

SUMMARY
B cell receptor (BCR) engagement induces naive B cells to differentiate and perform critical immune-regulatory functions. Acquisition of functional specificity requires that a cell survive, enter the cell cycle, and proliferate. We establish that quantitatively distinct Ca2+ signals triggered by variations in the extent of BCR engagement dynamically regulate these transitions by controlling nuclear factor κB (NF-κB), NFAT, and mTORC1 activity. Weak BCR engagement induces apoptosis by failing to activate NF-κB-driven anti-apoptotic gene expression. Stronger signals that trigger more robust Ca2+ signals promote NF-κB-dependent survival and NFAT-, mTORC1-, and c-Myc-dependent cell-cycle entry and proliferation. Finally, we establish that CD40 or TLR9 costimulation circumvents these Ca2+-regulated checkpoints of B cell activation and proliferation. As altered BCR signaling is linked to autoimmunity and B cell malignancies, these results have important implications for understanding the pathogenesis of aberrant B cell activation and differentiation and therapeutic approaches to target these responses.
Graphical Abstract
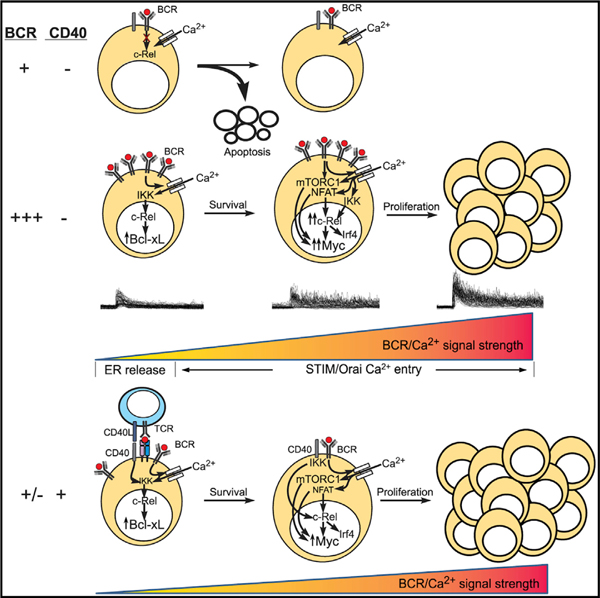
In Brief
Berry et al. establish that variations in the strength of BCR engagement are encoded as quantitatively distinct calcium signals that tune B cell fates by dynamically regulating NF-κB, NFAT, and mTORC1 activity. Targeting calcium signaling may thereby serve as an effective treatment strategy for regulating normal and pathological B cell activation.
INTRODUCTION
Quantitatively and qualitatively distinct signals generated by engagement of the B cell receptor (BCR) and costimulatory receptors on mature B cells control their survival, metabolic reprogramming, cell-cycle entry, and proliferation (Kouskoff et al., 1998; Casola et al., 2004; Pittner and Snow, 1998). Indeed, the mechanisms of BCR signal transduction have been extensively studied, yet relatively little is known about how differences in the affinity and avidity of BCR engagement are encoded within the cell and precisely how these signals are then decoded to regulate these key cell-fate transitions (Dal Porto et al., 2004; Kurosaki et al., 2010; Yam-Puc et al., 2018). Also unknown are the mechanisms by which costimulatory or co-activating signals impact the gain of BCR signaling to fine-tune a cell’s fate. Previous efforts point to a relationship between the affinity and the avidity of antigen binding to the BCR and the amplitude, duration, and periodicity of Ca2+ signals, and these studies reveal that distinct dynamics drive distinct fates of immature and mature B cells (Benschop et al., 1999; Hemon et al., 2017; Healy et al., 1997; Scharenberg et al., 2007; Nitschke et al., 1997; Cornall et al., 1998; Jellusova and Nitschke, 2012; Müller and Nitschke, 2014; Hoek et al., 2006). Indeed, mutations in signal transduction proteins downstream of the BCR, notably those that mobilize Ca2+, can lead to altered B cell activation and differentiation, skewed humoral immune responses, autoimmune disease, and B cell malignancies (reviewed in Baba and Kurosaki, 2016). Thus, Ca2+ serves as a central molecular switch for encoding and transducing differences in BCR signaling with significant biological and pathological consequences.
Despite the well-established importance of Ca2+ in the antigen-induced responses of mature B cells, current understanding is also clouded by conflicting reports regarding the consequences of variations in BCR-induced Ca2+ signals. Findings from a recent study suggest that in the absence of costimulation, BCR-derived Ca2+ signals in mature B cells initiate mitochondrial dysfunction resulting in apoptosis (Akkaya et al., 2018). However, others have described a dose-dependent relationship between BCR signal strength and Ca2+ signals, cell survival, and proliferation (Matsumoto et al., 2011; Mao et al., 2016; Tang et al., 2017). Furthermore, the absolute role or requirement for Ca2+ seems to vary with the stage of mature B cell differentiation (Matsumoto et al., 2011). For example, in germinal center (GC) B cells, the coupling between the BCR and Ca2+ is disrupted, and these cells rely principally on costimulatory signals to drive class switch recombination and affinity maturation (Luo et al., 2018; Khalil et al., 2012). These costimulatory pathways, namely those triggered by CD40 and Toll-like receptor (TLR) engagement, are generally thought to be Ca2+ independent, suggesting that Ca2+-dependent steps of B cell differentiation may be circumvented in some cases by costimulatory signals.
Among the mechanisms that critically regulate B cell activation and differentiation, several exhibit Ca2+ sensitivity. These include nuclear factor kB (NF-κB) (reviewed in Berry et al., 2018; Gerondakis and Siebenlist, 2010) and NFAT (Peng et al., 2001), which control the expression of diverse genes involved in cell survival and differentiation, mTORC1 (Li et al., 2016; Zhou et al., 2015), which regulates metabolic reprogramming, and c-Myc (Lindsten et al., 1988), which drives proliferative expansion (Stine et al., 2015; Saxton and Sabatini, 2017). In T cells, Ca2+ orchestrates a shift in cellular metabolism from oxidative phosphorylation to glycolysis by controlling the “master” regulators c-Myc and mTORC1 (Vaeth et al., 2017). However, the mechanisms by which the strength of antigen-receptor-induced quantitatively distinct Ca2+ signals tune steps that control B cell survival, metabolic reprogramming, cell-cycle entry, and proliferation are largely unexplored.
Consequently, we dissected the mechanisms by which Ca2+, and specific properties of BCR-induced Ca2+ signals, regulate mature B cell survival, cell-cycle entry, and proliferation. We identified a relationship between the strength of BCR engagement and amplitude and periodicity of resulting Ca2+ signals. Further, we established how BCR-induced Ca2+ signals are decoded to regulate NF-κB-dependent steps of cell survival and mTORC1-and c-Myc-dependent cell-cycle entry and proliferation. Finally, we show how CD40 or TLR9 signaling can circumvent Ca2+-regulated steps of B cell activation to promote enhanced survival and proliferation. Importantly, given that enhanced or constitutive BCR signaling is linked to autoimmunity and B cell malignancies (Hemon et al., 2017), understanding these mechanisms that control early B cell differentiation has important implications for preventing aberrant B cell differentiation and developing therapeutics to modify the course of human disease.
RESULTS
Ca2+ Signals Encode BCR Signal Strength to Regulate B Cell Activation
The strength of antigen-induced signaling in B lymphocytes can be modulated by varying the concentration of agonist anti-BCR antibody (Stadanlick et al., 2008; Galibert et al., 1996; Hao and August, 2005). Indeed, progressive increases in BCR signal strength are encoded as quantitatively distinct intracellular Ca2+ signals (Figure 1A). Therefore, we varied BCR signal strength in this way to determine its relationship with Ca2+ and B cell activation, including survival, cell-cycle entry, and proliferation. The lowest signal strength (1 µg/mL anti-BCR) tested accentuated cell death relative to untreated cells (Figures 1B and 1C). Subsequent increases in BCR signal strength triggered a progressive increase in cell viability (Figures 1B and 1C), cell numbers (Figure 1D), the proportion of cells that divide (Figure 1E), and the number of divisions per cell (Figure 1F). The proportion of cells progressing beyond mid-G1 of the cell cycle, as indicated by Ki67 expression, also increased with BCR signal strength; although, at the highest anti-BCR concentration, only approximately half of B cells actively cycle at 36 h (Figure 1G). Collectively, these data indicate that the strength of BCR signaling dictates whether a B cell will live or die, the efficiency of cell-cycle entry, and the extent to which they proliferate.
Figure 1. Ca2+ Signals Encode BCR Signal Strength to Regulate B Cell Activation.
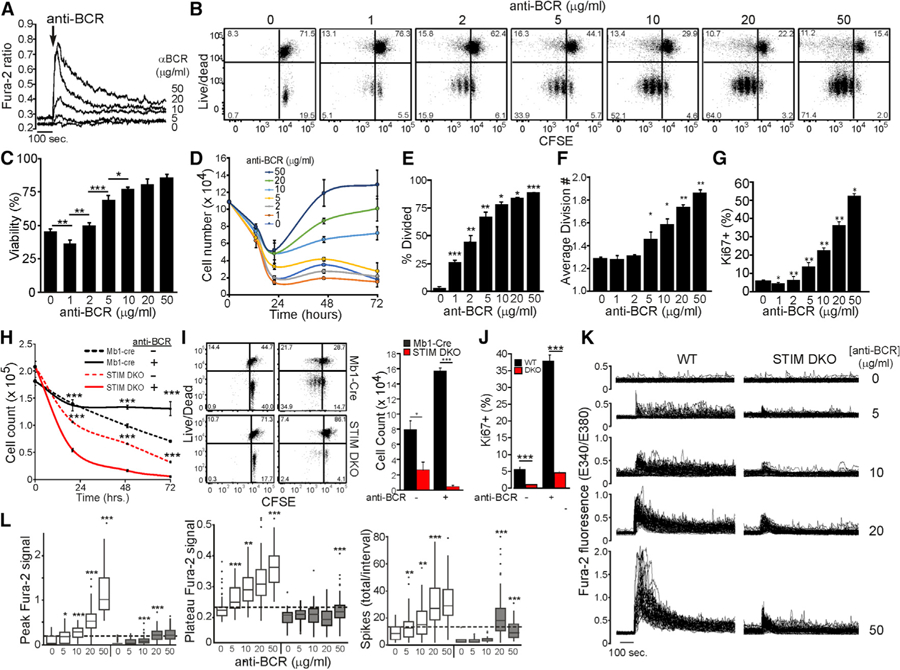
(A) Fura-2 ratiometric imaging of intracellular Ca2+ in WT CD23+ B cells stimulated with anti-BCR as indicated. Average results are representative of three independent experiments with >50 cells per condition.
(B) CFSE-labeled WT CD23+ B cells were stimulated in complete media with anti-BCR for 72 h as indicated. Carboxyfluorescein succinimidyl ester (CFSE) dilution and live/dead plots are representative of triplicate experiments.
(C and D) Viability of WT CD23+ B cells as determined by the proportion of live/dead cells at 22 h (C) and total number of live cells at each initial anti-BCR concentration for each time point (D) (mean ± SD from triplicate wells).
(E and F) The proportion of divided cells (E) and average division number (F) for data shown in (B) (mean ± SD from triplicate wells).
(G) Ki67 expression in WT CD23+ B cells stimulated for 32 h with anti-BCR as indicated (mean percentage ± SD from triplicate wells; statistical comparisons to adjacent [lower] concentration of anti-BCR).
(H) Total cell counts at indicated times are plotted for each condition (mean cell number ± SD of triplicate wells). CFSE-labeled CD23+ B cells from Stim1wt/ wtStim2wt/wtMb1cre+ (Mb1-Cre) and Stim1fl/flStim2fl/flMb1cre+ (STIM DKO) mice were unstimulated or stimulated for 72 h with anti-BCR. (I)C FSE dilution versus live/dead plot (left) is representative of results from triplicate wells (right) at 72 h.
(J) Ki67 expression (mean ± SD of triplicate wells) 32 h after anti-BCR stimulation of WT and STIM DKO B cells.
(K) Fura-2 ratiometric imaging of cytoplasmic [Ca2+] in WT and STIM DKO B cells stimulated with anti-BCR as indicated. Each line depicts the response of a single cell.
(L) Boxplot representation of mean initial peak (upper left), average sustained concentration (between 10 and 15 min, upper right), and total spikes (lower left) in WT and STIM DKO B cells. Results are representative of three independent experiments with >50 cells per genotype/treatment, and statistical comparisons were made to adjacent (lower) concentration of anti-BCR. For all figures, *p < 0.05, **p < 0.01, ***p < 0.001.
Given that Ca2+ drives the expression of fate-specific genes (Healy et al., 1997, 1998; Dolmetsch et al., 1997; Matsumoto et al., 2011; Benschop et al., 1999), we implemented a genetic approach to examine the impact of variations in Ca2+ signal strength on BCR-induced gene expression. Specifically, we generated mice with a B-cell-specific (Mb1-Cre driven) deletion of Stim1 and Stim2 to preclude BCR-induced Orai-dependent Ca2+ entry. B cells from Stim1/Stim2 double-knockout (DKO) mice develop normally in vivo but exhibit defects in survival and proliferation in vitro (Matsumoto et al., 2011; Mao et al., 2016). Consistent with these previous reports, we observed a greater rate and extent of death of Stim1fl/flStim2fl/flMb1cre+ (STIM DKO) by unstimulated B cells that was enhanced by BCR engagement (Figure 1H). Notably, most STIM DKO B cells died without proliferating (Figure 1I). Among the viable STIM DKO cells, relatively few entered the cell cycle (Figure 1J) and then divided only once (Figure 1I, STIM DKO, anti-BCR, lower right panel). Interestingly, this impairment in survival, cell-cycle entry, and proliferation by STIM DKO cells is comparable to the response of wild-type (WT) B cells to the lowest BCR strength tested, implying that a quantitative feature of Ca2+ signals, such as the initial peak amplitude, the steady-state concentration, or the frequency of spikes, tunes these responses to the BCR.
To better understand how Ca2+ regulates B cell fates, we quantified the impact of BCR signal strength on Ca2+ dynamics in mature WT and STIM DKO B cells. As anticipated, we observed a robust dose-dependent relationship between the strength of BCR engagement and the pattern of Ca2+ signaling (Figure 1K). Specifically, the amplitude of the initial peak Ca2+ rise increased with BCR signal strength in both WT and STIM DKO cells, although the range was greatly attenuated in the absence of Ca2+ entry (Figure 1L). Furthermore, in WT cells, BCR strength also induced a dose-dependent increase in steady-state Ca2+ levels and spike frequency. By contrast, in STIM DKO cells, while higher concentrations of anti-BCR elicited a modest increase in spike frequency, Ca2+ levels rapidly decayed to pre-stimulation levels at all BCR strengths. Collectively, these results indicate that Ca2+ entry is needed to support sustained signaling and that quantitative properties of Ca2+ signals reflective of the strength of BCR engagement control B cell activation by regulating cell survival, cell-cycle entry, and proliferation.
STIM/Orai-Dependent Ca2+ Signals Drive Bcl-xL-Dependent Rescue from Apoptosis
We next focused on the mechanism by which BCR stimulation, and specifically Ca2+, controls cell survival. Regardless of genotype or stimulus, nearly all nonviable B cells stained positive for Annexin V, suggesting that death of STIM DKO B cells occurs through apoptosis (Figure 2A). Indeed, a pan-caspase inhibitor (zVAD-fmk) attenuated caspase-3 cleavage and cell death (Figures 2B and 2C). Initiation of intrinsic apoptosis is dictated by a competitive equilibrium between Bcl-2 family members, including pro-apoptotic proteins and anti-apoptotic Bcl-2-like proteins (Enders et al., 2003). To evaluate whether Ca2+ acts to inhibit intrinsic apoptosis, we quantified its impact on the expression of four anti-apoptotic and seven pro-apoptotic genes. BCR stimulation of WT B cells led to a dramatic increase in the expression of anti-apoptotic genes Bcl2la1 (encoding A1) and Bcl2l1 (encoding Bcl-xL), and the induction of both genes was significantly attenuated in STIM DKO B cells (Figures 2D and S1A). Notably, BCR stimulation of WT B cells also led to a modest upregulation of several pro-apoptotic genes, including Bak, Bax, Bbc3 (encoding Puma), Bid, Bim, and Pmaip1 (encoding Noxa). Despite the observed apoptotic phenotype of STIM DKO B cells, upregulation of four of six pro-apoptotic genes was significantly decreased in the absence of Ca2+ entry. This result suggests the dominant impact of BCR-induced Ca2+ entry is to promote survival through anti-apoptotic gene expression. Indeed, BCR stimulation led to significant upregulation of Bcl-xL protein expression (Figure 2E), and the strength of BCR engagement correlated directly with the extent of its expression and viability of WT, but not DKO, cells (Figure 2F). Confirming that Bcl-xL induction by Ca2+ is sufficient to rescue B cells from apoptosis, heterologous Bcl-xL expression substantially improved cell survival in the absence of extracellular Ca2+ (Figure S1B). Thus, BCR-induced Ca2+ signals regulate cell survival by controlling the extent of Bcl-xL protein upregulation.
Figure 2. STIM/Orai-Dependent Ca2+ Signals Promote B Cell Survival through Induction of Bcl-xL Expression.
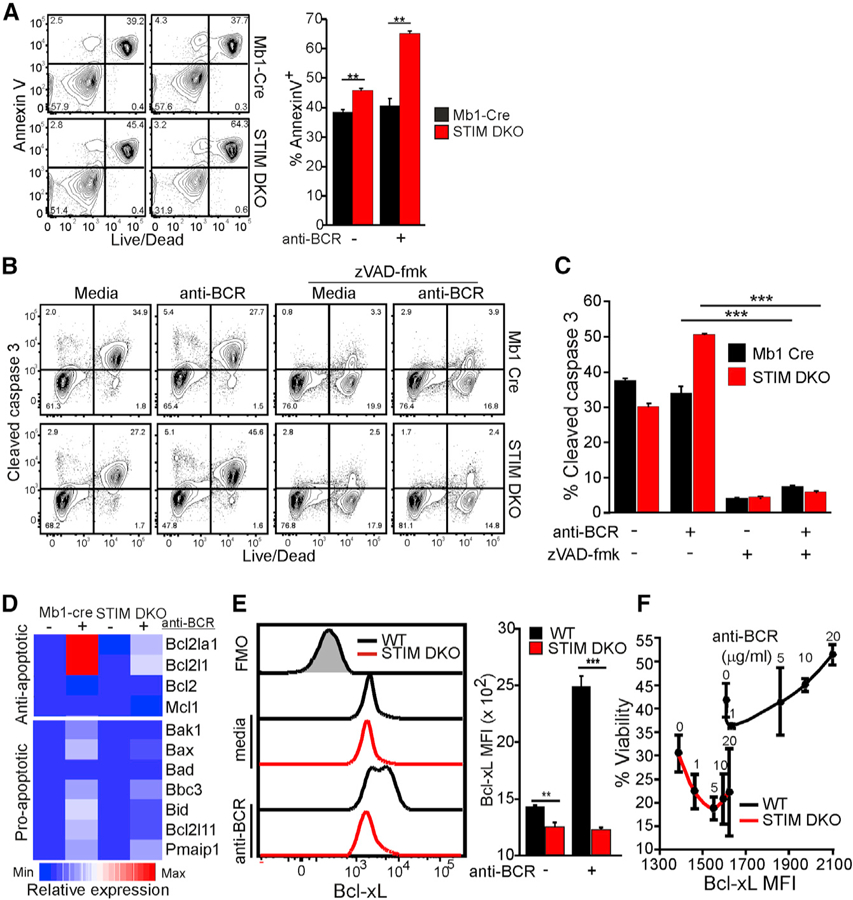
WT and STIM DKO CD23+ B cells were cultured in complete media in the absence or presence of anti-BCR.
(A) Annexin V and live/dead staining at 24 h of culture. (Left) Representative plot (right) and quantification of mean percentage of Annexin-V-stained cells (±SD from triplicate wells) are shown. (B and C) Flow cytometric analysis of intracellular cleaved caspase-3 at 12 h. The impact of the pan-caspase inhibitor zVAD-fmk (100 µM) on B cell viability was determined 12 h after anti-BCR stimulation.
(B) Representative cytometry plot (B) and percentage (mean ± SD) of cleaved caspase-3-stained cells from triplicate wells (C).
(D) Heatmap shows relative gene expression of anti-and pro-apoptotic genes based upon qRT-PCR analysis following culture as indicated for 6 h. Average expression from triplicate wells relative to Mb1-cre unstimulated (controls).
(E) Intracellular Bcl-xL expression (Fluorescence minus one (FMO), gray shaded histogram, left) in anti-BCR stimulated WT and STIM DKO B cells at 6 h (right, Bcl-xL MFI ± SD from triplicate wells).
(F) Bcl-xL mean fluorescence intensity (6 h) in CD23+ B cells from WT (black line) and STIM DKO (red line) mice stimulated with anti-BCR (at indicated concentration) plotted versus viability (24 h, % live cells ± SD from triplicate wells and duplicate cultures). For all figures, *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S1.
Apoptosis of primary B cells can also occur through extrinsic (death receptor) mechanisms that are initiated by ligation of receptors, including TNF-related apoptosis-inducing ligand (TRAIL), Fas, and tumor necrosis factor (TNF), linked to the activation of caspase-8 (Green and Llambi, 2015; Dickens et al., 2012). To evaluate whether BCR-induced Ca2+ signals also regulate extrinsic apoptosis, B cells were treated with the caspase 8 selective inhibitor z-IETD-fmk. Z-IETD-fmk did not rescue STIM DKO cells from BCR-induced apoptosis, indicating that cell-extrinsic apoptosis is unlikely to be involved in STIM DKO B cell death (Figure S1C). Importantly, although caspase-8 is a mediator of death-receptor-driven extrinsic apoptosis, it can also promote survival by preventing receptor-interacting serine/threonine protein kinase-3 (Ripk3)-dependent necroptosis (Philip et al., 2016). Thus, to definitively assess the role of caspase-8, we evaluated the kinetics of in vitro survival of Ripk3−/− and Ripk3−/−Casp8−/− B cells in the presence and absence of extracellular Ca2+. Although the viability of Rip3k−/−Casp8−/− B cells was marginally increased relative to WT cells in Ca2+-depleted media, loss of caspase-8 and Rip3k did not restore Ca2+-dependent survival (Figure S1D). Given that neither caspase-8 nor Rip3k expression impact Ca2+-dependent rescue, we conclude that BCR-induced Ca2+ signals do not promote survival by antagonizing extrinsic apoptosis.
Ca2+-Dependent Regulation of c-Rel Activation Promotes Bcl-xL Upregulation
To address how Ca2+ controls Bcl-xL expression, we focused on the NF-κB family member c-Rel because of its established role in BCR-induced Bcl2l1 and Bcl2la1 transcription (Sen, 2006; Hsia et al., 2002; Tumang et al., 1998; Grumont et al., 1998; Owyang et al., 2001). Consistent with a mechanistic link between Ca2+ and c-Rel activity, BCR-induced Bcl2l1 (Figure 3A) and Bcl-xL (Figure 3B) expression and cell survival (Figure 3C) were similarly impaired in c-Rel KO and STIM DKO B cells. We next asked if BCR-induced Ca2+ signals control the activation of c-Rel, and we found that phosphorylation of IKKβ and its target, IκBα, and degradation of IκBα were inhibited in STIM DKO B cells (Figure 3D). Consistent with a block in IKK complex activation, both c-Rel and p65 nuclear localization were inhibited in STIM DKO B cells (Figure 3E), as was the expression of classical NF-κB target genes, including Nfkbia (encoding IκBα) and Nfkb2 (encoding p100) (Figure 3F). Activation of preexisting p65 and c-Rel is critical during the initial phase of NF-κB-driven gene expression; however, de novo c-Rel induction is required to promote sustained NF-κB activity in lymphocytes (Venkataraman et al., 1995; Bajpai et al., 2000; Petro et al., 2000, 2002). Indeed, cells with higher c-Rel expression expressed more Bcl-xL (Pearson coefficient ᑭ = 0.48, p = 0.0; Figure 3G). Moreover, Ca2+ regulates de novo transcription of Rel, as this and c-Rel protein induction were restrained in STIM DKO B cells (Figures 3H and 3I).
Figure 3. Ca2+ Entry Regulates NF-κB-Dependent Anti-apoptotic Gene Expression.
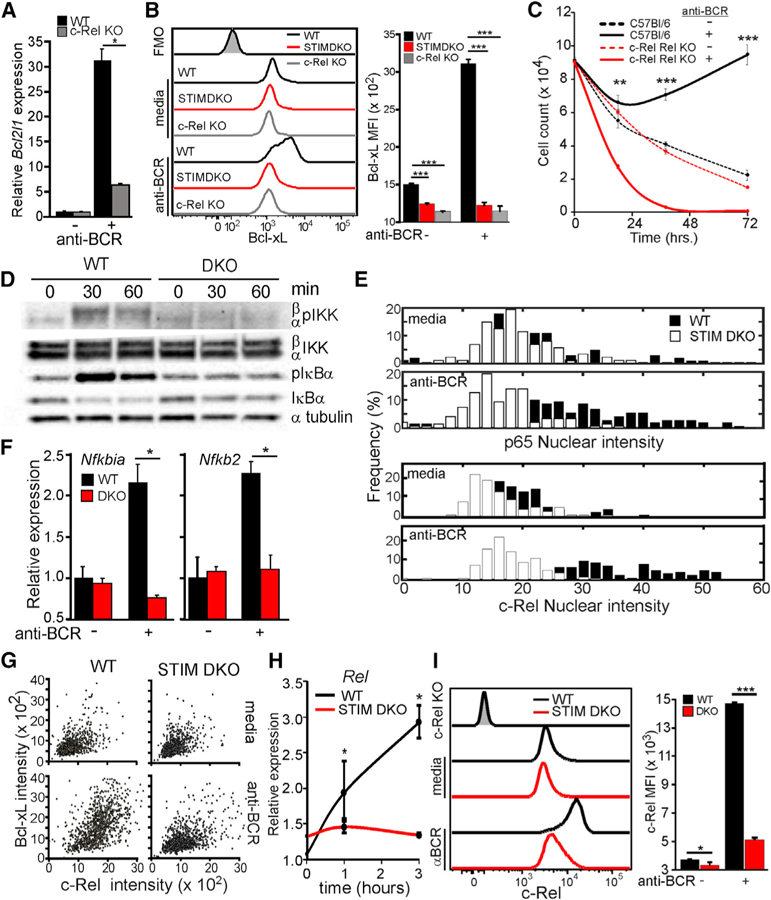
(A) Bcl2l1 expression after 6 h of anti-BCR stimulation in CD23+ B cells from WT and c-Rel KO mice (mean ± 95% confidence interval; *, statistically significant).
(B) Intracellular Bcl-xL protein levels (relative to FMO, gray shaded histogram) in CD23+ B cells from WT (black line), STIM DKO (gray line), and c-Rel KO (red line) mice 6 h after anti-BCR stimulation (left) and mean (median flourescence intensity (MFI) ± SD from triplicate wells) Bcl-xL fluorescence intensity (right).
(C) CD23+ B cells from WT (solid line) and c-Rel KO (dashed line) mice were cultured for indicated times in media alone or media containing anti-BCR. The total number of live cells was enumerated, and mean cell number (±SD of triplicate wells) is plotted for the indicated time points.
(D) Immunoblot of anti-BCR-dependent NF-κB activation in CD23+ WT and STIM DKO B cells at indicated time points.
(E) p65 (top) and c-Rel (bottom) nuclear localization in unstimulated (media) and anti-BCR stimulated (2 h) WT (black) and STIM DKO (white) B cells. Overlay of average c-Rel and p65 nuclear cell intensity (>70 cells per group) is shown.(F) Nfkbia (IκBα) and Nfkb2 (p100) mRNA expression in CD23+ B cells from WT and STIM DKO mice cultured for 3 h in the presence or absence of anti-BCR (mean ± 95% confidence interval; *, statistically significant).
(G) Scatterplot of c-Bcl-xL and Rel expression in individual WT (left) and STIM DKO (right) B cells cultured in the absence (media) and presence of anti-BCR for 20 h.
(H) Time course of anti-BCR-induced Rel expression in CD23+ B cells from WT and STIM DKO mice (mean ± 95% confidence interval; *, statistically significant).
(I) c-Rel expression in WT (black line) and STIM DKO (red line) B cells cultured for 12 h with anti-BCR. (Left) Mean c-Rel MFI (±SD from triplicate wells, right). For all figures except where indicated, *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S2.
We next asked how these two distinct waves of c-Rel activity (i.e., activation of pre-existing c-Rel and de novo Rel expression) are regulated. Previous studies indicate that activation of existing c-Rel is regulated by calcineurin (Cn)-dependent dephosphorylation of the CARD11-BCL-10-MALT1 (CBM) complex upstream of IKK activation (Frantz et al., 1994; Liu et al., 2016; Palkowitsch et al., 2011). However, various studies suggest that de novo Rel induction is controlled by both NFAT and NF-κB (Grumont et al., 2004; Nolz et al., 2007). Consistent with this model, we found that the Cn inhibitors cyclosporine A (CsA) and FK506 attenuated BCR-induced IκBα phosphorylation and degradation (Figure S2A), Bcl2l1 mRNA (Figure S2B) and protein expression (Figure S2C), and B cell survival (Figure S2D). However, Cn also regulates NFAT activation and was required for c-Rel induction in B cells (Figure S2E). Consequently, we used IKKβ-deficient B cells to distinguish between the roles of NF-κB and NFAT in de novo c-Rel induction (Shaw et al., 1988; Flanagan et al., 1991). We found that c-Rel induction was comparable in WT and IKKβ knockout (KO) (isolated from mb1-Cre x Ikk2fl/fl mice) B cells and in both was attenuated by FK506 (Figure S2F). Together, these data establish that Ca2+ regulates c-Rel function by two distinct mechanisms. First, it controls IKK activity and the activation of existing c-Rel to initiate expression of NF-κB target genes, including Bcl2l1. Second, it promotes Cn-induced, NFAT-dependent de novo c-Rel expression.
Ca2+-Dependent mTORC1 Activation and Myc Expression Promotes Cell-Cycle Entry
In addition to its role in Bcl-xL expression and cell survival, c-Rel regulates cell-cycle entry by driving expression of proteins such as cyclin E and E2F3a (Cheng et al., 2003). However, entry into and progression through the cell cycle also requires a shift in cellular metabolism from oxidative phosphorylation to aerobic glycolysis (Pearce et al., 2013). We therefore asked if Ca2+ regulates key determinants of this metabolic reprogramming and proliferation, namely mechanistic target of rapamycin (mTOR) and c-Myc (Grumont et al., 2002; Wang et al., 2011; DeBerardinis et al., 2008; Newsholme et al., 1985). BCR engagement elicited phosphorylation of the mTORC1 target S6 (Figure 4A) and increased c-Myc expression (Figure 4B) in ~40% of WT cells but significantly fewer (~12%) STIM DKO B cells. Consistent with the established role for mTORC1 in c-Myc translation (Csibi et al., 2014), the induction of pS6 and c-Myc were highly correlated. Virtually all cells with high levels of pS6 express high levels of c-Myc (Figure 4C). Indeed, the mTORC1-specific inhibitor rapamycin and the pan-mTOR (mTORC1 and mTORC2) inhibitor Torin-1 both blocked BCR-induced c-Myc protein expression (Figure S3A). Furthermore, Ca2+ activation of mTORC1 is physiologically significant, as inhibition of mTORC1 and STIM deficiency similarly inhibited cell growth (Figures 4D and 4E), cell-cycle entry (Figure 4F), and proliferation (Figure 4G versus Figure 1).
Figure 4. Ca2+ Is Required for mTORC1-and Myc-Dependent Cell-Cycle Entry.

(A and B) Intracellular phosphorylated S6 (pS6) (A) and c-Myc protein expression (B) in WT and STIM DKO at 4 h of stimulation with anti-BCR (mean percentage ± SD from triplicate wells).
(C) pS6 expression in Myc+ and Myc— B cells (top, from B) and Myc expression in phospho-S6 ‘‘high’’ and phospho-S6 ‘‘low’’ populations (bottom, from A).
(D) Cell size (Forward scatter area or FSC-A) of anti-BCR-stimulated WT and STIM DKO B cells (32 h) and impact of the mTORC1 inhibitor rapamycin (25 nM).
(E) Cell size from (D) (mean FSC-A ± SD from triplicate wells).
(F) Ki67 expression (mean ± SD of triplicate wells) in WT and STIM DKO B cells following 32 h of anti-BCR and the impact of rapamycin (25 nM).
(G) CFSE dilution analysis of the proliferative dynamics of anti-BCR-stimulated WT B cells in the absence or presence of rapamycin (25 nM) or torin-1 (100 nM). Results are representative of triplicate measurements.
(H) Time course of Myc expression in CD23+ B cells from WT (black line) and STIM DKO (red line) mice stimulated with anti-BCR (mean ± 95% confidence interval; *, statistically significant).
(I) c-Myc expression in CD23+ B cells from Ikk2fl/fl 3 Mb1cre— (WT, black line) and Ikk2fl/fl 3 Mb1cre+ (IKKβ KO, red line) mice stimulated with anti-BCR (6 h) in the absence or presence of FK506 (1 µM). Mean percentage of Myc+ cells (±SD of triplicate measurements) is shown.
(j) Immunoblot of mTORC1 activity in B cells stimulated with anti-BCR in the presence and absence of extracellular Ca2+.
(K) Densiometric analysis of phospho-TSC2-Thr1462 and phospho-S6 (from J). For all figures, *p < 0.05, **p < 0.01, ***p < 0.001.
See also Figure S3.
Although Ca2+-dependent regulation of mTORC1 likely impacts c-Myc translation, Ca2+ has been reported to drive Myc transcription (Vaeth et al., 2017; Lindsten et al., 1988; Nolz et al., 2007; Mognol et al., 2012). Indeed, BCR-induced Myc gene expression is attenuated in STIM DKO B cells (Figure 4H). Given that Cn/NFAT and c-Rel are known regulators of Myc expression, we next explored their roles in c-Myc induction using IKKβ KO and c-Rel KO mice (Grumont et al., 2002, 2004). Interestingly, we found that BCR-induced c-Myc induction is normal in IKKβ KO B cells but blocked by FK506 and removal of extracellular Ca2+ (Figure 4I). BCR-induced c-Myc expression is also significantly attenuated in c-Rel KO B cells (Figure S3B), indicating that both c-Rel and NFAT play primary and potentially redundant roles in c-Myc expression in mature B cells. Together, these results reveal that Ca2+ controls c-Myc expression by orchestrating its NFAT-and c-Rel-dependent transcription and mTORC1-regulated translation.
To address how Ca2+ regulates mTORC1 activity, we focused on Akt, which activates mTORC1 by inactivating the inhibitory TSC1-TSC2 complex (Inoki et al., 2002; Li et al., 2016; Divolis et al., 2016; Cao et al., 2017). Interestingly, BCR-induced Akt Thr308 and TSC2 Thr1462 phosphorylation with kinetics that corresponded to the phosphorylation of S6 (Figures 4J and 4K), and both TSC2 and S6 phosphorylation were transient in the absence of extracellular Ca2+. Consistent with Akt mediating Ca2+ control of mTORC1 activity, we also examined Foxo1 localization, which is likewise regulated by Akt (Brunet et al., 1999). We found that BCR-induced Foxo1 export from the nucleus is attenuated in STIM DKO B cells (Figure S3C, left panels), WT B cells loaded with the intracellular Ca2+ chelator 1,2-bis-(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetra(acetoxymethyl) (BAPTA), and WT B cells stimulated in Ca2+-free medium (Figure S3C, right panels). Thus, Ca2+ dependent Akt activation coordinately controls both TSC2 phosphorylation and cytoplasmic localization of Foxo1.
Interestingly, despite the known role for Ca2+ in Akt activation (Divolis et al., 2016; Deb et al., 2004; Conus et al., 1998), we observed higher levels of BCR-induced Akt Thr308 phosphorylation under Ca2+-free conditions than in the presence of Ca2+ (Figure 4J) and no relationship between Ca2+ and Akt Thr473 phosphorylation (Figure S3D). Furthermore, PDK1 (which phosphorylates Akt Thr308; Sarbassov et al., 2005; Shao et al., 2006) is constitutively phosphorylated and exhibited no sensitivity to BCR stimulation or Ca2+ (Figure 4J). Thus, while Ca2+ regulates Akt-dependent TSC2 phosphorylation, Akt Thr308 phosphorylation negatively correlates with mTORC1 activity, suggesting that Ca2+ control of mTORC1 is independent of Akt phosphorylation. We therefore asked if the Ca2+-regulated proteins CaM and Cn, which interact with Akt and the mTORC1 complex (Ni et al., 2007; Li et al., 2016; Zhou et al., 2015; Vaeth et al., 2017), might regulate their activity. Indeed, the CaM inhibitor W7 and the Cn inhibitor FK506 both attenuated BCR-induced S6 phosphorylation (Figure S3E). As Cn activity is regulated by CaM binding, we conclude that Ca2+-dependent control of mTORC1 in naive B cells is mediated by Cn-dependent regulation of Akt activity.
Ca2+ Signals Dynamically Tune mTORC1, NF-κB, and NFAT Activity
Given the relationship between the strength of BCR engagement and quantitative properties of Ca2+ dynamics (Figures 1K and 1L), we next asked if quantitative differences in BCR-induced Ca2+ signals might tune each of these key regulators of B cell activation. We began to dissect this relationship by examining the kinetics of Ca2+-dependent S6K phosphorylation (pS6K). In WT B cells, phosphorylation of S6K was evident 30 min after stimulation and increased for at least 90 additional minutes (Figure 5A). By contrast, in STIM DKO B cells, BCR engagement induced a transient increase in pS6K that peaked at 60 min and then decayed. The transient activation of mTORC1 in STIM DKO cells suggests that mTORC1 activation may be sensitive to transient changes in cytoplasmic Ca2+ generated by endoplasmic reticulum (ER) release in the absence of Ca2+ entry (Figure 1K). To further examine if ER Ca2+ release is responsible for this transient mTORC1 activity in STIM DKO B cells, we loaded WT B cells with the high-affinity Ca2+ chelator BAPTA and then cultured these cells in Ca2+-free medium. Under these conditions, BCR engagement failed to induce any detectable phosphorylation of S6 or S6K (Figure 5B). Further evidence that the amplitude and duration of BCR-induced changes in cytoplasmic Ca2+ directly tune mTORC1 activity is our observation that increasing the strength of BCR engagement produced a progressive and rapid increase in S6 phosphorylation (Figure 5C). Importantly, although muted relative to WT cells, the induction of pS6 in STIM DKO cells also exhibited a graded increase (Figure 1K, right panel) that parallels the graded impact of BCR signal strength on the extent of Ca2+ release from the ER in STIM DKO cells (see Figures 1K and 1L). Altogether, these results establish that mTORC1 activity is dynamically tuned by quantitative properties of BCR-induced Ca2+ signals and that the BCR-induced Ca2+ release from the ER can induce at least transient activation of mTORC1 in the absence of sustained Ca2+ entry through Orai channels.
Figure 5. BCR-Induced Ca2+ Signals Tune mTORC1, NF-κB, and NFAT Activity.

(A) Immunoblot analysis of S6 kinase phosphorylation (pS6K) in WT and STIM DKO B cells following stimulation with anti-BCR for times indicated.
(B) pS6K and S6 phosphorylation (pS6) following anti-BCR stimulation of WT B cells cultured in media containing 0 mM Ca2+ and 2 mM Ca2+ and in cells loaded with membrane-permeant Ca2+ chelator (BAPTA) in 0 mM Ca2+ medium.
(C) Anti-BCR dose dependence of pS6 in WT and STIM DKO at 6 h (mean percentage ± SD from triplicate wells are representative of two independent experiments).
(D) Boxplot representation of initial peak (left), average plateau/sustained concentration (from 10 to 15 min after anti-BCR, middle), and total Ca2+ spikes (20-min interval, right) in WT B cells stimulated in media containing indicated [Ca2+]. Results are representative of three independent experiments with >50 cells per genotype and treatment.
(E) Extracellular Ca2+ dependence of Rel, Nfkbia, Myc, and Irf4 mRNA expression 4 h after anti-BCR stimulation in media containing 0, 0.05, 0.2, 0.5, and 2.0 mM Ca2+ (mean ± 95% confidence interval; *, statistically significant).
(F) Anti-BCR dose-dependent expression of c-Myc, Irf4, and c-Rel protein in WT and STIM DKO at 6 h (mean percentage ± SD of triplicate measurements; representative of at least two independent experiments0.
For (C), (D), and (F), statistical significance was determined relative to preceding concentration of anti-BCR or extracellular [Ca2+]. For all figures, *p < 0.05, **p < 0.01, ***p < 0.001.
We next sought to define the relationships among BCR signaling strength, Ca2+ dynamics (e.g., maximum peak, plateau, and spike frequency), and gene expression. To do this, we modulated the quantitative properties of intracellular Ca2+ signals by varying the extracellular [Ca2+] at a fixed level (10 µg/mL anti-BCR) of BCR engagement (Figure 5D). This approach allowed us to distinguish between the impact of changing Ca2+ dynamics and confounding effects driven by other BCR-linked signaling pathways. We then examined the impact of these quantitatively distinct Ca2+ signals on the expression of NF-κB and NFAT target genes, including Nfkbia, Irf4 (a previously described c-Rel target gene Grumont and Gerondakis, 2000), Myc, and Rel. For each of these genes (Figure 5E), we observed a direct relationship between extracellular Ca2+ and the level of expression. A graded increase in c-Myc, Irf4, and c-Rel protein was also observed when the BCR signal strength was varied at a fixed extracellular Ca2+ concentration, and like phosphorylation of S6, expression of each was muted at each signal strength in STIM DKO cells (Figure 5F). Together, these results establish a links among the strength of BCR engagement, quantitative properties of Ca2+ signals, and the pattern of NF-κB, NFAT, and mTORC1 activation. Furthermore, our results demonstrate that high-strength BCR signals can compensate to a limited extent for the absence of Ca2+ entry by modulating the amount of Ca2+ released from the ER.
Costimulatory Signals Bypass Ca2+ Signals to Promote Survival and Proliferation
Although BCR-induced Ca2+ signals drive cell survival, cell-cycle entry, and proliferation, under physiological conditions, these fates require licensing by co-activating signals. Among the best characterized is the CD40 ligand on T lymphocytes that cooperatively regulates B cell survival and proliferation during authentic immune responses (Luo et al., 2018). Therefore, we asked how CD40 signaling impacts the requirement for BCR-induced Ca2+ entry in cell survival and proliferation. Consistent with a previous study of STIM DKO B cells (Matsumoto et al., 2011), CD40 engagement alone or in conjunction with the BCR dramatically improved the viability of Mb1-cre+ and STIM DKO B cells (Figure 6A versus Figure 1H) by preventing caspase-3-mediated intrinsic apoptosis (Figures 6B and 6C). Furthermore, CD40 co-engagement dramatically increased cell growth (Figure 6D) and the proportion of STIM DKO B cells that proliferate relative to BCR stimulation alone (Figures 6E and 6F). Notably, CD40 co-engagement failed to fully rescue cell-cycle entry (Figure 6G) and proliferation of STIM DKO B cells (Figure 6H). Importantly, this rescue is independent of Ca2+, as CD40 engagement has no impact on Ca2+ signaling dynamics (Figure S4A). Interestingly, stimulation with the TLR9 agonist CpG, which we previously found to mobilize cytoplasmic Ca2+ in B cells via scavenger-receptor-B1-mediated activation of TRPC3 channels (Zhu et al., 2009), also rescued WT and STIM DKO B cells from death (Figures S4B and S4C) and rescued STIM DKO cells from impaired proliferative expansion (Figures S4B and S4D). Thus, while alternative mechanisms of mobilizing Ca2+ could account for the ability of CpG to fully rescue defects in survival and proliferation of STIM DKO cells, CD40 costimulation bypasses the Ca2+-dependent regulation of survival and proliferation (Zhu et al., 2009).
Figure 6. CD40 Costimulation Promotes B Cell Survival and Proliferation.

(A) D23+ B cells from WT (black) and STIM DKO (red) mice were cultured for indicated times in media containing anti-CD40 ± anti-BCR. The total number of live cells was enumerated, and mean cell numbers (±SD of triplicate wells) are plotted for the indicated time points.
(B) Intracellular cleaved caspase-3 in live and dead anti-BCR-stimulated WT and STIM DKO B cells (24 h).
(C) Frequency of cells containing cleaved caspase-3 from (B) (mean percentage ± SD of triplicate measurements).
(D) Cell size (FSC-A) analysis of WT and STIM DKO B cells stimulated with anti-BCR and anti-CD40 (results from triplicate wells and two independent experiments).
(E) CFSE dilution and live/dead analysis of WT and STIM DKO CD23+ B cells stimulated as indicated for 72 h. Plots are representative of results from triplicate wells.
(F) Total live cells from (E) (mean ± SD from triplicate wells).
(G) Ki67 expression in WT and STIM DKO B cells following 32-h stimulation with anti-BCR in the absence or presence of anti-CD40 (mean percentage ± SD of Ki67+ cells from triplicate wells from at least two independent experiments).
(H) Percentage of divided cells and mean division number for data shown in (E) (mean ± SD from triplicate wells).
For all figures, *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4.
CD40-Dependent Canonical NF-κB Activation Rescues B Cells from Apoptosis
Given the established requirement for NF-κB in B cell survival, we next asked whether CD40-mediated rescue of STIM DKO B cells from apoptosis reflects its ability to bypass Ca2+-dependent steps of NF-κB activation. Indeed, CD40 and BCR co-engagement induced robust IKKβ and IκBα phosphorylation and IκBα degradation in STIM DKO B cells (Figure 7A). Furthermore, CD40 costimulation rescued p65 and c-Rel nuclear translocation (Figure 7B) and the expression of the NF-κB-dependent genes Nfkbia and Nfkb2 (Figure 7C). While CD40 significantly increased Bcl2l1 mRNA (Figure 7D) and Bcl-xL protein expression (Figure 7E), costimulation failed to fully rescue expression in STIM DKO B cells. Although CD40 activates both canonical and non-canonical NF-κB signaling (Coope et al., 2002), BLyS, which is a potent agonist of only non-canonical NF-κB (Stadanlick et al., 2008), improved the survival of unstimulated B cells but did not induce Bcl-xL expression (Figure 7E) or rescue viability of BCR-stimulated STIM DKO cells (Figure 7F). Thus, CD40-induced activation of canonical NF-κB is the primary mechanism by which CD40 rescues STIM DKO B cells from apoptosis, but CD40 costimulation fails to fully rescue Bcl2l1 gene expression.
Figure 7. Ca2+-Independent Activation of Canonical NF-κB and mTORC1 by CD40 Promotes B Cell Survival and Proliferation.
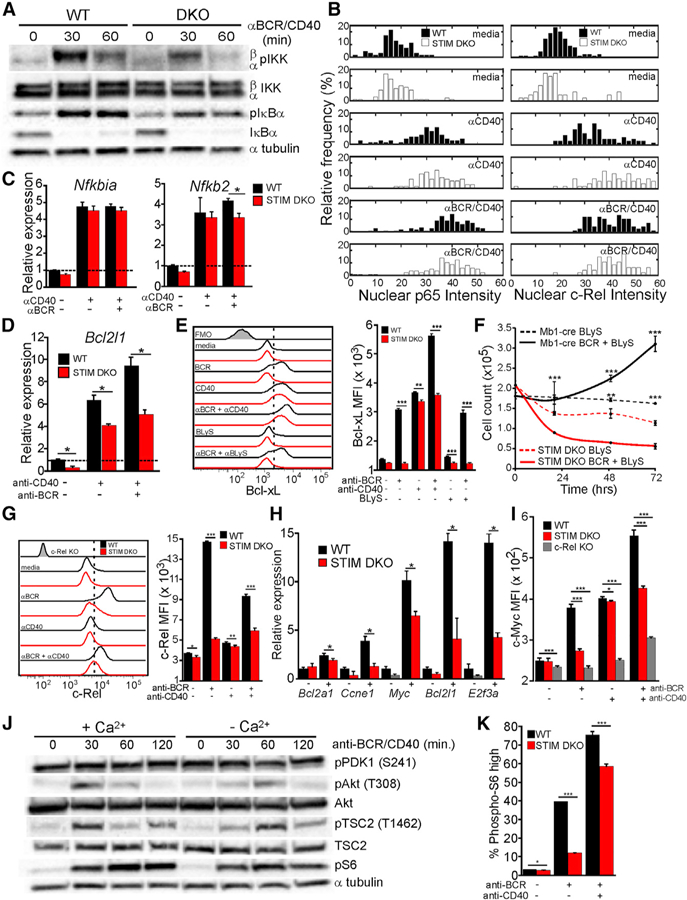
(A) Immunoblot analysis of canonical NF-κB activation in CD23+ WT and STIM DKO B cells stimulated with anti-BCR and anti-CD40 at indicated time points.
(B) NF-κB p65 (left) and c-Rel (right) nuclear intensity in WT (black) and STIM DKO (white) B cells (>70 cells per group) stimulated as indicated.
(C and D) (C) Nfkbia (IκBα) and Nfkb2 (p100) and (D) Bcl2l1 (Bcl-xL) mRNA expression in CD23+ B cells from WT and STIM DKO mice stimulated for 3 h with anti-CD40 alone or anti-CD40 and anti-BCR (mean ± 95% confidence interval; *, statistically significant).
(E) Bcl-xL expression in WT (black line) and STIM DKO (red line) B cells after 6 h of anti-BCR in the absence or presence of BLyS (100 ng/mL) and the absence or presence of anti-CD40 (left) and plot of mean Bcl-xL MFI (±SD) from triplicate wells (right).
(F) Impact of BLyS (100 ng/mL) on the viability of unstimulated and anti-BCR-stimulated WT and STIM DKO B cells (mean cell number ± SD of triplicate wells).
(G) c-Rel expression in WT (black line) and STIM DKO (red line) B cells stimulated as indicated for 12 h. Average MFI (±SD) of triplicate wells (right) is shown.
(H) Expression of c-Rel target genes Bcl2a1 (A1), Ccne1 (Cyclin E), Myc (c-Myc), Bcl2l1, and E2f3a (E2f3) in WT (black) and STIM DKO B cells (red) 24 h following stimulation with anti-BCR and anti-CD40 (mean ± 95% confidence interval; *, statistically significant).
(I) c-Myc expression in WT (black line), STIM DKO (red line), and c-Rel KO (gray) B cells following 20 h of culture with no stimulus (media), anti-BCR, or anti-BCR/CD40.
(J) Immunoblot of mTORC1 activity in B cells stimulated with anti-BCR and anti-CD40 in Ca2+ containing (2 mM) or Ca2+-free medium.
(K) Plot of phospho-S6 (pS6)-positive WT (black) and STIM DKO (red) B cells at 20 h of anti-BCR or anti-BCR/CD40 stimulation (mean percentage ± SD from triplicate wells). For all figures except where indicated, *p < 0.05, **p < 0.01, ***p < 0.001.
Ca2+ Entry Is Required for Efficient c-Rel-and NFAT-Dependent Gene Expression
Finally, we asked if the ability of CD40 to partially rescue cell-cycle entry and proliferation reflects its ability to bypass the requirement for Ca2+ in NFAT and mTORC1 activation and c-Rel expression. While CD40 drives robust IKK-dependent activation of preexisting c-Rel (Figure 7B), it did not efficiently rescue de novo c-Rel expression (Figure 7G) or the expression of the known anti-apoptotic and pro-proliferative c-Rel targets Bcl21l, Bcl2la1, Myc, Ccne1 (encoding cyclin E), and E2f3a (encoding E2F3) (Figure 7H) in STIM DKO cells (Tumang et al., 1998). The level of c-Myc induction by antigen is especially critical, as it determines the kinetics and extent of lymphocyte proliferation (Heinzel et al., 2017). Notably, c-Myc protein expression was comparably lower in CD40 costimulated STIM DKO and c-Rel KO cells than in WT cells (Figure 7I), indicating that CD40 is unable to fully restore c-Rel-and NFAT-dependent steps of c-Myc expression despite its ability to increase TSC2 and S6 phosphorylation in the absence of Ca2+ (Figure 7J) and pS6 in STIM DKO B cells (Figure 7K).
DISCUSSION
The critical role of B cells in adaptive immunity reflects their capacity to produce antibodies that opsonize or neutralize pathogens and perform immune-regulatory functions. Although a repertoire of B cells with virtually infinite antigen specificity is generated during development, initial interactions of naive B cells with antigen and costimulatory ligands dictate cell fates and functions critical for appropriate and effective immunity. Foremost among the gaps in our understanding of these processes are (1) how variations in the affinity and avidity of antigen/antigen receptor interactions and costimulatory or coactivating signals are encoded and (2) how these signals are decoded at key developmental/differentiation stages.
Results herein establish that variations in the strength of BCR engagement are encoded as dynamic and quantitatively distinct fluctuations in the concentration of cytoplasmic Ca2+. We find that the strength of BCR engagement is encoded in quantitative properties of intracellular Ca2+ signals, specifically their peak amplitude, steady-state concentration, and spike frequency. Subsequently, these variations in Ca2+ dynamics control critical steps that regulate B cell survival, cell-cycle entry, and proliferation. Mechanistically, we found that BCR-induced STIM/Orai-dependent Ca2+ entry promotes cell survival by activating the IKK complex, thereby initiating c-Rel/p65 nuclear localization and the transcription of anti-apoptotic genes (i.e., Bcl2l1 and Bcl2la1). Ca2+ entry also promotes Bcl-xL upregulation by increasing expression of c-Rel through NFAT-dependent transcription of Rel. ER release alone was insufficient to trigger IKK activation, c-Rel/p65 nuclear localization, de novo Rel expression, and NF-κB-dependent expression of Bcl2l1 and Bcl2la1. However, CD40 stimulation or costimulation rescued STIM DKO B cells from apoptosis by activating the IKK complex. While not addressed directly here, CD40 has previously been shown to activate NF-κB signaling through TNF receptor-associated factor (TRAF)-dependent mechanisms (Jabara et al., 2002; Ahonen et al., 2002) and our data indicate that this occurs independently of intracellular Ca2+ signals.
We further demonstrated that Ca2+ orchestrates cell-cycle entry and proliferation via mechanisms distinct from those that control cell survival. Specifically, we found that Ca2+ tunes the activity of mTORC1 and c-Myc; both are reported to coordinate metabolic reprogramming required for cell-cycle entry and proliferation (Boothby and Rickert, 2017; Waters et al., 2018; Grumont et al., 2002; Vaeth et al., 2017). Optimal c-Myc expression required NFAT-and c-Rel-dependent Myc transcription and mTORC1-dependent Myc translation. Similarly, robust and persistent activation of mTORC1 required STIM/Orai-mediated Ca2+ entry, as ER release induced only transient mTORC1 activity. Loss of Ca2+ entry effectively decreases the strength of BCR signaling, but CD40 costimulation can circumvent the requirement for Ca2+ in NF-κB-and mTORC1-dependent steps of survival, cycle entry, and proliferation. Collectively, our data support a model in which the strength of BCR signaling is encoded in patterns of Ca2+ dynamics, which are decoded by varying the extent of NF-κB, NFAT, and mTORC1 activation.
Recent studies have identified a role for NFAT in B cell function and specifically demonstrated that STIM/Orai-dependent NFAT-driven gene expression regulates Il10 induction by regulatory B cells to limit autoimmunity (Matsumoto et al., 2011; Tang et al., 2017). Here, we focused on the role for Ca2+ in NF-κB/c-Rel-dependent functions of B cells. While the general role of c-Rel-dependent gene expression in B cell survival, cell-cycle entry, and proliferation is established (Grumont et al., 1998; Tumang et al., 1998; Owyang et al., 2001; Hsia et al., 2002; Gilmore and Gerondakis, 2011; Cheng et al., 2003; Heise et al., 2014), we show that Ca2+ entry regulates c-Rel-dependent gene expression first by promoting IKK complex activation and then by driving NFAT-dependent de novo Rel transcription. Consistent with a direct functional link between Ca2+ entry and c-Rel is the similar impact of c-Rel and STIM deficiency on B cell viability and proliferative dynamics in vitro. This linkage reflects the requisite role for c-Rel in Bcl2l1 and Bcl2la1 expression and G1/S progression through its regulation of c-Myc, cyclin E, and E2f3 expression (Cheng et al., 2003; Grumont et al., 2002). Curiously, quantitative differences in c-Rel expression impacted the extent of c-Rel target gene expression, indicating that increases in c-Rel expression in the absence of adequate IKK complex activation may be sufficient to induce or maintain c-Rel target gene expression (Sen, 2006). Our finding that c-Myc expression is normal in IKKβ KO B cells supports this concept as c-Rel expression levels alone can impact binding at the Myc promoter to regulate the efficiency of its induction (Duyao et al., 1990; Grumont et al., 2002). Interestingly, c-Rel KO mice exhibit defects in B cell development, germinal center maintenance, and antibody production (Heise et al., 2014; Gerondakis and Siebenlist, 2010; Gilmore and Gerondakis, 2011) that are not observed in STIM DKO mice (Matsumoto et al., 2011; Mao et al., 2016). Thus, the escape by STIM DKO B cells from these additional defects may reflect the ability of CD40 and TLR9 costimulation to bypass the Ca2+ requirement for c-Rel and p65 activation in vivo.
Our work also clarifies the role of Ca2+ entry in B cell apoptosis. Previous studies linked BCR-induced Ca2+ signals, in the absence of costimulation to mitochondrial dysfunction and cell death (Akkaya et al., 2018). In agreement, our data indicate that weak BCR signaling, induced with low concentrations of anti-BCR or by loss of STIM proteins, cause mitochondrial-dependent intrinsic apoptosis. However, our data also reveal that higher-strength BCR/Ca2+ signals induce sufficient expression of anti-apoptotic genes to prevent apoptosis. Furthermore, at low BCR/Ca2+ signal strengths, we find that CD40 signaling prevents mitochondrial-dependent apoptosis by activating the IKK complex. Thus, the extent of B cell survival is tuned by BCR signal strength and costimulatory signals.
A paradox highlighted by our results is that mice with a B-cell-specific deletion of Stim1 and Stim2 exhibit no apparent defect in antibody production despite decreased survival and proliferation potential in vitro (Matsumoto et al., 2011; Mao et al., 2016). During T-cell-dependent (TD) immune responses, T-cell-derived signals (CD40 ligand and cytokines) facilitate B cell expansion in germinal centers. The discrepancy between the in vivo and in vitro phenotypes could reflect maturation stage-specific requirements for BCR-induced Ca2+ signals or the presence of factors that bypass Ca2+-dependent mechanisms of activation. Interestingly, germinal center B cells exhibit a cell-intrinsic block in BCR-induced Ca2+ signals (Khalil et al., 2012). This correlates with the reported inability of germinal center B cells to efficiently activate NF-κB, mTORC1, and c-Myc in response to BCR engagement (Luo et al., 2018). CD40 costimulation, acting independently of Ca2+, likely compensates for this rewiring of BCR signaling to facilitate optimal NF-κB, mTORC1, and c-Myc activation (Luo et al., 2018). Thus, the capacity of CD40 to drive optimal signaling of both germinal center B cells and STIM DKO B cells represents a potential mechanism to circumvent the requirement for Ca2+ entry in TD antibody responses. During T-cell-independent (TI) immune responses, TI antigens induce antibody production without the help of T cells. TI antigens include type 1 (TI-1), which act as polyclonal B cell activators (e.g., CpG and lipopolysaccharide), and type 2 (TI-2), which are polysaccharides that directly crosslink the BCR (e.g., Ficoll) (Mond et al., 1995). Consistent with our in vitro TLR9 stimulation assays, TI-1 immune responses are normal in STIM DKO mice (Matsumoto et al., 2011; Mao et al., 2016). In contrast, TI-2 responses are BCR mediated and would be expected to be Ca2+-entry dependent. However, TI-2 responses appear normal in STIM DKO mice. While not addressed here, TI-2 immune responses may require quantitatively smaller Ca2+ signals than those generated by STIM/Orai-mediated Ca2+ entry. Indeed, ablation of all three IP3 receptors (IP3R-TKO) and the attendant loss of BCR-induced ER release leads to decreased immunoglobulin M (IgM) antibody production in response to NP-Ficoll immunization (Tang et al., 2017). Additional studies are thus warranted to evaluate the cell-intrinsic role for Ca2+ release and entry in distinct B cell lineages, including marginal zone B cells, which are the primary mediators of the TI-2 antibody response (Mond et al., 1995).
The notion that the strength of antigen receptor engagement is reflected in distinct patterns of Ca2+ signaling is not new. Seminal efforts more than 20 years ago established how NFAT and NF-κB are tuned to quantitative differences in antigen-receptor-induced Ca2+ signals (Dolmetsch et al., 1998; Healy et al., 1998; Li et al., 1998). Building upon this framework, we focused on developing a more comprehensive understanding of the mechanisms by which variations in Ca2+ signals, acting on NFAT, NF-κB, and mTORC1, regulate key steps of B cell activation (survival, cell-cycle entry, and proliferation). Furthermore, we determined how costimulatory signals, which can be generated by T cell help (CD40 ligand) or bacterial pathogen-associated molecular patterns (PAMPs) (e.g., CpG) to validate antigen authenticity, lower the BCR/Ca2+ threshold required to drive efficient B cell activation. It is our expectation that this new framework can be used to develop rational approaches for modulating diseases that reflect dysregulation of B cell survival and proliferation associated with immune-mediated disease and cancer.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bruce D. Freedman (bruce@vet.upenn.edu). STIM1fl/fl, STIM2fl/fl, Mb1-cre single transgenic mice are available from Jackson Laboratories.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal studies were carried out in compliance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania and in accordance with the recommendations in the Guide and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the IACUC of the University of Pennsylvania, Philadelphia, PA. C57BL/6 mice (WT) were obtained from the Jackson Laboratory. Stim1fl/fl and Stim2fl/fl mice (Oh-Hora et al., 2008) were a generous gift from Dr. Anjana Rao (UCSD). Rel−/− mice (Liou et al., 1999) were obtained from Dr. Hsiou-chi Liou. Mb1-cre mice (Hobeika et al., 2006) were obtained from Dr. Michael Atchison (University of Pennsylvania) and Dr. Montserrat Anguera (University of Pennsylvania). Ikk2fl/fl mice (Greten et al., 2004) were obtained from Dr. Michael Karen (UCSD). Bcl2l1Tg (Grillot et al., 1996) mice were obtained from Dr. Michael Cancro. 6-to 16 week old age and sex matched mice were used for all experiments.
METHOD DETAILS
Cell culture and stimulation
Naive splenic B cells were purified by positive selection using CD23 coated microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) followed by magnetic selection of labeled cells (B cell purity > 95%). All primary mouse B cells were cultured in RPMI 1640 medium (BioWhittaker, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS, Hyclone, Thermoscientific, Logan, Utah), 2 mM L-glutamine, penicillin (50 U/ml), streptomycin (50 U/ml), pyruvate (1 mM), MEM NEAA (100 uM), 2-ME (55 uM), HEPES (10 mM). For experiments in which B cells were stimulated in Ca2+ free media, 0.5 mM EGTA was added to complete RPMI media.-For in vitro stimulation assays, BCR was engaged using soluble 10 mg/mL anti-mouse F(ab′)2 antibody (Jackson ImmunoResearch, West Grove, PA) unless indicated otherwise. For CD40 engagement, LEAF purified anti-CD40 antibody (HM40–3, BioLegend, San Diego, CA) was used at a final concentration of 2 ug/mL. CpG (ODN1826: 5′-TCC ATG ACG TTC CTG ACG TT-3′) was synthesized and high-performance liquid chromatography purified by Integrated DNA Technologies (Coralville, IA). CpG was used at a final concentration of 1 µM. BLyS (R&D Systems, Minneapolis, MN) was used at final concentration of 100 ng/mL.
Flow Cytometry and FACS
Cell viability, cell size, and intracellular protein expression were measured by flow cytometry. For cell counting, 123count eBeads Counting Beads (Thermo Fisher) were used according to the manufacturer’s instructions. For all flow cytometry experiments, cells were transferred to a round bottom 96 well plate, washed with FACS buffer (DPBS, 0.5% Bovine Serum Albumin, 2 mM EDTA), and stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (L34957; Thermo Fisher) or LIVE/DEAD Fixable eF780 Dead Cell Stain (65–0865; Thermo Fisher) and Fc block (anti-CD16/32) prior to additional staining or fixation. Following additional processing (see below) cells were collected on an LSRFortessa or FACSCanto (BD Biosciences) and data were analyzed with FlowJo 9.9.6 software. Cells were gated on lymphocytes (by forward scatter (FSC-A) and side scatter (SSC-A), singlets (by FSC-W versus FSC-H and SSC-W versus SSC-H), and viability dye excluding cells. For analysis of in vitro stimulated B and T cells, cells were fixed with 4% PFA for 10 minutes followed by permeabilization with 1X Permeabilization Buffer (Thermo Scientific). Cells were stained overnight with antibodies to Myc PE (clone: D84C12, Cell Signaling), phospho-S6 PE-Cy7 (clone: D57.2.2E, Cell Signaling), c-Rel eF660 (clone: 1RE-LAH5, Thermo Scientific) or c-Rel PE (clone: REA397, Miltenyi Biotec), Irf4 PE-Vio770 (clone: REA201, Miltenyi Biotec), and/or Cleaved Caspase-3 (Asp175) (clone: 5A1E, Cell Signaling). For experiments with Bcl-xL, cells were stained for Bcl-xL (clone: 54H65, Cell Signaling) following by Alex Fluor 488 goat anti-rabbit secondary antibody (Thermo Fisher). For CFSE dilution assays, cells were washed 2X with DPBS before staining with 5 µM CFSE (Biolegend) for 6 minutes followed by the addition of FBS and 2 additional washes with complete RPMI. Labeled B cells were incubated for 72 hours in complete RPMI. CFSE dilution, viability, and cell counts were then assessed by flow cytometry. For AnnexinV apoptosis assays, cells were stained with L/D eF780 Viability dye and Annexin V FITC (Thermo Fisher) in binding buffer as described by the manufacturer.
Immunofluorescence analysis of c-Rel, p65, and Foxo1 localization
For immunofluorescence analysis, B cells were adhered to coverslips coated with Cell-Tak (BD Biosciences, Franklin Lakes, NJ) after stimulation. Cells were fixed with 4% paraformaldehyde for 10 minutes, permeabilized with 0.2% Triton X-100 for 5 minutes, treated with 5% BSA for 1 hour, stained with primary antibody O/N at 4C, and stained with secondary antibody at RT for 45 minutes. Nuclei were then labeled with Hoechst 33342 (Life Technologies, Cat #H3570, 4 µg/ml), washed 3 3 5 minutes in 1% BSA in PBS, and mounted in Fluoromount B (Fisher). Images of p65, c-Rel, and Foxo1 localization were obtained with an inverted Leica SP5-II inverted (DMI6000 based) confocal microscope mounted on a Leica DMI4000 microscope (Leica Microsystems, Wetzlar, Germany) and imaging parameters were optimized independently for each channel to maintain fluorescence within the linear range while maximizing intensity resolution. Images analyzed using custom Macros in ImageJ to obtain nuclear intensities of p65, c-Rel, and Foxo1.
Immunoblot analysis
All cells were harvested and lysed using NP-40 lysis buffer consisting of 50 mM Tris-HCl (pH 7.5), 20mM EDTA, 1% NP-40 and complete inhibitors (1mM Sodium Orthovanadate, 1mM PMSF, 10 µg/ml Leupeptin, 5 µg/ml Aprotinin). Protein concentrations in cell lysates were determined using the Bio-Rad reagent (Bio-Rad Laboratories, Hercules, CA) and quantified in a Cary 50 Bio UV-visible Spectrophotometer. Proteins were resolved by SDS–polyacrylamide gel electrophoresis (4%–15%, Bio-Rad, Hercules, CA) then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA). Membranes were probed with respective primary anti-mouse antibodies including S6 Ribosomal Protein (54D2) Mouse mAb, Phospho-Tuberin/TSC2 (Thr1462) Antibody, Tuberin/TSC2 (D93F12) XP® Rabbit mAb #4308, Phospho-Akt (Thr308) (244F9) Rabbit mAb, Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb, Akt (pan) (C67E7) Rabbit mAb, Phospho-PDK1 (Ser241) (C49H2) Rabbit mAb, Phospho-p70 S6 Kinase (Thr389) (108D2) Rabbit mAb, p70 S6 Kinase (49D7) Rabbit mAb, Phospho-IKKα/β (Ser176/180) (16A6) Rabbit mAb, IKKβ (D30C6) Rabbit mAb, IKKα, Phospho-IκBα (Ser32) (14D4) Rabbit mAb, anti-IκBα (L35A5) Mouse mAb, anti-alpha-tubulin, Tuberin/TSC2 (D93F12) XP® Rabbit mAb #4308. Blots were then incubated with Protein A HRP, anti-rabbit HRP, or anti-mouse HRP secondary antibodies (Thermo Fisher). Blots were developed with enhanced chemiluminescence using pierce ECL Western Blotting Substrate (Pierce, Rockford, IL). All immunoblots presented are from a single experiment representative of at least three independent experiments.
Quantitative Real-Time PCR (qRT-PCR)
cDNA was synthesized from RNA isolated (RNeasy Plus Micro Kit, QIAGEN) from cells with a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). cDNA was amplified with on a 6500 Fast Real-Time PCR System (Applied Biosciences, Warrington, United Kingdom) using Power SYBR Green PCR Master Mix (Applied Biosciences). Ct values were obtained in triplicate for each target including Nfkbia, Nfkb2, Myc, Rel, Irf4, Bcl2l1, Bcl2l11, Bcl2a1, Bcl2, Mcl1, Bak1, Bax, Bad, Bbc3, Bid, Pmaimp1, Ccne1, E2f3, Ubc, and B2M (See Table S1). Data were analyzed with the instrument software v1.3.1 (Applied Biosystems, Warrington, United Kingdom). All plots show relative expression based on difference between stimulated and unstimulated samples. For all plots mean ± 95% confidence interval is shown.
Calcium Imaging
Mouse B cells (5 million/mL) were loaded with 3 µM fura-2 acetoxymethyl ester (Molecular Probes, Eugene, OR) in external solution containing 145mM NaCl, 4.5mM KCl, 2mM CaCl2, 1mM MgCl2, 10mM glucose, 10mM HEPES, 2mM glutamine, and 2% fetal bovine serum (Hyclone, ThermoScientific, Logan, Utah) for 10 minutes at 25°C. Cells were adhered to coverslips coated with Cell-Tak (BD Biosciences, Franklin Lakes, NJ), mounted on the stage of a Leica DMI6000 microscope configured with a Photometrics Evolve 512 Camera (Tucson, AZ) using an Olympus 40x oil objective (Shinjuku, Tokyo, Japan), and images were acquired with MetaFluor software (Molecular Devices, Downingtown, PA). The cells were then perfused with the balanced salt solution prior to addition of 10 µg/ml anti-IgM. Ca2+ mobilization was analyzed by plotting the emission ratio of 340/380-nm excitation for each cell. Each plot is the averaged ratio from at least 30 cells and is representative of at least 3 independent experiments. Analysis of Fura-2 ratios was completed in MATLAB (Mathworks, Massachusetts, USA) using custom written scripts. Maximum Fura-2 ratios were identified by calculating the maximum value within the first 5 minutes of recording and subtracting the average Fura-2 ratio from the first 0.5 minutes for each cell. The plateau value was measured by calculating the median Fura-2 ratio for each cell during the final 5 minutes of recording (10 min – 15 minutes). Spike interval number was calculated by detecting local minima and maxima and determined the absolute magnitude between the two local extrema. A fura-2 ratio “spike” threshold of 0.075 was identified as most accurate for spike detection. The total number of spikes were determined throughout the recording. Data visualization was performed using the statistical computing environment, R (v3.5.1), and RStudio (v1.1.456).
QUANTIFICATION AND STATISTICAL ANALYSIS
Significance for all statistical tests was determined at p values < 0.05 and is shown as * for p < 0.05, ** for p < 0.01, and *** for p < 0.001 in all figures except for qPCR data in which 95% confidence intervals were used. All data were assessed for normality using probability plots and the Kolmogorov-Smirnov test for normality. Normal distributions were compared using either two-tailed Student’s t test or Welch’s t test (depending on equality of variance) and non-normal data were compared using Wilcoxon rank sum test. The correlation between c-Rel and Bcl-xL expression was determined by calculating the Pearson correlation coefficient in WT anti-BCR stimulated samples. Unless otherwise indicated, results are representative of at least three independent experiments.
DATA AND CODE AVAILABILITY
This study did not generate/analyze relevant datasets.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-FoxO1 (C29H4) Rabbit mAb | Cell Signaling | Cat#2880S; RRID:AB_2106495 |
| anti-c-Myc PE (D84C12) | Cell Signaling | Cat#14819 RRID:AB_2798629 |
| S6 Ribosomal Protein (54D2) Mouse mAb | Cell Signaling | Cat#2317 RRID:AB_2238583 |
| Phospho-Tuberin/TSC2 (Thr1462) Antibody | Cell Signaling | Cat#3611 RRID:AB_329855 |
| Phospho-Akt (Thr308) (244F9) Rabbit mAb | Cell Signaling | Cat#4056 RRID:AB_331163 |
| Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb | Cell Signaling | Cat#4060S |
| Akt (pan) (C67E7) Rabbit mAb | Cell Signaling | Cat#4691 RRID:AB_915783 |
| Phospho-PDK1 (Ser241) (C49H2) Rabbit mAb | Cell Signaling | Cat#3438 RRID:AB_2161134 |
| Phospho-p70 S6 Kinase (Thr389) (108D2) Rabbit mAb | Cell Signaling | Cat#9234 RRID:AB_2269803 |
| p70 S6 Kinase (49D7) Rabbit mAb | Cell Signaling | Cat#2708 RRID:AB_10694087 |
| Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb | Cell Signaling | Cat#9664 RRID:AB_2070042 |
| Phospho-IKKα/β (Ser176/180) (16A6) Rabbit mAb | Cell Signaling | Cat#2697 RRID:AB_2079382 |
| IKKβ (D30C6) Rabbit mAb | Cell Signaling | Cat#8943 RRID:AB_11024092 |
| Phospho-IκBα (Ser32) (14D4) Rabbit mAb | Cell Signaling | Cat#2859 RRID:AB_561111 |
| IκBα (L35A5) Mouse mAb (Amino-terminal Antigen) #4814 | Cell Signaling | Cat#4814S RRID:AB_390781 |
| anti-alpha-tubulin | Sigma Aldrich | Cat#T1568 |
| anti-Ki67 | Millipore Sigma | Cat#AB9260 |
| Tuberin/TSC2 (D93F12) XP® Rabbit mAb | Cell Signaling | Cat#4308 RRID:AB_10547134 |
| anti-phospho-S6 PE-Cy7 (D57.2.2E) | Cell Signaling | Cat#34411S RRID:AB_2799051 |
| anti-c-Rel eF660 (1RELAH5) | Thermo Scientific | Cat#50–6111-80 RRID:AB_2574261 |
| anti-c-Rel PE (REA397) | Miltenyi Biotec | Cat#130–124-715 RRID:AB_2651454 |
| Anti-IRF-4-PE-Vio770 | Miltenyi Biotec | Cat#130–100-909 RRID:AB_2652519 |
| anti-Bcl-xL (54H6) | Cell Signaling | Cat# 2764S RRID:AB_2228008 |
| Alex Fluor 488 goat anti-rabbit secondary antibody | Thermo Fisher | Cat#A11008 RRID:AB_143165 |
| AffiniPure F(ab′)2 Fragment Goat Anti-Mouse IgM, Mu Chain Specific | Jackson ImmunoResearch | Cat#115–006-020 RRID:AB_2338469 |
| LEAF Purified anti-mouse CD40 antibody (HM40–3) | BioLegend | Cat#102908 RRID:AB_312951 |
| Purified anti-mouse CD16/32 Antibody | BioLegend | Cat#101302 RRID:AB_312801 |
| Protein A HRP | Cell Signaling | Cat#12291 |
| Goat anti-Rabbit IgG (H+L) Secondary Antibody, HRP | Thermo Fisher | Cat#65–6120 RRID:AB_2533967 |
| Goat anti-Mouse IgG (H+L) Secondary Antibody, HRP | Thermo Fisher | Cat#62–6520 RRID:AB_88369 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CFSE | Biolegend | Cat#423801 |
| Fura-2AM | Molecular Probes | CAS 108964–32-5 |
| FK506 (tacrolimus) | Tocris Bioscience | Cat#3631/10 |
| Cyclosporine A | Tocris Bioscience | Cat#1101 |
| Torin-1 | Tocris Bioscience | Cat#4247 |
| Rapamycin (Sirolimus) | Selleck Chemical | Cat#S1039 |
| W-7 hydrochloride | Tocris Bioscience | Cat#0369 |
| Fluoromount G | Molecular Probes | CAS 108964–32-5 |
| eBioscience Permeabilization Buffer (10X) | Thermo Fisher | Cat#00–8333-56 |
| Paraformaldehyde Aqueous Solution —16% | Electron Microscopy Systems | CAS #30525–89-4 |
| Corning Cell-Tak Cell and Tissue Adhesive | Fisher Scientific | Cat#CB40240 |
| Hoechst 33342 | Life Technologies | Cat#H3570 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain | Thermo Fisher | Cat#L34957 |
| LIVE/DEAD Fixable eF780 Dead Cell Stain | Thermo Fisher | Cat#65–0865 |
| Annexin V, FITC conjugate | Thermo Fisher | Cat#A13199 |
| Critical Commercial Assays | ||
| CD23 MicroBeads, mouse | Miltenyi Biotec | Cat#130–098-784 |
| RNeasy Micro Kit | QIAGEN | Cat#74034 |
| PERFECTA SYBR SMX L-ROX | VWR | Cat#101414–168 |
| High-Capacity RNA-to-cDNA Kit | Applied Biosystems | Cat#4387406 |
| AccuStart II GelTrack PCR SuperMix, QuantaBio | VWR | Cat#76047–140 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Bcl-xL Tg: B6.Bcl2l1Tg mice | Grillot et al., 1996 | N/A |
| Mouse: Stim1fl/flStim2fl/fl: B6(Cg)-Stim1tm1Rao/J Stim2tm1Rao/J | Oh-Hora et al., 2008 | N/A |
| Mouse: Mb1-cre(Stim1fl/flStim2fl/fl): B6(Cg)-Stim1tm1Rao/J xStim2tm1Rao/J x B6.C(Cg)-Cd79atm1(cre) Reth/EhobJ | This paper | N/A |
| Mouse: Ikk2fl/fl | Greten et al., 2004 | N/A |
| Mouse: Mb1-cre(Ikk2fl/fl) | This paper | N/A |
| Mouse: C57BL/6J | Jackson Laboratory | N/A |
| Mouse: Ripk3−/−Casp8−/− | Oberst et al., 2011 | N/A |
| Mouse: Mb1-cre+: B6.C(Cg)-Cd79atm1(cre)Reth/EhobJ | Hobeika et al., 2006 | N/A |
| Mouse: Rel−/− | Liou et al., 1999 | N/A |
| Oligonucleotides | ||
| qRT-PCR Primers | See Table S1 for all primers | N/A |
| Software and Algorithms | ||
| ImageJ | ImageJ | https://imagej.net/ImageJ |
| Flow Jo_v9 | Flow Jo | https://www.flowjo.com |
| Rstudio_v1.1.456 | Rstudio | https://rstudio.com/ |
| R_v3.5.1 | R | https://www.r-project.org/ |
| Bioconductor_v3.10 | Bioconductor | https://www.bioconductor.org/install/ |
| Matlab_R2017b | MATLAB | https://www.mathworks.com/products/matlab.html |
Highlights.
BCR signal strength is encoded as quantitatively distinct intracellular Ca2+ signals
Ca2+ dynamics are decoded by NF-κB, NFAT, and mTORC1 to drive cell fates
BCR-induced Ca2+ signals are required for maximal B cell survival and proliferation
CD40 compensates for weak BCR/Ca2+ signals to rescue NF-κB-and mTORC1-dependent fates
ACKNOWLEDGMENTS
We thank Dr. Hsiou-chi Liou (Cornell University) for providing the Rel−/− mice, Dr. Michael Atchison (University of Pennsylvania) and Dr. Montserrat Anguera (University of Pennsylvania) for providing mb1-cre mice, and Dr. Michael Karin (UCSD) for providing IKK2fl/fl mice. We thank A.A.D., L.C., A.P., and M.C.A. for assisting with methodology and supplying valuable resources. These studies were funded by the NIH (grants R56AI125415 and RO1AI60921 to B.D.F., grants R01HL096642 and R01AR066567 to M.J.M., and AI128530 to I.E.B.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.03.038.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahonen C, Manning E, Erickson LD, O’Connor B, Lind EF, Pullen SS, Kehry MR, and Noelle RJ (2002). The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nat. Immunol 3, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akkaya M, Traba J, Roesler AS, Miozzo P, Akkaya B, Theall BP, Sohn H, Pena M, Smelkinson M, Kabat J, et al. (2018). Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat. Immunol 19, 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba Y, and Kurosaki T (2016). Role of calcium signaling in B cell activation and biology. Curr. Top. Microbiol. Immunol 393, 143–174. [DOI] [PubMed] [Google Scholar]
- Bajpai UD, Zhang K, Teutsch M, Sen R, and Wortis HH (2000). Bruton’s tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J. Exp. Med 191, 1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop RJ, Melamed D, Nemazee D, and Cambier JC (1999). Distinct signal thresholds for the unique antigen receptor-linked gene expression programs in mature and immature B cells. J. Exp. Med 190, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry CT, May MJ, and Freedman BD (2018). STIM-and Orai-mediated calcium entry controls NF-κB activity and function in lymphocytes. Cell Calcium 74, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothby M, and Rickert RC (2017). Metabolic regulation of the immune humoral response. Immunity 46, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, and Greenberg ME (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. [DOI] [PubMed] [Google Scholar]
- Cao Q, Yang Y, Zhong XZ, and Dong XP (2017). The lysosomal Ca2+ release channel TRPML1 regulates lysosome size by activating calmodulin. J. Biol. Chem 292, 8424–8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC, and Rajewsky K (2004). B cell receptor signal strength determines B cell fate. Nat. Immunol 5, 317–327. [DOI] [PubMed] [Google Scholar]
- Cheng S, Hsia CY, Leone G, and Liou HC (2003). Cyclin E and Bcl-xL cooperatively induce cell cycle progression in c-Rel−/−B cells. Oncogene 22, 8472–8486. [DOI] [PubMed] [Google Scholar]
- Conus NM, Hemmings BA, and Pearson RB (1998). Differential regulation by calcium reveals distinct signaling requirements for the activation of Akt and p70S6k. J. Biol. Chem 273, 4776–4782. [DOI] [PubMed] [Google Scholar]
- Coope HJ, Atkinson PG, Huhse B, Belich M, Janzen J, Holman MJ, Klaus GG, Johnston LH, and Ley SC (2002). CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J 21, 5375–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, and Goodnow CC (1998). Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity 8, 497–508. [DOI] [PubMed] [Google Scholar]
- Csibi A, Lee G, Yoon SO, Tong H, Ilter D, Elia I, Fendt SM, Roberts TM, and Blenis J (2014). The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol 24, 2274–2280. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, and Cambier J (2004). B cell antigen receptor signaling 101. Mol. Immunol 41, 599–613. [DOI] [PubMed] [Google Scholar]
- Deb TB, Coticchia CM, and Dickson RB (2004). Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem 279, 38903–38911. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7, 11–20. [DOI] [PubMed] [Google Scholar]
- Dickens LS, Powley IR, Hughes MA, and MacFarlane M (2012). The ‘complexities’ of life and death: death receptor signalling platforms. Exp. Cell Res 318, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Divolis G, Mavroeidi P, Mavrofrydi O, and Papazafiri P (2016). Differential effects of calcium on PI3K-Akt and HIF-1a survival pathways. Cell Biol. Toxicol 32, 437–449. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, and Healy JI (1997). Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386, 855–858. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, and Lewis RS (1998). Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Buckler AJ, and Sonenshein GE (1990). Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc. Natl. Acad. Sci. USA 87, 4727–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enders A, Bouillet P, Puthalakath H, Xu Y, Tarlinton DM, and Strasser A (2003). Loss of the pro-apoptotic BH3-only Bcl-2 family member Bim inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J. Exp. Med 198, 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan WM, Corthésy B, Bram RJ, and Crabtree GR (1991). Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature 352, 803–807. [DOI] [PubMed] [Google Scholar]
- Frantz B, Nordby EC, Bren G, Steffan N, Paya CV, Kincaid RL, Tocci MJ, O’Keefe SJ, and O’Neill EA (1994). Calcineurin acts in synergy with PMA to inactivate I kappa B/MAD3, an inhibitor of NF-kappa B. EMBO J 13, 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galibert L, Burdin N, Barthélémy C, Meffre G, Durand I, Garcia E, Garrone P, Rousset F, Banchereau J, and Liu YJ (1996). Negative selection of human germinal center B cells by prolonged BCR cross-linking. J. Exp. Med 183, 2075–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerondakis S, and Siebenlist U (2010). Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol 2, a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore TD, and Gerondakis S (2011). The c-Rel transcription factor in development and disease. Genes Cancer 2, 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, and Llambi F (2015). Cell death signaling. Cold Spring Harb. Perspect. Biol 7, a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, and Karin M (2004). IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296. [DOI] [PubMed] [Google Scholar]
- Grillot DA, Merino R, Pena JC, Fanslow WC, Finkelman FD, Thompson CB, and Nunez G (1996). bcl-x exhibits regulated expression during B cell development and activation and modulates lymphocyte survival in transgenic mice. J. Exp. Med 183, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, Rourke IJ, O’Reilly LA, Strasser A, Miyake K, Sha W, and Gerondakis S (1998). B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF-kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J. Exp. Med 187, 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont RJ, Strasser A, and Gerondakis S (2002). B cell growth is controlled by phosphatidylinosotol 3-kinase-dependent induction of Rel/NF-kappaB regulated c-myc transcription. Mol. Cell 10, 1283–1294. [DOI] [PubMed] [Google Scholar]
- Grumont RJ, and Gerondakis S (2000). Rel Induces Interferon Regulatory Factor 4 (IRF-4) Expression in Lymphocytes. J. Exp. Med 191, 1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumont R, Lock P, Mollinari M, Shannon FM, Moore A, and Gerondakis S (2004). The mitogen-induced increase in T cell size involves PKC and NFAT activation of Rel/NF-kappaB-dependent c-myc expression. Immunity 21, 19–30. [DOI] [PubMed] [Google Scholar]
- Hao S, and August A (2005). Actin depolymerization transduces the strength of B-cell receptor stimulation. Mol. Biol. Cell 16, 2275–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, and Goodnow CC (1997). Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 6, 419–428. [DOI] [PubMed] [Google Scholar]
- Healy JI, Dolmetsch RE, Lewis RS, and Goodnow CC (1998). Quantitative and qualitative control of antigen receptor signalling in tolerant B lymphocytes. Novartis Found. Symp 215, 137–144, discussion 144–145, 186–190. [DOI] [PubMed] [Google Scholar]
- Heinzel S, Binh Giang T, Kan A, Marchingo JM, Lye BK, Corcoran LM, and Hodgkin PD (2017). A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat. Immunol 18, 96–103. [DOI] [PubMed] [Google Scholar]
- Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, and Klein U (2014). Germinal center B cell maintenance and differentiation are controlled by distinct NF-κB transcription factor subunits. J. Exp. Med 211, 2103–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemon P, Renaudineau Y, Debant M, Le Goux N, Mukherjee S, Brooks W, and Mignen O (2017). Calcium signaling: from normal B cell development to tolerance breakdown and autoimmunity. Clin. Rev. Allergy Immunol 53, 141–165. [DOI] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. USA 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KL, Antony P, Lowe J, Shinners N, Sarmah B, Wente SR, Wang D, Gerstein RM, and Khan WN (2006). Transitional B cell fate is associated with developmental stage-specific regulation of diacylglycerol and calcium signaling upon B cell receptor engagement. J. Immunol 177, 5405–5413. [DOI] [PubMed] [Google Scholar]
- Hsia CY, Cheng S, Owyang AM, Dowdy SF, and Liou HC (2002). c-Rel regulation of the cell cycle in primary mouse B lymphocytes. Int. Immunol 14, 905–916. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, and Guan KL (2002). TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol 4, 648–657. [DOI] [PubMed] [Google Scholar]
- Jabara H, Laouini D, Tsitsikov E, Mizoguchi E, Bhan A, Castigli E, Dedeoglu F, Pivniouk V, Brodeur S, and Geha R (2002). The binding site for TRAF2 and TRAF3 but not for TRAF6 is essential for CD40-mediated immunoglobulin class switching. Immunity 17, 265–276. [DOI] [PubMed] [Google Scholar]
- Jellusova J, and Nitschke L (2012). Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front. Immunol 2, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Cambier JC, and Shlomchik MJ (2012). B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science 336, 1178–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V, Famiglietti S, Lacaud G, Lang P, Rider JE, Kay BK, Cambier JC, and Nemazee D (1998). Antigens varying in affinity for the B cell receptor induce differential B lymphocyte responses. J. Exp. Med 188, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Shinohara H, and Baba Y (2010). B cell signaling and fate decision. Annu. Rev. Immunol 28, 21–55. [DOI] [PubMed] [Google Scholar]
- Li W, Llopis J, Whitney M, Zlokarnik G, and Tsien RY (1998). Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 392, 936–941. [DOI] [PubMed] [Google Scholar]
- Li RJ, Xu J, Fu C, Zhang J, Zheng YG, Jia H, and Liu JO (2016). Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 5, e19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, June CH, and Thompson CB (1988). Multiple mechanisms regulate c-myc gene expression during normal T cell activation. EMBO J 7, 2787–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, and Liou ML (1999). c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int. Immunol 11, 361–371. [DOI] [PubMed] [Google Scholar]
- Liu X, Berry CT, Ruthel G, Madara JJ, MacGillivray K, Gray CM, Madge LA, McCorkell KA, Beiting DP, Hershberg U, et al. (2016). T cell receptor-induced nuclear factor kB (NF-κB) signaling and transcriptional activation are regulated by STIM1-and Orai1-mediated calcium entry. J. Biol. Chem 291, 8440–8452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Weisel F, and Shlomchik MJ (2018). B cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center b cells. Immunity 48, 313–326.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Zhang J, Han Y, Luan C, Hu Y, Hao Z, and Chen M (2016). Deficient for endoplasmic reticulum calcium sensors Stim1 and Stim2 affects aberrant antibody affinity maturation in B cells. Oncotarget 7, 60885–60895. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, and Baba Y (2011). The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity 34, 703–714. [DOI] [PubMed] [Google Scholar]
- Mognol GP, de Araujo-Souza PS, Robbs BK, Teixeira LK, and Viola JP (2012). Transcriptional regulation of the c-Myc promoter by NFAT1 involves negative and positive NFAT-responsive elements. Cell Cycle 11, 1014–1028. [DOI] [PubMed] [Google Scholar]
- Mond JJ, Lees A, and Snapper CM (1995). T cell-independent antigens type 2. Annu. Rev. Immunol 13, 655–692. [DOI] [PubMed] [Google Scholar]
- Müller J, and Nitschke L (2014). The role of CD22 and Siglec-G in B-cell tolerance and autoimmune disease. Nat. Rev. Rheumatol 10, 422–428. [DOI] [PubMed] [Google Scholar]
- Newsholme EA, Crabtree B, and Ardawi MS (1985). The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep 5, 393–400. [DOI] [PubMed] [Google Scholar]
- Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, Kuro-O M, Rothermel BA, and Hill JA (2007). FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc. Natl. Acad. Sci. USA 104, 20517–20522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke L, Carsetti R, Ocker B, Köhler G, and Lamers MC (1997). CD22 is a negative regulator of B-cell receptor signalling. Curr. Biol 7, 133–143. [DOI] [PubMed] [Google Scholar]
- Nolz JC, Fernandez-Zapico ME, and Billadeau DD (2007). TCR/CD28-stimulated actin dynamics are required for NFAT1-mediated transcription of c-rel leading to CD28 response element activation. J. Immunol 179, 1104–1112. [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, and Green DR (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, and Rao A (2008). Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol 9, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owyang AM, Tumang JR, Schram BR, Hsia CY, Behrens TW, Rothstein TL, and Liou HC (2001). c-Rel is required for the protection of B cells from antigen receptor-mediated, but not Fas-mediated, apoptosis. J. Immunol 167, 4948–4956. [DOI] [PubMed] [Google Scholar]
- Palkowitsch L, Marienfeld U, Brunner C, Eitelhuber A, Krappmann D, and Marienfeld RB (2011). The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J. Biol. Chem 286, 7522–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, and Jones RG (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454.24115444 [Google Scholar]
- Peng SL, Gerth AJ, Ranger AM, and Glimcher LH (2001). NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 14, 13–20. [DOI] [PubMed] [Google Scholar]
- Petro JB, Rahman SM, Ballard DW, and Khan WN (2000). Bruton’s tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J. Exp. Med 191, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petro JB, Castro I, Lowe J, and Khan WN (2002). Bruton’s tyrosine kinase targets NF-kappaB to the bcl-x promoter via a mechanism involving phospholipase C-gamma2 following B cell antigen receptor engagement. FEBS Lett 532, 57–60. [DOI] [PubMed] [Google Scholar]
- Philip NH, DeLaney A, Peterson LW, Santos-Marrero M, Grier JT, Sun Y, Wynosky-Dolfi MA, Zwack EE, Hu B, Olsen TM, et al. (2016). Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced cytokine production independent of cell death. PLoS Pathog 12, e1005910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittner BT, and Snow EC (1998). Strength of signal through BCR determines the fate of cycling B cells by regulating the expression of the Bcl-2 family of survival proteins. Cell. Immunol 186, 55–62. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Scharenberg AM, Humphries LA, and Rawlings DJ (2007). Calcium signalling and cell-fate choice in B cells. Nat. Rev. Immunol 7, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R (2006). Control of B lymphocyte apoptosis by the transcription factor NF-kappaB. Immunity 25, 871–883. [DOI] [PubMed] [Google Scholar]
- Shao Z, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, Wang YM, Kyriakis J, Mohanlal R, Kuida K, et al. (2006). c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circ. Res 98, 111–118. [DOI] [PubMed] [Google Scholar]
- Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, and Crabtree GR (1988). Identification of a putative regulator of early T cell activation genes. Science 241, 202–205. [DOI] [PubMed] [Google Scholar]
- Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ 3rd, Brezski RJ, Treml LS, Jordan KA, Monroe JG, et al. (2008). Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat. Immunol 9, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, metabolism, and cancer. Cancer Discov 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Wang H, Lin Q, Fan F, Zhang F, Peng X, Fang X, Liu J, and Ouyang K (2017). Loss of IP3 receptor-mediated Ca2+ release in mouse B cells results in abnormal B cell development and function. J. Immunol 199, 570–580. [DOI] [PubMed] [Google Scholar]
- Tumang JR, Owyang A, Andjelic S, Jin Z, Hardy RR, Liou ML, and Liou HC (1998). c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur. J. Immunol 28, 4299–4312. [DOI] [PubMed] [Google Scholar]
- Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E, Berberich-Siebelt F, et al. (2017). Store-operated Ca2+ entry controls clonal expansion of T cells through metabolic reprogramming. Immunity 47, 664–679.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman L, Burakoff SJ, and Sen R (1995). FK506 inhibits antigen receptor-mediated induction of c-rel in B and T lymphoid cells. J. Exp. Med 181, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LR, Ahsan FM, Wolf DM, Shirihai O, and Teitell MA (2018). Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience 5, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam-Puc JC, Zhang L, Zhang Y, and Toellner KM (2018). Role of B-cell receptors for B-cell development and antigen-induced differentiation. F1000Res 7, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Clister TL, Lowry PR, Seldin MM, Wong GW, and Zhang J (2015). Dynamic visualization of mTORC1 activity in living cells. Cell Rep 10, 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Liu X, Treml LS, Cancro MP, and Freedman BD (2009). Mechanism and regulatory function of CpG signaling via scavenger receptor-B1 in primary B lymphocytes. J. Biol. Chem [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze relevant datasets.