8-TBG-D8-hGALNS, striped bar.
Untreated MPS IVA mice exhibited GAG storage vacuoles in heart valves and muscle. AAV8-TBG-hGALNS or AAV8-TBG-D8-hGALNS provided nearly complete clearance in these heart lesions of treated KO and MTOL mice (Figures 5A and 5B).
Figure 5.

Correction of Heart Pathology in MPS IVA Mice Treated with AAV8 Vectors
(A and B) Correction of vacuolization was assessed by toluidine blue staining analysis using light microscopy of (A) heart valve and (B) heart muscle of MPS IVA mice treated with AAV8 vectors. Heart pathology in knockout (KO) and tolerant (MTOL) mice were compared with wild-type, untreated MPS IVA, and treated MPS IVA with AAV8 vectors with or without bone-targeting signal. The arrows indicate the location of disease-related vacuoles. (a) Untreated KO, (b) untreated MTOL, (c) KO with AAV8-D8-hGALNS, (d) MTOL with AAV8-D8-hGALNS, (e) KO with AAV8-hGALNS, (f) MTOL with AAV8-hGALNS, and (g) wild-type (WT). Scale bars, 20 μm.
Circulating Anti-hGALNS Antibodies
To investigate the possibility of a humoral response to hGALNS, we performed an enzyme-linked immunosorbent assay (ELISA) using plasma from wild-type and treated and untreated MPSIVA mice. KO mice treated with both AAV vectors showed the presence of significantly higher levels of circulating anti-hGALNS antibodies when compared to untreated KO mice (0.50 ± 0.38 or 0.62 ± 0.43 optical density [OD] unit for KO mice treated with AAV8-TBG-hGALNS or AAV8-TBG-D8-hGALNS) (Figure S6). In AAV-hGALNS MTOL mice, these antibodies were not detected.
Discussion
In this study, administration of the AAV8-GALNS vector into two MPS IVA mouse models demonstrated some differences in response but overall provided (1) a continuous high level of GALNS activity in the circulation; (2) GALNS activity in target tissues, including bone and heart; (3) significant reduction of KS accumulation in blood and tissues; (4) consequent amelioration of bone pathology; and (5) significant reversal of heart pathology. The levels of KS in blood and urine correlate with clinical severity of MPS IVA in childhood, and therefore they are a useful prognostic biomarker of the disease.55, 56, 57, 58 Blood KS levels directly reflect the turnover of proteoglycans from cartilage, the primary tissue where KS is synthesized during growth. Urinary KS levels were substantially reduced after ERT,59,60, while blood KS levels were not changed after this therapy.17,18 The reduction in urinary KS levels seen in MPS IVA patients receiving ERT treatment does not correlate with improvements in bone pathology and skeletal symptoms. Blood KS could be a more predictive biomarker for evaluation of the therapeutic efficacy in bone lesions of patients with MPS IVA.4,17 We measured plasma mono-sulfated KS levels in KO and MTOL mice biweekly for 12 weeks until the endpoint of this study. Two weeks after injection of AAV-GALNS vectors, mono-sulfated KS levels in the plasma were normalized to the wild-type levels, and these levels were maintained for at least another 10 weeks. In our previous study where MPS IVA mice were treated with ERT, the KS levels in the plasma were not normalized even after 12 weekly infusions,19 suggesting that high and sustained circulating levels of GALNS enzyme produced by gene therapy may be necessary to normalize plasma KS levels and to penetrate cartilage lesions in MPS IVA mice.
The improvement in bone pathology was evident in MTOL mice when treated at 4 weeks of age and assessed 12 weeks after vector administration. This was not the case with the KO mice. This could be because the KO mice have a more severe phenotype at the time of treatment compared to MTOL mice. At 4 weeks of age, plasma mono-sulfated KS levels in MTOL mice were indistinguishable from those of wild-type mice, whereas the KO mice had higher levels of plasma mono-sulfated KS. Throughout the study, plasma mean mono-sulfated KS levels in KO mice were substantially higher than those in MTOL mice. In the heart, more accumulation of storage material in valvular cells and muscle was observed in KO mice, compared to that in MTOL mice, by pathological assessment, and more GAG accumulation was observed in the liver of KO mice by quantification using mass spectrometry.
Heart and cardiovascular involvement are life-threatening to MPS IVA patients and cause severe morbidity and mortality. One-third of patients die of heart disease.61 In patients with MPS IVA, the small left ventricle reduces the stroke volume and increases heart rate. The diastolic filling pattern is impaired because of the relatively thickened ventricle.62 In MPS IVA mice treated with ERT, the cardiac lesions are not improved, as storage materials remained in heart valves.19 Our findings suggest that gene therapy has the potential to improve cardiac function. Liver-targeted AAV8 gene therapy led to high hGALNS activity in the heart and subsequent near-complete clearance of storage material in heart valves and cardiac muscle of MPS IVA mice. Overall, these data indicate that sustained high levels of hGALNS activity in circulation could provide cross-correction and improvement in cardiac lesions. Even though we observed higher circulating levels of hGALNS activity in AAV8-TBG-D8-hGALNS-treated mice, especially in the MTOL mice, it did not lead to any significantly better therapeutic effect on bone and heart pathology when compared to AAV8-TBG-hGALNS treatment.
This study provides the first comprehensive results on in vivo AAV gene therapy for MPS IVA. We demonstrate improvements in bone and heart pathology as well as KS accumulation. However, the present data suggest that i.v. injection of AAV8 vectors expressing hGALNS with 5 × 1013 GC/kg at 4 weeks of age could not provide complete restoration of bone pathology in the growth plate and articular cartilage lesions of both MPS IVA murine models during the 12-week monitoring period. Tessitore et al.49 showed that i.v. administration of 4.1 × 1013 GC/kg AAV8 vector significantly improved skull abnormalities and tibia length in the MPS VI rat model. Cotugno et al.50 also showed that i.v. administration of 6.0 × 1013 GC/kg AAV8 vector significantly improved, but did not completely correct, bone length in the MPS VI feline model. Thus, the current dose and the starting age may not be enough to restore skeletal lesions completely. A dose-finding study should be conducted to determine the optimal dose/age to treat MPS IVA mice. To obtain more significant improvement in these lesions, early treatment starting at the newborn period should be evaluated since significant bone damage might already occur by 4 weeks of age
In conclusion, systemic administration of liver-targeted AAV8 vector expressing hGALNS has the potential for treating MPS IVA. Supraphysiological levels of enzyme activity in circulation were associated with partial correction of GALNS deficiency in skeletal and cardiovascular lesions. These data support the translation of AAV gene therapy toward clinical development for MPS IVA.
Materials and Methods
Expression Cassette Design and AAV Vector Production
The expression cassettes carrying the native and D8 containing GALNS transgenes were designed for packaging into the AAV8 vector (Figure 1). The bone-targeting signal, D8 sequences, was inserted after the N-terminal signal peptide of hGALNS. The design included a liver-specific TBG promoter along with a rabbit β-globin polyadenylation tail (RBG pA). We used a codon-optimized hGALNS sequence (GenScript, Piscataway, NJ, USA) for both vectors for the mouse studies. We confirmed the GALNS enzymatic activity of these expression cassette plasmids in a transfection experiment using Huh-7 cells. We determined the activity levels in both cell lysate and supernatant 48 h post-transfection (Figures S1A and S1B). The GALNS activity levels from the codon-optimized construct were similar to that produced by the native hGALNS coding sequence.
AAV8-TBG-hGALNS and AAV8-TBG-D8-hGALNS vectors were generated following a scaled-down version of the proprietary GMP vector production protocols at REGENXBIO (Rockville, MD, USA). Briefly, HEK293 cells (RGX293) were triple transfected with the helper plasmid, AAV8 capsid plasmid, and the transgene plasmid containing the hGALNS/D8-hGALNS. The packaged vectors were purified from the cell culture supernatant using affinity chromatography and tittered using the Droplet Digital PCR (Bio-Rad, Hercules, CA, USA) method.
Murine Models and In Vivo Study Design
We had previously described the development of two MPS IVA murine models, MPS IVA knockout mice (Galns−/−)27 and MPS IVA mice tolerant to hGALNS protein (Galnstm(hC79S.mC76S)slu)28 in the C57BL/6 background. The GALNS KO mouse model (KO, Galns−/−) was developed by targeted disruption of the GALNS gene.27 The mouse model tolerant to hGALNS (MTOL, Galnstm(hC79S.mC76S)slu) contains a transgene expressing hGALNS in intron 1 and an active site mutation (C76S) adjacent to exon 2, thereby introducing both the inactive hGALNS coding sequence with C79S active site mutation.28 Both mouse models had no detectable enzyme activity in blood and tissues and showed the accumulation of storage materials primarily within reticuloendothelial Kupffer cells, heart valves and muscle, and chondrocytes, including growth plate and articular cartilage. Genotyping for the experimental cohorts was done by PCR on day 14. Homozygous MPS IVA mice at 4 weeks of age, both male and female, were treated with either AAV8 vector i.v. at a uniform dose of 5 × 1013 GC/kg. Another cohort of MPS IVA mice, as well as unaffected C57BL/6 littermates, were administered with phosphate-buffered saline (PBS). The total dose volume administration was approximately 100 μL per mouse. All animal care and experiments were approved by the Institutional Animal Care and Use Committee of Nemours/Alfred I. duPont Hospital for Children.
Blood and Tissue Collection
Approximately 100 μL of blood was collected in tubes with EDTA (Becton Dickinson, Franklin Lakes, NJ, USA) every other week from all animals in the study. The blood was centrifuged at 8,000 rpm for 10 min, and plasma separated was kept at −20°C until performing the GALNS enzyme assay and GAG assay. At 16 weeks of age, mice were euthanized in a CO2 chamber and perfused with 20 mL of 0.9% saline. Liver, kidney, lung, spleen, heart, and knee joint were collected and stored at −80°C until processing for the GALNS enzyme assay and GAG assay. Additionally, various tissue samples were collected and stored in 10% neutral buffered formalin for histopathology analysis.
GALNS Activity Assay
GALNS activities in plasma and tissues were determined as described previously.33 Frozen tissues were homogenized with homogenization buffer consisting of 25 mmol/L Tris-HCl (pH 7.2) and 1 mmol/L phenylmethylsulfonyl fluoride by using a homogenizer. Tissue lysate or plasma and 22 mM 4-methylumbelliferyl-β-galactopyranoside-6-sulfate (Research Products International, Mount Prospect, IL, USA) in 0.1 M NaCl/0.1 M sodium acetate (pH 4.3) were incubated at 37°C for 16 h. Then, 10 mg/ml β-galactosidase from Aspergillus oryzae (Sigma-Aldrich, St. Louis, MO, USA) in 0.1 M NaCl/0.1 M sodium acetate (pH 4.3) was added to reaction sample, and additional incubation was at 37°C for 2 h. The sample was transferred to stop solution (1 M glycine, NaOH [pH 10.5]), and the plate was read at excitation 366 nm and emission 450 nm on a PerkinElmer Victor X4 plate reader (PerkinElmer, Waltham, MA, USA). The activity was expressed as nanomoles of 4-methylumbelliferone released per hour per microliter of plasma or milligram of protein. Protein concentration was determined by a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
Extraction of GAGs from Tissue
GAG extraction from various mouse tissues was modified from that developed by Mochizuki et al.63 Briefly, excised tissues were frozen in liquid nitrogen and homogenized with acetone using a homogenizer. The obtained powder was dried under a centrifuge vacuum. The defatted tissue powder was suspended in 0.5 M NaOH and incubated at 50°C for 2 h to remove GAG chains from its core protein. After neutralization with 1 M HCl, NaCl was added to a final concentration of 3 M. Insoluble materials were removed by centrifugation, and the pH of the supernatant was adjusted below 1.0 with 1 M HCl. Precipitated nucleotides were removed by centrifugation, and the supernatant was neutralized with 1 M NaOH. The crude GAGs were precipitated by the addition of 2 vol of ethanol containing 1.3% potassium acetate. After centrifugation, the precipitate was dissolved in distilled water.
GAG Assay
Blood and tissue GAG levels were measured by liquid chromatography-tandem mass spectrometry as described previously.64, 65, 66, 67, 68 Briefly, 50 mM Tris-HCl (pH 7.0) and sample were added into a 96-well Omega 10K MWCO (molecular weight cutoff) filter plate (Pall Corporation, Port Washington, NY, USA) on a 96-well receiver plate. Samples were centrifuged for 15 min at 2,500 × g. The filter plate was transferred to a new receiver plate, and a cocktail mixture of 50 mM Tris-HCl (pH 7.0), 5 μg/mL chondrosine as internal standard (IS), 1 mU heparitinase, and 1 mU keratanase II was added to the filter plate. Samples were incubated at 37°C in a water bath overnight. Then, the samples were centrifuged for 15 min at 2,500 × g. The apparatus consisted of a 1290 Infinity LC system with a 6460 triple quad mass spectrometer (Agilent Technologies, Palo Alto, CA, USA). Disaccharides were separated on a Hypercarb column (2.0 mm inner diameter [i.d.], 50 mm long, 5-μm particles; Thermo Fisher Scientific, Waltham, MA, USA) kept at 60°C. The mobile phase was a gradient elution of 5 mM ammonium acetate (pH 11.0) (solution A) to 100% acetonitrile (solution B). The flow rate was 0.7 mL/min, and the gradient was as follows: 0 min, 100% solution A; 1 min, 70% solution A; 2 min, 70% solution A; 2.20 min, 0% solution A; 2.60 min, 0% solution A; 2.61 min, 100% solution A; 5 min, 100% solution A. The mass spectrometer was operated with electrospray ionization in the negative ion mode (Agilent Jet Stream technology). Specific precursor and product ions, m/z, were used to quantify each disaccharide, respectively (IS, 354.3→193.1; mono-sulfated KS, 462→97; HS-0S, 378.3→175.1). The injection volume was 10 μL, with a running time of 5 min per sample.
Toluidine Blue Staining and Pathological Assessment
Toluidine blue staining was performed as described previously.28 Briefly, the knee joint and mitral heart valve were collected from MPS IVA and wild-type mice at 16 weeks of age to evaluate levels of storage granules by light microscopy. Tissues were fixed in 2% paraformaldehyde, 4% glutaraldehyde in PBS and post-fixed in osmium tetroxide and embedded in Spurr’s resin. Then, toluidine blue-stained 0.5-μm-thick sections were examined. To evaluate chondrocyte cell size (vacuolization) in the growth plate of femur or tibia, approximately 300 chondrocytes in the proliferative area were measured in each mouse by ImageJ software, and the results were expressed as fold change from the wild-type group.
Detection of Antibodies against GALNS by an ELISA
An indirect ELISA method was used to detect antibodies against hGALNS in plasma of treated and untreated mice as described previously.69 Briefly, a 96-well microtiter plate was coated overnight with 2 μg/mL purified rhGALNS (R&D Systems, Minneapolis, MN, USA) in 15 mM Na2CO3, 35 mM NaHCO3, and 0.02% NaN3 (pH 9.6). The wells were washed three times with Tris-buffered saline (TBS)-T (10 mM Tris [pH 7.5], 150 mM NaCl, 0.05% Tween 20) and then blocked for 1 h at room temperature with 3% bovine serum albumin in PBS (pH 7.2). After washing three times with TBS-T, a 100-fold dilution of mouse plasma in TBS-T was added to the wells and incubated at 37°C for 2.5 h. The wells were washed four times with TBS-T, and then TBS-T containing a 1:1,000 dilution of peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) (Thermo Fisher Scientific, Waltham, MA, USA) was added to the wells and incubated at room temperature for 1 h. The wells were washed three times with TBS-T and twice with TBS (10 mM Tris [pH 7.5], 150 mM NaCl). Peroxidase substrate (ABTS solution, Invitrogen, Carlsbad, CA, USA) was added (100 μL per well) and plates were incubated at room temperature for 30 min. The reaction was stopped with the addition of 1% SDS, and the plates were read at OD410 on a PerkinElmer Victor X4 plate reader (PerkinElmer, Waltham, MA, USA).
AAV Vector Genome Biodistribution
DNA was purified from the liver by using the Gentra Puregene kit according to the instruction manual (QIAGEN, Germantown, MD, USA). Digital PCR (dPCR) analysis on genomic DNA extracted from frozen liver samples of mice was performed using specific primers and probe sequences for RBG pA, which are as follows: forward primer, 5′-GCCAAAAATTATGGGGACAT-3′; reverse primer, 5′-ATTCCAACACACTATTGCAATG-3′; and probe, 5′-6FAM-ATGAAGCCCCTTGAGCATCTGACTTCT-QSY-3′. Genomic DNA was tested non-fragmented as well as fragmented using either enzymatic digestion or an M220 Focused-ultrasonicator (Covaris, Woburn, MA, USA). The RBG TaqMan assay (FAM labeled) (Thermo Fisher Scientific, Waltham, MA, USA) was used for quantitative dPCR analysis of the AAV vector. The concentration of DNA for the AAV chip for the liver samples processed was between 0.5 and 2 ng per 16-μL reaction, dependent on the DNA concentration needed to bring the AAV copies/μL into the detectable range of the instrument. The Tfrc TaqMan copy number reference assay (VIC labeled) was obtained from Thermo Fisher Scientific. 40 ng of genomic DNA per 16-μL reaction was used for Tfrc dPCR. Each reaction was loaded onto a separate QuantStudio chip (QuantStudio 3D digital PCR 20K chip kit v2, A26316; Thermo Fisher Scientific, Waltham, MA, USA).
The PCR amplification profile was conducted with an ABI GeneAmp 9700 PCR thermal cycler with dual flat blocks (Applied Biosystems, Waltham, MA, USA) as follows: 96°C for 10 min; 39 cycles of 60°C for 2 min and 98°C for 30 s; 60°C for 2 min; 10°C final hold. After PCR amplification, chips were read on the QuantStudio 3D instrument to obtain the number of wells positive for the VIC and FAM channels, as well as the number of wells without DNA and empty wells. Data analysis and chip quality were assessed using the QuantStudio 3D analysis suite. All chips were between 25% and 75% empty wells, ensuring suitability for quantitation. Copies/μL for both Tfrc and AAV were determined and normalized using the dilution factor for AAV sample input. Using Tfrc results as a reference for two copies, the number of copies of AAV per mouse genome was calculated.
Statistical Analysis
All data were expressed as means and standard deviations (SD). Multiple comparison tests were performed by one-way ANOVA with the Bonferroni’s post hoc test using GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA). The statistical significance of difference was considered as p < 0.05.
Author Contributions
K.S. performed experiments in the mouse model and wrote the first draft of the manuscript. K.S., S.K., and M.S. analyzed data and made figures. K.S. and S.T. performed a pathological assessment in murine models. S.K.-M., O.D., and S.T. helped design the construction of the vector. K.S., S.K.-M., J.T.B., O.D., and S.T. designed experiments and interpreted all data. S.K.-M., J.T.B., O.D., and S.T. edited the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
S.K.-M., J.T.B., and O.D. are employees of REGENXBIO. S.K.-M., O.D., S.T., and K.S. declare competing financial interests: they have U.S. Provisional Patent Application nos. 62/711,238, 62/756,880, and 62/799,834. The patent applications cover the underlying concept of AAV gene therapy to treat mucopolysaccharidosis IVA described in the manuscript. The remaining authors declare no competing interests.
Acknowledgments
This work was supported by grants from National MPS Society Research Grant; the Austrian MPS Society; the Carol Ann Foundation; the Angelo R. Cali & Mary V. Cali Family Foundation; the Vain and Harry Fish Foundation; the Bennett Foundation; the Jacob Randall Foundation; and Nemours Funds. S.T. was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under grant number P30GM114736. This work was also supported by a sponsored research agreement from REGENXBIO.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.015.
Contributor Information
Olivier Danos, Email: odanos@regenxbio.com.
Shunji Tomatsu, Email: stomatsu@nemours.org.
Supplemental Information
References
- 1.Brailsford J.F. Chondro-osteo-dystrophy, roentgenopgraphic & clinical features of a child with dislocation of vertebrae. Am. J. Surg. 1929;7:404–410. [PubMed] [Google Scholar]
- 2.Morquio L. Sur une forme de dystrophie osseuse familial. Arch. Méd. Enfants Paris. 1929;32:129–135. [Google Scholar]
- 3.Neufeld E.F., Muenzer J. The mucopolysaccharidoses. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- 4.Khan S., Alméciga-Díaz C.J., Sawamoto K., Mackenzie W.G., Theroux M.C., Pizarro C., Mason R.W., Orii T., Tomatsu S. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol. Genet. Metab. 2017;120:78–95. doi: 10.1016/j.ymgme.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peracha H., Sawamoto K., Averill L., Kecskemethy H., Theroux M., Thacker M., Nagao K., Pizarro C., Mackenzie W., Kobayashi H. Molecular genetics and metabolism, special edition: diagnosis, diagnosis and prognosis of Mucopolysaccharidosis IVA. Mol. Genet. Metab. 2018;125:18–37. doi: 10.1016/j.ymgme.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sawamoto K., Alméciga-Díaz C.J., Mason R.W., Orii T., Tomatsu S. Mucopolysaccharidosis type IVA: clinical features, biochemistry, diagnosis, genetics, and treatment. In: Tomatsu S., editor. Mucopolysaccharidoses Update. Nova Science Publishers; 2018. pp. 235–272. [Google Scholar]
- 7.Tomatsu S., Averill L.W., Sawamoto K., Mackenzie W.G., Bober M.B., Pizarro C., Goff C.J., Xie L., Orii T., Theroux M. Obstructive airway in Morquio A syndrome, the past, the present and the future. Mol. Genet. Metab. 2016;117:150–156. doi: 10.1016/j.ymgme.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melbouci M., Mason R.W., Suzuki Y., Fukao T., Orii T., Tomatsu S. Growth impairment in mucopolysaccharidoses. Mol. Genet. Metab. 2018;124:1–10. doi: 10.1016/j.ymgme.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montaño A.M., Tomatsu S., Gottesman G.S., Smith M., Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J. Inherit. Metab. Dis. 2007;30:165–174. doi: 10.1007/s10545-007-0529-7. [DOI] [PubMed] [Google Scholar]
- 10.Tomatsu S., Mackenzie W.G., Theroux M.C., Mason R.W., Thacker M.M., Shaffer T.H., Montaño A.M., Rowan D., Sly W., Alméciga-Díaz C.J. Current and emerging treatments and surgical interventions for Morquio A syndrome: a review. Res. Rep. Endocr. Disord. 2012;2012:65–77. doi: 10.2147/RRED.S37278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pizarro C., Davies R.R., Theroux M., Spurrier E.A., Averill L.W., Tomatsu S. Surgical reconstruction for severe tracheal obstruction in Morquio A syndrome. Ann. Thorac. Surg. 2016;102:e329–e331. doi: 10.1016/j.athoracsur.2016.02.113. [DOI] [PubMed] [Google Scholar]
- 12.Tomatsu S., Alméciga-Díaz C.J., Montaño A.M., Yabe H., Tanaka A., Dung V.C., Giugliani R., Kubaski F., Mason R.W., Yasuda E. Therapies for the bone in mucopolysaccharidoses. Mol. Genet. Metab. 2015;114:94–109. doi: 10.1016/j.ymgme.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomatsu S., Sawamoto K., Shimada T., Bober M.B., Kubaski F., Yasuda E., Mason R.W., Khan S., Alméciga-Díaz C.J., Barrera L.A. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opin. Orphan Drugs. 2015;3:1279–1290. doi: 10.1517/21678707.2015.1086640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Do Cao J., Wiedemann A., Quinaux T., Battaglia-Hsu S.F., Mainard L., Froissart R., Bonnemains C., Ragot S., Leheup B., Journeau P., Feillet F. 30 Months follow-up of an early enzyme replacement therapy in a severe Morquio A patient: about one case. Mol. Genet. Metab. Rep. 2016;9:42–45. doi: 10.1016/j.ymgmr.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doherty C., Stapleton M., Piechnik M., Mason R.W., Mackenzie W.G., Yamaguchi S., Kobayashi H., Suzuki Y., Tomatsu S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019;64:625–635. doi: 10.1038/s10038-019-0604-6. [DOI] [PubMed] [Google Scholar]
- 16.Yasuda E., Suzuki Y., Shimada T., Sawamoto K., Mackenzie W.G., Theroux M.C., Pizarro C., Xie L., Miller F., Rahman T. Activity of daily living for Morquio A syndrome. Mol. Genet. Metab. 2016;118:111–122. doi: 10.1016/j.ymgme.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan S.A., Mason R.W., Giugliani R., Orii K., Fukao T., Suzuki Y., Yamaguchi S., Kobayashi H., Orii T., Tomatsu S. Glycosaminoglycans analysis in blood and urine of patients with mucopolysaccharidosis. Mol. Genet. Metab. 2018;125:44–52. doi: 10.1016/j.ymgme.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujitsuka H., Sawamoto K., Peracha H., Mason R.W., Mackenzie W., Kobayashi H., Yamaguchi S., Suzuki Y., Orii K., Orii T. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019;19:100455. doi: 10.1016/j.ymgmr.2019.100455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomatsu S., Montaño A.M., Ohashi A., Gutierrez M.A., Oikawa H., Oguma T., Dung V.C., Nishioka T., Orii T., Sly W.S. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum. Mol. Genet. 2008;17:815–824. doi: 10.1093/hmg/ddm353. [DOI] [PubMed] [Google Scholar]
- 20.Qi Y., Musson D.G., Schweighardt B., Tompkins T., Jesaitis L., Shaywitz A.J., Yang K., O’Neill C.A. Pharmacokinetic and pharmacodynamic evaluation of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome. Clin. Pharmacokinet. 2014;53:1137–1147. doi: 10.1007/s40262-014-0173-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawamoto K., Suzuki Y., Mackenzie W.G., Theroux M.C., Pizarro C., Yabe H., Orii K.E., Mason R.W., Orii T., Tomatsu S. Current therapies for Morquio A syndrome and their clinical outcomes. Expert Opin. Orphan Drugs. 2016;4:941–951. doi: 10.1080/21678707.2016.1214572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinen Y., Higa T., Tomatsu S., Suzuki Y., Orii T., Hyakuna N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol. Genet. Metab. Rep. 2014;1:31–41. doi: 10.1016/j.ymgmr.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tomatsu S., Sawamoto K., Alméciga-Díaz C.J., Shimada T., Bober M.B., Chinen Y., Yabe H., Montaño A.M., Giugliani R., Kubaski F. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des. Devel. Ther. 2015;9:1937–1953. doi: 10.2147/DDDT.S68562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yabe H., Tanaka A., Chinen Y., Kato S., Sawamoto K., Yasuda E., Shintaku H., Suzuki Y., Orii T., Tomatsu S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016;117:84–94. doi: 10.1016/j.ymgme.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J., Luan Z., Jiang H., Fang J., Qin M., Lee V., Chen J. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis—a ten-year report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016;22:2104–2108. doi: 10.1016/j.bbmt.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Taylor M., Khan S., Stapleton M., Wang J., Chen J., Wynn R., Yabe H., Chinen Y., Boelens J.J., Mason R.W. Hematopoietic stem cell transplantation for mucopolysaccharidoses: past, present, and future. Biol. Blood Marrow Transplant. 2019;25:e226–e246. doi: 10.1016/j.bbmt.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tomatsu S., Orii K.O., Vogler C., Nakayama J., Levy B., Grubb J.H., Gutierrez M.A., Shim S., Yamaguchi S., Nishioka T. Mouse model of N-acetylgalactosamine-6-sulfate sulfatase deficiency (Galns−/−) produced by targeted disruption of the gene defective in Morquio A disease. Hum. Mol. Genet. 2003;12:3349–3358. doi: 10.1093/hmg/ddg366. [DOI] [PubMed] [Google Scholar]
- 28.Tomatsu S., Gutierrez M., Nishioka T., Yamada M., Yamada M., Tosaka Y., Grubb J.H., Montaño A.M., Vieira M.B., Trandafirescu G.G. Development of MPS IVA mouse (Galnstm(hC79S.mC76S)slu) tolerant to human N-acetylgalactosamine-6-sulfate sulfatase. Hum. Mol. Genet. 2005;14:3321–3335. doi: 10.1093/hmg/ddi364. [DOI] [PubMed] [Google Scholar]
- 29.Tomatsu S., Montaño A.M., Dung V.C., Ohashi A., Oikawa H., Oguma T., Orii T., Barrera L., Sly W.S. Enhancement of drug delivery: enzyme-replacement therapy for murine Morquio A syndrome. Mol. Ther. 2010;18:1094–1102. doi: 10.1038/mt.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomatsu S., Montaño A.M., Oikawa H., Dung V.C., Hashimoto A., Oguma T., Gutiérrez M.L., Takahashi T., Shimada T., Orii T., Sly W.S. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: early treatment rescues bone lesions? Mol. Genet. Metab. 2015;114:195–202. doi: 10.1016/j.ymgme.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neufeld E.F. In: Enzyme Replacement Therapy. Lysosomal Disorders of the Brain. Platt F.M., Walkley S.U., editors. Oxford University Press; 2004. pp. 327–338. [Google Scholar]
- 32.Krivit W., Peters C., Shapiro E.G. Bone marrow transplantation as effective treatment of central nervous system disease in globoid cell leukodystrophy, metachromatic leukodystrophy, adrenoleukodystrophy, mannosidosis, fucosidosis, aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly syndromes, and Gaucher disease type III. Curr. Opin. Neurol. 1999;12:167–176. doi: 10.1097/00019052-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Toietta G., Severini G.M., Traversari C., Tomatsu S., Sukegawa K., Fukuda S., Kondo N., Tortora P., Bordignon C. Various cells retrovirally transduced with N-acetylgalactosoamine-6-sulfate sulfatase correct Morquio skin fibroblasts in vitro. Hum. Gene Ther. 2001;12:2007–2016. doi: 10.1089/104303401753204571. [DOI] [PubMed] [Google Scholar]
- 34.Alméciga-Díaz C.J., Rueda-Paramo M.A., Espejo A.J., Echeverri O.Y., Montaño A., Tomatsu S., Barrera L.A. Effect of elongation factor 1α promoter and SUMF1 over in vitro expression of N-acetylgalactosamine-6-sulfate sulfatase. Mol. Biol. Rep. 2009;36:1863–1870. doi: 10.1007/s11033-008-9392-3. [DOI] [PubMed] [Google Scholar]
- 35.Alméciga-Díaz C.J., Montaño A.M., Tomatsu S., Barrera L.A. Adeno-associated virus gene transfer in Morquio A disease—effect of promoters and sulfatase-modifying factor 1. FEBS J. 2010;277:3608–3619. doi: 10.1111/j.1742-4658.2010.07769.x. [DOI] [PubMed] [Google Scholar]
- 36.Alméciga-Díaz C.J., Montaño A.M., Barrera L.A., Tomatsu S. Tailoring the AAV2 capsid vector for bone-targeting. Pediatr. Res. 2018;84:545–551. doi: 10.1038/s41390-018-0095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.US Food and Drug Administration . 2017. FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss.https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss [Google Scholar]
- 38.US Food and Drug Administration . 2019. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality.https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease [Google Scholar]
- 39.Mattar C.N., Nathwani A.C., Waddington S.N., Dighe N., Kaeppel C., Nowrouzi A., Mcintosh J., Johana N.B., Ogden B., Fisk N.M. Stable human FIX expression after 0.9G intrauterine gene transfer of self-complementary adeno-associated viral vector 5 and 8 in macaques. Mol. Ther. 2011;19:1950–1960. doi: 10.1038/mt.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greig J.A., Limberis M.P., Bell P., Chen S.J., Calcedo R., Rader D.J., Wilson J.M. Non-clinical study examining AAV8.TBG.hLDLR vector-associated toxicity in chow-fed wild-type and LDLR+/− rhesus macaques. Hum. Gene Ther. Clin. Dev. 2017;28:39–50. doi: 10.1089/humc.2017.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattar C.N.Z., Gil-Farina I., Rosales C., Johana N., Tan Y.Y.W., McIntosh J., Kaeppel C., Waddington S.N., Biswas A., Choolani M. In utero transfer of adeno-associated viral vectors produces long-term factor IX levels in a cynomolgus macaque model. Mol. Ther. 2017;25:1843–1853. doi: 10.1016/j.ymthe.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Nichols T.C., Read M.S., Bellinger D.A., Verma I.M. Sustained expression of therapeutic level of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol. Ther. 2000;1:154–158. doi: 10.1006/mthe.2000.0031. [DOI] [PubMed] [Google Scholar]
- 43.Mingozzi F., Liu Y.L., Dobrzynski E., Kaufhold A., Liu J.H., Wang Y., Arruda V.R., High K.A., Herzog R.W. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ziegler R.J., Lonning S.M., Armentano D., Li C., Souza D.W., Cherry M., Ford C., Barbon C.M., Desnick R.J., Gao G. AAV2 vector harboring a liver-restricted promoter facilitates sustained expression of therapeutic levels of α-galactosidase A and the induction of immune tolerance in Fabry mice. Mol. Ther. 2004;9:231–240. doi: 10.1016/j.ymthe.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 45.Sondhi D., Peterson D.A., Giannaris E.L., Sanders C.T., Mendez B.S., De B., Rostkowski A.B., Blanchard B., Bjugstad K., Sladek J.R., Jr. AAV2-mediated CLN2 gene transfer to rodent and non-human primate brain results in long-term TPP-I expression compatible with therapy for LINCL. Gene Ther. 2005;12:1618–1632. doi: 10.1038/sj.gt.3302549. [DOI] [PubMed] [Google Scholar]
- 46.Dobrzynski E., Fitzgerald J.C., Cao O., Mingozzi F., Wang L., Herzog R.W. Prevention of cytotoxic T lymphocyte responses to factor IX-expressing hepatocytes by gene transfer-induced regulatory T cells. Proc. Natl. Acad. Sci. USA. 2006;103:4592–4597. doi: 10.1073/pnas.0508685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao O., Dobrzynski E., Wang L., Nayak S., Mingle B., Terhorst C., Herzog R.W. Induction and role of regulatory CD4+CD25+ T cells in tolerance to the transgene product following hepatic in vivo gene transfer. Blood. 2007;110:1132–1140. doi: 10.1182/blood-2007-02-073304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mingozzi F., Hasbrouck N.C., Basner-Tschakarjan E., Edmonson S.A., Hui D.J., Sabatino D.E., Zhou S., Wright J.F., Jiang H., Pierce G.F. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110:2334–2341. doi: 10.1182/blood-2007-03-080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tessitore A., Faella A., O’Malley T., Cotugno G., Doria M., Kunieda T., Matarese G., Haskins M., Auricchio A. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol. Ther. 2008;16:30–37. doi: 10.1038/sj.mt.6300325. [DOI] [PubMed] [Google Scholar]
- 50.Cotugno G., Annunziata P., Tessitore A., O’Malley T., Capalbo A., Faella A., Bartolomeo R., O’Donnell P., Wang P., Russo F. Long-term amelioration of feline mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol. Ther. 2011;19:461–469. doi: 10.1038/mt.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishioka T., Tomatsu S., Gutierrez M.A., Miyamoto K., Trandafirescu G.G., Lopez P.L., Grubb J.H., Kanai R., Kobayashi H., Yamaguchi S. Enhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol. Genet. Metab. 2006;88:244–255. doi: 10.1016/j.ymgme.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Montaño A.M., Oikawa H., Tomatsu S., Nishioka T., Vogler C., Gutierrez M.A., Oguma T., Tan Y., Grubb J.H., Dung V.C. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol. Genet. Metab. 2008;94:178–189. doi: 10.1016/j.ymgme.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 53.Oldberg A., Franzén A., Heinegård D. The primary structure of a cell-binding bone sialoprotein. J. Biol. Chem. 1988;263:19430–19432. [PubMed] [Google Scholar]
- 54.Kasugai S., Fujisawa R., Waki Y., Miyamoto K., Ohya K. Selective drug delivery system to bone: small peptide (Asp)6 conjugation. J. Bone Miner. Res. 2000;15:936–943. doi: 10.1359/jbmr.2000.15.5.936. [DOI] [PubMed] [Google Scholar]
- 55.Tomatsu S., Okamura K., Taketani T., Orii K.O., Nishioka T., Gutierrez M.A., Velez-Castrillon S., Fachel A.A., Grubb J.H., Cooper A. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr. Res. 2004;55:592–597. doi: 10.1203/01.PDR.0000113767.60140.E9. [DOI] [PubMed] [Google Scholar]
- 56.Tomatsu S., Montaño A.M., Oguma T., Dung V.C., Oikawa H., de Carvalho T.G., Gutiérrez M.L., Yamaguchi S., Suzuki Y., Fukushi M. Validation of keratan sulfate level in mucopolysaccharidosis type IVA by liquid chromatography-tandem mass spectrometry. J. Inherit. Metab. Dis. 2010;33(Suppl 3):S35–S42. doi: 10.1007/s10545-009-9013-x. [DOI] [PubMed] [Google Scholar]
- 57.Hintze J.P., Tomatsu S., Fujii T., Montaño A.M., Yamaguchi S., Suzuki Y., Fukushi M., Ishimaru T., Orii T. Comparison of liquid chromatography-tandem mass spectrometry and sandwich ELISA for determination of keratan sulfate in plasma and urine. Biomark. Insights. 2011;6:69–78. doi: 10.4137/BMI.S7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martell L.A., Cunico R.L., Ohh J., Fulkerson W., Furneaux R., Foehr E.D. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis. 2011;3:1855–1866. doi: 10.4155/bio.11.172. [DOI] [PubMed] [Google Scholar]
- 59.Hendriksz C.J., Burton B., Fleming T.R., Harmatz P., Hughes D., Jones S.A., Lin S.P., Mengel E., Scarpa M., Valayannopoulos V., STRIVE Investigators Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014;37:979–990. doi: 10.1007/s10545-014-9715-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hendriksz C.J., Parini R., AlSayed M.D., Raiman J., Giugliani R., Solano Villarreal M.L., Mitchell J.J., Burton B.K., Guelbert N., Stewart F. Long-term endurance and safety of elosulfase alfa enzyme replacement therapy in patients with Morquio A syndrome. Mol. Genet. Metab. 2016;119:131–143. doi: 10.1016/j.ymgme.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 61.Lavery C., Hendriksz C. Mortality in patients with Morquio syndrome A. JIMD Rep. 2015;15:59–66. doi: 10.1007/8904_2014_298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kampmann C., Abu-Tair T., Gökce S., Lampe C., Reinke J., Mengel E., Hennermann J.B., Wiethoff C.M. Heart and cardiovascular involvement in patients with mucopolysaccharidosis type IVA (Morquio-A syndrome) PLoS ONE. 2016;11:e0162612. doi: 10.1371/journal.pone.0162612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mochizuki H., Yoshida K., Shibata Y., Kimata K. Tetrasulfated disaccharide unit in heparan sulfate: enzymatic formation and tissue distribution. J. Biol. Chem. 2008;283:31237–31245. doi: 10.1074/jbc.M801586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oguma T., Tomatsu S., Okazaki O. Analytical method for determination of disaccharides derived from keratan sulfates in human serum and plasma by high-performance liquid chromatography/turbo-ionspray ionization tandem mass spectrometry. Biomed. Chromatogr. 2007;21:356–362. doi: 10.1002/bmc.760. [DOI] [PubMed] [Google Scholar]
- 65.Oguma T., Tomatsu S., Montano A.M., Okazaki O. Analytical method for the determination of disaccharides derived from keratan, heparan, and dermatan sulfates in human serum and plasma by high-performance liquid chromatography/turbo ionspray ionization tandem mass spectrometry. Anal. Biochem. 2007;368:79–86. doi: 10.1016/j.ab.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 66.Shimada T., Tomatsu S., Yasuda E., Mason R.W., Mackenzie W.G., Shibata Y., Kubaski F., Giugliani R., Yamaguchi S., Suzuki Y. Chondroitin 6-sulfate as a novel biomarker for mucopolysaccharidosis IVA and VII. JIMD Rep. 2014;16:15–24. doi: 10.1007/8904_2014_311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimada T., Tomatsu S., Mason R.W., Yasuda E., Mackenzie W.G., Hossain J., Shibata Y., Montaño A.M., Kubaski F., Giugliani R. Di-sulfated keratan sulfate as a novel biomarker for mucopolysaccharidosis II, IVA, and IVB. JIMD Rep. 2015;21:1–13. doi: 10.1007/8904_2014_330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kubaski F., Mason R.W., Nakatomi A., Shintaku H., Xie L., van Vlies N.N., Church H., Giugliani R., Kobayashi H., Yamaguchi S. Newborn screening for mucopolysaccharidoses: a pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017;40:151–158. doi: 10.1007/s10545-016-9981-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomatsu S., Orii K.O., Vogler C., Grubb J.H., Snella E.M., Gutierrez M., Dieter T., Holden C.C., Sukegawa K., Orii T. Production of MPS VII mouse (Gustm(hE540A x mE536A)Sly) doubly tolerant to human and mouse β-glucuronidase. Hum. Mol. Genet. 2003;12:961–973. doi: 10.1093/hmg/ddg119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.