

Abstract
The formation of silver and Au@Ag core@shell nanoparticles via reduction of AgNO3 by trisodium citrate was followed using in situ X-ray absorption near-edge structure (XANES) spectroscopy and time-resolved UV–visible (UV–vis) spectroscopy. The XANES data were analyzed through linear combination fitting, and the reaction kinetics were found to be consistent with first-order behavior with respect to silver cations. For the Au@Ag nanoparticles, the UV–vis data of a lab-scale reaction showed a gradual shift in dominance between the gold- and silver-localized surface plasmon absorbance bands. Notably, throughout much of the reaction, distinct gold and silver contributions to the UV–vis spectra were observed; however, in the final product, the contributions were not distinct.
Introduction
Metal nanoparticles are a popular research area because of their unique properties, which enable myriad applications across a broad range of fields. Silver and silver-containing nanoparticles, in particular, are highly desirable because of their extremely strong localized surface-plasmon resonance (LSPR).1,2 Gold nanoparticles also display surface plasmon resonance and, in addition, have high chemical stability, particularly in biological environments. Consequently, researchers have sought to combine these properties by preparing gold/silver heterostructures.3,4 Such structures have also been investigated for their catalytic activities5,6 and to gain underlying theoretical understanding.7−10
Generally, metal nanoparticles are prepared via “bottom-up” methods—very often, these are wet-chemistry methods, in which colloidal nanoparticles are prepared through reduction of a metal salt.2 Understanding these formation processes is an important first step toward controllable synthetic routes, which will allow the properties of the final particles to be optimized as desired; consequently, these mechanisms have been the focus of substantial research efforts.1,2,11,12
Citrate reduction is one of the oldest and most widely used methods for preparing metal colloids; it was first reported for preparation of gold colloids13 but has since been extended to other metals, such as silver.14 The overall mechanism of the synthesis is a complex mixture of chemical considerations, arising from the chemical mechanism of the reduction, and kinetic considerations, arising from the particle nucleation and growth processes.15−18 Trisodium citrate alone takes on up to three separate roles in the process: reducing agent, capping agent, and pH control.19 While a reasonable amount of work has been reported examining the mechanism of the citrate reduction of chloroauric acid,15−18 far less has been reported on the reduction of silver nitrate (see Rycenga et al.1 and Pacioni et al.2). In particular, the studies mentioned above have focused on varying the synthesis, rather than understanding the kinetics of the reaction and the nature of the metal ions present throughout the course of the reaction; therefore, we focused our attention on investigating these two aspects. In addition, we investigated the seeded growth of core@shell nanoparticles and compared this to the nucleation/growth of the pure silver colloids.
While it is anticipated that the basics of the citrate reduction mechanism will be shared between different metals and precursors, there exist major differences between such systems that merit individual study. Notably, in this case, silver nitrate needs to be reduced by just one oxidation state, compared to the three oxidation-state reduction required for chloroauric acid. Additionally, the high stability20 of Ag+ eliminates the possibility of the surface-catalyzed disproportionation route proposed for the gold system.21,22
X-ray absorption near-edge structure (XANES) spectroscopy is a powerful, element-specific tool for probing the oxidation state and coordination environment of a chosen element, while achieving a time resolution suitable for in situ studies. In situ and ex situ X-ray absorption spectroscopy (XAS) measurements can be combined with other techniques to provide advanced characterization of samples and processes.23,24 XAS has previously been used to study the citrate reduction of gold. Most such studies have been performed by quenching and analyzing aliquots removed from the reaction mixture, rather than being true in situ studies.25 Additionally, as identified by Plech et al.,26 beam-induced reduction is a significant danger during this type of experiment (although pure silver nitrate, in the absence of any reducing or capping agents, has been found to be stable in a synchrotron beam27). Such beam-induced reduction is a well-known phenomenon that has been identified in a range of other systems.28 Additionally, the concentrations required for good quality XAS data reach the bounds for stable colloid formation in the Turkevich process (ca. 1–2 mM maximum).16,29 Because of these challenges, XANES has been underutilized for mechanistic studies of citrate reduction.
Previously, Harada and Ikegami used in situ XAS to investigate the reduction of gold nanoparticles.30 Nishimura et al.31 used XANES to follow the formation of silver nanoparticles and determine the role of NaOH in a different synthesis using sodium acrylate as the reducing/capping agent, by removing and quenching aliquots from the reaction. Henglein and Giersig32 used γ-radiolysis to decouple the reducing and capping functions of trisodium citrate and investigate only its capping properties. Abécassis et al. performed coupled in situ XANES and small-angle X-ray scattering on the reduction of AuCl3 by sodium borohydride, identifying the presence of transient monomer intermediates and investigating the effect of ligand choice on reaction kinetics.33
Herein, we present time-resolved, in situ Ag K-edge XANES studies of the reduction of silver nitrate by trisodium citrate. First, a pure silver colloid is studied; this is followed by the reduction of silver onto a gold seed to form an Au@Ag core@shell colloid. Linear combination fitting (LCF) is used to quantify the proportion of reduced/oxidized silver as a function of time, and kinetic conclusions are drawn. UV–visible (UV–vis) spectroscopy is utilized to simultaneously investigate the gold and silver components of the core@shell system. The results of these analyses are then discussed in light of the known mechanism for the citrate reduction of silver nitrate to metallic silver, and the equivalent, but better understood, reduction of gold chloride to metallic gold.
Results and Discussion
Figure 1 shows XANES recorded during the first 40 min of the reduction of AgNO3. The spectra show a clear progression from Ag-nitrate-like to Ag-foil-like as the reaction progresses. This change was quantified using LCF (Figure 2). The reaction was found to follow a first-order rate equation with respect to silver cations (eq 1)—the curves shown in Figure 2 correspond to the fitting of eq 1 to the data, adjusted-R2 = 0.90 (vs 0.72 for a linear, zero-order fit). The rate coefficient, k, was found to be 0.09 ± 0.03 s–1.
| 1 |
Here, [Ag+]t is the mole fraction of Ag+ (dimensionless) at time t (s). [Ag+]eqm is the “equilibrium” mole fraction of Ag+ (dimensionless) (i.e., the mole fraction of unreacted Ag+ that remains at the end of the reaction). [Ag+]0 is the mole fraction of Ag+ (dimensionless) at t = 0 (s). k is the rate coefficient (s–1).
Figure 1.
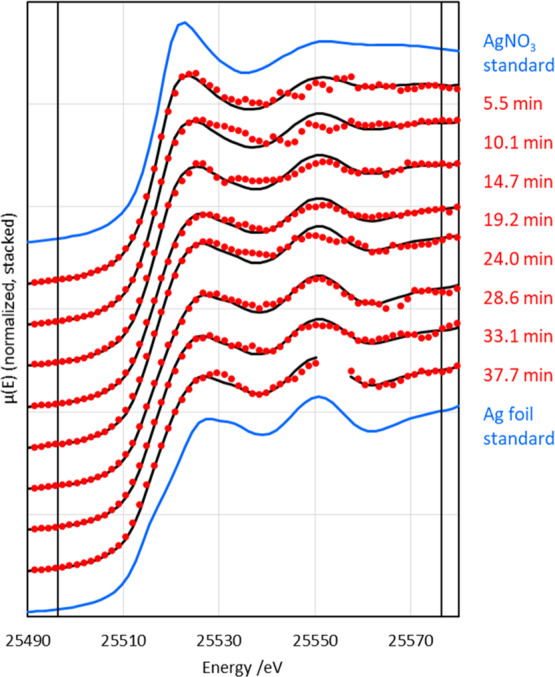
Smoothed (boxcar, kernel = 3) in situ Ag K-edge XANES throughout the citrate reduction of AgNO3 (red) with corresponding LCF fits (black) and standards (blue). The vertical black lines indicate the fitting region. Uncertainty in the time is ±13.8 s. (Unsmoothed XANES data are given in Figure S1. The gap present in the 37.7 min XANES is due to a measurement glitch (see Figure S1), and this region was not included in the fitting.)
Figure 2.

Results of LCF fitting of XANES from ca. 5 min to ca. 40 min to Ag0 and AgNO3 standards, with first-order rate equations (with respect to silver cations) fitted to the data. Error bars give the statistical uncertainty in the LCF fitting results, as estimated by Athena. Uncertainty in the time is ±13.8 s.
The eightfold excess of citrate used in this experiment means that pseudo-first-order behavior (steady-state approximation of [citrate]) is plausible; however, a beam-induced unimolecular decomposition is also a plausible explanation for this observation. Given that silver nitrate, in the absence of stabilizing agents, has been shown to be stable in the beam (the authors of that work attribute this to the reduction potential of silver; they were able to stimulate beam-induced reduction through the addition of one of 2-propanol, polyethylene glycol, or polyvinylpyrrolidone),27 and that similar beam-induced reduction of HAuCl4 has been shown to be zero-order,34 beam-induced reduction seems less likely. Additionally, a mixture of HAuCl4 and AgNO3 (forming the particularly photosensitive AgCl, which is less stable than AgNO3) left in the beam for a prolonged period remained stable (see Figure S2), suggesting that the beam alone, at the intensity used, was incapable of reducing the silver precursor. Further, the observed timescale of the reaction in-beam (ca. 40 min) is similar to that which would be expected in the lab (ca. 1 h14). This suggests that it is the citrate reduction, and not beam-induced reduction, being followed here. Altogether, this suggests that the citrate reduction of silver proceeds via a process that is pseudo-first-order with respect to silver cations.
In their seminal study, Turkevich et al.35 found that the nucleation of gold particles during reduction by acetone dicarboxylate (DCA, a decomposition product of trisodium citrate) displayed first-order behavior. This was attributed to unimolecular decomposition of a DCA–gold(I) complex, it being the rate-determining step. In reactions using trisodium citrate, they35 observed autocatalytic behavior during the nucleation period (immediately after the induction period), which they ascribed to the formation of DCA from trisodium citrate. This is not present in the data reported herein, most likely because these experiments followed the method reported by Ojea-Jiménez et al.36/Sivaraman et al.37 in which DCA has time to form before the metal salt is added. The observation of pseudo-first-order behavior (with respect to silver cations) here, therefore, is consistent with the reduction of silver nitrate by trisodium citrate via a route involving [Ag+–DCA] complex formation, which is similar to that reported for gold.
Depending on the extent of the conversion of citrate to DCA, direct interaction of Ag+ with citrate cannot be ruled out. In the citrate reduction of gold, the initial reduction from Au3+ to Au+ has been proposed to occur through direct reduction by citrate as it oxidizes to DCA;38 however, a directly equivalent reaction could not occur for silver, which only requires a reduction of one in its oxidation state (from Ag+ to Ag0, compared to Au3+ to Au0via Au+).
Another mechanism entailing direct interaction between citrate and Ag+ has been proposed for radiolysis-induced reduction of silver cations. This proceeds via a multistep process whereby the radiolysis reduces Ag+ to Ag0, which then combines with additional Ag+ to form an (Ag)2+ dimer, which complexes with citrate to form [Ag2+–citrate–] before decomposing.39 If an [Ag2+–citrate–] intermediate were present in the reaction performed here, an induction period for the initial silver reduction and dimer formation would be expected. There is also no clear route for the initiation of the initial reduction step in this case (ref (39) initiated the reaction through radiolysis). Consequently, the results in Figure 2 are not consistent with this mechanism.
In order to confirm this conclusion, experimental results that provide direct evidence of an [Ag+–DCA] complex are desirable. Although, in principle, XAS can identify such species, because of the low concentration of the reaction and short data collection times the data quality does not permit such an analysis here. Experimental techniques that could provide additional or direct evidence of such a complex are likely to be a fruitful area for further work; these techniques include in situ infrared spectroscopy,40−42 isotopic experiments,43 and, noting the difficulty of identifying a transient intermediate using ex situ techniques, nuclear magnetic resonance spectroscopy.44 Computational approaches may also be informative.21,38
Beyond the nucleation phase, the reduction kinetics deviate from those reported for gold.35 In the citrate reduction of gold, a surface-catalyzed disproportionation step21,22,45 renders that reaction autocatalytic;25 however, autocatalytic behavior is not observed in Figure 2. This is due to the unfavorableness of the higher oxidation states of silver20 preventing such a reaction. Although disproportionation of Ag+ to Ag0 and Ag2+ has occasionally been reported,46,47 these reports concern situations that are not relevant to this work. Additionally, the presence of an appreciable quantity of Ag2+ would be evident in the XANES and be expected to shift the Ag K-edge to high energy (up to 10 eV).48
A number of studies of various routes for the synthesis of silver nanoparticles49−51 (including that discussed with regard to the [Ag2+–citrate–] complex above39) also propose an autocatalytic mechanism, whereby Ag0 formed early in the reaction dimerizes with Ag+. As Figure 2 does not display any autocatalytic behavior, such a step cannot be rate-limiting in this reaction. Beyond the nucleation phase, the Ag0 present is likely to be part of the nanoparticles, and not available for dimer formation. It is possible that a surface reaction (e.g., surface-mediated electron transfer) occurs (in particular, to explain the decomposition of any Ag complex present), although as it cannot be rate-determining, an alternative step (such as complex formation) would be required to be rate-determining to explain the first-order character of Figure 2. Indeed, there are other explanations that are at least as likely to explain complex decomposition (e.g., the further thermal decomposition of citrate and/or DCA52).
All these facts suggest that the chemical mechanism of the growth phase of the reaction proceeds by the same unimolecular decomposition as the nucleation phase; importantly, this is consistent with the observation of only first-order character in Figure 2.
Figure 3 shows in situ Ag K-edge XANES of the reduction of AgNO3 onto gold cores. As in the pure Ag reduction, there is a clear progression from Ag+ to Ag0. Composition results, determined through LCF fitting, again display first-order characteristics (with respect to silver cations) (Figure 4). The rate coefficient was found to be 0.030 ± 0.006 s–1. The reason for the lower rate coefficient in this case is not immediately apparent. Possible explanations include the presence of silver chloride, which is less soluble than silver nitrate and, therefore, less available for participation in the reaction, and the possibility that some of the trisodium citrate added for the silver reduction coordinates with the gold nanoparticles present in the colloid. Additionally, the Cl– ions may etch some of the reduced silver shell after deposition, effectively reducing the apparent reduction rate.4
Figure 3.
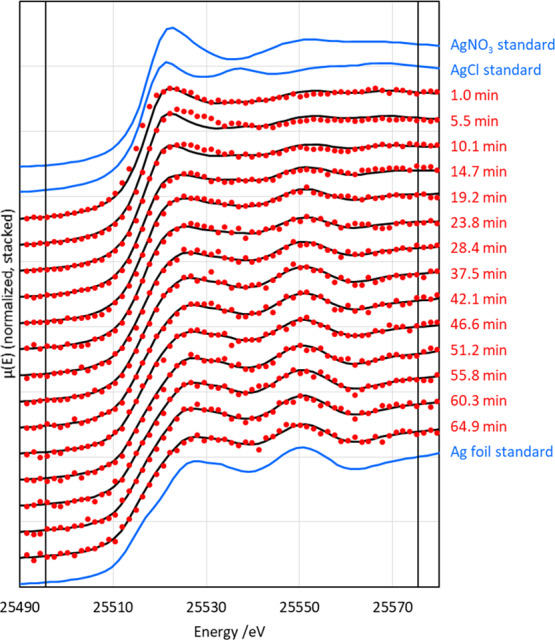
In situ Ag K-edge XANES of the reduction of Ag onto gold seeds by trisodium citrate (red), LCF fits to the XANES (black), and standards used for fitting (blue). The vertical black lines indicate the fitting region. Uncertainty in the time is ±13.8 s.
Figure 4.

Results of LCF fitting of XANES data in Figure 3 to Ag0, AgNO3, and AgCl standards, with first-order rate equations (with respect to silver cations) fitted to the data. Error bars give the statistical uncertainty in the LCF fitting results, as estimated by Athena. Uncertainty in the time is ±13.8 s.
On examining Figure 4, it can be seen that the reactant and product concentrations remained virtually unchanged over the first five, or so, minutes of the reaction. This corresponds to the induction period required to oxidize the trisodium citrate to DCA; unlike in the pure silver reduction, the trisodium citrate was not preheated in this case.a We note that in the case of reduction in the presence of preformed gold cores, the silver grows on the existing cores,7,53 and there is no nucleation phase; therefore, the induction period cannot be attributed to nucleation or prenucleation processes.
Solvated in the dispersant, there are various byproducts of the initial preparation of the gold seeds—including Na+, Cl–, and the decomposition products of citrate—which may become involved in the reaction. One particularly likely consequence of this is the formation of AgCl, either from a substitution reaction between AgNO3 and Cl– or from etching of the reduced silver by the Cl–;4 this is observed in the LCF results as anticipated. The presence of insoluble AgCl has the potential to substantially retard the reaction or even to prevent a substantial amount of the silver from reacting. However, it appears that both the silver nitrate and the silver chloride participate in the reaction at similar rates. In Figure 4, about half of the Ag+ species are present as silver nitrate and half as silver chloride; once the reaction is underway, they appear to be consumed at similar rates, although a slight preference for AgCl is apparent before ca. 20 min.
The participation of silver chloride in the reaction is unexpected, but it may occur either because the concentration of silver chloride is below its solubility limit (or because, as the very low solvated portion is consumed, some of the solid phase dissolves to compensate) or as a heterogeneous reaction between the solid and solution phases. Previous work has shown that insoluble silver salts can participate in aqueous reduction in some reactions.31 Additionally, the solubility of silver chloride is known to be greater in chloride-rich environments, where AgCl2– complexes may form, and at higher temperatures.54 Our previous ex situ characterization of this system has demonstrated that the seeds are reduced gold and the final product comprises a reduced silver shell on a reduced gold core;7 consequently, no silver chloride is present in the final product. The EXAFS metal–metal distances determined in that paper were consistent with the face-centered cubic structures of gold and silver. The microscopy and extended X-ray absorption fine structure (EXAFS) results therein also confirm the core@shell nature of the colloid.
Both reactions appear to only go to 80 or 90% completion (Figures 2 and 4). To shed further light on this, the final four scans of the sequential reduction reaction (by which point, the fraction of reduced/oxidized silver was fairly constant; the pure silver reduction did not produce spectra of sufficient quality to extract the EXAFS) were merged and the EXAFS extracted. The EXAFS and Fourier transform are shown in Figure 5. Ag0 is the only major component visible; however, the data are not of sufficient quality for EXAFS modeling to confirm this. Indeed, a small oxidized component may simply be indistinguishable from noise. The peak at ca. 2 Å (without phase correction) in the Fourier transform does display a very slight shift toward high angstrom (ca. 0.03 Å with respect to the foil standard), which could be indicative of an AgCl component.
Figure 5.

(a) Fourier transform (not phase-corrected) of the Ag K-edge EXAFS of the final four scans of the sequential reduction reaction (merged). (b) EXAFS of (a).
A similar observation of incomplete reduction was observed for gold by Mikhlin et al.,55 who ascribed it to the presence of a hydrodynamic shell of unreacted precursor around the particles. Over time, this reduces onto the surface of the particles, explaining why it has not been identified during the previous ex situ characterization of these systems. The presence of high-oxidation-state surface sites interacting with the citrate anions was also considered, but these would not explain the absence of such an effect in the ex situ studies.
Figure 6 shows time-resolved UV–vis spectra for an Au@Ag synthesis under laboratory conditions. The weak peak at ca. 520 nm corresponds to the gold seeds. By 1 min, this peak has been blue-shifted to ca. 500 nm, suggesting the deposition of a thin shell of silver influencing the gold LSPR, but not yet thick enough to manifest as a distinct feature. A shallow, broad peak at ca. 600 nm is also present—this may be a consequence of large, reduced or partially reduced agglomerates, as identified during the reduction of gold.56 By 1 min, two distinct peaks (ca. 400 nm and ca. 490 nm) are visible; this suggests that the silver is now thick enough to show a distinct LSPR absorption of its own (400 nm), but thin enough that the gold is able to maintain a distinct absorption (490 nm).
Figure 6.
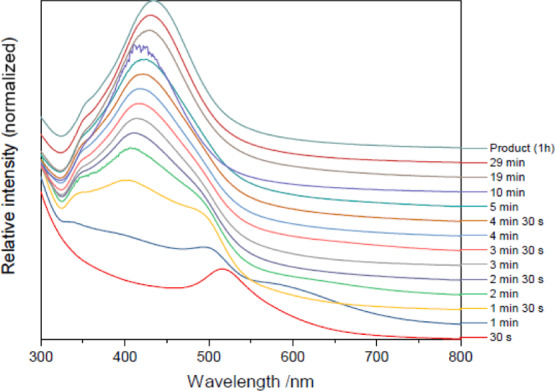
In situ UV–vis spectra of the reduction of AgNO3 onto gold seeds by trisodium citrate. (Normalized/stacked).
From this point, the gold peak becomes a steadily diminishing shoulder to the silver peak until, after 1 h, a single, silver-dominant peak at ca. 435 nm is present. This wavelength is consistent with theoretical and experimental studies of core@shell Au@Ag LSPR behavior, confirming core@shell formation.57 There is an additional, low-intensity, low-wavelength peak at ca. 355 nm (present from 1 min onward), suggesting that some pure-silver nucleation occurs but that these nuclei do not grow. The linked behavior of the LSPR peaks also confirms the bimetallic nature of the system throughout all but the very early stage of the reaction.
Despite the different conditions of the in situ and ex situ reactions, this is consistent with the observation from the XANES data that reduction continues throughout the reaction process (i.e., that the growth period does not merely consist of processes such as Ostwald ripening). This indicates that the continual reduction of silver is a feature of this reaction over a wide range of concentrations.
Conclusions
The reduction of AgNO3 by trisodium citrate, both alone and in the presence of gold seeds, has been found to be a first-order or pseudo-first-order process (with respect to silver cations). This is interpreted as a consequence of a unimolecular decomposition rate-determining step, as previously reported for the reduction of HAuCl4 by trisodium citrate. As with gold, the order of precursor addition affects the presence of an induction period at the beginning of the reaction, while citrate is oxidized to DCA. The reduction of silver onto gold seeds has been found to be slower than the monometallic reduction, probably because of the presence of AgCl in the reaction. UV–vis spectroscopy of a synthesis under typical laboratory conditions has confirmed the prolonged nature of the chemical aspects of the reduction onto gold seeds under these conditions also, which is in accordance with the previously cited literature.
Methods
Ag K-edge in situ X-ray absorption experiments were performed on beamline B18 at the Diamond Light Source.58 Measurements were performed in quick-EXAFS mode, and in situ data were recorded using a 13-element Ge fluorescence detector. Simultaneously, with each in situ measurement, silver-foil reference data were recorded in transmission mode. Standard compounds for LCF analysis (Ag foil, AgNO3, AgCl) were also recorded in transmission mode. The AgNO3 and AgCl standard spectra were measured by preparing pellets with fumed silica as a binding agent. An Si(311) monochromator and Pt-coated harmonic rejection mirror were used for these measurements. The time taken to scan the XANES region of the XAS spectrum was 13.8 s; therefore, this value is taken for the uncertainty in the time of each XANES spectrum.
Reactions were carried out in a bespoke in situ XAS cell, described in more detail in Figures S3 and S4; for these experiments, the cell was equipped with a syringe driver and magnetic stirrer. The temperatures reported here are estimated from the temperatures measured by a thermocouple at the base of the cell; the temperature reported by this thermocouple had previously been calibrated to the temperature of the reaction chamber. After the reactions, some staining was visible on the windows of the in situ XAS cell because of the intense color of the colloids. Fresh windows were used for each reaction.
The pure silver colloid was prepared by injecting an aqueous solution of AgNO3 (0.35 cm3, 20 mM) into a 79 °C aqueous solution of trisodium citrate (0.35 cm3, 160 mM), with stirring. The mixture was then maintained at 79 °C, with stirring, for 40 min and XAS data were recorded continuously (5 min per scan). This protocol is based on that published by Lee and Meisel;14 however, we have used substantially higher concentrations (ca. 10 mM Ag total vs ca. 1 mM Ag total) to increase the signal/noise ratio of the data.
The Au@Ag colloid was prepared by injecting a mixture of aqueous solutions of AgNO3 (0.26 cm3, 20 mM) and trisodium citrate (0.26 cm3, 160 mM) into a 79 °C aqueous gold colloid [0.09 cm3, synthesis given shortly, and deionized water (0.08 cm3)], with stirring. The mixture was then maintained at 79 °C, with stirring, for 1 h and XAS data were recorded throughout. This protocol is based on that published by Anh et al.,53 again at increased concentration.
The gold colloid used as seeds for the sequential reduction was prepared by adding HAuCl4(aq) (2.5 cm3, 20 mM) to a boiling solution of trisodium citrate (47.98 cm3, 7.2 mM) and refluxed and stirred for 1 h.36 (Because our previous work7—see also Figure S5—showed that the gold cores remain unchanged throughout the reaction, we did not perform Au edge experiments; a transmission electron micrograph of the gold seed colloid is given in Figure S6.)
UV–vis spectroscopy was carried out using a Shimadzu UV1800 spectrophotometer; data were collected using quartz cuvettes, diluting samples as necessary, and with a reference channel containing solely a water-filled cuvette. Aliquots were removed from the reaction at specific time intervals and immediately quenched on ice; UV–vis spectra were then recorded as soon as possible. The reaction was carried out as follows: HAuCl4(aq) (90 cm3, 0.57 mM) was added to a boiling solution of trisodium citrate (50 cm3, 13.5 mM) and refluxed and stirred for 1 h to prepare a gold colloid for use as seeds. The as-prepared gold-seed colloid (20 cm3) was then heated to the boil, and AgNO3(aq) (4.82 cm3, 13.3 mM) and trisodium citrate (3.38 cm3, 20 mM) were added. The reaction was then refluxed and stirred for 1 h.
XAS data were analyzed using Demeter 0.9.25;59 the results were first aligned, normalized, and deglitched as required, and then, LCF was performed within −20 to + 60 eV of the edge. The spectra from the pure silver colloid were smoothed (boxcar, kernel = 3) prior to analysis. First-order rate curves were fit to the LCF results using the commercial software package OriginPro 2017.
The crystalline nature of the nanoparticles produced was verified by performing high-resolution transmission electron microscopy (HRTEM) and X-ray diffraction (XRD) measurements on samples prepared ex situ. Full details and results are given in Figures S7 and S8.
Acknowledgments
Optical spectroscopy was performed at the UK Catalysis Hub (EPSRC grants EP/K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1, and EP/M013219/1) at the Research Complex at Harwell. XAS experiments were performed on beamline B18 at the Diamond Light Source (proposal SP-9949). IG thanks UCL/JAIST/EPSRC for a studentship through the M3S IDC at UCL (EP/G036675/1). This work was derived from IG’s PhD thesis (available at https://discovery.ucl.ac.uk/id/eprint/10040860).
Glossary
Nomenclature
- [x]
concentration of species x
- DCA
acetone dicarboxylate
- EXAFS
extended X-ray absorption fine structure
- LCF
linear combination fitting
- XANES
X-ray absorption near-edge structure
- XAS
X-ray absorption spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00697.
In situ XANES of pure silver reduction prior to data smoothing, beam stability study, in situ XAS cell used in this work, effect of reaction on gold cores, TEM of gold seed colloid, TEM and HRTEM of bimetallic nanocrystals, XRD pattern of bimetallic nanoparticles, TEMs of product of a similar colloidal synthesis in the in situ cell and under lab conditions (PDF)
Author Contributions
I.J.G., A.J.D., and G.S. performed the XANES measurements, I.J.G. and G.S. analyzed the XANES data, I.J.G. performed the UV–vis measurements, I.P.P., S.M., and G.S. supervised the work. All authors have contributed to and given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Footnotes
This was a necessary consequence of the experimental setup: one set of reactants could be preheated and one set could be injected at ambient temperature. Injecting ambient AgNO3 + citrate into preheated Au0 was the most desirable combination as it avoided possible side reactions occurring between premixed HAuCl4 and AgNO3, the formation of a pure Ag colloid during the preheating step, and the risks to the stability of the gold colloid that would occur with the addition of excess citrate.
Supplementary Material
References
- Rycenga M.; Cobley C. M.; Zeng J.; Li W.; Moran C. H.; Zhang Q.; Qin D.; Xia Y. Controlling the Synthesis and Assembly of Silver Nanostructures for Plasmonic Applications. Chem. Rev. 2011, 111, 3669–3712. 10.1021/cr100275d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacioni N. L.; Borsarelli C. D.; Rey V.; Veglia A. V.. Synthetic Routes for the Preparation of Silver Nanoparticles. In Silver Nanoparticle Applications; Alarcon E. I., Griffith M., Udekwu K. I., Eds.; Springer International Publishing: Cham, CH, 2015; pp 13–46. [Google Scholar]
- Mott D. M.; Anh D. T. N.; Singh P.; Shankar C.; Maenosono S. Electronic Transfer as a Route to Increase the Chemical Stability in Gold and Silver Core–Shell Nanoparticles. Adv. Colloid Interface Sci. 2012, 185–186, 14–33. 10.1016/j.cis.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Shankar C.; Dao A. T. N.; Singh P.; Higashimine K.; Mott D. M.; Maenosono S. Chemical Stabilization of Gold Coated by Silver Core–Shell Nanoparticles via Electron Transfer. Nanotechnology 2012, 23, 245704. 10.1088/0957-4484/23/24/245704. [DOI] [PubMed] [Google Scholar]
- Yen C.-W.; Lin M.-L.; Wang A.; Chen S.-A.; Chen J.-M.; Mou C.-Y. CO Oxidation Catalyzed by Au–Ag Bimetallic Nanoparticles Supported in Mesoporous Silica. J. Phys. Chem. C 2009, 113, 17831–17839. 10.1021/jp9037683. [DOI] [Google Scholar]
- Wang A.-Q.; Chang C.-M.; Mou C.-Y. Evolution of Catalytic Activity of Au–Ag Bimetallic Nanoparticles on Mesoporous Support for CO Oxidation. J. Phys. Chem. B 2005, 109, 18860–18867. 10.1021/jp051530q. [DOI] [PubMed] [Google Scholar]
- Godfrey I. J.; Dent A. J.; Parkin I. P.; Maenosono S.; Sankar G. Structure of Gold–Silver Nanoparticles. J. Phys. Chem. C 2017, 121, 1957–1963. 10.1021/acs.jpcc.6b11186. [DOI] [Google Scholar]
- Deng L.; Hu W.; Deng H.; Xiao S.; Tang J. Au–Ag Bimetallic Nanoparticles: Surface Segregation and Atomic-Scale Structure. J. Phys. Chem. C 2011, 115, 11355–11363. 10.1021/jp200642d. [DOI] [Google Scholar]
- Gomes J. F.; Garcia A. C.; Pires C.; Ferreira E. B.; Albuquerque R. Q.; Tremiliosi-Filho G.; Gasparotto L. H. S. Impact of the AuAg NPs Composition on Their Structure and Properties: A Theoretical and Experimental Investigation. J. Phys. Chem. C 2014, 118, 28868–28875. 10.1021/jp509437a. [DOI] [Google Scholar]
- Gould A. L.; Logsdail A. J.; Catlow C. R. A. Influence of Composition and Chemical Arrangement on the Kinetic Stability of 147-Atom Au–Ag Bimetallic Nanoclusters. J. Phys. Chem. C 2015, 119, 23685–23697. 10.1021/acs.jpcc.5b03577. [DOI] [Google Scholar]
- Coskun S.; Aksoy B.; Unalan H. E. Polyol Synthesis of Silver Nanowires: An Extensive Parametric Study. Cryst. Growth Des. 2011, 11, 4963–4969. 10.1021/cg200874g. [DOI] [Google Scholar]
- Lin G.; Lu W.; Cui W.; Jiang L. A Simple Synthesis Method for Gold Nano- and Microplate Fabrication Using a Tree-Type Multiple-Amine Head Surfactant. Cryst. Growth Des. 2010, 10, 1118–1123. 10.1021/cg9008976. [DOI] [Google Scholar]
- Hauser E. A.; Lynn J. E.. Chapter 2: Preparation of Colloidal Systems. Experiments in Colloid Chemistry; McGraw-Hill Book Co.: New York, NY, USA, 1940; pp 7–27. [Google Scholar]
- Lee P. C.; Meisel D. Adsorption and Surface-Enhanced Raman of Dyes on Silver and Gold Sols. J. Phys. Chem. 1982, 86, 3391–3395. 10.1021/j100214a025. [DOI] [Google Scholar]
- Wuithschick M.; Birnbaum A.; Witte S.; Sztucki M.; Vainio U.; Pinna N.; Rademann K.; Emmerling F.; Kraehnert R.; Polte J. Turkevich in New Robes: Key Questions Answered for the Most Common Gold Nanoparticle Synthesis. ACS Nano 2015, 9, 7052–7071. 10.1021/acsnano.5b01579. [DOI] [PubMed] [Google Scholar]
- Kimling J.; Maier M.; Okenve B.; Kotaidis V.; Ballot H.; Plech A. Turkevich Method for Gold Nanoparticle Synthesis Revisited. J. Phys. Chem. B 2006, 110, 15700–15707. 10.1021/jp061667w. [DOI] [PubMed] [Google Scholar]
- Thanh N. T. K.; Maclean N.; Mahiddine S. Mechanisms of Nucleation and Growth of Nanoparticles in Solution. Chem. Rev. 2014, 114, 7610–7630. 10.1021/cr400544s. [DOI] [PubMed] [Google Scholar]
- Shi L.; Buhler E.; Boué F.; Carn F. How Does the Size of Gold Nanoparticles Depend on Citrate to Gold Ratio in Turkevich Synthesis? Final Answer to a Debated Question. J. Colloid Interface Sci. 2017, 492, 191–198. 10.1016/j.jcis.2016.10.065. [DOI] [PubMed] [Google Scholar]
- Ji X.; Song X.; Li J.; Bai Y.; Yang W.; Peng X. Size Control of Gold Nanocrystals in Citrate Reduction: The Third Role of Citrate. J. Am. Chem. Soc. 2007, 129, 13939–13948. 10.1021/ja074447k. [DOI] [PubMed] [Google Scholar]
- Cotton S. A.Silver and Gold. Chemistry of Precious Metals; Blackie Academic & Professional: London, UK, 1997; pp 273–327. [Google Scholar]
- Kumar S.; Gandhi K. S.; Kumar R. Modeling of Formation of Gold Nanoparticles by Citrate Method. Ind. Eng. Chem. Res. 2007, 46, 3128–3136. 10.1021/ie060672j. [DOI] [Google Scholar]
- Rodríguez-González B.; Mulvaney P.; Liz-Marzán L. M. An Electrochemical Model for Gold Colloid Formation via Citrate Reduction. Z. Phys. Chem. 2007, 221, 415–426. 10.1524/zpch.2007.221.3.415. [DOI] [Google Scholar]
- Kongmark C.; Coulter R.; Cristol S.; Rubbens A.; Pirovano C.; Löfberg A.; Sankar G.; van Beek W.; Bordes-Richard E.; Vannier R.-N. A Comprehensive Scenario of the Crystal Growth of γ-Bi2MoO6 Catalyst during Hydrothermal Synthesis. Cryst. Growth Des. 2012, 12, 5994–6003. 10.1021/cg301070e. [DOI] [Google Scholar]
- Nishimura S.; Dao A. T. N.; Mott D.; Ebitani K.; Maenosono S. X-Ray Absorption Near-Edge Structure and X-Ray Photoelectron Spectroscopy Studies of Interfacial Charge Transfer in Gold–Silver–Gold Double-Shell Nanoparticles. J. Phys. Chem. C 2012, 116, 4511–4516. 10.1021/jp212031h. [DOI] [Google Scholar]
- Polte J.; Ahner T. T.; Delissen F.; Sokolov S.; Emmerling F.; Thünemann A. F.; Kraehnert R. Mechanism of Gold Nanoparticle Formation in the Classical Citrate Synthesis Method Derived from Coupled In Situ XANES and SAXS Evaluation. J. Am. Chem. Soc. 2010, 132, 1296–1301. 10.1021/ja906506j. [DOI] [PubMed] [Google Scholar]
- Plech A.; Kotaidis V.; Siems A.; Sztucki M. Kinetics of the X-Ray Induced Gold Nanoparticle Synthesis. Phys. Chem. Chem. Phys. 2008, 10, 3888–3894. 10.1039/b716599d. [DOI] [PubMed] [Google Scholar]
- Wang C.-H.; Liu C.-J.; Wang C.-L.; Chien C.-C.; Hwu Y.; Liu R.-S.; Yang C.-S.; Je J.-H.; Lin H.-M.; Margaritondo G. Intense X-Ray Induced Formation of Silver Nanoparticles Stabilized by Biocompatible Polymers. Appl. Phys. A 2009, 97, 295–300. 10.1007/s00339-009-5377-x. [DOI] [Google Scholar]
- Martis V.; Nikitenko S.; Sen S.; Sankar G.; van Beek W.; Filinchuk Y.; Snigireva I.; Bras W. Effects of X-Rays on Crystal Nucleation in Lithium Disilicate. Cryst. Growth Des. 2011, 11, 2858–2865. 10.1021/cg200004y. [DOI] [Google Scholar]
- Zabetakis K.; Ghann W. E.; Kumar S.; Daniel M.-C. Effect of High Gold Salt Concentrations on the Size and Polydispersity of Gold Nanoparticles Prepared by an Extended Turkevich–Frens Method. Gold Bull. 2012, 45, 203–211. 10.1007/s13404-012-0069-2. [DOI] [Google Scholar]
- Harada M.; Ikegami R. In Situ Quick X-Ray Absorption Fine Structure and Small-Angle X-Ray Scattering Study of Metal Nanoparticle Growth in Water-in-Oil Microemulsions during Photoreduction. Cryst. Growth Des. 2016, 16, 2860–2873. 10.1021/acs.cgd.6b00173. [DOI] [Google Scholar]
- Nishimura S.; Mott D.; Takagaki A.; Maenosono S.; Ebitani K. Role of Base in the Formation of Silver Nanoparticles Synthesized Using Sodium Acrylate as a Dual Reducing and Encapsulating Agent. Phys. Chem. Chem. Phys. 2011, 13, 9335. 10.1039/c0cp02985h. [DOI] [PubMed] [Google Scholar]
- Henglein A.; Giersig M. Formation of Colloidal Silver Nanoparticles: Capping Action of Citrate. J. Phys. Chem. B 1999, 103, 9533–9539. 10.1021/jp9925334. [DOI] [Google Scholar]
- Abécassis B.; Testard F.; Kong Q.; Francois B.; Spalla O. Influence of Monomer Feeding on a Fast Gold Nanoparticles Synthesis: Time-Resolved XANES and SAXS Experiments. Langmuir 2010, 26, 13847–13854. 10.1021/la1020274. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y.; Nakagawa T.; Seino S.; Kugai J.; Yamamoto T. A.; Nitani H.; Niwa Y. X-Ray-Induced Reduction of Au Ions in an Aqueous Solution in the Presence of Support Materials and in Situ Time-Resolved XANES Measurements. J. Synchrotron Radiat. 2014, 21, 1148–1152. 10.1107/S1600577514012703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkevich J.; Stevenson P. C.; Hillier J. A Study of the Nucleation and Growth Processes in the Synthesis of Colloidal Gold. Discuss. Faraday Soc. 1951, 11, 55–75. 10.1039/df9511100055. [DOI] [Google Scholar]
- Ojea-Jiménez I.; Bastús N. G.; Puntes V. Influence of the Sequence of the Reagents Addition in the Citrate-Mediated Synthesis of Gold Nanoparticles. J. Phys. Chem. C 2011, 115, 15752–15757. 10.1021/jp2017242. [DOI] [Google Scholar]
- Sivaraman S. K.; Kumar S.; Santhanam V. Monodisperse sub-10 nm gold nanoparticles by reversing the order of addition in Turkevich method—The role of chloroauric acid. J. Colloid Interface Sci. 2011, 361, 543–547. 10.1016/j.jcis.2011.06.015. [DOI] [PubMed] [Google Scholar]
- Ojea-Jiménez I.; Campanera J. M. Molecular Modeling of the Reduction Mechanism in the Citrate-Mediated Synthesis of Gold Nanoparticles. J. Phys. Chem. C 2012, 116, 23682–23691. 10.1021/jp305830p. [DOI] [Google Scholar]
- Pillai Z. S.; Kamat P. V. What Factors Control the Size and Shape of Silver Nanoparticles in the Citrate Ion Reduction Method?. J. Phys. Chem. B 2004, 108, 945–951. 10.1021/jp037018r. [DOI] [Google Scholar]
- Floate S.; Hosseini M.; Arshadi M. R.; Ritson D.; Young K. L.; Nichols R. J. An In-Situ Infrared Spectroscopic Study of the Adsorption of Citrate on Au(111) Electrodes. J. Electroanal. Chem. 2003, 542, 67–74. 10.1016/S0022-0728(02)01451-1. [DOI] [Google Scholar]
- Wulandari P.; Li X.; Tamada K.; Hara M. Conformational Study of Citrates Adsorbed on Gold Nanoparticles Using Fourier Transform Infrared Spectroscopy. J. Nonlinear Opt. Phys. Mater. 2008, 17, 185–192. 10.1142/S0218863508004032. [DOI] [Google Scholar]
- Wulandari P.; Nagahiro T.; Fukada N.; Kimura Y.; Niwano M.; Tamada K. Characterization of Citrates on Gold and Silver Nanoparticles. J. Colloid Interface Sci. 2015, 438, 244–248. 10.1016/j.jcis.2014.09.078. [DOI] [PubMed] [Google Scholar]
- Ojea-Jiménez I.; Romero F. M.; Bastús N. G.; Puntes V. Small Gold Nanoparticles Synthesized with Sodium Citrate and Heavy Water: Insights into the Reaction Mechanism. J. Phys. Chem. C 2010, 114, 1800–1804. 10.1021/jp9091305. [DOI] [Google Scholar]
- Doyen M.; Bartik K.; Bruylants G. UV-Vis and NMR study of the formation of gold nanoparticles by citrate reduction: Observation of gold-citrate aggregates. J. Colloid Interface Sci. 2013, 399, 1–5. 10.1016/j.jcis.2013.02.040. [DOI] [PubMed] [Google Scholar]
- Gammons C. H.; Yu Y.; Williams-Jones A. E. The disproportionation of gold(I) chloride complexes at 25 to 200 °C. Geochim. Cosmochim. Acta 1997, 61, 1971–1983. 10.1016/S0016-7037(97)00060-4. [DOI] [Google Scholar]
- Kestner M. O.; Allred A. L. Ligand-Induced Disproportionation of Silver(I). J. Am. Chem. Soc. 1972, 94, 7189. 10.1021/ja00775a074. [DOI] [Google Scholar]
- Seoung D.; Lee Y.; Cynn H.; Park C.; Choi K.-Y.; Blom D. A.; Evans W. J.; Kao C.-C.; Vogt T.; Lee Y. Irreversible Xenon Insertion into a Small-Pore Zeolite at Moderate Pressures and Temperatures. Nat. Chem. 2014, 6, 835–839. 10.1038/nchem.1997. [DOI] [PubMed] [Google Scholar]
- Mazej Z.; Michałowski T.; Goreshnik E. A.; Jagličić Z.; Arčon I.; Szydłowska J.; Grochala W. The First Example of a Mixed Valence Ternary Compound of Silver with Random Distribution of Ag(i) and Ag(Ii) Cations. Dalton Trans. 2015, 44, 10957–10968. 10.1039/C5DT00740B. [DOI] [PubMed] [Google Scholar]
- Harada M.; Inada Y.; Nomura M. In Situ Time-Resolved XAFS Analysis of Silver Particle Formation by Photoreduction in Polymer Solutions. J. Colloid Interface Sci. 2009, 337, 427–438. 10.1016/j.jcis.2009.05.035. [DOI] [PubMed] [Google Scholar]
- Mehta S. K.; Chaudhary S.; Gradzielski M. Time Dependence of Nucleation and Growth of Silver Nanoparticles Generated by Sugar Reduction in Micellar Media. J. Colloid Interface Sci. 2010, 343, 447–453. 10.1016/j.jcis.2009.11.053. [DOI] [PubMed] [Google Scholar]
- Zaheer Z.; Rafiuddin Nucleation and Growth Kinetics of Silver Nanoparticles Prepared by Glutamic Acid in Micellar Media. Int. J. Chem. Kinet. 2012, 44, 680–691. 10.1002/kin.20711. [DOI] [Google Scholar]
- Barbooti M. M.; Al-Sammerrai D. A. Thermal Decomposition of Citric Acid. Thermochim. Acta 1986, 98, 119–126. 10.1016/0040-6031(86)87081-2. [DOI] [Google Scholar]
- Anh D. T. N.; Singh P.; Shankar C.; Mott D.; Maenosono S. Charge-Transfer-Induced Suppression of Galvanic Replacement and Synthesis of (Au@Ag)@Au Double Shell Nanoparticles for Highly Uniform, Robust and Sensitive Bioprobes. Appl. Phys. Lett. 2011, 99, 073107. 10.1063/1.3626031. [DOI] [Google Scholar]
- Forbes G. S. The Solubility of Silver Chloride in Chloride Solutions and the Existence of Complex Argentichloride Ions. J. Am. Chem. Soc. 1911, 33, 1937–1946. 10.1021/ja02225a008. [DOI] [Google Scholar]
- Mikhlin Y.; Karacharov A.; Likhatski M.; Podlipskaya T.; Zubavichus Y.; Veligzhanin A.; Zaikovski V. Submicrometer Intermediates in the Citrate Synthesis of Gold Nanoparticles: New Insights into the Nucleation and Crystal Growth Mechanisms. J. Colloid Interface Sci. 2011, 362, 330–336. 10.1016/j.jcis.2011.06.077. [DOI] [PubMed] [Google Scholar]
- Chow M. K.; Zukoski C. F. Gold Sol Formation Mechanisms: Role of Colloidal Stability. J. Colloid Interface Sci. 1994, 165, 97–109. 10.1006/jcis.1994.1210. [DOI] [Google Scholar]
- Sinzig J.; Quinten M. Scattering and Absorption by Spherical Multilayer Particles. Appl. Phys. A: Solids Surf. 1994, 58, 157–162. 10.1007/BF00332172. [DOI] [Google Scholar]
- Dent A. J.; Cibin G.; Ramos S.; Smith A. D.; Scott S. M.; Varandas L.; Pearson M. R.; Krumpa N. A.; Jones C. P.; Robbins P. E. B18: A Core XAS Spectroscopy Beamline for Diamond. J. Phys.: Conf. Ser. 2009, 190, 012039. 10.1088/1742-6596/190/1/012039. [DOI] [Google Scholar]
- Ravel B.; Newville M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.