

Abstract
Six sesquiterpenoids with unprecedented macrocyclic humulene-type structures, namely, dolichocarpols A-E (1–5) with ether-bridged bicyclic rings between C-10 and C-2, C-10 and C-7, C-10 and C-6 (×2), and C-6 and C-3 and dolichocarpol F (6) cyclized between C-7 and C-2 and with an ether bridge between C-10 and C-3, were isolated from Anaxagorea dolichocarpa roots. The structures of the compounds were elucidated by NMR, MS, and IR data. Absolute configurations of compounds 1–3 and 6 were established on the basis of data from electronic circular dichroism, whereas relative configurations of compounds 4 and 5 were suggested by the NOESY spectrum. In addition, a putative biosynthetic pathway of the compounds is proposed. Furthermore, the cytotoxicity of 3, 4, and 6 against HCT-116 (human colorectal carcinoma) and L929 (murine fibroblast) was evaluated.
Introduction
Sesquiterpenoids are considered to be the most diverse group of terpenoids containing a large variety of chemical structures that present different forms of derivation and cyclization from farnesyl diphosphate (FPP).1,2 The humulene-type, an 11-membered macrocyclic sesquiterpenoid, is broadly found in plants, and previous reports have demonstrated various kinds of biological activities such as antitumor, anti-inflammatory, antibacterial, antiviral, and other bioactivities.3−5
The genus Anaxagorea (Annonaceae) comprises 30 species distributed in the neotropics and tropical Asia.6 Previous phytochemical studies with plants of this genus revealed the presence of xanthones, flavonoids,7−9 neolignans,10 terpenoids, and alkaloids,11−13 with some of them exhibiting remarkable biological activities. The analysis of the essential oil from the leaves of Anaxagorea brevipes has revealed the presence of mono- and sesquiterpenes such as α-, β-, and γ-eudesmol, guaiol, and caryophyllene oxide. In addition, its biological evaluation showed antimicrobial activity against Gram-positive bacteria and yeast and antiproliferative activity against breast, lung, and prostate cancer cell lines.11 An interesting sesquiterpene called nordine besides copyrine alkaloids has been isolated from A. javanica, and some of these compounds have been assayed for nitric oxide (NO) inhibition.13A. dolichocarpa, widely distributed in South America,14 is found in Brazil mainly in the North and Northeast regions.15 Previous works exploring different parts of this plant reported the isolation of aporphine and azaphenanthrene alkaloids,16,17 the identification of mono- and sesquiterpenes,12,18 the phenolic quantification and determination of its antioxidant activity,19 and a phytochemical screening and cytotoxicity evaluation of its extract.20
The gradual emergence of active agents derived from the plant kingdom into the fight against cancer has gained importance in the last decade.21 These compounds have the potential to be harnessed for preventative strategies, standalone treatments, or complementary therapeutics in combination with traditional pharmacological interventions.22 In addition, the literature has shown exciting data with respect to potential of natural compounds and their analogues in cancer prevention and treatment.23
In a continuing effort to isolate active compounds from plants in Brazil’s semiarid area and motivated by the promising bioactivities and diversity of sesquiterpenoid structures, the ethanol extract of A. dolichocarpa roots was investigated. Thus, we describe here the isolation and structural elucidation of six humulene-type sesquiterpenoids (1–6). Furthermore, compounds 3, 4, and 6 were tested for cytotoxicity.
Results and Discussion
The roots of A. dolichocarpa were dried and extracted with ethanol three times at room temperature. The ethanolic extract was suspended in MeOH-H2O (7:3) and sequentially partitioned with n-hexane, chloroform, and ethyl acetate (EtOAc). The n-hexane- and chloroform-soluble phases were fractionated by silica gel chromatography column and medium-pressure liquid chromatography, respectively. The obtained fractions were then purified by preparative HPLC to yield the unprecedented macrocyclic humulene-type sesquiterpenoids dolichocarpols A (1) and E (5) from the hexane phase and dolichocarpols B-D (1–4) and F (6) from the chloroform phase. The structures were proposed after 1D- and 2D-NMR, IR, HRESIMS, ESIMS/MS, and experimental and calculated electronic circular dichroism (ECD) data analysis.
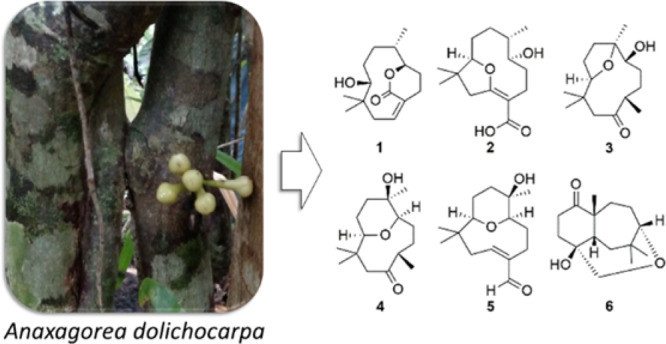
Compound 1 was isolated as a colorless oil. Its IR spectrum showed a hydroxyl group (3466 cm–1) and a carbonyl group (1706 cm–1). The molecular formula was determined as C15H24O3 by HRESIMS based on the ion at m/z 275.1614 [M + Na]+ (calcd for C15H24O3Na, 275.1618, Δ = 1.2 ppm), corresponding to four indices of hydrogen deficiency. Of these, the 13C NMR and DEPT spectra of 1 showed that one was associated with an olefinic group (δC 134.3, 133.2) and another with a carbonyl group (δC 168.8). Additionally, the spectrum showed signals assigned to two oxygenated carbons (δC 83.2, 82.3), one non-hydrogenated carbon (δC 39.3), one methine carbon (δC 37.7), five methylene carbons (δC 40.2, 31.7, 28.9, 27.3, 22.8), and three methyl carbons (δC 25.9, 22.0, 20.4) (Table 1). In the 1H NMR spectrum, methyl groups could be inferred from two signals for tertiary methyl protons at δH 1.08 (s) and 0.82 (s) and one secondary methyl at 0.81 (d, J = 6.4 Hz), an olefinic proton at δH 5.84 (m), and two oxymethine proton at δH 4.58 (ddd, J = 9.2, 6.0, 2.0 Hz) and 3.10 (dl, J = 8.4 Hz) (Table 2). The comparison of these 1D NMR spectral data with those of nordine isolated from A. javanica(13) suggested that compound 1 was a macrocyclic humulene-type sesquiterpenoid as shown in Chart 1. In the HMBC spectrum, the correlations observed from H-10 to C-14, C-15, C-1, C-8, and C-9 and from H2-1 to C-2 and C-3 localized a Δ2(3) olefinic double bond (Figure 1). Moreover, correlations from H-6 to C-7, C-8, C-13, and C-12 and from H-2 and H2-4 to C-3 and C-12 in combination with COSY correlations between H-8, H-7, and H-6 indicated an unusual ester fusion between C-6 and C-3. Also, in the COSY spectrum, the correlations between H2-1, H-2 and H2-4, H2-5 established two pairs of allylic protons to C-2 and C-3. The ESIMS/MS spectrum of 1 exhibited ion fragments at m/z 257 [M - H2O + Na]+ and 217 [M - C3H6O + Na]+. The relative configuration was deduced from NOESY experiments. The correlation between H-10, H3-14, and H-2 indicated that these protons are cofacial adopting an α-orientation and assigning a β-orientation of OH-10 (Figure 2). In addition, the correlation from H-2 to H3-14 suggested the E-configuration of the Δ2(3) double bond, which is in agreement with an X-ray study that assigns the E-configuration to the Δ2(3) double bond of the humulene skeleton.24,25 The negative specific rotation ([α]D25 −41, CHCl3) when compared with other humulenes suggests the α-orientation to H3-13 in this type of sesquiterpenoid, and the NOESY cross-peak from H3-13 to H-6 revealed that they are cofacial with an α-orientation.26 The ECD experimental spectra showed a negative Cotton effect at λ 234 nm (Δε −0.89) and a positive one at λ 255 nm (Δε +0.43); thus, the comparison of these with the calculated data allowed determination of the absolute configuration of 1 as (6R,7S,10R) (Figure 3). According to these lines of evidence, the structure of 1 was defined as humulene-type sesquiterpenoid named dolichocarpol A.
Table 1. 13C (100 and 125 MHz) NMR Spectroscopic Data for Compounds 1–6 in CDCl3.
| no | 1 | 2a | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 40.2 | 45.3 | 47.1 | 55.4 | 39.4 | 33.6 |
| 2 | 134.3 | 173.0b | 213.5 | 213.2 | 152.8 | 52.7 |
| 3 | 133.2 | 101.8b | 47.0 | 49.4 | 147.8 | 74.1 |
| 4 | 28.9 | 22.2 | 30.6 | 32.2 | 18.1 | 33.0 |
| 5 | 22.8 | 33.6 | 37.5 | 26.4 | 26.3 | 32.9 |
| 6 | 82.3 | 67.5 | 75.5 | 84.6 | 83.5 | 215.9 |
| 7 | 37.7 | 31.7 | 84.0 | 69.4 | 69.9 | 50.5 |
| 8 | 31.7 | 32.2 | 27.7 | 32.8 | 34.3 | 29.9 |
| 9 | 27.3 | 26.1 | 29.8 | 20.9 | 20.6 | 23.7 |
| 10 | 83.2 | 92.3 | 85.6 | 73.4 | 72.7 | 82.6 |
| 11 | 39.3 | 40.0 | 39.6 | 39.9 | 36.1 | 39.5 |
| 12 | 168.8 | 170.6b | 19.3 | 20.7 | 195.7 | 68.3 |
| 13 | 20.4 | 14.2 | 20.9 | 24.7 | 23.7 | 30.6 |
| 14 | 25.9 | 29.2 | 27.7 | 27.3 | 23.7 | 33.2 |
| 15 | 22.0 | 21.9 | 27.6 | 21.1 | 22.2 | 29.2 |
13C NMR (125 MHz).
13C NMR (200 MHz).
Table 2. 1H (400 and 500 MHz) NMR Spectroscopic Data for Compounds 1–6 in CDCl3.
| no | 1 | 2a | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 2.88, td (13.6, 0.8) | 2.97, d (19.5) | 2.52, d (12.4) | 2.34, dd (10.8) | 3.12, t (11.2) | 1.91, dd (16.0, 4.8) |
| 1.96, m | 2.76, d (19.5) | 1.84, d (12.4) | 1.99, dd (10.8) | 1.88, dd (11.2, 7.2) | 1.66, dd (16.0, 2.8) | |
| 2 | 5.84, m | 6.52, dd (10.8, 7.2) | 2.29, m | |||
| 3 | 2.74, m | 2.71, dd (9.6, 6.8) | ||||
| 4 | 2.51, m | 2.39, m | 1.89, m | 1.86, m | 2.61, dd (10.8, 1.2) | 2.01, dt (13.2, 4.0) |
| 2.51, m | 2.39, m | 1.37, m | 1.49, m | 2.20, dd (13.2, 8.0) | 1.74, ddd (13.6, 10.8, 5.6) | |
| 5 | 2.18, m | 1.62, m | 2.05, m | 1.77, m | 1.85, m | 2.80, ddd (16.4, 13.2, 5.2) |
| 1.99, m | 1.62, m | 1.62, m | 1.53, m | 1.52, m | 2.29, m | |
| 6 | 4.58, ddd (9.2, 6.0, 2.0) | 3.57, dd (11.0, 4.0) | 3.50, dd (5.2, 2.4) | 3.52, m | 3.58, dd (12.4, 5.6) | |
| 7 | 2.22, m | 2.56, m | ||||
| 8 | 1.85, m | 1.57, m | 1.73, m | 1.69, m | 1.69, m | 2.29, m |
| 1.34, m | 1.57, m | 1.62, m | 1.48, m | 1.56, m | 1.45, ddd (14.8, 12.4, 7.6) | |
| 9 | 1.34, m | 1.57, m | 2.09, m | 1.53, m | 1.49, m | 1.93–1.91, m |
| 1.19, m | 1.45 | 1.21, m | 1.40, m | 1.30, m | ||
| 10 | 3.10, dl (8.4) | 4.07, d (12.0) | 3.48, dd (6.8, 4.0) | 3.68, m | 2.90, dd (11.2, 1.6) | 3.52, dd (6.4, 2,4) |
| 11 | ||||||
| 12 | 0.96, d (7.2) | 0.96, d (6.8) | 9.37, d (0.8) | 3.66, d (14.0) | ||
| 3.26, dd (14.0, 1.6) | ||||||
| 13 | 0.81, d (6.4) | 0.84, d (7.0) | 1.09, s | 0.99, s | 0.98, s | 1.09, s |
| 14 | 1.08, s | 1.09, s | 1.24, s | 0.91, s | 0.84, s | 1.23, s |
| 15 | 0.82, s | 1.07, s | 0.85, s | 1.03, s | 1.00, s | 1.05, s |
1H NMR (500 MHz).
Chart 1. Chemical Structures of Isolated Compounds 1–6 from Roots of A. dolichocarpa.

Figure 1.
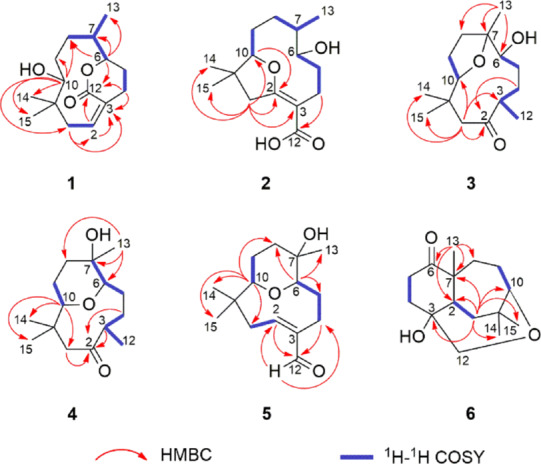
Key HMBC and 1H-1H COSY correlations of compounds 1–6.
Figure 2.

Key 1H-1H NOESY correlations of compounds 1–6.
Figure 3.
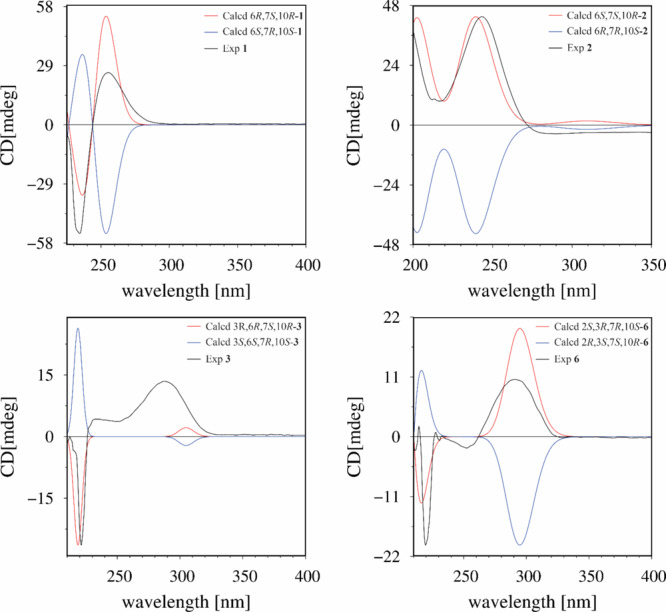
Experimental and calculated ECD spectra of compounds 1–3 and 6.
Compound 2 was obtained as a colorless oil. Its IR spectrum showed a hydroxyl group (3401 cm–1) and a carbonyl group (1671 cm–1). The molecular formula was established as C15H24O4 by HRESIMS m/z 291.1566 [M + Na]+ (calcd for C15H24O4Na, 291.1567, Δ = 0.1 ppm), indicating four indices of hydrogen deficiency. The 1H NMR spectrum of 2 was similar to that of 1 except for the absence of an olefinic proton, thus still having three methyl groups and five methylene groups besides two oxymethine protons and one methine proton (Table 2). The 13C NMR spectrum (125 MHz/CDCl3) exhibited 12 carbon resonances, corresponding to two oxygenated carbons (δC 92.3, 67.5), one non-hydrogenated carbon (δC 40.0), one methine carbon (δC 31.7), five methylene carbons (δC 45.3, 33.6, 32.2, 26.1, 22.2), and three methyl carbons (δC 29.2, 21.9, 14.2) (Table 1). Other three non-hydrogenated carbons (δC 173.0, 170.6, 101.8) were detected in the 200 MHz (CDCl3) 13C NMR spectrum (Figure S21, Supporting Information). The HSQC spectrum revealed that the chemical shifts at δC 173.0, 101.8, and 170.6 were observed instead of those at δC 134.3, 133.2, and 168.8 assigned to C-2, C-3, and C-12 of compound 1, respectively, and a double bond with the ether bridge between C-10 and C-2. In the 1H and 13C NMR spectra of 2 (Tables 1 and 2), the upfield chemical shifts observed for H-10 and C-10 and downfield chemical shifts of H-6 and C-6 supported the opening of the ester bridge C-6 and C-3 to form a C-6 hydroxyl. Similarly, the absence of H-2 and C-2 and downfield chemical shifts for C-2, C-3, and C-12 suggested an ether bridge between C-10 and C-2. In the HMBC spectrum, the correlation from H2-1 to C-15, C-14, C-10, C-3, and C-2, together with the correlation from H-10 to C-2 and from H2-4 to C-12, corroborated the connectivity between C-10 and C-2 (Figure 1). In the 1H–1H COSY spectrum of 2, as expected, the correlation H-2/H2-1 was not displayed, leaving those between H-10 and H2-9, between H-8, H-7, and H3-13, and between H-6, H2-5, and H2-4. The ESIMS/MS spectrum of 2 exhibited ion fragments at m/z 251 [M - H2O + H]+, m/z 233 [M - 2H2O + H]+, 215 [M - 3H2O + H] +, and 205 [M - H2O – HCOOH + H]+ compatible with a carboxyl group. The insertion of this group at C-3 is compatible with the chemical shift attributed to C-2. The NOESY correlation observed between H-10 and H3-14 indicated an α-orientation to these protons (Figure 2). The multiplicity of the H-6 signal (dd) together with the absence of the 1H–1H COSY correlation with H-7 suggested that the dihedral angle between these protons is close to 90°. Thus, considering this evidence, the NOESY correlation between H-6 and H-7 indicated that these protons are cofacial with the β-orientation, so OH-6 and H3-13 adopted an α-orientation, with a negative specific rotation ([α]D25 −15, CHCl3). The (6S,7S,10R) absolute configuration of 2 was determined by ECD experimental, with positive Cotton effects at λ 201 nm (Δε +1.55) and λ 243 nm (Δε +1.92), and calculated data (Figure 3). Therefore, the structure of compound 2, named dolichocarpol B, was established as shown.
Compound 3 was also purified as a colorless oil. The IR spectrum showed a hydroxyl group (3458 cm–1) and a carbonyl group (1703 cm–1). The molecular formula of dolichocarpol C (3) was deduced as C15H26O3 by HRESIMS at m/z 277.1773 [M + Na]+ (calcd for C15H26O3Na, 277.1774, Δ = 0.3 ppm), differing from compound 1 by the absence of the double bond. The 1D NMR data of compound 3 were similar to those of 1 and 2 but with an additional methyl carbon (δC 19.3) and a ketone carbonyl group (δC 213.5) instead of carbonyl ester or acid at C-3 (Tables 1 and 2). In the COSY spectrum, the combination of vicinal homonuclear coupling correlations observed between H-3, H2-4, and H3-12, between H-10 and H-9, and between H-6 and H2-5 indicated an alteration in the junction of the carbon skeleton, compatible with a new bicyclic fusion, and defined the chemical shifts of the methine proton H-3 and oxymethine protons H-10 and H-6 (Figure 1). The HMBC cross-peak correlation from H2-1 to C-15, C-14, C-10, and C-3 and from H3-12, H2-4, and H2-1 to C-2 established that the ether bridge in 3 continued through C-10 but unlike the previous compounds. Additionally, the correlation from H3-13 to C-8, C-7, and C-6 defined the ether bridge between C-10 and C-7. Compound 3 was acetylated, and only one acetate was obtained at C-6 (δH 4.61, dd, J = 5.2, 2.4, H-6). The HMBC spectrum of acetylated 3 (Figure S21, Supporting Information) showed correlations from H-6 to C-13, C-8, C-7, C-5, and C-4. This finding confirmed the bridge between C-10 and C-7, and the chemical shift at δC 84.0 was unequivocally assigned to C-7. The ESIMS/MS spectrum exhibited ion fragments at m/z 237 [M - H2O + H]+, 219 [M - 2H2O + H]+, and 201 [M - 3H2O + H]+. The analysis of specific rotation ([α]D25 −20, CHCl3), 1H NMR, and NOESY spectra of 3 recorded in both chloroform and pyridine defined the correlations between H-3, H-6, and H3-13 and between H-10 and H3-14, all with the same α-orientation, while H3-12 and OH-6 adopted a β-orientation (Figure 2). The absolute configuration was established as (3R,6R,7S,10R) by comparing a negative Cotton effect at λ 221 nm (Δε −0.22) and a positive one at λ 287 nm (Δε +0.11) from experimental ECD with the calculated spectra. In view of the spectral data, including those of the acetylated derivative, the structure of 3 was determined as described and named dolichocarpol C.
Compound 4 was isolated as a colorless oil. Its IR spectrum showed a hydroxyl group (3461 cm–1) and a carbonyl group (1705 cm–1). The C15H26O3 molecular formula was determined by HRESIMS data, m/z 277.1772 [M + Na]+ (calcd for C15H26O3Na, 277.1774, Δ = 0.6 ppm). The 1H and 13C NMR data of 4 (Tables 1 and 2) were similar to those of 3, including a carbonyl (δC 213.2) and two oxymethine carbons (δC 73.4, 84.6). However, a significant alteration occurred at the quaternary carbinolic carbon C-7 (δC 69.4, Δ = +14.4 ppm). The HMBC spectrum displayed identical correlations from H2-1, H-3, and H-4 to C-2, as in 3 (Figure 1). The cross-peaks from H-10 to C-15, C-14, and C-1 and from H3-13 to C-8, C-7, and C-6 combined with vicinal homonuclear coupling correlations in COSY between H-10 and H2-9, between H-7, H-6, and H2-5, and between H-3, H2-4, and H3-12 suggested another type of fusion in the carbon skeleton of 4. Analysis of the HSQC spectrum allowed an unequivocal establishment of the chemical shifts of C-10 (δC 73.4) and C-6 (δC 84.6). Thus, the changes in the chemical shifts were compatible with an ether bridge between C-10 and C-6 instead of C-10 and C-7 (Tables 1 and 2). As expected, compound 4 could not be acetylated. The ESIMS/MS spectrum of 4 exhibited ion fragments at m/z 237 [M - H2O + H]+, 219 [M - 2H2O + H]+, and 201 [M - 3H2O + H]+. The negative specific rotation ([α]D25 −12, CHCl3) and the NOESY correlations observed from H-10 to H3-14 and H-3 and from H-14 to H-1α suggest that these protons are cofacial in the α-orientation, whereas the correlations from H3-15 to H-1β indicate a β-orientation (Figure 2). Furthermore, the correlation between H-6 and H3-13 accompanied by previous data from the other compounds suggested the α-orientation for these protons and revealed the relative configuration shown in Figure 2. Thus, the structure of compound 4 was suggested as shown and named dolichocarpol D.
Compound 5 was obtained as a colorless oil. Its IR spectrum showed a hydroxyl group (3321 cm–1) and a carbonyl group (1659 cm–1). The 1D NMR data of compound 5 (Tables 1 and 2) were similar to those of compounds 1, 2, and 4, altered by the presence of the formyl proton (δH 9.37, d, J = 0.8), as confirmed by HRESIMS at m/z 275.1622 [M + Na]+ (calcd for C15H24O3Na, 275.1618, Δ = −1.6 ppm), consistent with a molecular formula of C15H24O3. The general characteristics observed in the 1H and 13C NMR spectra of 5 were parallel to those of the previous compounds. 2D NMR–HSQC analysis confirmed the chemical shifts for H-10 (δH 2.90, dd, J = 11.2, 1.6 Hz)/C-10 (δC 72.7), H-6 (δH 3.58, dd, J = 12.4, 5.6 Hz)/C-6 δC 83.5), and H-2 (δH 6.52, dd, J = 10.8, 7.2 Hz)/C-2 (δC 152.8). Thus, the ether bridge between C-10 and C-6 was maintained. The localization of the double bond in carbons C-2 and C-3 was defined by HMBC, through the identical sequence of the correlations from H-2 and H-4b to C-12 and from H-12 to C-2 and C-4, like in compound 1 (Figure 1). In addition, the cross-peaks from H-10 to C-15, C-14, C-8, and C-1 and from H-6 to C-13, C-8, and C-5 corroborated ether bridges C-10 and C-6. The 1H–1H COSY spectrum exhibited the vicinal coupling between H-2 and H2-1, between H-10 and H-9, and between H-6, H-5, and H-4. The negative specific rotation ([α]D25 −9, CHCl3) and NOESY spectrum showed that the α-orientation was maintained for H-2, H-10, and H3-14 as well as for H3-13 and H-6, and the relative configuration is shown in Figure 2. From the analysis of the spectral data and comparison with the analogous compounds, the structure of 5 was proposed and named dolichocarpol E.
Compound 6 was isolated as a light brown oil. The IR spectrum showed a hydroxyl group (3444 cm–1) and a carbonyl group (1701 cm–1). Its molecular formula was determined as C15H24O3 by HRESIMS analysis on the basis of ion m/z 275.1616 [M + Na]+ (calcd for C15H24O3Na, 275.1618, Δ = 0.8 ppm), corresponding to four indices of hydrogen deficiency. Unlike for compounds 1–5, the 1H NMR spectrum of 6 exhibited only one oxymethine proton at δH 3.52 (dd, J = 6.4, 2.4 Hz) and in addition two oxymethylene protons at δH 3.66 (d, J = 14.0 Hz) and 3.26 (dd, J = 14.0, 1.6 Hz). Besides these, there were one methine proton at δH 2.29 (m) and three methyl protons at δH 1.23 (s), δH 1.09 (s), and 1.05 (s) (Table 1). The 13C NMR and DEPT spectra showed a carbonyl group (δC 215.9), three non-hydrogenated carbons including one oxygenated (δC 74.1, 50.5, 39.5), methylene carbons (δC 33.6, 33.0, 32.9, 29.9, 23.7), and methyl carbons (δC 33.2, 30.6, 29.2). The cross-peaks in HMBC from H3-13 to C-8, C-7, C-6, and C-2 suggested a C-C fusion not found in other sesquiterpenoids (1–5) (Figure 1). Another correlation equally important from H2-1 to C-15, C-14, C-10, C-7, C-3, and C-2 defined a new fusion between C-7 and C-2. The HSQC spectrum exhibited the correlation from H3-13 to C-13, indicating its insertion in the quaternary carbon C-7 (δC 50.5), neighboring the carbonyl carbon. Likewise, the HMBC correlation from H2-12 to C-10, C-3, and C-2, combined with the sequence of correlations of vicinal homonuclear coupling in COSY between H-10 and H2-9, between H2-1 and H-2, and between H2-4 and H2-5, established the ether bridge between C-10 and C-3 via methylene carbon C-12. The ESIMS/MS spectrum showed an ion fragment at m/z 215 [M - C2H4O2 + Na]+. The positive specific rotation ([α]D25 +10, CHCl3) indicates a different orientation for H3-13 from those of the compounds shown previously. Furthermore, the NOESY correlation between H-2, H3-13, H-8β, H3-15, and H-1β suggested that all the protons were cofacial, thus assigned to the β-orientation (Figure 2). The Cotton effects observed at λ 219 nm (Δε −0.18) and at λ 291 nm (Δε +0.09) allowed establishment of the absolute configuration of 6 as (2S,3R,7R,10S) by the comparison of experimental ECD with the calculated data (Figure 3). On the basis of these findings, the structure of compound 6 was defined as a new humulene-type sesquiterpene, named dolichocarpol F.
A putative biosynthetic pathway toward compounds 1–6 is shown in Figure 4. Starting from 10-hydroxyhumula-2E,6E-diene, oxidations occurred producing the hydroxylated intermediate at C-10 and also the epoxidized intermediate at the double bonds Δ2,3 and Δ6,7. Through successive oxidations and different fusions and cyclizations, compounds 1–6 were produced.
Figure 4.

Putative biosynthetic pathway toward compounds 1–6.
The cytotoxicity of the selected sesquiterpenoids (3, 4, and 6) against HCT-116 and L929 cell lines was evaluated. Compound 3 significantly inhibited the proliferation of HCT-116 human colon cancer cells at a concentration of 50 μM with an inhibitory rate of 26.63 ± 1.21% (Figure 5). No selectivity was observed for 3 toward tumor cells, considering the inhibitory rate against the L929 murine fibroblast nontumor cell line (27.91 ± 0.20%). Compounds 4 and 6 showed weak cytotoxic effects against HCT-116 cells with inhibitory rates under 20%. Moreover, compound 4 exhibited the highest cytotoxicity against L929 nontumor cells (30.76 ± 3.52%).
Figure 5.
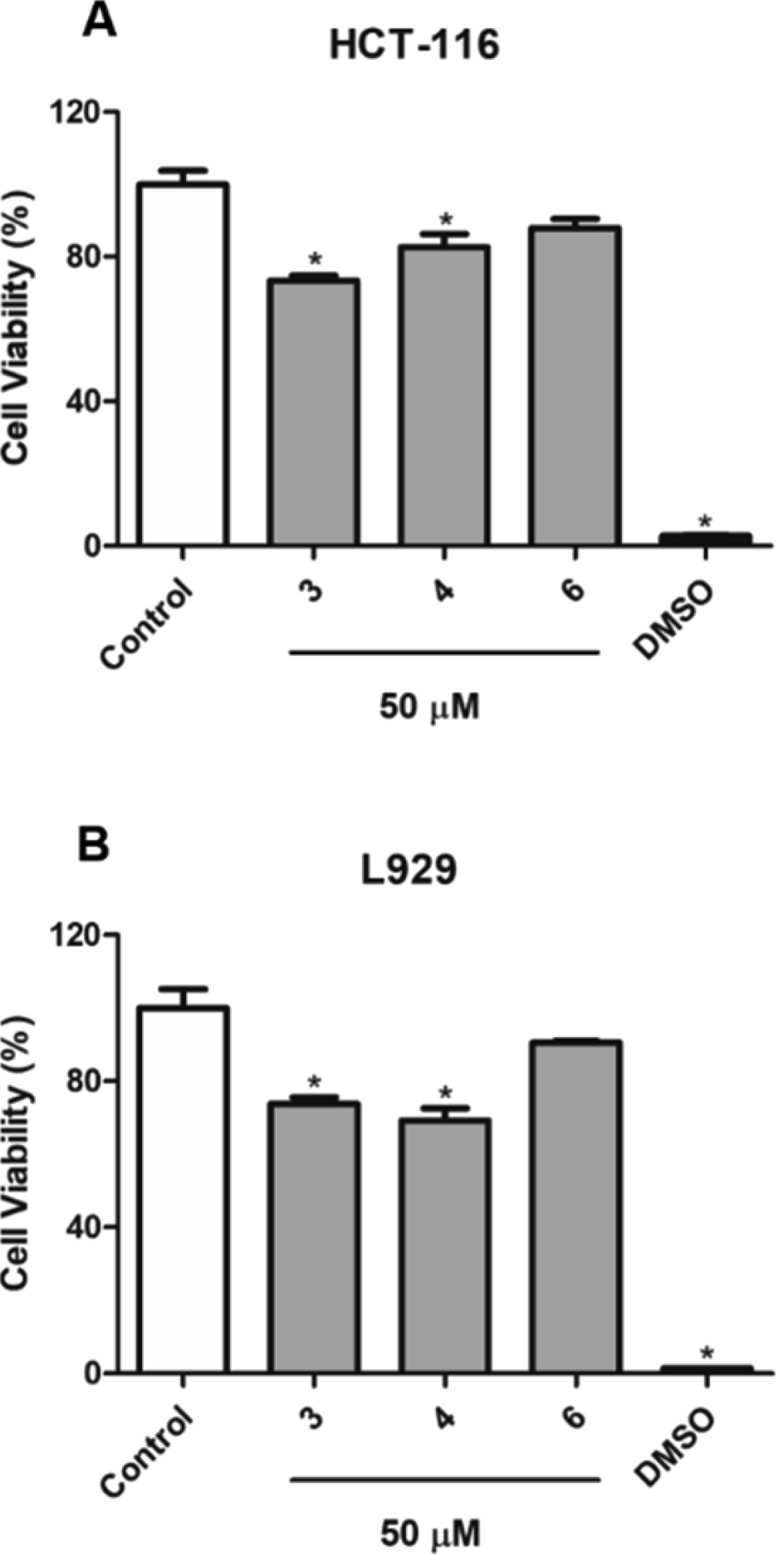
Cytotoxicity of compounds 3, 4, and 6 against HCT-116 (A) and L929 (B) cells lines, after 72 h of treatment. The values are presented as mean ± standard error for four independent replicates at a concentration of 50 μM (*p < 0.05 compared with untreated cells).
Experimental Section
General Experiment Procedures
Optical rotations were measured on a Jasco P-2000 polarimeter (Easton, MD, USA) at 25 ° C in CHCl3. FTIR spectra were acquired on Bruker Vertex 70 (Bruker, Billerica, MA, USA) and PerkinElmer Frontier (Waltham, MA, USA) spectrometers. 1D- and 2D-NMR spectra were recorded on a Bruker AVANCE III HD 800 MHz spectrometer (800 and 200 MHz for 1H and 13C, respectively), Varian NMR spectrometer (500 and 125 MHz for 1H and 13C, respectively), and Bruker AVANCE III HD spectrometer (400 and 100 MHz for 1H and 13C, respectively) using CHCl3 (δH 7.24 and δC 77.0), the residual solvent, as an internal standard. High-resolution electrospray ionization mass spectrometry (HRESIMS) and tandem (MSn) analyses were carried out using Bruker spectrometers of models micrOTOFII and Ion Trap-amaZonX (Billerica, MA, USA), respectively, both operating in the positive mode. Column chromatography (CC) was performed on a silica gel 60 (60–200 μm, 70–230 mesh, SiliaCycle, Quebec, Canada), and medium-pressure liquid chromatography (MPLC) was performed on a silica gel 60 (40–63 μm, 230–400 mesh, SiliCycle). Thin-layer chromatography (TLC) was carried out using precoated silica gel F-254 aluminum sheets (SiliCycle), and the compound spots were observed under UV light at 254 and 366 nm, staining with iodine vapor. Analytical high-performance liquid chromatography (HPLC) was performed on a Prominence Shimadzu instrument equipped with a SPD-M20A diode array detector and a reversed-phase Phenomenex Gemini C18 column (250 mm × 4.6 mm ID filled with 5 μm particles). For preparative HPLC, a Shimadzu apparatus with an SPD-M10A VP diode array detector and an ACE C18 column (250 mm × 21.2 mm and 5 μm particles) was used with a flow rate of 8.0 mL/min.
Plant Material
The roots of A. dolichocarpa were collected from Cruz do Espírito Santo, Paraíba, Brazil (7°09′43.7″S, 35°02′11.1″W) on April 2018. Access registration in the National Management System of Genetic Patrimony and Associated Traditional Knowledge (SISGEN) was obtained under number AE4B71A. A voucher specimen (AGRA & GÓES 5543) was identified by M. F. Agra and deposited at Herbarium Prof. Lauro Pires Xavier (JPB), Federal University of Paraiba (UFPB), Brazil.
Extraction and Isolation
The air-dried and powdered roots of A. dolichocarpa (1.72 kg) were extracted with ethanol for 72 h (3 × 4 L) at room temperature. The extract was concentrated under reduced pressure at 40 ° C to afford 52.6 g of crude extract (CE). Part of CE (49.3 g) was suspended in MeOH-H2O (7:3) and sequentially partitioned with hexane, chloroform, and EtOAc. An aliquot of the hexane-soluble portion (8.12 g) was subjected to CC over silica gel, eluting with a gradient of hexane-EtOAc and EtOAc-MeOH, which after TLC analysis were grouped to give the fractions H-1 to H-6. Fraction H-2 (95.0 mg) was subjected to preparative HPLC with the following gradient elution: solvent A = H2O; solvent B = MeOH; elution system = 0–40 min (20–70% B); 40–50 min (70% B); and 50–65 min (70–100% B), yielding compound 5 (2.0 mg). Fraction H-4 (85.8 mg) was also subjected to preparative HPLC using gradient elution with H2O–MeOH 5 to 100% of MeOH in 75 min to afford compound 1 (1.0 mg). The chloroform-soluble fraction (6.70 g) was separated by MPLC over silica gel using a gradient elution with hexane–EtOAc and EtOAc–MeOH, and the fractions obtained were combined according to the TLC profile to afford C1–C4. The fraction C-2 (197.3 mg) was purified by preparative HPLC with the following elution gradient: solvent A = H2O; solvent B = MeOH; elution system = 0–45 min (0–65% B); 45–70 min (65% B); and 70–105 min (65–100% B). This resulted in compounds 2 (0.5 mg), 3 (12.1 mg), 4 (6.8 mg), and 6 (6.0 mg).
Dolichocarpol A (1): colorless oil; [α]D25 −41 (c 0.1, CHCl3); ECD (CHCl3) λ (Δε) 234 (−0.89), 255 (+0.43); IR (film) νmax 3466, 2955, 2931, 2868, 1706, 1468, 1402, 1213, 1104 cm–1; 1H and 13C NMR data, see Tables 1 and 2; positive-ion HRESIMS m/z 275.1614 [M + Na]+ (calcd for C15H24O3Na, 275.1618, Δ = 1.2 ppm); positive-ion ESIMS/MS fragments m/z 257, 217.
Dolichocarpol B (2): colorless oil; [α]D25 −15 (c 0.1, CHCl3); ECD (CHCl3) λ (Δε) 201 (+1.55), 243 (+1.92); IR (film) νmax 3401, 2958, 2923, 2869, 1703, 1459, 1366, 1214, 1082, 1047 cm–1; 1H and 13C NMR data, see Tables 1 and 2; positive-ion HRESIMS m/z 291.1566 [M + Na]+ (calcd for C15H24O4Na, 291.1567, Δ = 0.1 ppm); positive-ion ESIMS/MS fragments m/z 251, 233, 215, 205.
Dolichocarpol C (3): colorless oil; [α]D25 −20 (c 0.1, CHCl3); ECD (CHCl3) λ (Δε) 221 (−0.22), 287 (+0.11); IR (film) νmax 3458, 2956, 2924, 2873, 1703, 1458, 1367, 1215, 1048 cm–1; 1H and 13C NMR data, see Tables 1 and 2; positive-ion HRESIMS m/z 277.1773 [M + Na]+ (calcd for C15H26O3Na, 277.1774, Δ = 0.3 ppm); positive-ion ESIMS/MS fragments m/z 237, 219, 201.
Dolichocarpol D (4): colorless oil; [α]D25 −12 (c 0.1, CHCl3); IR (film) νmax 3461, 2960, 2930, 2875, 1705, 1457, 1363, 1214, 1049 cm–1; 1H and 13C NMR data, see Tables 1 and 2; positive-ion HRESIMS m/z 277.1772 [M + Na]+ (calcd for C15H26O3Na, 277.1774, Δ = 0.6 ppm); positive-ion ESIMS/MS fragments m/z 237, 219, 201.
Dolichocarpol E (5): colorless oil; [α]D25 −9 (c 0.1, CHCl3); IR (film) νmax 3321, 2975, 2929, 2883, 1659, 1453, 1383, 1270, 1082, 1045 cm–1; 1H and 13C NMR data, see Tables 1 and 2; positive-ion HRESIMS m/z 275.1622 [M + Na]+ (calcd for C15H24O3Na, 275.1618, Δ = −1.6 ppm).
Dolichocarpol F (6): colorless oil; [α]D25 +10 (c 0.1, CHCl3); ECD (CHCl3) λ (Δε) 219 (−0.18), 291 (+0.09); IR (film) νmax 3344, 2954, 2923, 2867, 1701, 1462, 1368, 1213, 1092 cm–1; 1H and 13C NMR data, see Tables 1 and 2; positive-ion HRESIMS m/z 275.1616 [M + Na]+ (calcd for C15H24O3Na, 275.1618, Δ = 0.8 ppm); positive-ion ESIMS/MS fragments m/z 215.
ECD Calculation
Conformational studies for compounds 1–6 were carried out on the Spartan’16 software using the MMFF94 molecular mechanics force field calculation. Conformers within a 10 kcal/mol energy window were generated and further optimized using density functional theory (DFT) calculation at the B3LYP/6-31G*(d) level. The conformers with over 1% of Boltzmann distribution were chosen for ECD calculation at the B3LYP/6-311+G(2d,p) level. The calculated ECD spectra were obtained by DFT and time-dependent DFT (TD-DFT) using Gaussian 09 and analyzed on SpecDis v1.71.
Cytotoxicity Assay
Human colon cancer HCT-116 cells and nontumor murine fibroblast L929 cells were cultured in an RPMI-1640 medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C with 5% CO2 in a humidified atmosphere. Cells were seeded in 96 well plates at a density of 3 × 105 cells per well. Following a 24 h period, cells were incubated with the sesquiterpenoids (3, 4, and 6) (50 μM) dissolved in DMSO (0.4%). After culturing for 72 h, 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL) was added and incubated for another 4 h. The deposited formazan was dissolved with dodecyl sulfate sodium salt (100 μL).27 Positive control was DMSO (20%). The optical densities were measured using a microplate spectrophotometer (Microplate reader BioTek Instruments, Sinergy HT, Winooski, VT, USA). Data were analyzed with GraphPad Prism 5.0 using the analysis of variance (one-way ANOVA).
Acknowledgments
The authors acknowledge the Brazilian agencies Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior-Brasil (CAPES) (Finance Code 001) and Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq) for financial support and fellowships. We are also thankful for collaborating with Rede Norte-Nordeste de Fitoterápicos (INCT-RENNOFITO), Centro Analítico de Instrumentação da Universidade de São Paulo (Central Analítica IQ-USP), and Instituto de Química - Câmpus de Araraquara - UNESP. Dr. A. Leyva (USA) helped with the English translation and editing of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00690.
HRESIMS, IR, NMR, and ESIMS/MS data of compounds 1–6 (PDF)
Author Contributions
The manuscript was written through contributions from all authors. All authors approved the final version of the manuscript. All authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Ludwiczuk A.; Skalicka-Woźniak K.; Georgiev M. I.. Terpenoids. In Pharmacognosy: Fundamentals, Applications and Strategies; McCreath S. B., Delgoda R., Eds.; Elsevier/Academic Press: Boston, 2017; pp 233–266. [Google Scholar]
- Azimova S. S.; Saidkhodzhaev A. I.. Natural Sesquiterpene Esters. In Natural Compounds; Azimova S. S., Saidkhodzhaev A. I., Eds.; Springer-Verlag New York: New York, 2013; pp 174–370. [Google Scholar]
- Jiao S. G.; Zhang R. F.; Li J. J.; Wuken S. N.; Zhang H. X.; Tu P. F.; Chai X. Y. Phytochemical and Pharmacological Progress on Humulane-Type Sesquiterpenoids. Zhongguo Zhongyao Zashi 2018, 43, 4380–4390. 10.19540/j.cnki.cjcmm.20180725.001. [DOI] [PubMed] [Google Scholar]
- Dai J.-R.; Cardellina J. H. II; Mahon J. B. M.; Boyd M. R. Zerumbone, an HIV-Inhibitory and Cytotoxic Sesquiterpene of Zingiber Aromaticum and Z. Zerumbet. Nat. Prod. Lett. 1997, 10, 115–118. 10.1080/10575639708043725. [DOI] [Google Scholar]
- Legault J.; Dahl W.; Debiton E.; Pichette A.; Madelmont J.-C. Antitumor Activity of Balsam Fir Oil: Production of Reactive Oxygen Species Induced by α-Humulene as Possible Mechanism of Action. Planta Med. 2003, 69, 402–407. 10.1055/s-2003-39695. [DOI] [PubMed] [Google Scholar]
- Gottsberger G. The Reproductive Biology of the Early-Divergent Genus Anaxagorea (Annonaceae), and Its Significance for the Evolutionary Development of the Family. Acta Bot. Brasilica 2016, 30, 313–325. 10.1590/0102-33062015abb0311. [DOI] [Google Scholar]
- Gonda R.; Takeda T.; Akiyama T. Studies on the Constituents of Anaxagorea Luzonensis A. Gray. Chem. Pharm. Bull. 2000, 48, 1219–1222. 10.1248/cpb.48.1219. [DOI] [PubMed] [Google Scholar]
- Sabphon C.; Sermboonpaisarn T.; Sawasdee P. Cholinesterase Inhibitory Activities of Xanthones from Anaxagorea Luzonensis A. Gray. J. Med. Plants Res. 2012, 6, 3781–3785. 10.5897/JMPR12.346. [DOI] [Google Scholar]
- Sabphon C.; Temkitthawon P.; Ingkaninan K.; Sawasdee P. Phosphodiesterase Inhibitory Activity of the Flavonoids and Xanthones from Anaxagorea Luzonensis. Nat. Prod. Commun. 2015, 10, 301–303. 10.1177/1934578X1501000222. [DOI] [PubMed] [Google Scholar]
- Puentes de Díaz A. M. Neolignans from Anaxagorea Clavata. Phytochemistry 1997, 44, 345–346. 10.1016/S0031-9422(96)00541-9. [DOI] [Google Scholar]
- de Alencar D. C.; Pinheiro M. L. B.; Pereira J. L. D. S.; De Carvalho J. E.; Campos F. R.; Serain A. F.; Tirico R. B.; Hernández-Tasco A. J.; Costa E. V.; Salvador M. J. Chemical Composition of the Essential Oil from the Leaves of Anaxagorea Brevipes (Annonaceae) and Evaluation of Its Bioactivity. Nat. Prod. Res. 2016, 30, 1088–1092. 10.1080/14786419.2015.1101103. [DOI] [PubMed] [Google Scholar]
- Andrade E. H. A.; Oliveira J.; Zoghbi M. d. G. B. Volatiles of Anaxagorea Dolichocarpa Spreng. & Sandw. and Annona Densicoma Mart. Growing Wild in the State of Pará, Brazil. Flavour Fragrance J. 2007, 22, 158–160. 10.1002/ffj.1776. [DOI] [Google Scholar]
- Husain K.; Zakaria S. M.; Lajis N. H.; Shaari K.; Ismail I. S.; Israf D. A.; Paetz C. Novel Sesquiterpene and Copyrine Alkaloids from Anaxagorea Javanica Blume. Phytochem. Lett. 2012, 5, 788–792. 10.1016/j.phytol.2012.09.003. [DOI] [Google Scholar]
- Scharaschkin T.; Doyle J. A. Phylogeny and Historical Biogeography of Anaxagorea (Annonaceae) Using Morphology and Non-Coding Chloroplast Sequence Data. Syst. Bot. 2005, 30, 712–735. 10.1600/036364405775097888. [DOI] [Google Scholar]
- Lobão A. Q.; de Araujo D. S. D.; Kurtz B. C. Annonaceae das restingas do estado do Rio de Janeiro, Brasil. Rodriguesia 2005, 56, 85–96. 10.1590/2175-78602005568706. [DOI] [Google Scholar]
- Hocquemiller R.; Rasamizafy S.; Moretti C.; Jacquemin H.; Cavé A. L’Anaxagoréine, Nouvel Alcaloïde Aporphinique Isolé de Deux Espèces d’Anaxagorea. Planta Med. 1981, 41, 48–50. 10.1055/s-2007-971672. [DOI] [PubMed] [Google Scholar]
- Lúcio A. S. S. C.; Da Silva Almeida J. R. G.; Barbosa-Filho J. M.; Pita J. C. L. R.; Branco M. V. S. C.; Formiga Melo Diniz M. d. F.; De Fátima Agra M.; Da-Cunha E. V. L.; Da Silva M. S.; Tavares J. F. Azaphenanthrene Alkaloids with Antitumoral Activity from Anaxagorea Dolichocarpa Sprague and Sandwith (Annonaceae). Molecules 2011, 16, 7125–7131. 10.3390/molecules16087125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier G.; Hadjiakhoondi A.; Charles B.; Leboeuf M.; Cavé A. Volatile Components of Anaxagorea Dolichocarpa Fruit. Biochem. Syst. Ecol. 1994, 22, 605–608. 10.1016/0305-1978(94)90073-6. [DOI] [Google Scholar]
- Da Silva Almeida J. R. G.; De Oliveira M. R.; Guimarães A. L.; De Oliveira A. P.; De Araújo Ribeiro L. A.; Lúcio A. S. S. C.; Quintans Júnior L. J. Phenolic Quantification and Antioxidant Activity of Anaxagorea Dolichocarpa and Duguetia Chrysocarpa (Annonaceae). Int. J. Pharma Bio Sci. 2011, 2, 367–374. [Google Scholar]
- da Silva Pinheiro R.; Rabelo S. V.; de Oliveira A. P.; Guimarães A. L.; de Moraes-Filho M. O.; da Costa M. P.; do Ó Pessoa C.; Lúcio A. S. S. C.; da Silva Almeida J. R. G. Phytochemical Screening and Evaluation of Cytotoxicity of Stem Bark Extracts of Anaxagorea Dolichocarpa and Duguetia Chrysocarpa (Annonaceae). Trop. J. Pharm. Res. 2016, 15, 793–798. 10.4314/tjpr.v15i4.18. [DOI] [Google Scholar]
- Dutt R.; Garg V.; Khatri N.; Madan A. K. Phytochemicals in Anticancer Drug Development. Anti-Cancer Agents Med. Chem. 2019, 19, 172–183. 10.2174/1871520618666181106115802. [DOI] [PubMed] [Google Scholar]
- Wang X.-J.; Chen J.-Y.; Fu L.-Q.; Yan M.-J. Recent Advances in Natural Therapeutic Approaches for the Treatment of Cancer. Aust. J. Chem. 2020, 32, 53–65. 10.1080/1120009X.2019.1707417. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Damodaran N. P.; Dev S. Studies in Sesquiterpenes—XXXVIII: Structure of humulene epoxide-I and humulene epoxide-II. Tetrahedron 1968, 24, 4123–4132. 10.1016/0040-4020(68)88175-X. [DOI] [Google Scholar]
- McPhail A. T.; Sim G. A. Sesquiterpenoids. Part IV. The Stereochemistry of Humulene: X-Ray Analysis of the Humulene–Silver Nitrate Adduct. J. Chem. Soc. B 1966, 112–120. 10.1039/J29660000112. [DOI] [Google Scholar]
- Zhang R.; Feng X.; Su G.; Mu Z.; Zhang H.; Zhao Y.; Jiao S.; Cao L.; Chen S.; Tu P.; et al. Bioactive Sesquiterpenoids from the Peeled Stems of Syringa Pinnatifolia. J. Nat. Prod. 2018, 81, 1711–1720. 10.1021/acs.jnatprod.7b01071. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.