

Abstract
Bioactive natural products often possess uniquely functionalized structures with unusual modes of action; however, the natural product itself is not always the active species. We discuss molecules that draw on protecting group chemistry or else require activation to unmask reactive centers, illustrating that nature is not only a source of complex structures but also a guide for elegant chemical transformations which provides ingenious chemical solutions for drug delivery.
Keywords: Natural products, Protecting groups, Prodrugs, Cytotoxicity, Mechanism of action
Introduction
Natural products (NPs) are small molecules produced by living organisms. The term “secondary metabolite” is often used to differentiate NPs from other molecules that are synthesized in the course of primary metabolism,[1] for example amino acids, nucleotides, neurotransmitters, etc. Secondary metabolites are not essential to the life of the producing organism, but thought to confer survival advantages.[1] A common strategy is to produce toxic compounds, in order to kill competing organisms or to deter predators. Clinically, such NPs have been used as antibiotics and as anticancer agents. In fact, the majority (65%) of anticancer drugs are either NPs or derived from an NP pharmacophore.[2]
NPs are products of natural selection over vast timescales and therefore are expected to be exquisitely optimized for their target. However, in the case of toxic compounds, some method of self-resistance must also co-evolve in order for the producing organism to survive. For NPs that operate through DNA alkylation or other types of damaging chemistry, an added consideration is that the compound should not be so reactive that its lifetime is too short to ever enter cells of the target organism(s). This concept is neatly illustrated by the parabolic relationship between reactivity and potency exhibited in a series of synthetic and natural duocarmycins.[3]
In nature these two problems are solved in a variety of ways, including the expression of efflux pumps[4] or use of carrier proteins (for example the apoproteins that bind 9-membered enediynes).[5] This article focuses on cytotoxic or antibacterial NPs that are effectively “prodrugs”, rendered inactive by protecting groups or otherwise requiring conversion before they can exert their effect. Through these examples it can be appreciated that nature can provide ingenious chemical solutions for drug delivery.
Protecting Groups in Self-Resistance – Macrolides and Bleomycin
In medicinal chemistry, protecting groups are often added to a critical part of a molecule to create an inactive species (a prodrug), which is then activated by metabolism or non-enzymatically in vivo.[6] The objective is either to alter the drug’s physical properties (for example to make it more lipophilic), or else to alter its pharmacokinetics. Very often, prodrugs are made by simple addition of groups such as acetyl or alkyl chains to the drug through amide or ester formation. Similarly, in nature there are examples where protecting groups are added to an NP, as a strategy for self-resistance. The macrolide antibiotics are an example of this. They are macrocyclic lactones which have a pendant desosamine sugar and inhibit bacterial protein synthesis through interaction with 23S rRNA.[7] The desosamine is essential for activity, because it forms crucial hydrogen bonds with the ribosome exit tunnel.[7] As one strategy for self-resistance, Streptomyces antibioticus glycosylates oleandomycin (1, Figure 1) at the 2′-OH of desosamine to give an inactive species.[8] Two proteins, OleI and OleN2 act as glycosyltransferases.[8] Upon glycosylation, 1 is transported out of the cell, the extracellular enzyme OleR deprotects it.[8] A similar process is seen in S. venezuelae, which produces methymycin (2, Figure 1) and neomethymycin (3). These compounds are glycosylated in the same position as for 1, and are deprotected by DesR, a protein that is likely excreted into the extracellular milieu.[9] It is worth noting, however, that antibiotic-producing organisms typically utilize multiple resistance strategies, for example S. venezuelae expresses two putative rRNA methyltransferases, that may confer resistance in a similar manner to the producers of carbomycin[10] and tylosin.[11]
Figure 1.
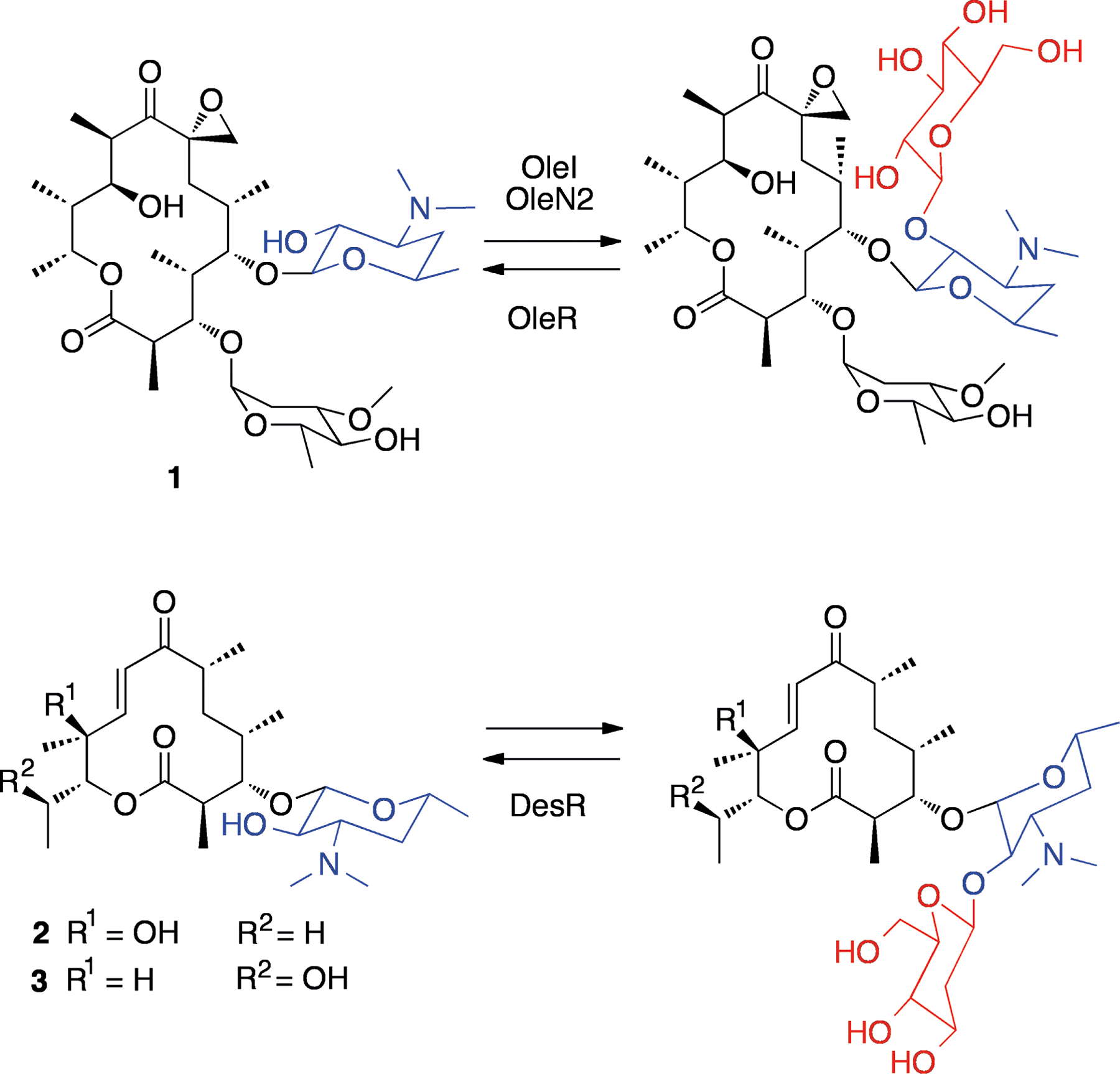
Structures of oleandomycin (1), methymycin (2) and neomethymycin (3), and their protected glycosylated forms. Desosamine units are shown in blue while protecting glucose units are shown in red.
Another example is bleomycin A2 (4, Figure 2), an antibiotic produced by Streptomyces verticillus that forms metal complexes and subsequently cleaves DNA strands in an O2 dependent manner.[12] The proposed structure of the complex of bleomycin A2 and Fe(II) is shown in Figure 2 (4, for a more comprehensive discussion on the structure of metal-bleomycin complexes, see the recent review by Pitié and Pratviel).[13] The producing organism expresses an N-acetyltransferase, BAT, that acts on the β-aminoalanine moiety of bleomycin, reducing its ability to bind Fe(II) (see Figure 2).[14] The crystal structure of this enzyme in complex with bleomycin and CoA was recently solved.[15] In addition to BAT, S. verticillus is known to produce another bleomycin resistance protein, BLMA. This protein binds tightly to metal-containing bleomycin, thus preventing it from damaging DNA.[16]
Figure 2.
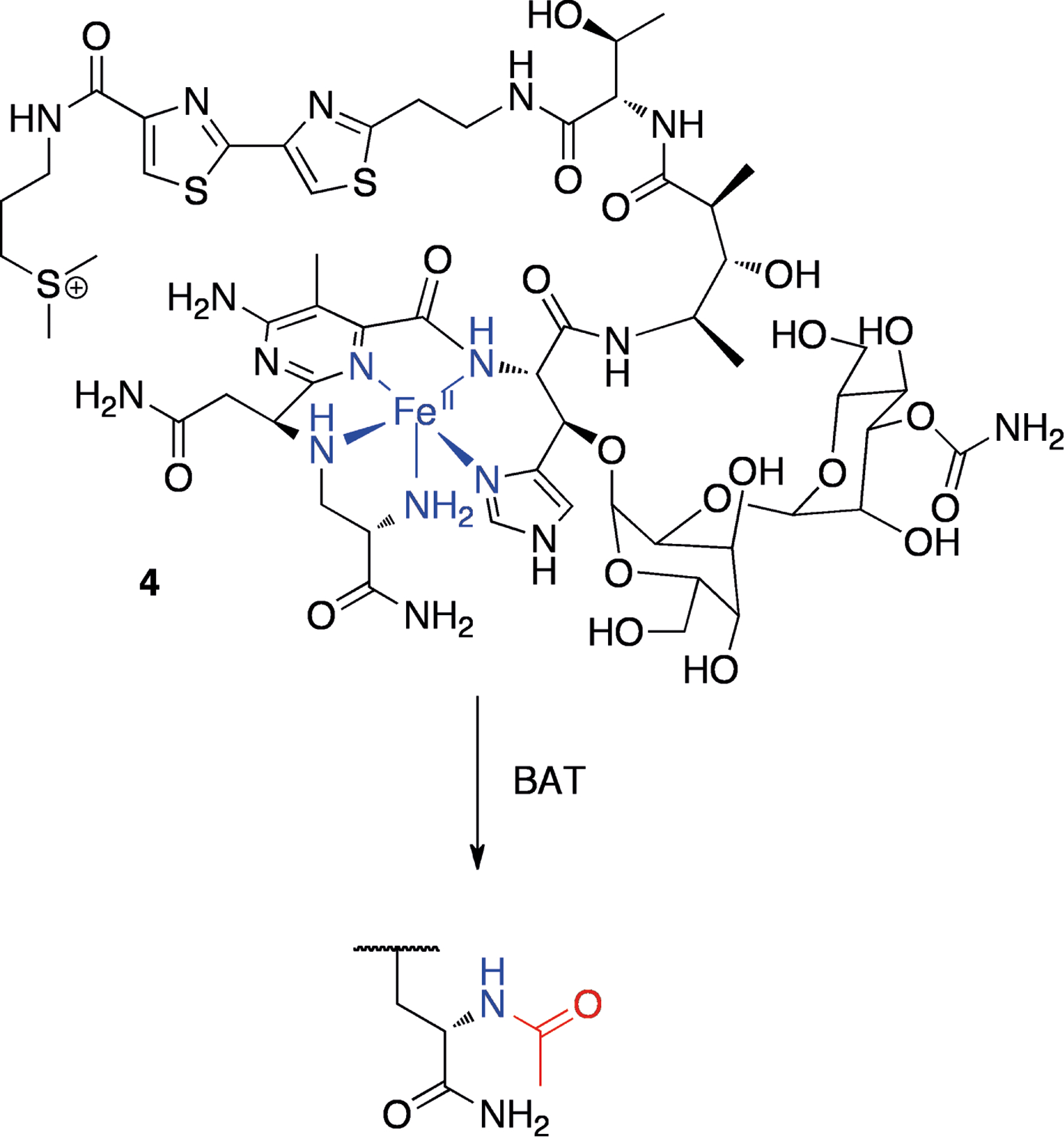
Proposed structure of the bleomycin A2-iron(II) complex (4, iron and coordingating groups shown in blue) and protection by bleomycin N-acetyl transferase (the added acetyl group is shown in red).
Sulfur Chemistry – HDAC Inhibitors
Several natural compounds have been described that inhibit eukaryotic histone deacetylase (HDAC) enzymes, containing disulfide bonds or a thioester (5–8, Figure 3). HDACs remove acetyl groups from histones leaving positively charged lysine residues that tightly bind to DNA, making it less accessible to the transcriptional machinery. In human cancers, HDACs are often overexpressed and are recruited to reduce the transcription of tumor suppressor genes and other cell growth regulators.[17] HDAC inhibitors are therefore thought to increase the expression of tumor supressor genes, and they are an emerging compound class in the treatment of cancers, with several agents either approved or in development.[18]
Figure 3.
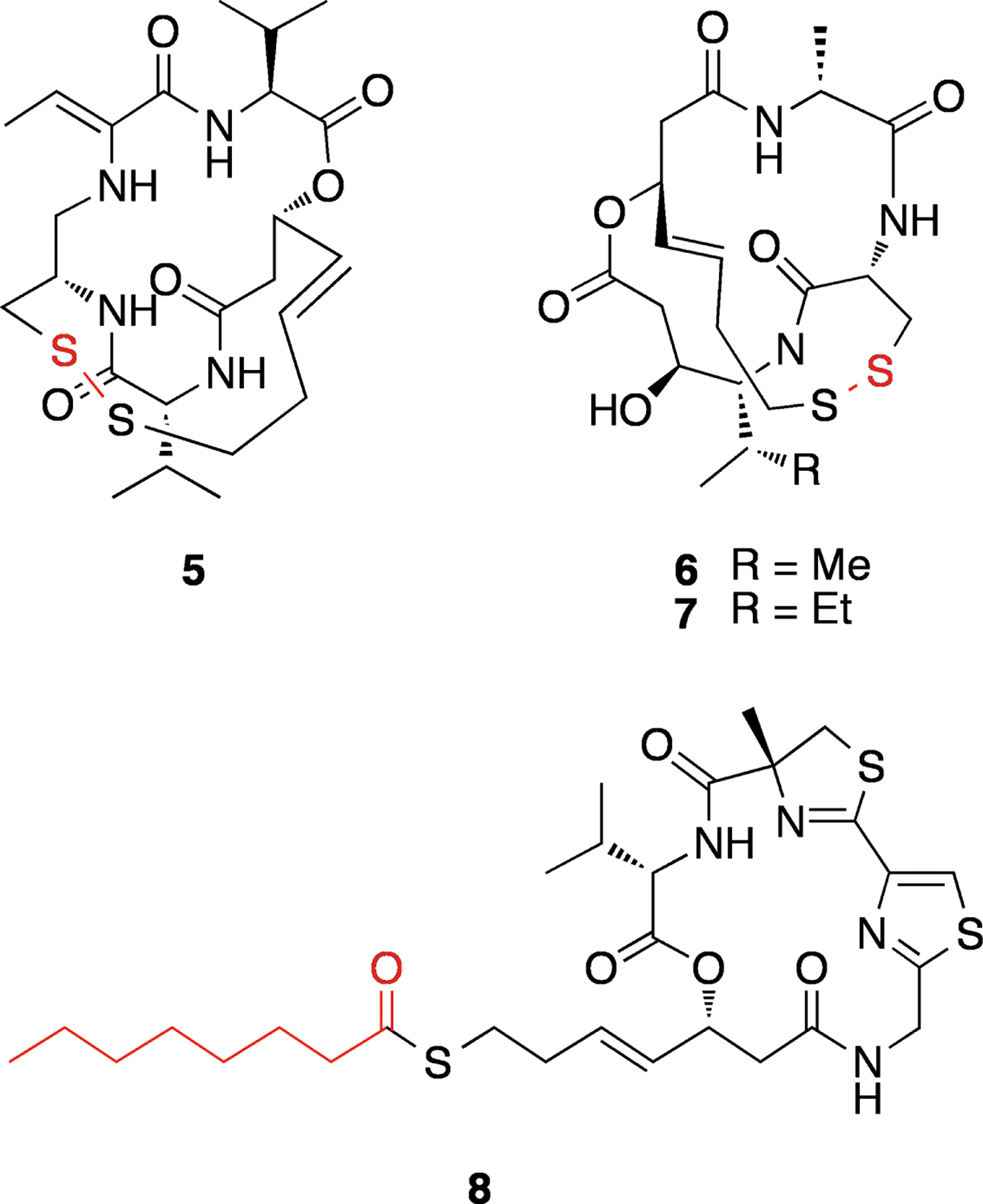
Structures of HDAC inhibitors FK228 (5), spiruchostatins A (6) and B (7), and largazole (8). Protecting groups or functionalities are shown in red.
Most HDACs are zinc metalloenzymes, and all respective NP HDAC inhibitors possess functional groups which can complex Zn2+. In several sulfur-containing HDAC inhibitors, however, this group is “masked” as either a disulfide bond or thioester (see Figure 3), which must be reduced to a thiol before binding to zinc. This group of compounds all liberate a (3S,4E)-3-hydroxy-7-mercapto-4-heptenoic acid side chain, which is involved in zinc binding, and possess a depsipeptide core.
FK228 (5) is a disulfide-containing member of this series, isolated from a culture of Chromobacterium violaceum (Figure 3).[19] This compound has also been referred to as FR901228, “depsipeptide”, NSC 630176 and romidepsin.[20] Furumai et al.[21] demonstrated that the reduced form of FK228 more potently inhibits HDAC1 in vitro (IC50 of 1.0 nM versus 36 nM for the parent compound), but that this form is less stable in growth medium and serum. FK228 therefore acts as a stable prodrug for the thiol form. It was additionally shown that cellular extracts are able to reduce FK228, and that this conversion is partly dependent on glutathione. Spiruchostatins A (6, also referred to as YM753[22]) and B (7) are also members of this class (Figure 3), originally isolated from Pseudomonas sp.[23] Similar to FK228, reduced spiruchostatin A is more potent than the unreduced form (IC50 0.62 nM), with the disulfide being essentially inactive against HDAC1 in vitro.[24]
Largazole (8), isolated from the marine cyanobacterium Symploca sp.[25] is another sulfur-containing HDAC inhibitor, but instead of a disulfide it possesses a thioester (Figure 3). It was demonstrated that largazole thiol is ~10 fold more potent against HDAC1, compared with the parent compound, and that therefore hydrolysis is probably required for inhibition.[26] Some workers have reported similar potency for largazole (8) and the free thiol in cellular systems,[26–27] while others have found the thiol to be less potent than the parent compound against other cell lines.[28] This perhaps indicates differences in cellular uptake of the thiol versus the protected compound in different cell lines, or else could reflect different extents of extracellular deprotection of 8 under different assay conditions.
Rearrangement – Leinamycin and 3-oxo-C12-HSL
Rather than possessing cleavable groups that protect a reactive center, some NPs undergo rearrangement in order to yield the active species. Such rearrangements can be spontaneous or precipitated under specific circumstances. Leinamycin (9, Scheme 1), isolated from Streptomyces sp.[29] is an example of a sulfur-containing compound that rearranges to a reactive species in the presence of thiols. It contains an unusual 1,3-dioxo-1,2-dithiolane moiety and has been found to have significant antitumor activity in mice.[29b] DNA adduct formation is thought to proceed through an episulfonium ion (10) as shown in Scheme 1.[30] Attack of the thiolate at the S2′ position is depicted, as suggested by recent theoretical calculations.[31] The episulfonium 10 would be able to react with guanine N7, followed by excision of guanine to produce the adduct 11 detected by Asai et al.[32] Additional evidence for the presence of 10 was found by the characterization of epoxide 12 in reaction mixtures. This epoxide is itself able to alkylate DNA through the episulfonium ion to give 11.[32] There is additional evidence that 9 could also generate reactive oxygen species (ROS) through the production of polysulfides,[33] as supported by the residual activity of the S-deoxy analog of leinamycin.[34]
Scheme 1.
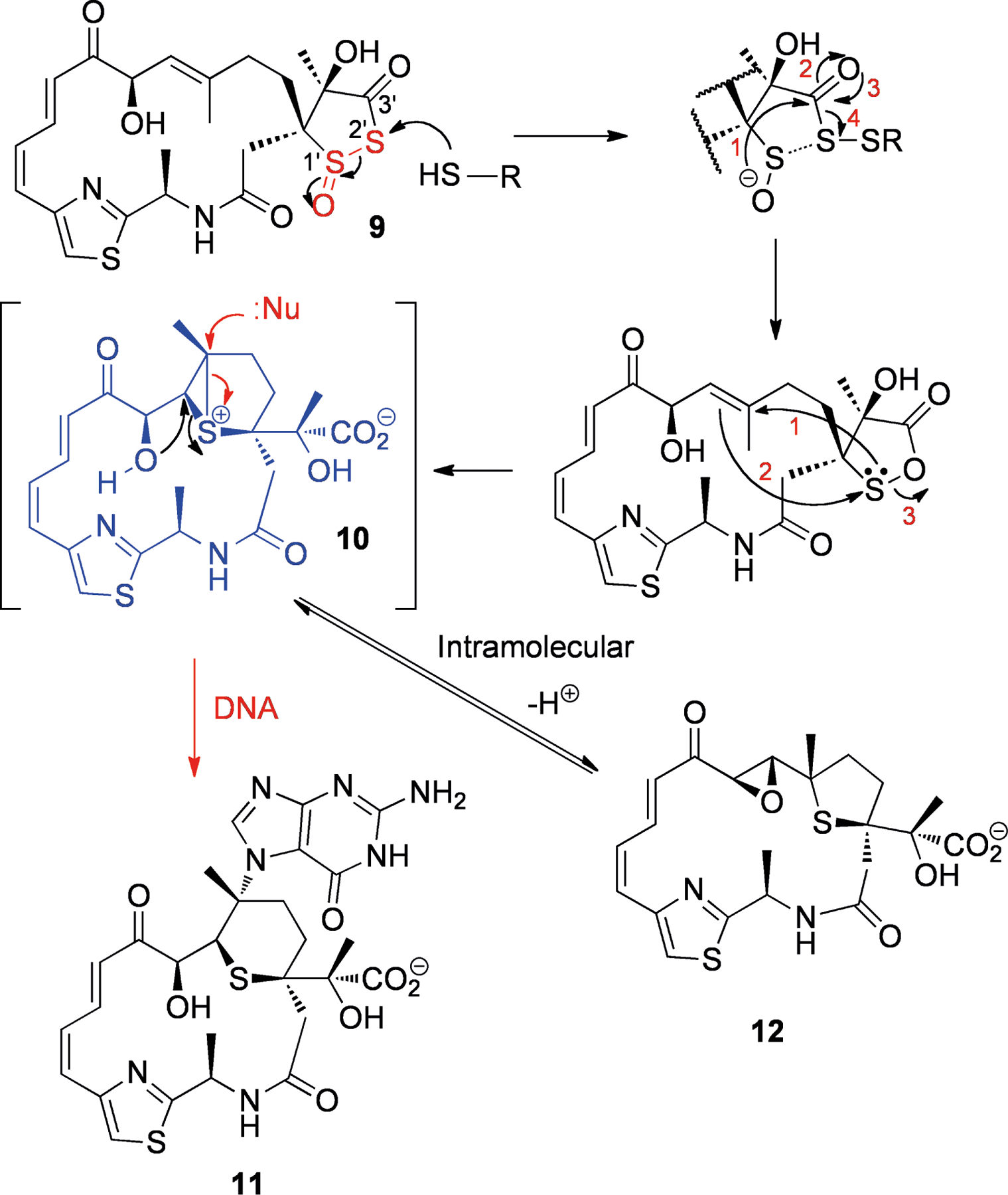
Structure of leinamycin (9) and activation by thiols to the episulfonium ion (10), which is either attacked by guanine N7 followed by base excision to give adduct 11 (red arrows) or undergoes intramolecular attack to produce the epoxide 12 (black arrows). The labile moiety is shown in red, while the reactive species is shown in blue.
The acylhomoserine lactone 3-oxo-C12-HSL (13, Scheme 2) is known to undergo spontaneous rearrangement to the tetramic acid form (15) under basic conditions (see Scheme 2).[35] Acylhomoserine lactones (AHLs), such as 13, are used by some gram negative bacteria to coordinate gene expression based on their own population (termed quorum sensing).[36] As extracellular AHL concentration and cell density increase, target genes (for example virulence factors) are upregulated. AHLs induce their own synthesis pathways, and therefore they are sometimes known as “autoinducers”.
Scheme 2.
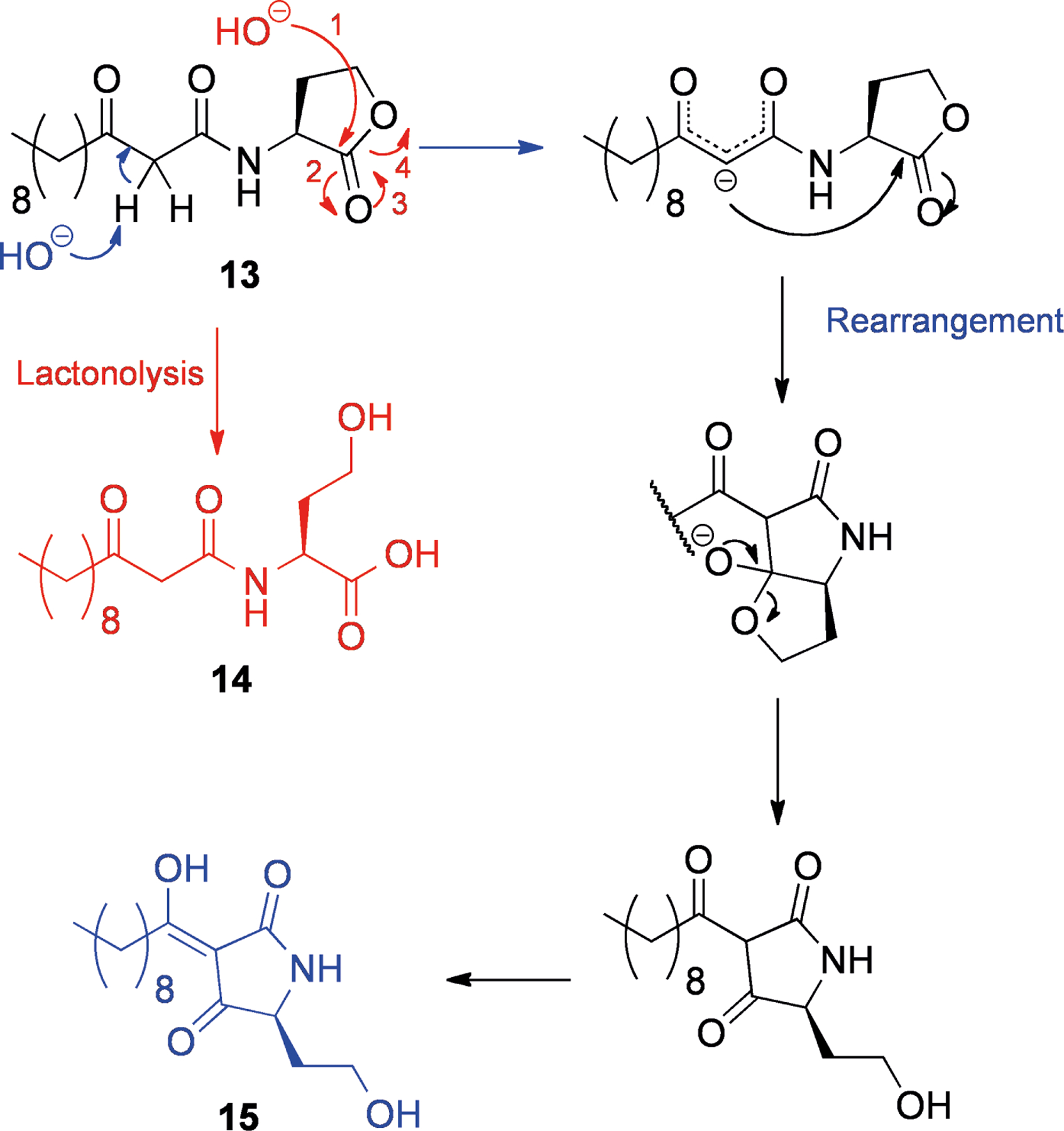
Conversion of 3-oxo-C12-HSL (13) to the ring-opened acid (14, red) and the tetramic acid (15, blue) forms.
The opportunistic pathogen Pseudomonas aeruginosa has several quorum sensing pathways, one of which responds to 13.[37] It is known that AHLs are degraded into the ring-opened forms (for example, 14, Scheme 2)[38] spontaneously at high pH.[39] In addition P. aeruginosa produces two enzymes, PvdQ[40] and QuiP,[41] that cleave the amide bond of AHLs, allowing for their catabolism and acting as a mechanism for quorum sensing downregulation.
Tetramic acid 15 (Scheme 2) appears to have distinct effects to 13.[35] Typically for a tetramic acid, 15 binds to iron, potentially acting as a siderophore for the P. aeruginosa, and it also possesses antibacterial activity against gram positive bacteria that may compete for the same resources.[42] Interestingly, although AHLs such as 13 have various immunological effects in mammals[43] and have been shown to be toxic to bone-marrow derived macrophages, 15 was not toxic to this cell line and did not stimulate macrophages.[42]
Conformational Activation – Duocarmycins
The duocarmycins are a group of NPs containing a cyclopropane ring that can alkylate DNA. Examples of this class include duocarmycin A (16),[44] yatakemycin (17)[45] and CC-1065 (18)[46] (Figure 4, all from species of Streptomyces). These compounds are very stable in solution, and yet they are able to effectively alkylate adenine N3 through attack of the cyclopropane (Scheme 3a).[47] It was suggested that the duocarmycins are activated by virtue of a conformational change upon binding to DNA in the minor groove.[47] This theory was supported by results that indicated the amide functionality had a significant stabilizing effect[48] and elegant X-ray crystallography studies of several model compounds (Scheme 3b, Table 1),[49] which showed that twisting of the bond adjacent to the amide linkage (bond c in Scheme 3b) reduced its double bond character, and concomitantly one of the cyclopropane bonds (bond b) became increasingly conjugated with the cyclohexenone (19–21). This also correlated with their respective half lives in aqueous buffer (see Table 1). Therefore it would seem that the duocarmycins are selectively activated upon binding to their target (DNA), and that activation does not require any chemical transformation.
Figure 4.
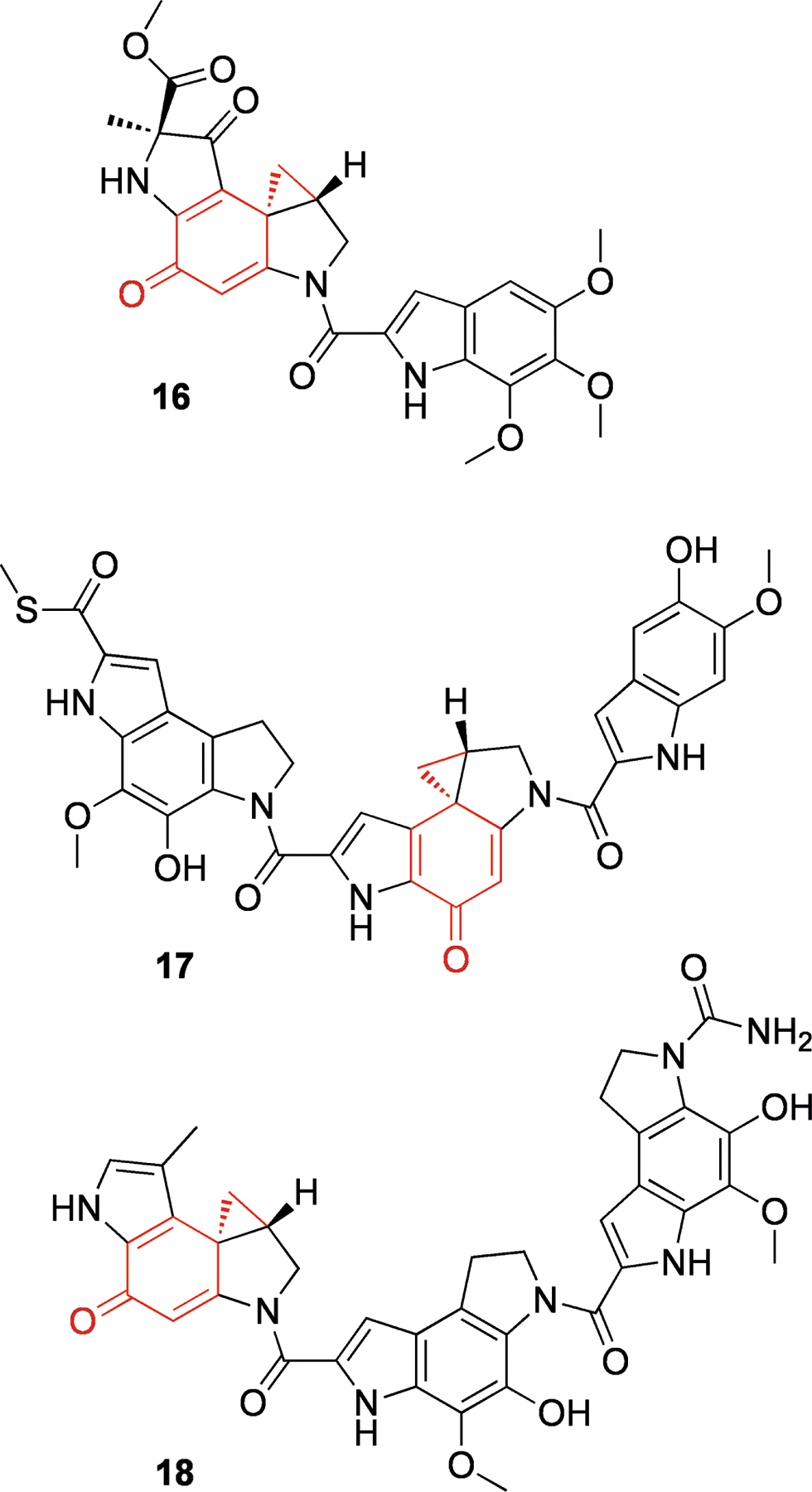
Structures of duocarmycin A (16), yatakemycin (17) and CC-1065 (18). The active center containing a semiquinone and a cyclopropane is shown in red.
Scheme 3.
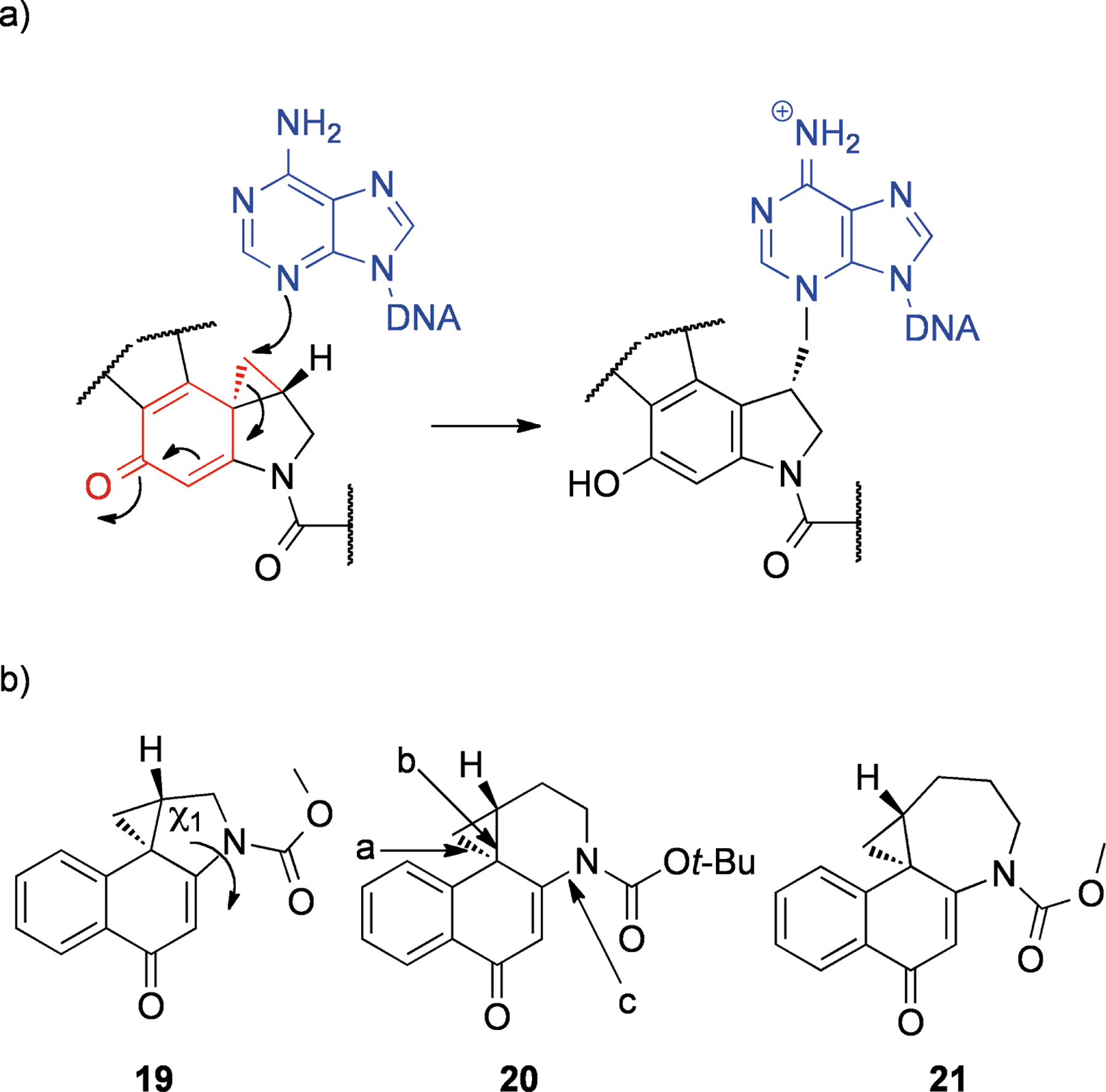
Putative mechanism of action of the duocarmycins: a) proposed mechanism of DNA alkylation with cyclopropane and semiquinone shown in red and DNA shown in blue; b) model compounds from Boger and Turnbull.[49]
Table 1.
Properties of duocarmycin model compounds.[49]
| X-ray bond lengths, Å | 19 | 20 | 21 |
|---|---|---|---|
| a | 1.521 | 1.528 | 1.565 |
| b | 1.544 | 1.543 | 1.525 |
| c | 1.390 | 1.415 | 1.428 |
| X-ray dihedral angle χ1 | 21.2° | 34.2° | 86.4° |
| t1/2 (pH 3) | 133 h | 2.1 h | 0.028 h |
In situ Activation – Salinosporamide A
Salinosporamide A (22, Scheme 4) is a potent inhibitor of the 20S proteasome that was isolated from the marine actinomycete Salinaspora tropica.[50] After binding in the active site of the 20S proteasome, the β-lactone is attacked by Thr-1 to form an adduct, similar to natural substrates (see Scheme 4).[51] Under normal circumstances, the free amine of Thr-1 would recruit a water molecule to achieve the release of the substrate. With salinosporamide A, however, this water molecule is replaced by the free OH from lactone cleavage. Attack of this hydroxyl by the Thr-1 amine results in a tetrahydrofuran ring by elimination of the chlorine atom. It is thought that the resulting species effectively excludes water from the vicinity of the Thr-1 amine, which in any case is already positively charged, preventing cleavage of the adduct.[51] Essentially salinosporamide A (22) is a prodrug that is activated by its target protein. An analog of this compound that lacks a leaving group was less cytotoxic,[52] demonstrating this moiety’s contribution to activity.
Scheme 4.
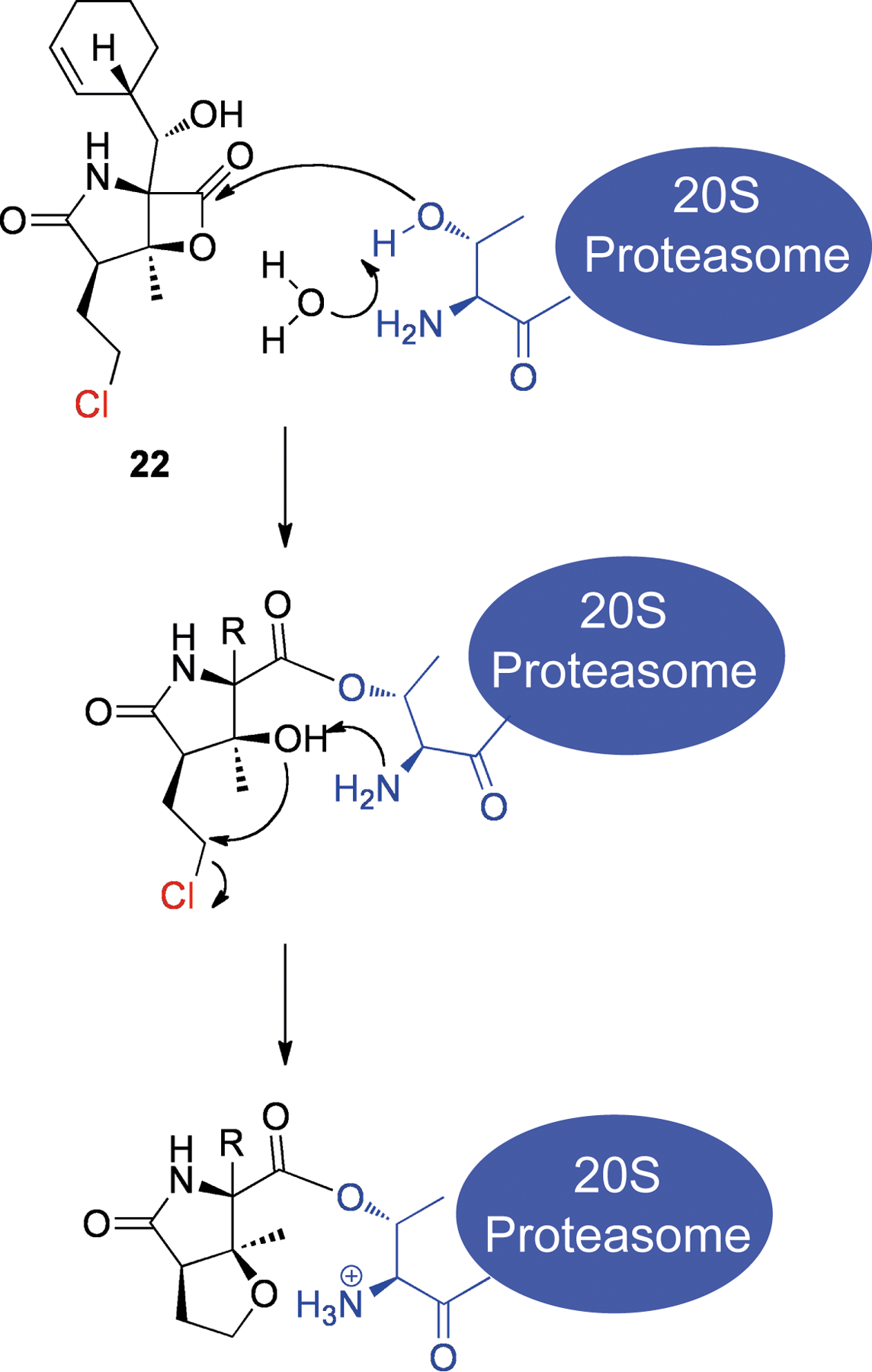
Structure of salinosporamide A (22) and mechanism of irreversible binding to the 20S proteasome. The chlorine leaving group is shown in red and Thr-1 of the 20S proteasome is shown in blue.
Iminium Synthons – Ecteinascidin 743 and Bioxalomycins
Ecteinascidin 743 (23, Figure 5) is an alkaloid isolated from the marine tunicate Ecteinascidia turbinata,[53] that is now an approved anti-cancer drug in Europe under the generic name trabectedin (and brand name Yondelis). This compound selectively alkylates N2 of guanine in the minor groove of the sequences 5′-PuGC or 5′-PyGG.[54] The hydroxyl at position 21 is essential for DNA binding,[55] and it is thought that in the minor groove, dehydration produces an iminium ion that is easily attacked by the N2 of guanine (see Scheme 5).[56] In the parent drug, N2 is most likely “protected” from protonation due to steric hindrance.[56] The other basic nitrogen (N12) is most likely protonated. Upon binding to target triplets, it is believed that the N12 proton is in close proximity to both 21-OH and a hydrogen bond acceptor from the opposite DNA strand (Scheme 5).[56–57] This configuration serves to weaken the 12N-H and 21C-O bonds and facilitate the loss of water to produce the iminium ion, which can subsequently be attacked by guanine N2.[56] The sequence selectivity of 23 is thought to be controlled by the network of hydrogen bonds possible in favored versus non-favored triplets,[58] and potentially through increased reversibility of adduct formation in non-favored sequences.[54]
Figure 5.
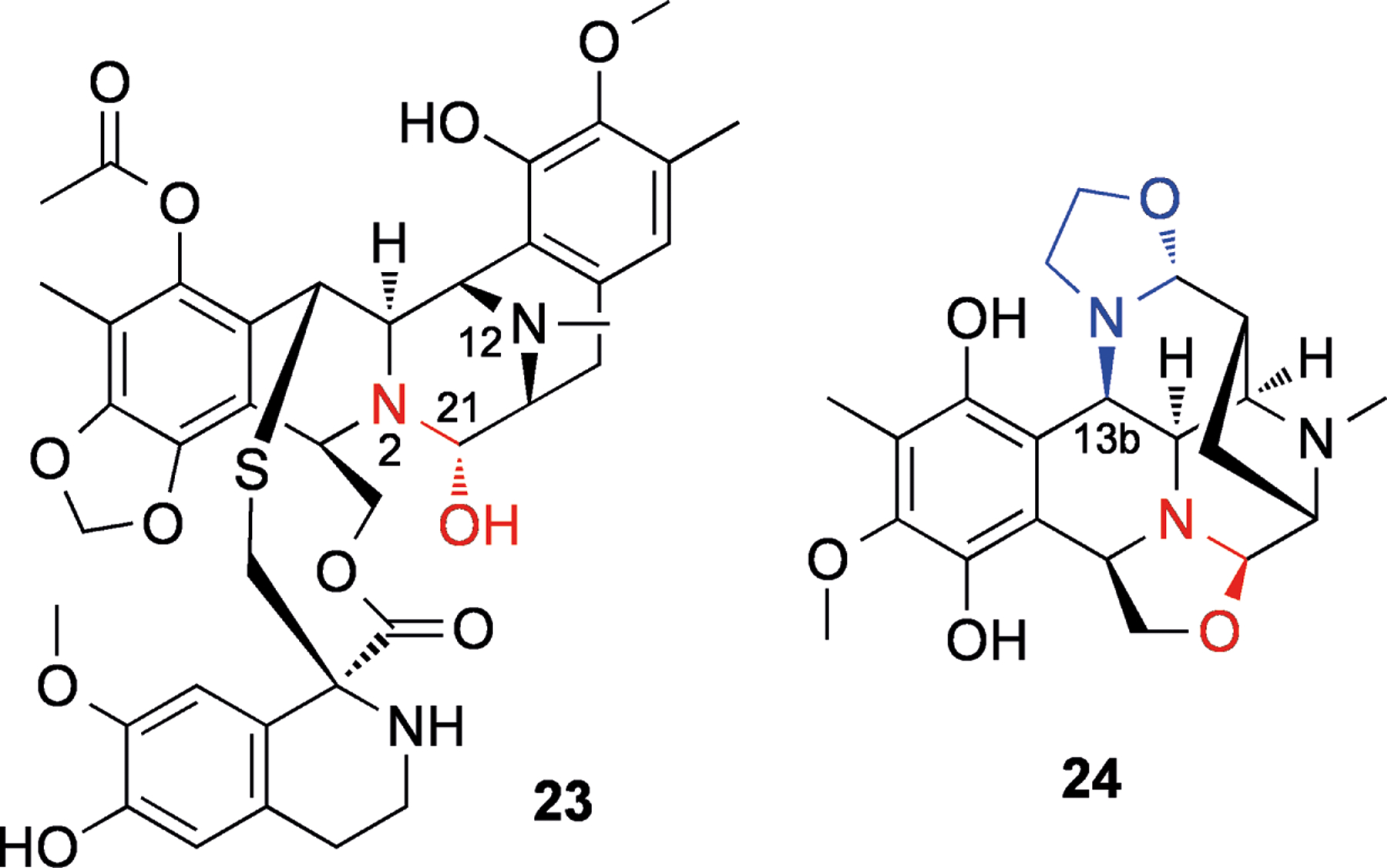
Structures of ecteinascidin 743 (23) and bioxalomycin α2 (24). Iminium synthons are shown in red, and the second leaving group in 24 is shown in blue.
Scheme 5.
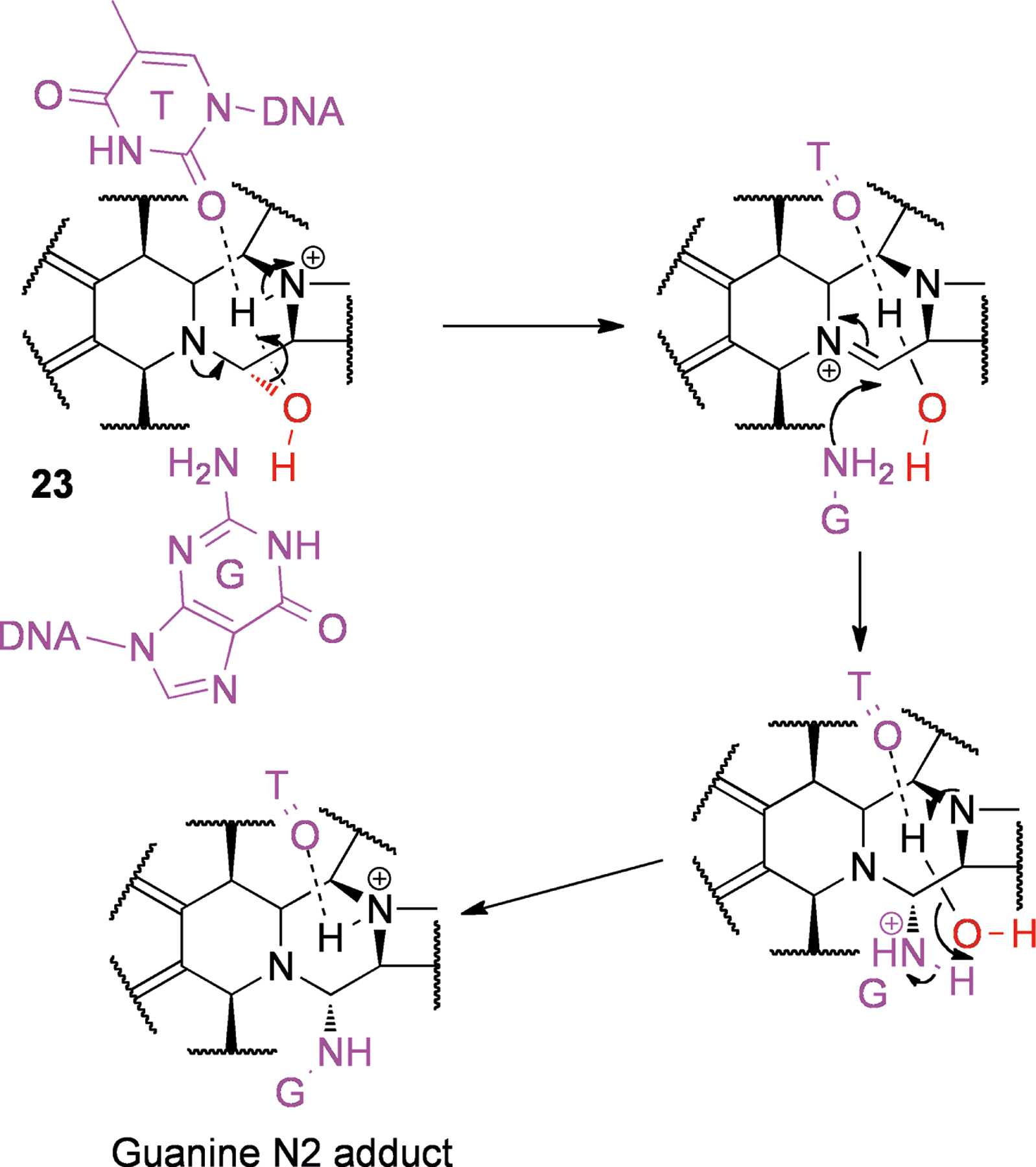
Proposed mechanism for adduct formation by ecteinascidin 743 (23) to the double stranded triplet 5′-AGC-3′ as an example. The leaving group is shown in red while DNA is shown in purple.
Many related structures have been reported and reviewed by Scott and Williams.[59] Notable amongst these are the bioxalomycins (e.g., 24, Figure 5), which were isolated from Streptomyces viridostaticus.[60] It has been shown that bioxalomycin α2 (24) is able to form interstrand adducts between guanines in 5′-CG-3′ duplex DNA.[61] The proposed mechanism involved the generation of an iminium ion (as with 23, Scheme 6). The second alkylation is thought to occur via conjugate addition after deprotection of the 13b position, with the northern oxazolidine acting as a leaving group (see Scheme 6).[59]
Scheme 6.
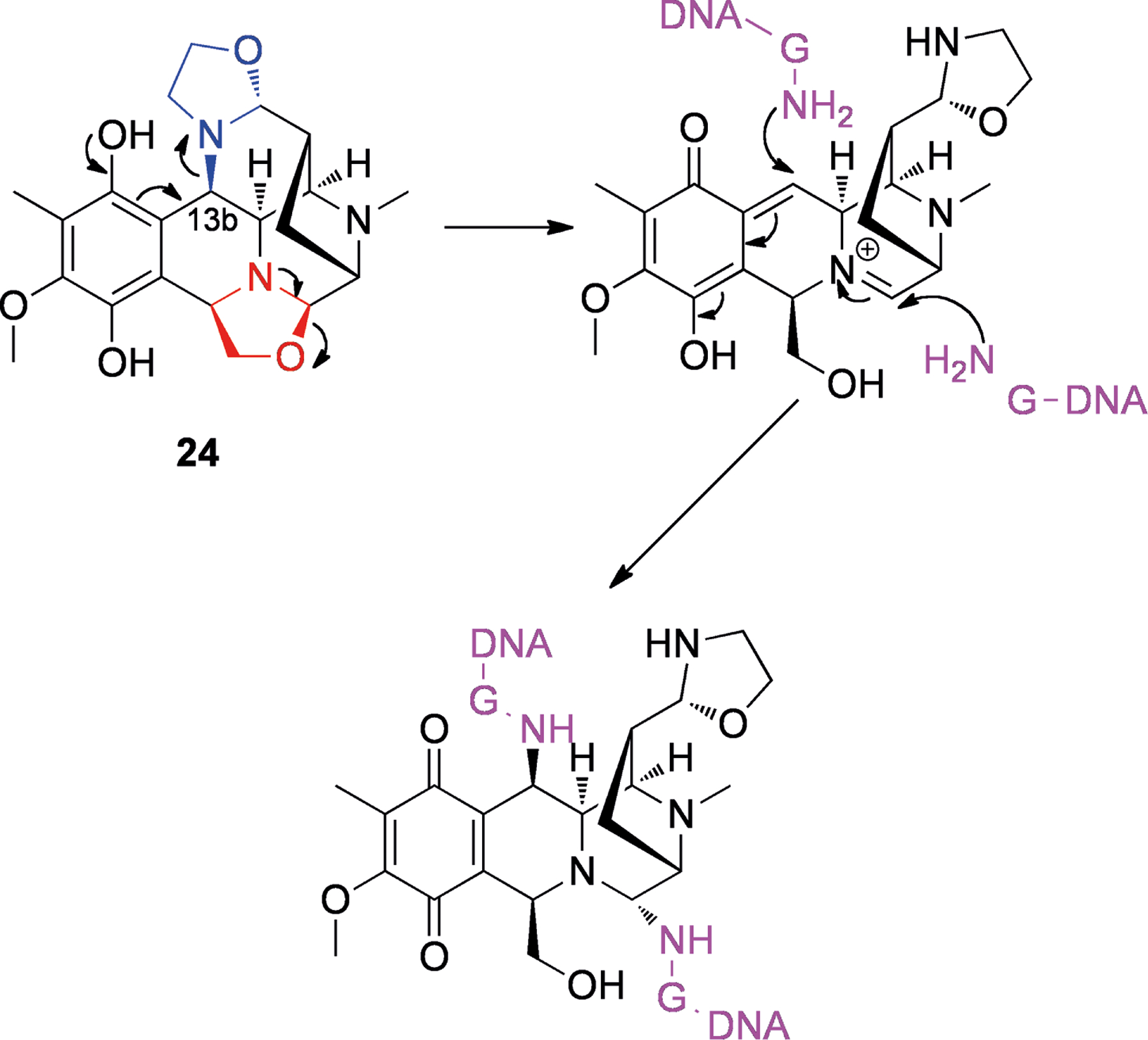
Proposed mechanism for formation of interstrand crosslink’s by bioxalomycin α2 (24) in 5′-CG-3′ duplexes. The iminium synthon is shown in red and the second leaving group is shown in blue, while DNA is shown in purple.
Reductive Activation of Nitrogen-Containing Leaving Groups – Diazoparaquinones and Mitomycin C
Similar to the bioxalomycins, there are many NPs whose activation involves quinones, with the oxidation state of this moiety acting as a molecular switch to trigger conversion to the active species. For instance, reduction of the quinone triggers the elimination of the diazo leaving group in the diazoparaquinones. The diazoparaquinone natural products are exemplified by kinamycin A (25, Figure 6), isolated from Streptomyces murayamaensis,[62] and lomaivitivins A (26) and B (27) from Micromonospora lomaivitiensis, an actinomycete symbiont of the marine ascidian Polysyncraton lithostrotum.[63] The lomaivitivins and the kinamycins are both able to cleave DNA.[63–64] There is some evidence that under reducing conditions (such as within solid tumors that are relatively hypoxic),[65] one-electron reduction of the quinone can lead to the production of a radical species[66] (Scheme 7a). This radical can attack DNA, leading to nicks and strand cleavage. Recently, evidence for an additional two-electron reduction pathway has come to light (Scheme 7b),[67] resulting in a quinone methide that could potentially be attacked by nucleophiles, for example guanines within DNA, leading to adducts. These studies were carried out under abiological conditions, but it was speculated that two-electron reduction could be carried out by the quinone reductase DT-diaphorase, while one-electron reduction could be carried out by cytochromes.[67]
Figure 6.
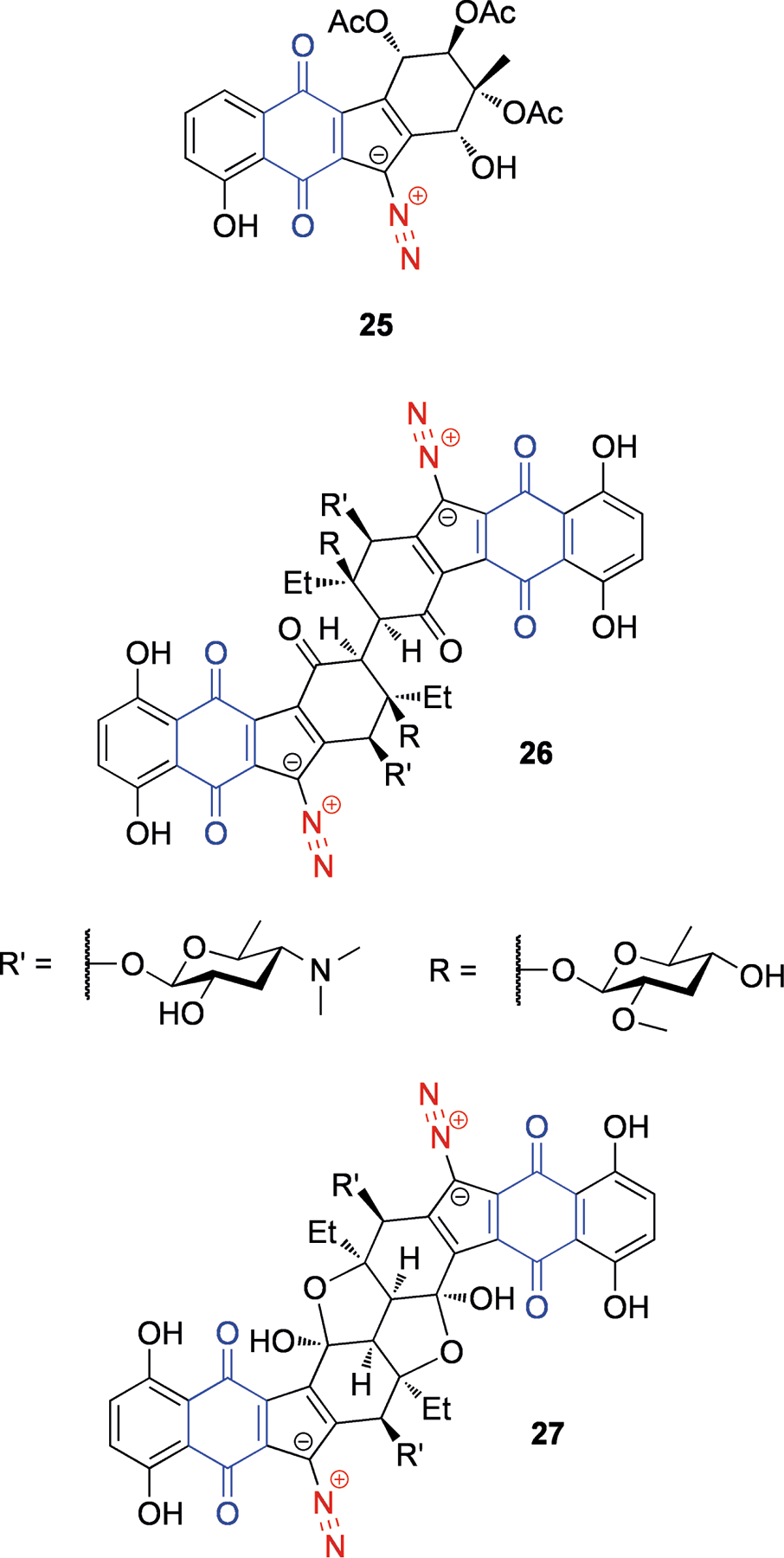
Structures of kinamycin A (25), lomaiviticins A (26) and B (27). The diazo leaving group is shown in red, and the quinone that participates in activation is shown in blue.
Scheme 7.
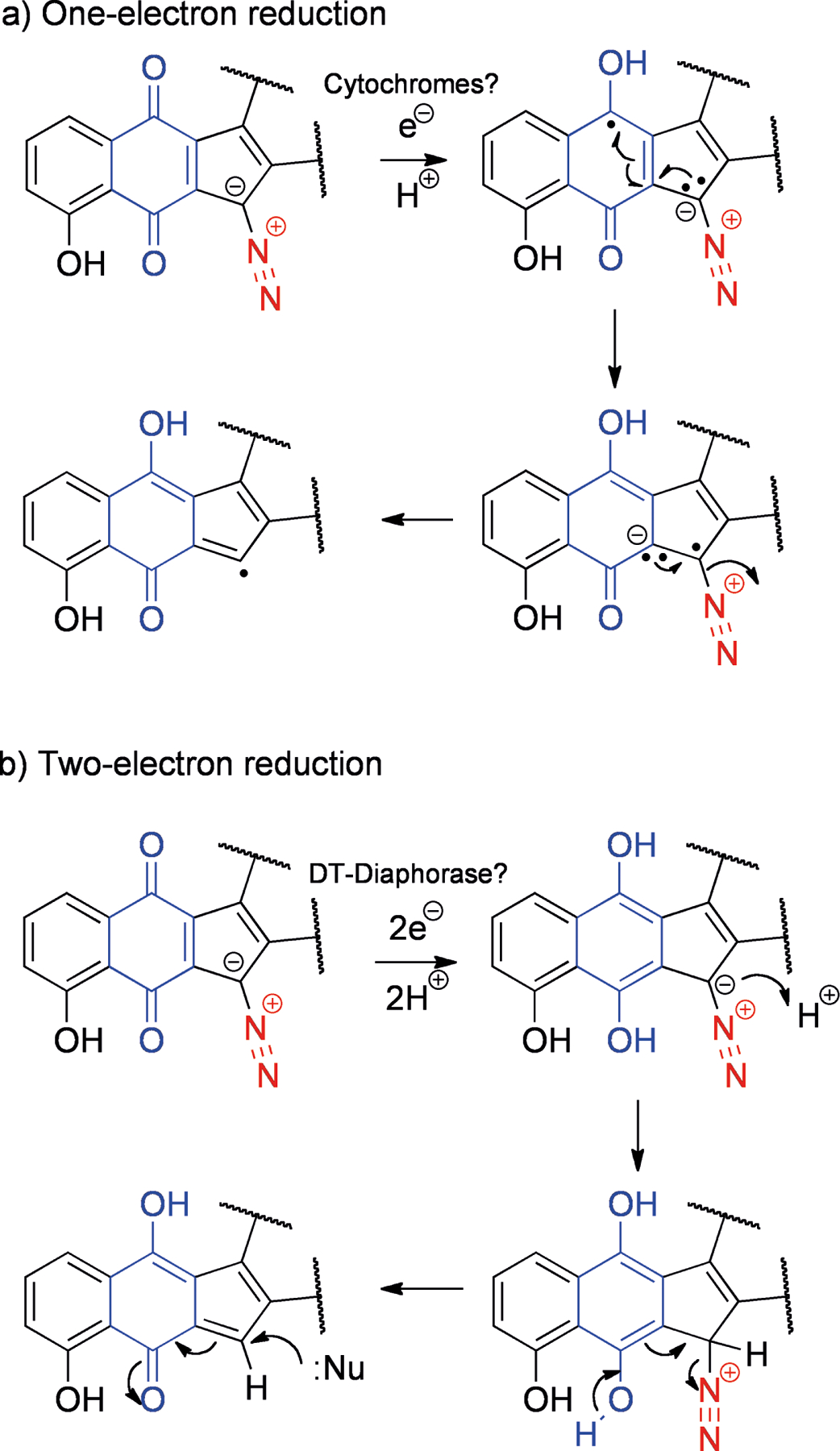
Proposed mechanisms for activation of kinamycin A (25) and related structures by a) one-electron reduction and b) two-electron reduction. Leaving groups are shown in red while participating quinones are shown in blue.
Mitomycin C (28, Figure 7) is an aziridine-containing compound that was isolated from Streptomyces caespitosus in 1958.[68] Its structure was determined in 1962,[69] and since then only four total syntheses of mitomycins have been published.[70] Although largely inert in the native form, upon reductive activation 28 can cross-link DNA strands by double-adduct formation[71] (Scheme 8). In the quinone form, the lone pair of the 4-position nitrogen is partially withdrawn into the delocalized system, but after reduction there is a driving force to the indole with irreversible ejection of the methoxy group. Following this step, a cascade of conversions can easily occur, with the addition of two nucleophiles, which can originate from opposite strands of DNA. This mechanism, proposed in 1964,[71] is still widely accepted, and the expected diadduct has been isolated and characterized.[72] However, the nature of the initial reduction (one- or two-electron) and the oxidation state of the quinone during each step are still subjects of debate.[73]
Figure 7.
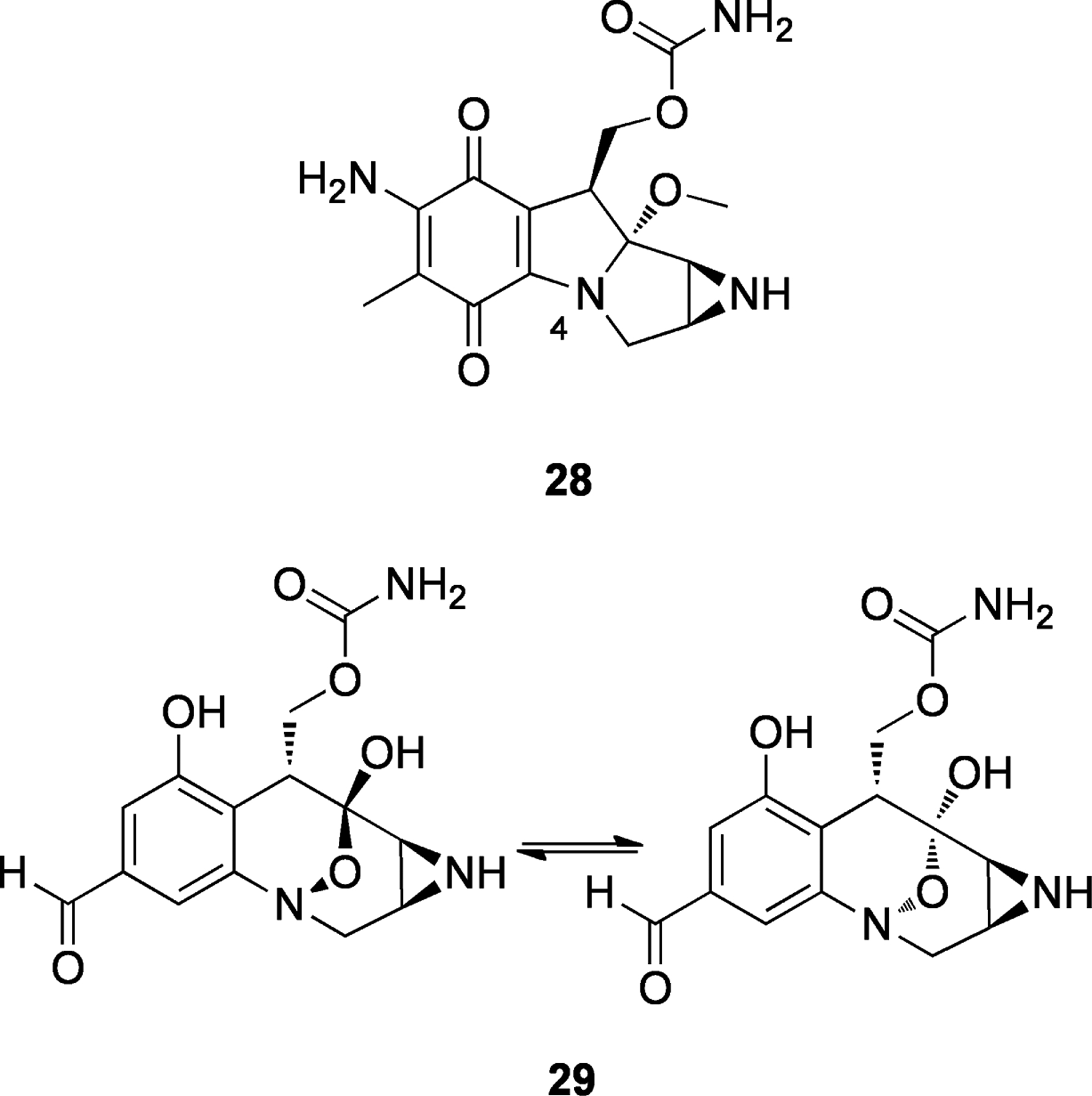
Structures of mitomycin C (28) and FR900482 (29).
Scheme 8.
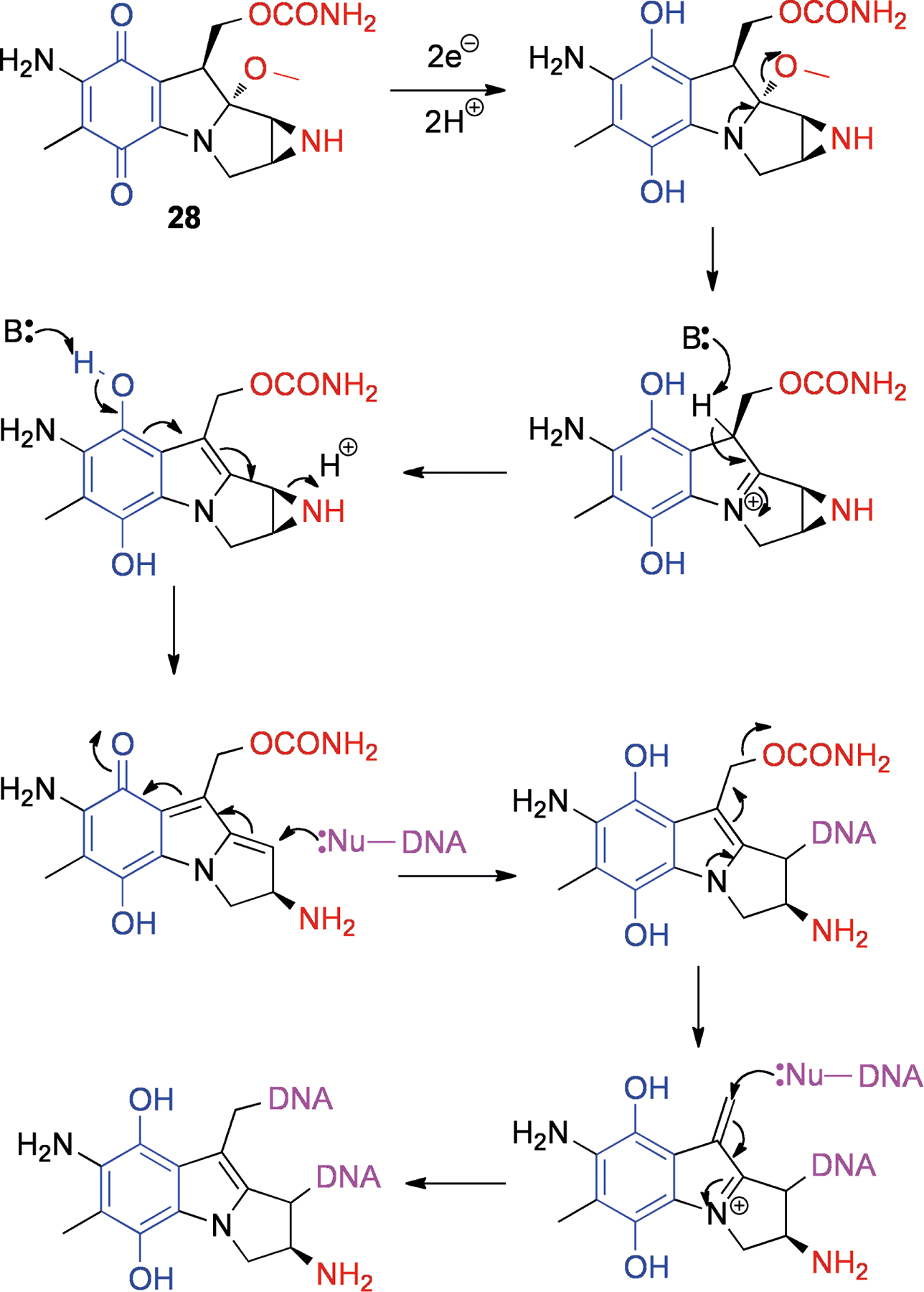
Reductive activation of mitomycin C (28), followed by two DNA alkylations. Leaving groups are shown in red, the participating quinone is shown in blue and DNA is shown in purple.
FR900482 (29, Figure 7), which was isolated from Streptomyces sandaensis,[74] uses a different pathway of reductive activation (Scheme 9). The compound interconverts between two stereoisomers through tautomeric conversion to the 8-membered hydroxylamine 30. If this species is reduced to the amine, rearrangement to a structure similar to the mitomycin C skeleton is possible (31). It can be easily envisioned that this species could form diadducts to DNA in an analogous manner to mitomycin C (28). Studies on the related structure FR66979 showed evidence of conversion to a mitomycin skeleton[75] and also the presence of the expected DNA diadduct.[76] Interestingly, FR900482 (29) showed lower toxicity in mice compared to mitomycin C (28).[77] This could be due to the absence of a quinone in the molecule, which in the case of mitomycin C (28) can give rise to reactive oxygen species (ROS) through redox cycling.[78]
Scheme 9.
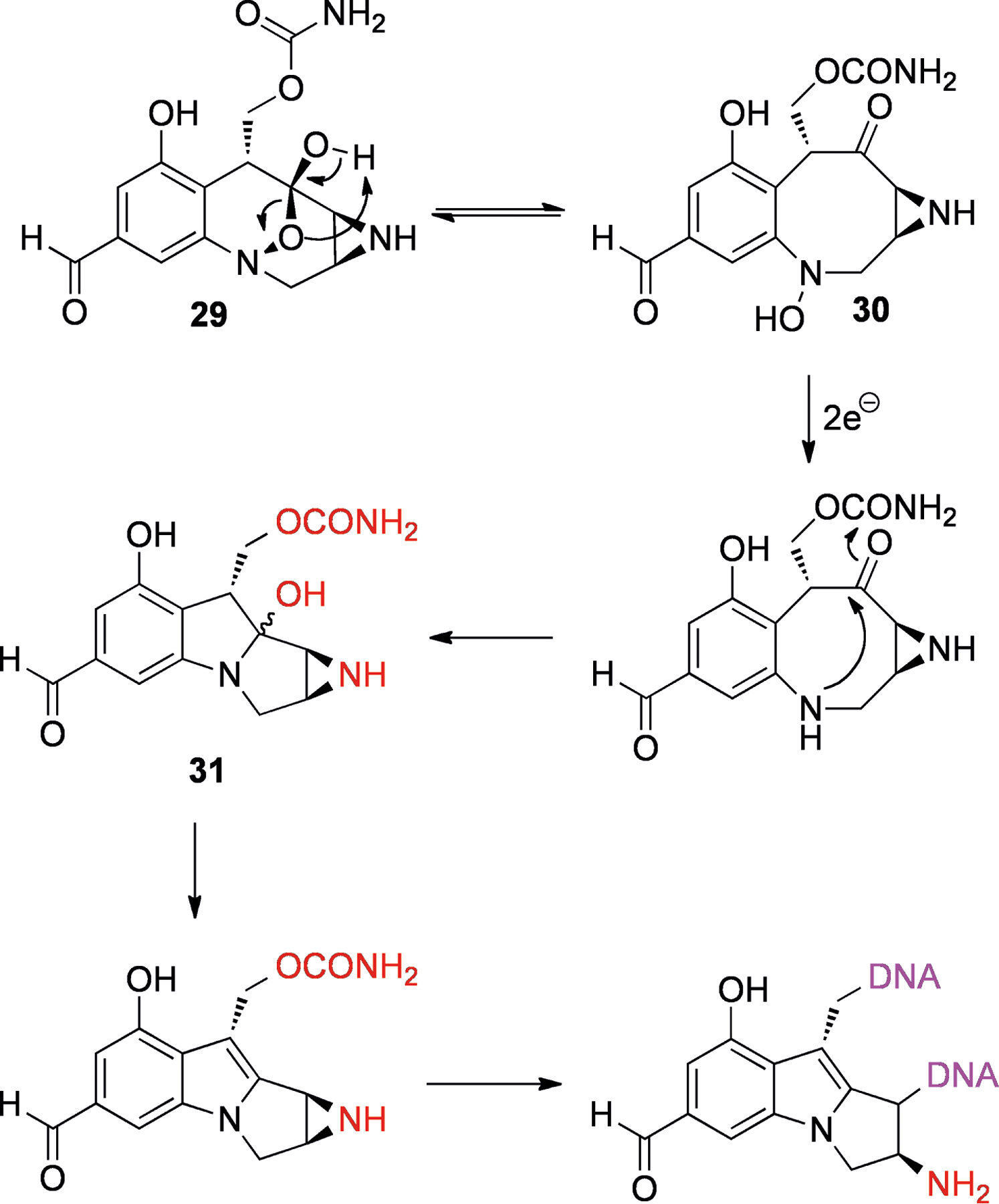
Reductive activation of FR900482 (29) to give a species with a mitomycin-type skeleton (31) which can bisalkylate DNA. Leaving groups are shown in red and DNA is shown in purple.
Conclusion
The preceding examples illustrate some of the ways in which protective chemistry has been used in nature to confer self-resistance, to achieve target selectivity and to deliver reactive species. However, a question that remains largely unanswered is how did such intricate activating mechanisms come about? The problems of drug design and drug delivery are usually considered and tackled separately by scientists. But in a complex and competitive natural environment, selective pressure favors NPs that solve both issues in parallel. As products of evolution rather than human “rational drug design”, natural compounds offer a complementary source of bioactive scaffolds. NPs often work through novel and sometimes surprising mechanisms of action, and they therefore serve as inpiration to synthetic and medicinal chemists, in terms of both structure and mechanism.
Acknowledgements
Natural products research in the authors’ laboratory is supported by the National Institutes of Health, NIGMS grant P41GM806210 and NCI grants R01CA138544 and R21CA133681.
References
- [1].Jenke-Kodama H, Müller R, Dittmann E, Prog. Drug Res 2008, 65, 121–140. [DOI] [PubMed] [Google Scholar]
- [2].Newman DJ, Cragg GM, J. Nat. Prod 2007, 70, 461–477. [DOI] [PubMed] [Google Scholar]
- [3].MacMillan KS, Boger DL, J. Med. Chem 2009, 52, 5771–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Méndez C, Salas JA, FEMS Microbiol. Lett 1998, 158, 1–8. [DOI] [PubMed] [Google Scholar]
- [5].Liang Z-X, Nat. Prod. Rep 2010, 27, 499–528. [DOI] [PubMed] [Google Scholar]
- [6].Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, Savolainen J, Nat. Rev. Drug Discov 2008, 7, 255–270. [DOI] [PubMed] [Google Scholar]
- [7].Schlünzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F, Nature 2001, 413, 814–821. [DOI] [PubMed] [Google Scholar]
- [8].Quirós LM, Aguirrezabalaga I, Olano C, Méndez C, Salas JA, Mol. Microbiol 1998, 28, 1177–1185. [DOI] [PubMed] [Google Scholar]
- [9].a) Zhao L, Beyer NJ, Borisova SA, Liu H-W, Biochem. 2003, 42, 14794–14804; [DOI] [PubMed] [Google Scholar]; b) Zhao L, Sherman DH, Liu H-W, J. Am. Chem. Soc 1998, 120, 9374–9375. [Google Scholar]
- [10].Zalacain M, Cundliffe E, Eur. J. Biochem 1990, 189, 67–72. [DOI] [PubMed] [Google Scholar]
- [11].Zalacain M, Cundliffe E, J. Bacteriol 1989, 171, 4254–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B, Chem. Rev 2005, 105, 739–758. [DOI] [PubMed] [Google Scholar]
- [13].Pitié M, Pratviel G, Chem. Rev 2010, 110, 1018–1059. [DOI] [PubMed] [Google Scholar]
- [14].Sugiyama M, Kumagai T, Shionoya M, Kimura E, Davies JE, FEMS Microbiol. Lett 1994, 121, 81–85. [DOI] [PubMed] [Google Scholar]
- [15].Oda K, Matoba Y, Noda M, Kumagai T, Sugiyama M, J. Biol. Chem 2010, 285, 1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sugiyama M, Kumagai T, Hayashida M, Maruyama M, Matoba Y, J. Biol. Chem 2002, 277, 2311–2320. [DOI] [PubMed] [Google Scholar]
- [17].Glozak MA, Seto E, Oncogene 2007, 26, 5420–5432. [DOI] [PubMed] [Google Scholar]
- [18].Tan J, Cang S, Ma Y, Petrillo RL, Liu D, J. Hematol. Oncol 2010, 3, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ueda H, Nakajima H, Hori Y, Fujita T, Nishimura M, Goto T, Okuhara M, J. Antibiot 1994, 47, 301–310. [DOI] [PubMed] [Google Scholar]
- [20].Cheng Y-Q, Yang M, Matter AM, Appl. Env. Microbiol 2007, 73, 3460–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Furumai R, Matsuyama A, Kobashi N, Lee K-H, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S, Cancer Res. 2002, 62, 4916–4921. [PubMed] [Google Scholar]
- [22].Shindoh N, Mori M, Terada Y, Oda K, Amino N, Kita A, Taniguchi M, Sohda K-Y, Nagai K, Sowa Y, Masuoka Y, Orita M, Sasamata M, Matsushime H, Furuichi K, Sakai T, Int. J. Oncol 2008, 32, 545–555. [PubMed] [Google Scholar]
- [23].Masuoka Y, Nagai A, Shin-ya K, Furihata K, Nagai K, Suzuki K-I, Hayakawa Y, Seto H, Tetrahedron Lett. 2001, 42, 41–44. [Google Scholar]
- [24].Crabb SJ, Howell M, Rogers H, Ishfaq M, Yurek-George A, Carey K, Pickering BM, East P, Mitter R, Maeda S, Johnson PWM, Townsend P, Shin-ya K, Yoshida M, Ganesan A, Packham G, Biochem. Pharmacol 2008, 76, 463–475. [DOI] [PubMed] [Google Scholar]
- [25].Taori K, Paul VJ, Luesch H, J. Am. Chem. Soc 2008, 130, 1806–1807. [DOI] [PubMed] [Google Scholar]
- [26].Ying Y, Taori K, Kim H, Hong J, Luesch H, J. Am. Chem. Soc 2008, 130, 8455–8459. [DOI] [PubMed] [Google Scholar]
- [27].Numajiri Y, Takahashi T, Takagi M, Shin-ya K, Doi T, Synlett 2008, 2483–2486.
- [28].a) Seiser T, Kamena F, Cramer N, Angew. Chem. Int. Ed 2008, 47, 6483–6485; [DOI] [PubMed] [Google Scholar]; b) Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM, J. Am. Chem. Soc 2009, 130, 11219–11222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Hara M, Asano K, Kawamoto I, Takiguchi T, Katsumata S, Takahashi K-I, Nakano H, J. Antibiot 1989, 42, 1768–1774; [DOI] [PubMed] [Google Scholar]; b) Hara M, Takahashi I, Yoshida M, Asano K, Kawamoto I, Morimoto M, Nakano H, J. Antibiot 1989, 42, 333–335. [DOI] [PubMed] [Google Scholar]
- [30].Asai A, Hara M, Kakita S, Kanda Y, Yoshida M, Saito H, Saitoh Y, J. Am. Chem. Soc 1996, 118, 6802–6803. [Google Scholar]
- [31].Breydo L, Gates KS, J. Org. Chem 2002, 67, 9054–9060. [DOI] [PubMed] [Google Scholar]
- [32].Asai A, Saito H, Saitoh Y, Bioorg. Med. Chem 1997, 5, 723–729. [DOI] [PubMed] [Google Scholar]
- [33].Mitra K, Kim W, Daniels JS, Gates KS, J. Am. Chem. Soc 1997, 119, 11691–11692. [Google Scholar]
- [34].Sivaramakrishnan S, Gates KS, Bioorg. Med. Chem. Lett 2008, 18, 3076–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kaufmann GF, Sartorio R, Lee S-H, Rogers CJ, Meijler MM, Moss JA, Clapham B, Brogan AP, Dickerson TJ, Janda KD, Proc. Natl. Acad. Sci. USA 2005, 102, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].McDougald D, Rice SA, Kjelleberg S, Anal. Bioanal. Chem 2007, 387, 445–453. [DOI] [PubMed] [Google Scholar]
- [37].Schuster M, Greenberg EP, Int. J. Med. Microbiol 2006, 296, 73–81. [DOI] [PubMed] [Google Scholar]
- [38].Cataldi TRI, Bianco G, Abate S, J. Mass Spec 2009, 44, 182–192. [DOI] [PubMed] [Google Scholar]
- [39].Yates EA, Philipp B, Buckley C, Atkinson S, Chhabra SR, Sockett RE, Goldner M, Dessaux Y, Cámara M, Smith H, Williams P, Infection Immun. 2002, 70, 5635–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang JJ, Han J-I, Zhang L-H, Leadbetter JR, Appl. Env. Microbiol 2003, 69, 5941–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huang JJ, Petersen A, Whiteley M, Leadbetter JR, Appl. Env. Microbiol 2006, 72, 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lowery CA, Park J, Gloeckner C, Meijler MM, Mueller RS, Boshoff HI, Ulrich RL, Barry CE, Bartlett DH, Kravchenko VV, Kaufmann GF, Janda KD, J. Am. Chem. Soc 2009, 131, 14473–14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Williams P, Microbiol. 2007, 153, 3923–3938. [DOI] [PubMed] [Google Scholar]
- [44].Takahashi I, Takahashi K-I, Ichimura M, Morimoto M, Asano K, Kawamoto I, Tomita F, Nakano H, J. Antibiot 1988, 41, 1915–1917. [DOI] [PubMed] [Google Scholar]
- [45].a) Igarashi Y, Futamata K, Fujita T, Sekine A, Senda H, Naoki H, Furumai T, J. Antibiot 2003, 56, 107–113; [DOI] [PubMed] [Google Scholar]; b) Tichenor MS, Kastrinsky DB, Boger DL, J. Am. Chem. Soc 2004, 126, 8396–8398. [DOI] [PubMed] [Google Scholar]
- [46].Martin DG, Biles C, Gerpheide SA, Hanka LJ, Krueger WC, McGovren JP, Mizsak SA, Neil GL, Stewart JC, Visser J, J. Antibiot 1981, 34, 1119–1125. [DOI] [PubMed] [Google Scholar]
- [47].Boger DL, Garbaccio RM, Bioorg. Med. Chem 1997, 5, 263–276. [DOI] [PubMed] [Google Scholar]
- [48].Boger DL, Turnbull P, J. Org. Chem 1998, 63, 8004–8011. [Google Scholar]
- [49].Boger DL, Turnbull P, J. Org. Chem 1997, 62, 5849–5863. [Google Scholar]
- [50].Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W, Angew. Chem. Int. Ed 2003, 42, 355–357. [DOI] [PubMed] [Google Scholar]
- [51].Groll M, Huber R, Potts BCM, J. Am. Chem. Soc 2006, 128, 5136–5141. [DOI] [PubMed] [Google Scholar]
- [52].Williams PG, Buchanan GO, Feling RH, Kauffman CA, Jensen PR, Fenical W, J. Org. Chem 2005, 70, 6196–6203. [DOI] [PubMed] [Google Scholar]
- [53].Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH, Martin DG, J. Org. Chem 1990, 55, 4512–4515. [Google Scholar]
- [54].Zewail-Foote M, Hurley LH, J. Am. Chem. Soc 2001, 123, 6485–6495. [DOI] [PubMed] [Google Scholar]
- [55].David-Cordonnier M-H, Gajate C, Olmea O, Laine W, de la Iglesia-Vicente J, Perez C, Cuevas C, Otero G, Manzanares I, Bailly C, Mollinedo F, Chem. Biol 2005, 12, 1201–1210. [DOI] [PubMed] [Google Scholar]
- [56].Moore BM, Seaman FC, Wheelhouse RT, Hurley LH, J. Am. Chem. Soc 1998, 120, 2490–2491. [Google Scholar]
- [57].Marco E, David-Cordonnier M-H, Bailly C, Cuevas C, Gago F, J. Med. Chem 2006, 49, 6925–6929. [DOI] [PubMed] [Google Scholar]
- [58].Seaman FC, Hurley LH, J. Am. Chem. Soc 1998, 120, 13028–13041. [Google Scholar]
- [59].Scott JD, Williams DH, Chem. Rev 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]
- [60].Zaccardi J, Alluri M, Ashcroft J, Bernan V, Korshalla JD, Morton GO, Siegel M, Tsao R, Williams DR, Maiese W, Ellestad GA, J. Org. Chem 1994, 59, 4045–4047. [Google Scholar]
- [61].Williams RM, Herberich B, J. Am. Chem. Soc 1998, 120, 10272–10273. [Google Scholar]
- [62].a) Hata T, Omura S, Iwai Y, Nakagawa A, Otani M, J. Antibiot 1971, 24, 353–359; [DOI] [PubMed] [Google Scholar]; b) Mithani S, Weeratunga G, Taylor NJ, Dmitrienko GI, J. Am. Chem. Soc 1994, 116, 2209–2210. [Google Scholar]
- [63].He H, Ding W-D, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT, J. Am. Chem. Soc 2001, 123, 5362–5363. [DOI] [PubMed] [Google Scholar]
- [64].a) Ballard TE, Melander C, Tetrahedron Lett. 2008, 49, 3157–3161; [Google Scholar]; b) O’Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB, Free Radical Biol. Med 2007, 43, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tomasz M, Chem. Biol 1995, 2, 575–579. [DOI] [PubMed] [Google Scholar]
- [66].Feldman KS, Eastman KJ, J. Am. Chem. Soc 2006, 128, 12562–12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Khdour O, Skibo EB, Org. Biomol. Chem 2009, 7, 2140–2154. [DOI] [PubMed] [Google Scholar]
- [68].Wakaki S, Marumo H, Tomioka K, Shimizu G, Kato E, Kamada H, Kudo S, Fujimoto Y, Antibiot. Chemother 1958, 8, 228–240. [PubMed] [Google Scholar]
- [69].Webb JS, Cosulich DB, Mowat JH, Patrick JB, Broschard RW, Meyer WE, Williams RP, Wolf CF, Fulmor W, Pidacks C, Lancaster JE, J. Am. Chem. Soc 1962, 84, 3185–3187. [Google Scholar]
- [70].Andrez J-C, Beilstein J Org. Chem 2009, 5, No. 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Iyer VN, Szybalski W, Science 1964, 145, 55–58. [DOI] [PubMed] [Google Scholar]
- [72].Tomasz M, Lipman R, Chowdary D, Pawlak J, Verdine GL, Nakanishi K, Science 1987, 235, 1204–1208. [DOI] [PubMed] [Google Scholar]
- [73].Rajski SR, Williams RM, Chem. Rev 1998, 98, 2723–2795. [DOI] [PubMed] [Google Scholar]
- [74].Uchida I, Takase S, Kayakiri H, Kiyoto S, Hashimoto M, J. Am. Chem. Soc 1987, 109, 4108–4109. [Google Scholar]
- [75].Paz MM, Hopkins PB, J. Am. Chem. Soc 1997, 119, 5999–6005. [Google Scholar]
- [76].Huang H, Pratum TK, Hopkins PB, J. Am. Chem. Soc 1994, 116, 2703–2709. [Google Scholar]
- [77].Hirai O, Shimomura K, Mizota T, Matsumoto S, Mori J, Kikuchi H, J. Antibiot 1987, 40, 607–611. [DOI] [PubMed] [Google Scholar]
- [78].Pritsos CA, Sartorelli AC, Cancer Res. 1986, 46, 3528–3532. [PubMed] [Google Scholar]