

Abstract
Src family kinases (SFKs) are a group of non-receptor tyrosine kinases that are characterized by their involvement in critical signal transduction pathways. SFKs are often found attached to membranes but little is known about the conformation of the protein in this environment. Here, solution nuclear magnetic resonance (NMR), neutron reflectometry (NR), and molecular dynamics (MD) simulations were employed to study the membrane interactions of the intrinsically disordered SH4 and Unique domains of the Src family kinase Hck. Through development of a procedure to combine the information from the different techniques, we were able produce a first-of-its-kind atomically detailed structural ensemble of a membrane-bound intrinsically disordered protein. Evaluation of the model demonstrated its consistency with previous work and provided insight into how SFK Unique domains act to differentiate the family members from one another. Fortuitously, the position of the ensemble on the membrane allowed the model to be combined with configurations of the multi-domain Hck kinase previously determined from small-angle solution X-ray scattering to produce full-length models of membrane-anchored Hck. The resulting models allowed us to estimate that the kinase active site is positioned about 65 ± 35 Å away from the membrane surface, offering the first estimations of the lengthscale associated with the concept of SFK subcellular localization.
Keywords: Src family kinase, Hck, intrinsically disordered protein, membrane
Introduction
Kinases are enzymes that regulate many key cellular processes via their roles in signal transduction pathways. c-Src kinase, the prototypical member of the Src family kinases [1], is a non-receptor tyrosine kinase vital for cell growth, proliferation, metabolism, differentiation, adhesion, and migration [2, 3, 4]. Over-activation of Src is often at the origin of several pathologies and anti-cancer drugs such as the kinase inhibitors dasatinib and bosutinib were designed to target Src specifically because of its importance in cellular signaling [5, 6].
Structurally, the members of the Src family of kinases share a common multi-domain architecture, consisting of SH3, SH2, and kinase (SH1) domains preceded by a ~80 residue region of low conservation called the Unique (U) domain and a membrane-targeting SH4 region at the N terminus (1). Crystal structures have shown that the catalytic activity of SFKs is tightly regulated by autoinhibition, with activation being achieved by displacing one or all of the interactions between the SH1 and the regulatory SH3 and SH2 domains [7, 8, 9].
While the structured domains (SH3, SH2 and Kinase) are highly homologous, the membrane anchoring SH4 and U domains share very little sequence similarity among the family. The SH4-U region is intrinsically disordered and ranges in size from about 60 to 90 amino acids. Intrinsically disordered proteins (IDP) are not expected to adopt a unique molecular conformation. The SH4 domain spans the first 10–15 residues and drives membrane association via three distinct features: N-terminal myristoylation, an SFK-dependent number of palmitoyl groups, and a lysine rich stretch of amino acids that augments lipid binding and directs SFKs to negatively charged membranes [10, 11, 12].
The functional redundancies and high degree of conservation in the regulatory and catalytic domains has left the specific roles of the eight human SFKs (Src, Yes, Fyn, Fgr, Hck, Lyn, Lck, and Blk) largely unknown. The search for distinguishing features has identified sub-cellular localization and sensitivity to various stimuli as promising candidates for further exploration [13, 14, 15]. In this regard, the SH4-U region is of high interest owing to its direct interaction with membranes and diversity among the otherwise highly conserved SFK sequences. Clear lines of evidence show that SH4-U domains influence SFK substrate specificity and function [16, 17, 18, 19, 20]. Localization of activated SFKs at the membrane through the SH4-U region is critical for the regulation of specific cellular processes, and may permit a tight selection over downstream substrates. Most notably, a Zn2+ binding site in the U domain of Lck has been found to couple the protein to CD-4 and CD-8 in an interaction required for proper T cell development and activation [19], and a lipid binding region on the Unique domain was identified that depends on phosphorylation state [21]. Further studies of Src detailed specific interactions between the SH4-U region and the SH3 domain, providing a potential mechanism for how the SH4-U communicates with the rest of the kinase [21, 22, 23]. This sparseness of detailed structural information on SFK SH4-U regions stems from difficulty in employing standard structural techniques to IDPs and is compounded with further challenges when investigating their membrane bound forms. Further development of procedures and continued efforts to examine SH4-U–membrane interactions and how they contribute to kinase function promises to answer many outstanding questions about roles SH4-U regions play in distinguishing the SFKs from one another.
Here we focus on Hematopoietic cell kinase (Hck), a phagocyte specific SFK proto-oncogene that plays a critical role in Bcr/Abl-chronic myeloid leukemia (CML); constitutive activation of Hck by direct interaction with the oncogene Bcr/Abl is required for the establishment of leucocyte transformation [24, 25, 26, 27]. Hck is expressed as two isoforms (generated by alternative initiations of translation of a single mRNA) with apparent molecular weights of 59 kDa (p59Hck) and 61 kDa (p61Hck) by SDS- PAGE [28]. The two isoforms differ by 21 residues at the N-terminal end (MGGRSSCEDPGCPRDEERAPR). Both isoforms may be myristoylated at the N-terminus (p59Hck more likely so than p61Hck) but p59Hck can also be palmitoylated on Cys3, leading to different localizations within the cell: p59Hck is mainly associated with the plasma membrane and p61Hck with lysosomal membranes [29, 30, 31, 32]. Interestingly, the sequence of the U domain of Hck is well conserved across different species, despite differences between SFK members (Fig. 2), arguing for its importance as a critical structural feature for regulating SFK function.
Figure 2:
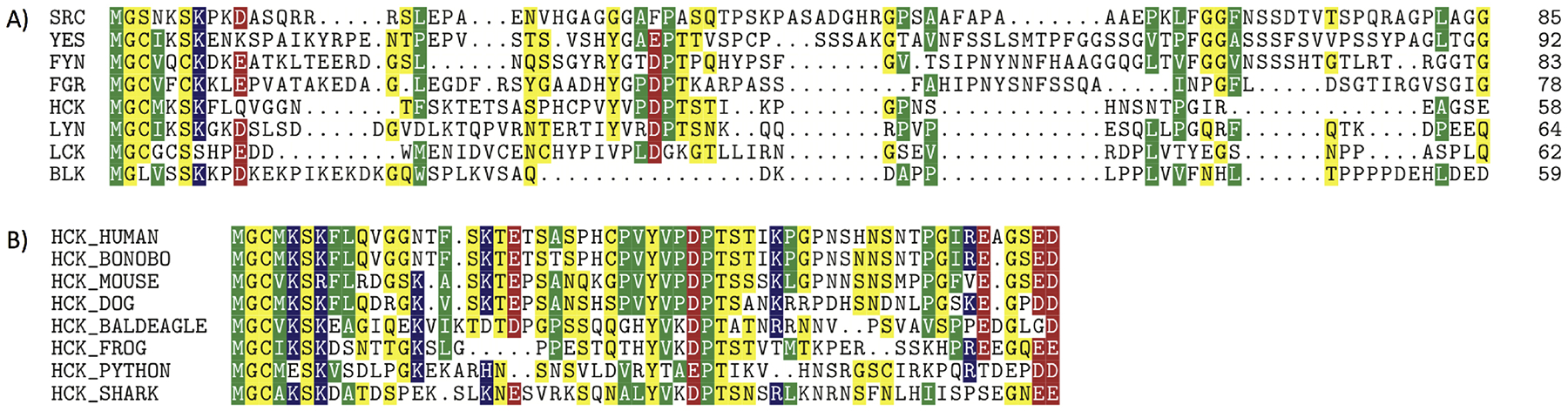
Sequence comparison of SFKs and Hck. (A) Low sequence conservation of human SFKs in the SH4-U region; acidic (−) X basic (+), polar uncharged, hydrophobic nonpolar (p59Hck isoform shown). (B) Observed sequence conservation for p59Hck across organisms.
From a wider perspective, records in the cBioPortal for Cancer Genomics [33] COSMIC [34] and the ICGC portal [35] indicate a cluster of cancer related mutations around amino acids (aa) 53–62: P53R (melanoma), D54Y (lung), T56K (melanoma), G62E (lung), as well as other mutations in the first 78aa: R4H (breast), S5P (liver), E16K (large intestine), G23R (stomach), S27F (melanoma), G33K (melanoma), S38* (cervical), E41K (cervical), and I73F (glioblastoma). We also note that several of these mutations introduce or modify charged residues in the sequence, which is likely to affect the association of the SH4-U with the membrane [10, 21].
Previous studies have provided a detailed picture of how the folded domains (SH3-SH2-Catalytic) behave in solution [8, 36, 37, 38]. Our main goal here is to expand this description by seeking to characterize the SH4-U domains bound to the membrane surface. As in our previous work [39], we study an unmyristoylated construct of the Hck SH4-U domains containing residues 2–79, hereafter referred to as HckSH4–U (previously referred to as p61HckSH4–U). Experimental characterization of the average spatial distribution of HckSH4–U at the membrane surface is carried out using nuclear magnetic resonance (NMR) and neutron reflectometry (NR) experiments. Restrained-ensemble molecular dynamics (re-MD) simulations are then utilized to generate statistical models of membrane-bound Hck.
The results confirm an interaction between the SH4 domain and acidic lipids, and yield an atomically detailed model of membrane bound HckSH4–U. Combination of the new model with a previously generated ensemble of Hck solution conformations [38] facilitated the production of the first full-length models of a membrane-anchored SFK and yielded new quantitative parameters to help better define the concept of “membrane localization” for an SFK.
Results
Surface Plasmon Resonance
Resh and coworkers have demonstrated that SFK SH4-Unique domains preferentially associate with acidic membranes [10, 40]. The binding of HckSH4–U to lipid membranes was therefore studied as a function of the chemistry and concentration of acidic phospholipids embedded in zwitterionic phosphatidylcholine (PC) using phosphatidic acid (PA), phosphatidylglycerol (PG) and phosphatidylserine (PS) as anionic lipid components of sparsely-tethered bilayer lipid membranes (stBLMs). stBLMs share many traits with physiological membranes, support a wide range of lipid compositions, and are structurally inert toward changes in environmental conditions, temperature, pH, and ionic strength [41]. Our screening for optimized experimental conditions to observe HckSH4–U membrane binding revealed a strong preference of the peptide for PA-containing membranes (Fig. S1), although, the overall responses are small in part due to modest size of the 78 amino acid HckSH4–U construct.
Nuclear Magnetic Resonance
Chemical shifts measured by solution 1H-15 N HSQC (heteronuclear single quantum coherence) NMR are sensitive probes of the backbone amide chemical environment. Perturbations to the chemical shifts (CSPs) induced by a binding partner subsequently yield a rich source of residue-specific information about the interaction. Technical challenges prohibit the use of biological membranes in solution NMR experiments and require the search for a mimic. Small, rapidly tumbling, disk-like bicelles can be formed by combining long chain lipids with a molar excess of short chain lipids (q < 1.0). Bicelles have found broad application in NMR [42, 43], including in studies of SrcSH4–U [21, 22], and were therefore chosen for characterization of membrane-HckSH4–U interactions.
The CSPs induced in HckSH4–U by PA or PG-containing or pure PC bicelle are shown in Fig. 3. The magnitude of the CSPs is sensitive to several factors including the strength of interaction, magnetic properties of the lipid head-groups, and in some cases, the number of bicelles present in the sample. Efforts to prepare the samples in near identical fashion were undertaken (see Methods), but the nature of the procedure makes it difficult to control lipid/bicelle concentrations and results in data that are only amenable to qualitative analysis. In all three cases HckSH4–U interacts with the bicelles using a region spanning residues Arg18 to Thr40. This region includes a lysine rich stretch of amino acids [10], plus two additional arginine residues upstream. The larger CSPs observed for the negatively charged head-groups (PA and PG) are consistent with an interpretation that HckSH4–U-membrane association is largely driven by electrostatics. The spectral perturbations in all three cases were small (Fig. S2) and, while minor structural organization in the binding region can not be ruled out, indicate that the protein remains intrinsically disordered. An additional analysis of peak intensities and volumes was carried out to search for signs of signal loss that might accompany HckSH4–U lipid association. No systematic changes were identified outside of a select few residues certered around Leu30, indicating that interactions outside of the binding region were unlikely. (Fig. S3).
Figure 3:
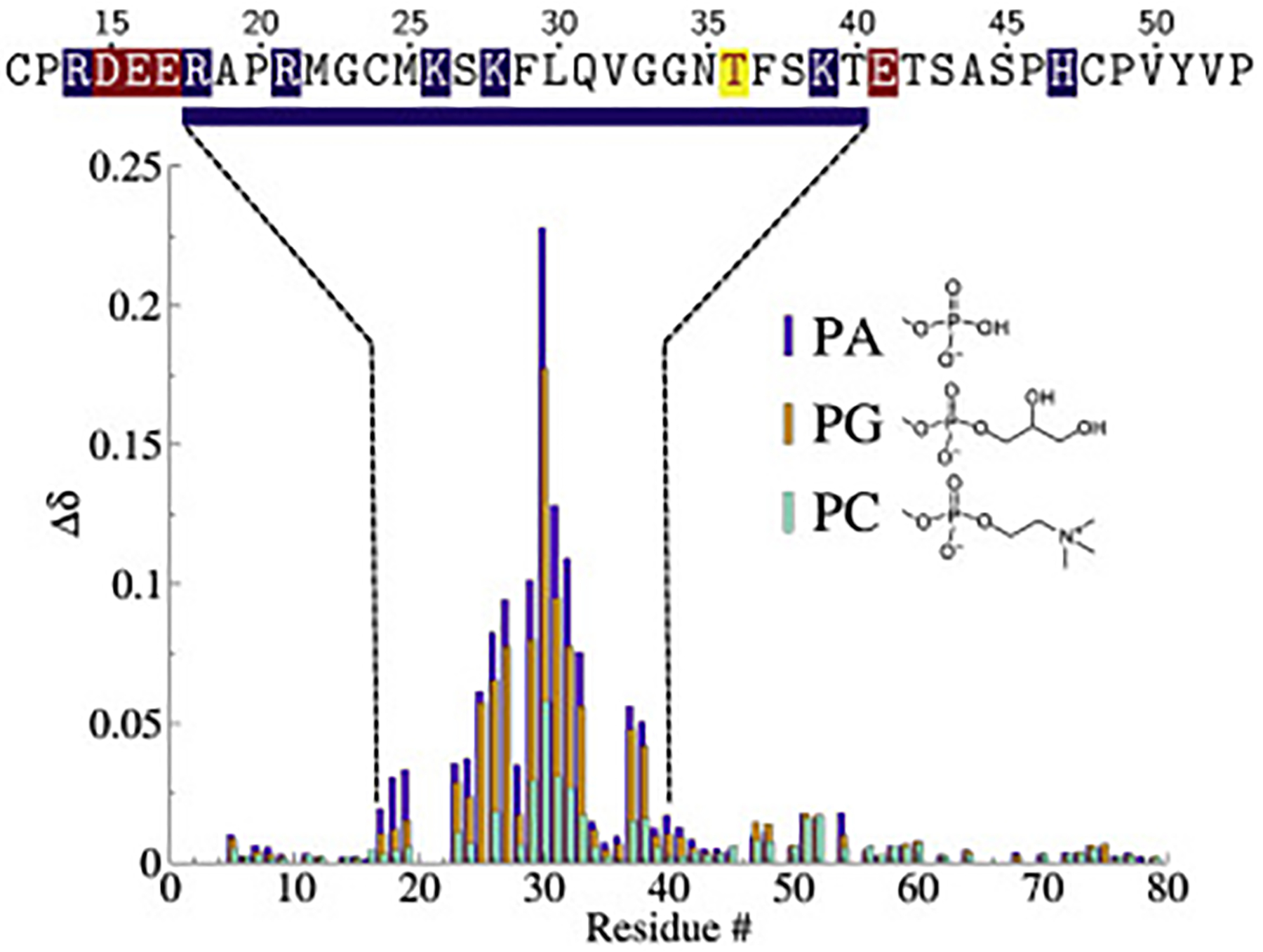
Chemical shift perturbations (CSPs, Δδ = [(Δδ1H)2 + (Δδ/515N2)]) of HckSH4–U with PA (purple), PG (orange), and pure PC (teal) bicelles. The sequence region experiencing larger CSPs is displayed above the data and highlights acidic (red) and basic (blue) residues near the lipid binding span. The enriched basic character spanning R18 to T40 suggests that the binding to negatively charged lipids is driven by electrostatics. The location of the phosphorylation site Thr36 is marked in yellow.
Neutron Reflectometry
Surface-sensitive scattering, a.k.a. reflectometry, provides long-range constraints on the organization of molecular architectures at interfaces. Neutron reflection (NR) is particularly well suited for the investigation of biological samples [44] and constitutes a valuable complement to NMR which provides local structural information. Our initial SPR characterizations showed that HckSH4–U binds in substantial interfacial concentrations to acidic stBLMs that contain 50 mol% of PA. We also observed that some protein binds loosely at the membrane surface, because a minor fraction of HckSH4–U could be rinsed off after incubation while the majority of the surface-bound protein remained stably associated.
Fig. 4 shows the material density distribution profiles of molecular components across the interface of an stBLM (50:50 PA:PC) incubated with HckSH4–U in a stably bound molecular layer after the rinseoff of loosely bound protein.
Figure 4:
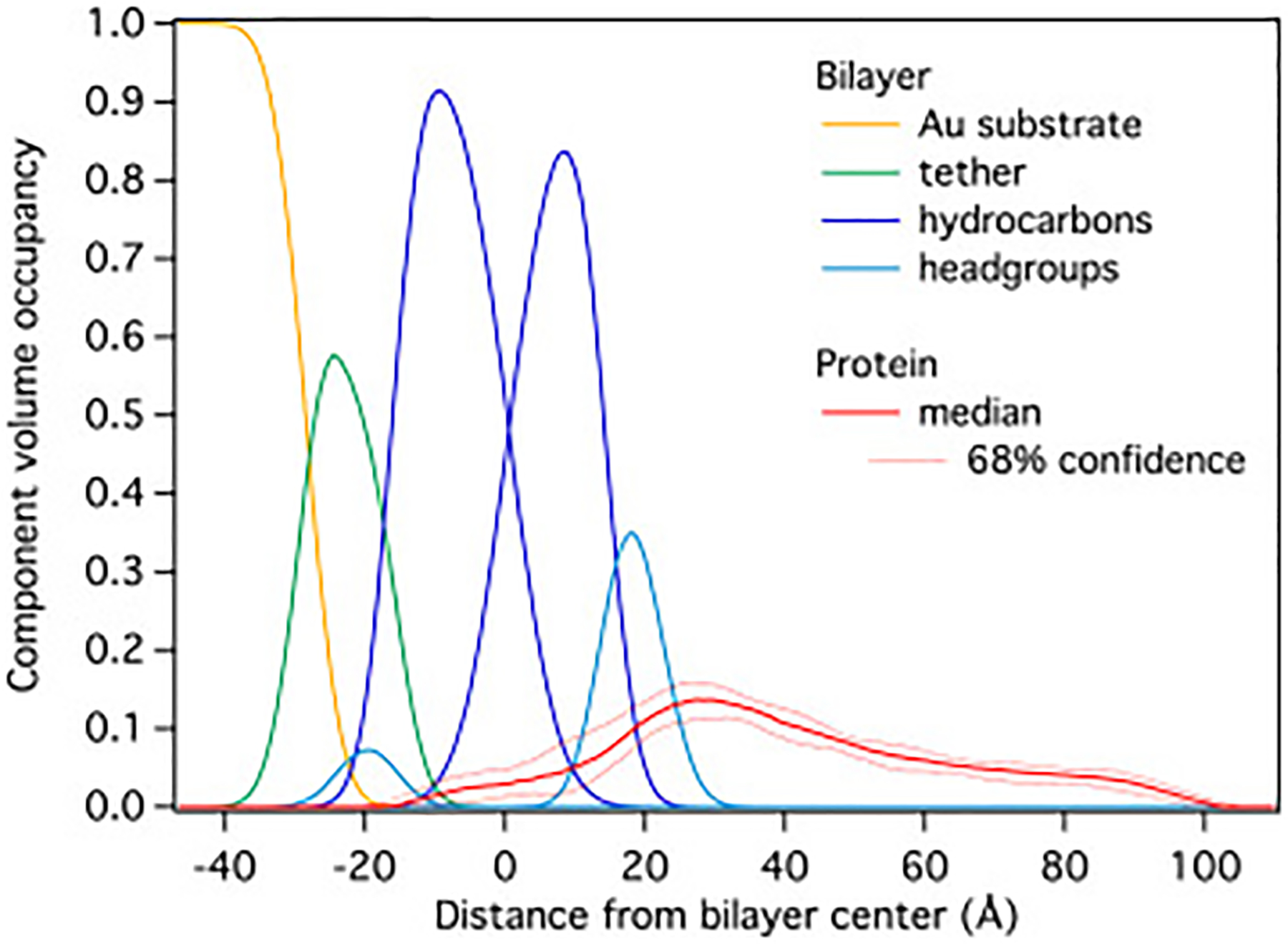
Component volume occupancy plot detailing HckSH4–U-bilayer interactions with a DOPC/DOPA (50:50) stBLM obtained from NR. Protein density co-localized with the hydrocarbon tails of the lipids indicates membrane insertion, while the peak near 30 Å suggests there are extensive interactions between HckSH4–U and the head-groups. The raw data yielding this protein distribution on the membrane are shown in the Supporting Information (Figs. S4 and S5).
Because the model is constructed such that all molecular components, including water, fill the available volume in the interfacial layer [45], this representation is denoted as component volume occupancy (CVO) representation [44]. The HckSH4–U density spans approximately 120 Å along the membrane normal and contains density inside of the stBLM, indicating partial insertion into the bilayer. The bulk of the density is located near the lipid head-groups in the substrate-distal bilayer leaflet and is suggestive of extensive surface interactions between HckSH4–U and the bilayer. The remainder of the density envelope extends to ~80 Å away from the bilayer. The deep penetration of HckSH4–U into the bilayer and low density of this region relative to the protein density found free in solution hints at a heterogeneous ensemble of conformations with varying levels of insertion.
Molecular Dynamics
Two different structural ensembles of HckSH4–U, seen at the bottom of the figure, were generated from 500 ps of re-MD in Generalized Born (GB) implicit solvent. The right panel has the N-termini oriented toward the bilayer and the left panel has the C-termini oriented toward the bilayer.
The use of neutron and X-ray reflectivity data in combination with MD simulation to produce atomic models of membrane-bound proteins has gained interest in recent years [46, 47, 44, 48]. For disordered proteins such as HckSH4–U, one must consider that the data cannot be simply mapped onto a unique three-dimensional structure, as multiple conformational states are expected to contribute to the observable data. The restrained ensemble MD (re-MD) approach, which is formally equivalent to the maximum entropy method for perturbing a statistical ensemble for the purpose of matching experimental observables while decreasing the impact of arbitrary biases [49, 50], is appealing in this situation. The simulation scheme consists of carrying out parallel MD simulations of N replicas of the basic system in the presence of a global biasing potential that approximately enforces agreement between the ensemble-average over the N replicas of a given property and its known experimental value. Because the ensemble comprises a large number of replicas, the biasing potential enforcing the ensemble-average property is only a small perturbation for any individual replica and avoids large unrealistic distortions.
Our methods for constructing a restraining potential from the one-dimensional NR density profiles and its implementation in the re-MD framework can be found in Methods. As mentioned previously, the NMR CSPs induced by complex formation are often not straightforward to interpret in a rigorous fashion and modeling these interactions is beyond the scope of this work. We were, however, able to qualitatively augment the simulations with our NMR results in order to resolve redundancies and create models that are consistent with all experimental data.
An illustration of the procedure is shown in Fig. 5. The ensemble on the left was produced from a starting conformation positioned with the N-terminus pointing towards the hypothetical location of the bilayer (i.e. the N-terminus is on the left in this figure at lower values of z in the CVO profile), and the ensemble on the right was produced from the same starting conformation, but oriented with the C-terminus pointing towards the bilayer. The structures are colored by the NMR backbone amide chemical shift perturbations (CSPs) induced by the presence of PA containing bicelles (purple bars in Fig. 3), and are scaled and positioned to match the protein density in the CVO plots generated from the NR data shown above the structures (same as Fig. 4 with dotted lines drawn to aid the eye). The ensemble on the left demonstrates a clear correspondence between the NMR and NR data, as the residues with higher CSP are positioned directly where the lipid head-groups appear in the NR data, whereas the ensemble on the right has these same residues out in solution. Therefore, the ensemble with the N-termini oriented towards the bilayer was used to setup the all-atom simulations.
Figure 5:
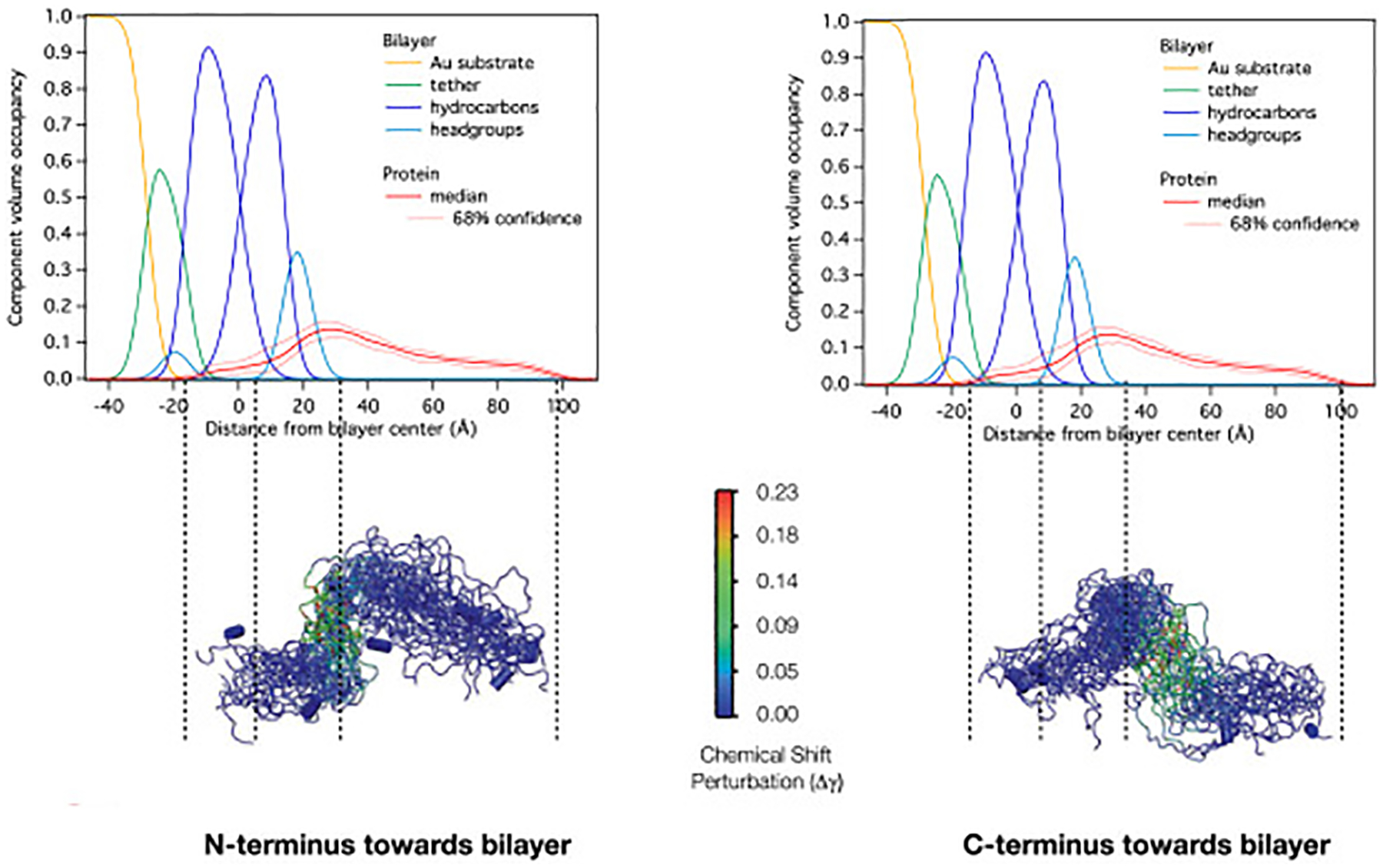
Qualitative use of NMR data for determining HckSH4–U re-MD starting orientations.
The results from the all-atom re-MD of HckSH4–U and a bilayer composed of 50:50 PA:PC are shown in Fig. 6. Out of 30 structures, 8 were found to be fully inserted into the membrane, 6 were found to be partially inserted, and the remaining 16 were found to have no density beyond the lipid head-groups (Figs. S6–S8). Segmental density profiles generated from the ensemble highlight general trends. The N-terminal segment spanning the first 16 residues appears to be responsible for insertion of HckSH4–U into the stBLM in the NR experiments. This requires the burial of 8 charged residues (3 Arg, 3 Glu, and 2 Asp) into the hydrophobic portion of the bilayer. Inspection of the ensemble suggests that this is possible via ”snorkeling” of the side chains to form interactions with the lipid head-groups and the formation of salt-bridges. The next 23 residues, identified by NMR as the stretch of amino acids that drive membrane association, are similarly interacting with the bilayer despite the absence of any restraints explicitly enforcing this interaction. The remaining 39 residues at the C-terminus of the construct extend into solution.
Figure 6:
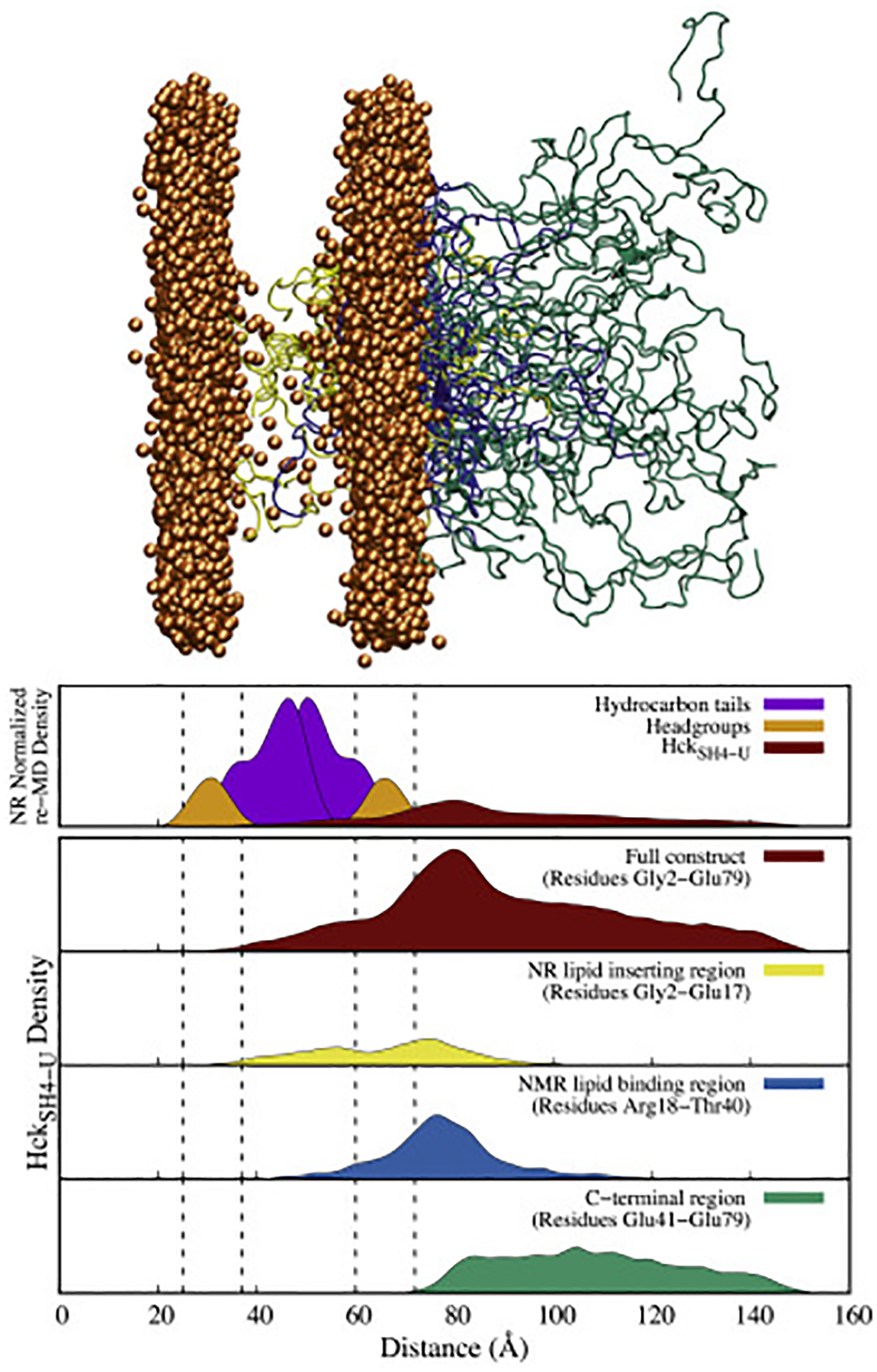
The ensemble of 30 HckSH4–U structures generated after 100 ns of NR restrained-ensemble MD in explicit solvent (top) and corresponding density profiles (bottom). The results indicate that N-terminal region of the protein is buried in the lipid bilayer in the NR studies and are consistent with the results from NMR which show the positively charged region spanning Arg18 to Thr40 interacting with the lipid head-groups. Note: The dashed lines in the density profiles indicate the position of the lipid head-groups in the MD simulation, and only the phosphorus atoms of the lipids are shown in the ensemble for clarity.
Interestingly, 14 of the 16 non-membrane-inserted structures had the positively charged amino terminus interacting with the PA head-groups in a conformation that mimics the anticipated conformation of the myristoylated form (Fig. S9). The generated ensemble is therefore able to capture all of the available data while maintaining consistency with the previously known features that drive SFK membrane binding.
Conformations of the full-length membrane-bound kinase
We previously used a Bayesian-based Monte Carlo procedure with coarse-grained (CG) simulations to construct statistical ensembles describing the assembly states of the SH3–SH2–Catalyic–C-tail domains (HckSAXS) of Hck in solution from small-angle X-ray scattering (SAXS) data [38]. The analysis, which involved determining the statistical weights of clusters of structures generated from CG simulation by matching the experimental SAXS profile, allowed the characterization of the relevant assembly states of HckSAXS upon the binding of different signaling peptides. Specifically, three assembly states were found to be of high relevance: one that captured the down-regulated state (cluster 1 in the original work), and two that were able to reproduce the SAXS data in the up-regulated state (clusters 5 and 6).
Combining the information about the assembly states of HckSAXS with the conformations of HckSH4–U determined here makes it possible to generate a set of full-length models of membrane-bound Hck in the up and down regulated states. While interactions between HckSAXS and the SH4-U are likely to alter the distribution, the ensemble of conformations can be used to estimate the plausible localization of the active site. In practice, the set of full-length models was generated by linking the C-terminus of HckSH4–U to the N-terminus of the HckSAXS construct in random orientations, and rejecting all models that position the protein inside the bilayer. The results of this modeling are displayed in Fig. 7.
Figure 7:
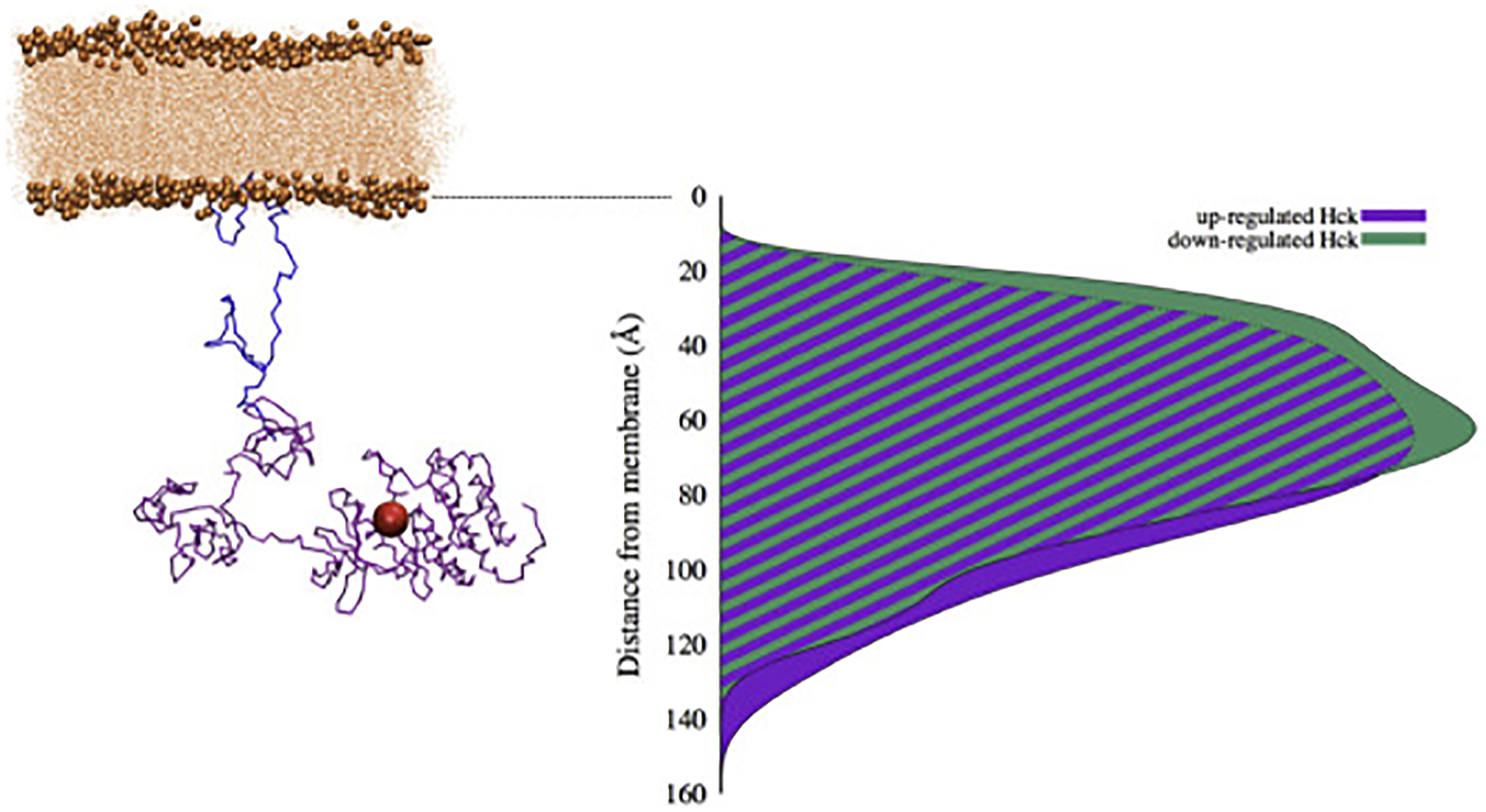
Model of membrane-anchored Hck. On the left is an illustrative example of up-regulated Hck conformation generated from re-MD and SAXS results. HckSH4–U is shown in blue and the folded domains and C-tail from SAXS (HckSAXS) in purple, with the position of the active site represented by a red sphere. B) Distributions for the up-regulated (purple) and down-regulated (green) SAXS clusters generated from the modeling procedure.
While one example of Hck in an up-regulated conformation is shown on the left in Fig. 7, it is important to note that the present procedure provides a statistical ensemble of conformations of the full-length model of membrane-bound Hck. Of primary interest in these models is the location of the catalytic site relative to the membrane, as this will govern the local spatial localization of the phosphorylation signaling relative to potential downstream substrates of Hck. In Fig. 7, the position of the active site is indicated by a red sphere. This distributions relative to the membrane surface can be calculated by averaging over the statistical ensemble of full-length membrane-bound Hck conformations. From this analysis, the active site of Hck is 65 Å away from the membrane surface, with variations on the order of ± 35 Å relative to the mean position. In going from the down- to the up-regulated form there is only a moderate shift in the position of the catalytic phosphorylation site, by which the distance to the membrane increases by about 10 Å.
Discussion
Model consistency and significance
While the HckSH4–U construct studied here is not myristoylated, it nonetheless can provide meaningful information about the membrane interactions, as shown by previous work by Pons and collaborators [21, 22, 23]. However, it is important to examine if the structures generated from the NR driven re-MD are consistent with the NMR data because multiple sources of information were used to produce a first-of-its kind structural ensemble of a lipid bound IDP. Reassuringly, the ensemble has the same basic stretch of amino acids interacting with the lipid head-groups as the NMR data. This occurred despite the absence of specific NMR derived restraints and suggests a consistency between the results from NR and NMR. Additionally, the fact that changes in NMR chemical shifts remain small indicate that no secondary structure was formed, which is similarly observed in the ensemble.
The lack of bicelle induced NMR CSPs in the N-terminal region of HckSH4–U (Fig. 3), however, is not likely consistent with the partial membrane insertion observed in the NR data. Furthermore, long-lived HckSH4–U-bicelle complexes expected to accompany insertion would result in line broadening or a loss of signal for residues near the protein-lipid interface due to an increase in rotational correlation time [51]. Analysis of the peak intensities and volumes of HckSH4–U in the presence and absence of bicelles indicates that there are no systematic changes to the N-terminal resonances as a result of lipid association (Fig. S3). We therefore conclude that insertion is not occurring to any appreciable extent in our NMR samples. Given that PA is known to create packing defects in lipid bilayers that facilitate penetration into the hydrophobic core [52], we propose that the insertion is a result of including PA in the stBLM and that the different morphology of the NMR bicelles did not promote the same phenomenon. We do not anticipate that this mode of binding is physiologically significant since membrane localization is further reduced in the absence of myristoylation [29], the p61Hck isoform that contains the buried residues is primarily located in the cytosol, and the SPR and NR experiments suggest that the inserted structures have high membrane affinity. We note, however, that the model retains significance because the portion of the construct buried in the ensemble contains the residues that would be adjacent to the membrane if the construct were lipidated (Fig. S9).
Comparison with previous work
Hck exists as two equally populated, but differently localized, isoforms that vary by a 21 residue extension at the N-terminus (termed p61Hck and p59Hck for the long and short isoforms, respectively) [29]. The extension, which is present in the construct studied here, lacks a lysine at the N-terminus and the palmitoylation site present in the shorter isoform. The lipid binding region of HckSH4–U observed in this work spans Arg18 to Thr40 and contains the lysine rich region corresponding to the SH4 of p59Hck. The lack of CSPs upstream of Arg18, and small signal change for Arg18 and Arg21 suggests that the 21 amino acid extension present in the longer isoform does not significantly contribute to lipid binding. This would indicate that the two isoforms interact with the membrane in a similar fashion and the extension may lead to distinct cellular localization due to its different lipidation pattern.
When compared to Src, both HckSH4–U and SrcSH4–U have a preference for negatively charged lipids, but in HckSH4–U the SH4 lysines were primarily responsible for the interaction as opposed to the ”RRR” motif found in Src. Further differences were observed in the Unique domain, as no additional residues in Hck were found to interact with the lipids. Because p61Hck and Src both lack palmitoylation sites and myristoylation alone is not sufficient to drive membrane interaction [10], more importance is placed on SH4-U for assisting membrane binding of these two proteins. The fewer lipid binding interactions found in HckSH4–U may weaken membrane affinity and could be responsible for the higher portion of p61Hck present in the cytosol (~ 70%) [29] than Src (~ 30%) [53].
Phosphorylation of SrcSH4–U has been shown to alter interactions with the membrane [21], raising the question of whether the same could be occurring in Hck. Indeed, phosphorylation has been reported on the Unique domain tyrosine in mouse Hck (Tyr51 in human Hck) and has been shown to impact Hck activity [54]. Given that Tyr51 is 11 residues downstream of the lipid binding site, phosphorylating this residue is unlikely to significantly impact membrane binding. Interestingly, it has been reported that the 28 C-terminal residues of the Hck Unique domain modulate the function of the Hck SH3 domain [55], hinting at a possible implication of Tyr51.
A search of the PhosphoSite Plus database (https://www.phosphosite.org) identifies a second phosphorylation site in the Unique domain at position Thr36. This residue is located within the lipid binding region (Fig. 3), suggesting that phosphorylation may affect membrane binding. The functional consequence of such a modification is unclear, however, as it may serve to detach a portion of the lipid binding region allowing for further kinase extension away from the membrane, or it may promote membrane dissociation alltogether as observed for a similar phosphorylation site in Src at Ser17 [56].
Biological significance
It has long been understood that SFKs do not display high specificity for their substrates [57], and that sub-cellular localization is an important mechanism for directing one given kinase toward a specific downstream target protein. Clearly, the biological signal can be locally enhanced toward specific membrane-bound targets if the enzymatic activity of a kinase is restricted to the vicinity of the bilayer. In this regard, it becomes of particular interest to clarify the length-scale associated with the concept of spatial localization. According to the present analysis, the active site of Hck is positioned about 65 Å away from the membrane surface (Fig. 7), and is considerably restricted within a microscopic volume near the membrane surface, with variations on the order of ± 35 Å relative to the mean position. The change in active site location accompanied by a change in assembly states of the multi-domain kinase is smaller, about 10 Å, but may also be functionally significant.
The characterization of the membrane anchor of Hck kinase allows us to attribute, for the first time, a physical meaning to the concept of spatial localization. Even if only a fraction of Hck is localized at the membrane through equilibrium partitioning with the cytoplasm, the effective local concentration of catalytic sites would clearly be very high, resulting in a biological phosphorylation signal that is sharply focused near the membrane surface. These results lead to the idea that the SH4-U region might perhaps play a more subtle functional role than that of a simple flexible membrane anchor and that the SH4-U region could be a critical element for controlling both the membrane localization and the specificity of the kinase toward downstream targets. Thus, an additional level of specificity may emerge through the localization of the kinase active site at a specific distance or range of distances from the membrane surface that may select membrane-associated protein targets according to their conformation. This hypothesis is supported by evidence showing that SH4-U domains influence SFK substrate specificity and function [16, 17, 18, 19, 20]. However, to what extent the average spatial distribution of a membrane-anchored SFK effects its ability to phosphorylate specific tyrosine residues in membrane-bound downstream targets remains unclear. This also raises a host of questions about the sensitivity of the signaling event to the local structural and dynamical features of the SH4-U region and how those might impact the accessibility to a specific site in a target protein. One possibility to explore these intriguing issues, would be to develop an assay measuring the functional potency of a member of a SFK to phosphorylate specific tyrosines in a given membrane-bound protein. For example, one could investigate the impact of point mutations in SH4-U on a downstream target or the outcome with different engineered chimeric constructs obtained by swapping SH4-U from different kinases.
Conclusion
We have produced a structural ensemble of the membrane bound SH4-U region of Hck, identifying specific interactions of the region with the lipids and providing an atomically detailed model that is consistent with previous work on SFKs. This difficult task was accomplished by using NR measurements as restraints in re-MD simulations supplemented with qualitative NMR data to remove ambiguity that could not be resolved with the NR or NMR data alone. We note that the present approach, while employed for a protein with significant conformational flexibility, has general utility and will be useful for capturing heterogeneity for globular proteins studied with NR. The nature of the ensemble enabled us to combine the results with previous work on Hck and produce a first estimate of the location of the active site of membrane-bound Hck at 65 Å away from the bilayer surface. Importantly, this suggests that the SH4-U region might perhaps play a subtler functional role than that of a simple flexible membrane anchor. Experiments could be designed specifically for the purpose of investigating how the average structural and dynamical features of the SH4-U region contributes to the ability of a SFKs to phosphorylate specific tyrosine residues in membrane-bound downstream targets.
Methods
NMR
NMR spectra were collected at 25 °C on a 600 MHz Bruker Avance III HD equipped with a triple resonance (TXI) probe. Uniformly 15N labeld HckSH4–U NMR samples were produced in 50 mM PIPES 5 mM TCEP at pH 6.7 containing either 5% or 10% D2O (Additional details on protein purification can be found in the SI Protein production and purification). The pH of 6.7 was selected to balance spectral quality and physiological conditions and was sufficient for transferring all but G3, H66, and N67 resonances from the previously reported assignments at low pH [39]. Control experiments for lipid binding were performed at pH 7.2 and yielded results consistent with those obtained at pH 6.7 (data not shown).
Bicelles were formed using the short chain lipid 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) and the long chain lipids 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2-dimyristoyl-sn-glycero-3-phosphate (DMPA), and 1,2-dimyristoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (DMPG). All lipid (Avanti Polar Lipids, Alabaster, AL) were resuspended in chloroform/methanol (65:35). The solubilized lipids were combined in a long chain to short chain ratio of 4:5 (q=0.8) using glass microliter syringes into glass vials on ice, then gently mixed and dried down with N2 gas. When DMPA or DMPG were used, they were prepared in the presence of DMPC at a molar ratio of 20:80 (PA/PG):PC. The lipid mixtures were placed under vacuum overnight to complete the removal of organic solvent. The dried mixtures were resuspended in 50 mM PIPES 5 mM TCEP at pH 6.7 to form a ~12.5%w/v lipid solution, and subject to several freeze-thaw cycles by submerging the samples in ice and a 42 °C water bath until the samples appeared translucent. HckSH4–U was then added to the sample to bring the final lipid concentration to ~ 8%w/v and subject to analysis by 31P NMR to ensure proper bicelle formation. For the measurements presented here, PC, PA, and PG samples were all prepared simultaneously using identical buffers and a single HckSH4–U preparation equally divided among the three samples.
SPR and neutron reflectometry
stBLMs were formed on glass (SPR) or Si (NR) substrates coated with an atomically flat gold layer (thickness ~450 Å or ~100 Å for SPR or NR, respectively) on top of a Cr bonding layer. After formation of a self-assembled monolayer (SAM) composed of the tether compound HC18 [Z20-(Z-octadec-9-enyloxy)-3,6,9,12,15,18,22-heptaoxatetracont-31-ene-1-thiolacetate] and β-mercaptoethanol [58]. Lipid vesicles of the desired composition in 1 M NaCl were allowed to incubate the SAM-covered substrate for about 2h. By rinsing with low salt buffer (50 mM NaCl, 10 mM Tris, 5 mM TCEP, pH 8), these were subsequently fused into single bilayers anchored to the substrate by incorporation of HC18 chains, which stabilizes a 15 Å aqueous film between substrate and bilayer that is bridged by hydrophilic hexa(ethyleneoxide) tethers. SPR and NR measurements were subsequently performed in the same buffer, for NR with distinct isotopic compositions, using H2O or D2O, in succession on the same sample. The binding of HckSH4–U to such bilayers was measured by SPR on a custom-built spectrometer (SPR Biosystems, Germantown, MD) as described previously [59]. Neutron reflectivities were recorded on the NG7 or Magik [60] reflectometers at the NIST Center for Neutron Research (NCNR) for momentum transfer values 0.01 ≤ qz ≤ 0.25 Å−1 in buffers based on either H2O or D2O at room temperature [61]. Following NR measurements of as-prepared DOPC stBLMs that contained 30 mol% or 50 mol% DOPA, HckSH4–U was allowed to incubate the membranes in concentrations between 10 μM and 300 μM for 1–3 h before rinsing the bilayers with protein-free buffer and initiating the measurement of the protein-loaded membranes. Adequate counting statistics were obtained after 5–7 h. Samples with 50 μM and 50% PA yielded the highest quality data (Figs. S4 and S5) and were used for re-MD.
The NR data were analyzed in terms of component volume occupancy (CVO) profiles, constructed as described previously [45, 61]. The resulting structural models of protein-free and protein-bearing stBLMs were annotated with the neutron scattering lengths of their molecular components considering the isotopic identities of water within the structure. They were then used to simultaneously refine their expected neutron reflectivities against the measured data sets using a Monte Carlo Markov Chain algorithm implemented in the garefl and Refl1D software [62] to globally refine the structural model. This procedure permitted a rigorous evaluation of the confidence intervals associated with the CVO profiles (only shown for the protein components).
Neutron reflectometry and restrained-ensemble molecular dynamics (re-MD)
Neutron reflectivity of a planar sample is physically controlled by the variations in the scattering cross-section per area as a function of z, represented as the neutron scattering length density (nSLD),
| (1) |
where ρi(z) is the density of particles of type i at the position z perpendicular to the planar system, and bi their scattering length. It is convenient to deconvolute the data into the contributions from the individual components by modeling the experimental nSLD profile in terms of CVO profiles for the membrane, solvent and protein in the system [44, 61],
| (2) |
where ai(z) is the total cross-sectional area occupied by particles of type i at position z, and ci is the nSLD per unit area ascribed to them. The result of the analysis of experimental data is shown for all the components in Fig. 4. As the CVO profile for the protein, aprot (z), depends on its net concentration per unit surface of the membrane, which is not directly known, we construct a normalized experimental profile for the protein to use as a reference in the re-MD simulations,
| (3) |
The protein nSLD profile from the MD system is calculated by assuming an elementary form factor modeled by a gaussian function with a standard deviation (σ) of 1.5 Å scaled by the atom specific scattering lengths (bi). The ensemble average nSLD density profile is obtained by summing over all N replicas in the re-MD simulations,
| (4) |
The profile from re-MD is then normalized
| (5) |
to be consistent with the experimental profile in Eq. 3. In practice, the z-axis for Pexp (z) and PMD (z) was discretized in equal increments Δz = 1 Å for convenience, with z = nΔz (normalization is achieved by summing the value of P over all bins, ). The calculated protein density envelope was restrained to match Pexp (z) of the protein from the NR experiment,
| (6) |
and turned into a one dimensional force by taking the derivative with respect to z
| (7) |
where K is the force constant restraining the ensemble of replicas, PMD (n) is the nth bin ensemble-average density profile generated from the re-MD, Pexp (n) is the nth bin density profile from NR (see SI), bi is the atom specific neutron scattering length density of the protein atom i, σ = 1.5 Å is the width ascribed to the gaussian atomic NR form factor, zi is the z-coordinate of the protein atom i, and Δz = 1.0 Å is the bin width used to calculate the density profile.
The multiple copy scripting interface was used to calculate the individual density profiles of each copy independently, then collect them together to create the ensemble average nSLD density profile from which the forces could be calculated for the re-MD simulations. The procedure was sufficiently efficient to allow for evaluation of the forces every 100 timesteps without an appreciable impact on performance. The calculation of the protein density profiles was too expensive to perform in tcl, so the operation was performed in C using SWIG to generate a tcl wrapper. Scripts for implementing the procedure in NAMD are available on GitHub at https://github.com/RouxLab/Restrained-ensemble-molecular-dynamics-simulations.
The choice of a starting conformation for the fitting routine is somewhat arbitrary. An initial structure of HckSH4–U was generated and subject to 1 ns of equilibration with the generalized Born (GB) implicit solvation model using NAMD [63]. Our procedure was to begin with a simple elongated model of the protein (end to end Cα distance of ~238 Å) and generate a structure compact enough to fit into the 123 Å SLD envelope. While this procedure avoids unrealistic distortions of the starting structure during the initial phases of the restraining procedure, the one-dimensional information is insufficient to unambiguously orient the protein within the SLD envelope, thus making the results dependent on the starting orientation. We reasoned that the relatively large envelope for a 78 amino acid protein favored an extended structure with its longest dimension parallel with the membrane normal, and therefore decided to orient the axis defining the terminal cα atoms of the protein along the long axis (see Fig. 4) of our system. We were then presented with the decision of whether to orient the N- or C-terminus towards the membrane. The resulting structure had a distance of 109 Å (approximate width of the NR protein density profile was 123 Å), which was aligned along the z-axis, and translated to position it within the NR protein density envelope (N-terminal and C-terminal Cα atoms to 40 Å and 148.9 Å, respectively). To distinguish the better orientation, re-MD of both orientations were carried with implicit solvent and were qualitatively checked against the NMR CSPs. The results clearly favored an orientation with the N-terminus pointing towards the membrane (Fig. 5). Using this initial configuration, a re-MD simulation with 30 replicas was carried out using the multiple copy framework of namd [64] and a restraining potential on each protein atom according to Eq. (6). Additional half-harmonic potentials at 29.0 and 152.0 Å were introduced to ensure that HckSH4–U remained within the NR protein density envelope.
The re-MD simulation was first carried out for 100 ps with GB implicit solvent (effective ion concentration of 0.3 M, solvent dielectric of 20) in order to generate an ensemble of structures that could be used as starting conformations for building all-atom systems. Approximately 50% POPA and 50% POPC were used to construct bilayers around these structures using the replacement method as implemented in charmm-gui [65]. In each all-atom system, the upper leaflets contained 200 lipids and the bottom leaflets were constructed with between 200 and 205 lipids. The fully solvated membrane-protein systems (ranging in size from 207,300 to 210,000 atoms - nearly 6.5 million atoms in total for the 30 replicas), were minimized using the charmm-gui protocol, simulated for 1 ns without restraints to relax the system, and then run with re-MD for 100 ns. Because the procedure was designed to fit the absolute positions along the z-axis, additional restraints were applied to the lipids in the colvars module of namd to restrict the motion of the bilayer in the z-direction.
Supplementary Material
Figure 1:
Schematic representation of a full-length membrane-associated SFK, SH4-U-SH3-SH2-[kinase domain], in the down-regulated inactive conformation.
Research Highlights.
Src family kinases are a group of signaling proteins that participate in many critical cellular pathways.
These enzymes are often found associated with the membrane but details of their conformation are not known due to difficulties in studying them in this environment.
Data from multiple techniques are combined using a new computational methodology to characterize the domains of a Src family kinase (Hck) that interact with the membrane.
Using this new information in concert with results from our previous work allows us to refine the concept of membrane localization for these proteins.
Acknowledgments
The authors would like to thank Lydia Blachowicz and Timofey Karginov for helpful discussion and assistance with sample production and Dr. Joseph Sachleben for assistance with NMR data acquisition. Dr. David Vanderah (Institute for Bioscience and Biotechnology Research, Rockville, MD) provided the tether lipid HC18. This work was supported by the National Institutes of Health via Grant R01-CA093577 from the National Cancer Institute, the U.S. Department of Commerce under grant 70NANB17H299, and resources provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory, under grant 1S10OD018495-01. Beamtime at the NIST Center for Neutron Research is gratefully acknowledged.
Certain commercial materials, equipment, and instruments are identified in this paper in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials, equipment, or instruments identified are necessarily the best available for the purpose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Boggon TJ, Eck MJ, Structure and regulation of Src family kinases, Oncogene 23 (48) (2004) 7918–7927. [DOI] [PubMed] [Google Scholar]
- [2].Thomas SM, Brugge JS, Cellular functions regulated by Src family kinases, Annu. Rev. Cell Dev. Biol 13 (1997) 513–609. [DOI] [PubMed] [Google Scholar]
- [3].Hubbard SR, Till JH, Protein tyrosine kinase structure and function, Annu. Rev. Biochem 69 (2000) 373–398. [DOI] [PubMed] [Google Scholar]
- [4].Mohapatra DP, Park KS, Trimmer JS, Dynamic regulation of the voltage-gated Kv2.1 potassium channel by multisite phosphorylation, Biochem. Soc. Trans 35 (Pt 5) (2007) 1064–1068. [DOI] [PubMed] [Google Scholar]
- [5].Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC, 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor, J. Med. Chem 49 (23) (2006) 6819–6832. [DOI] [PubMed] [Google Scholar]
- [6].Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C, In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells, Cancer Res. 66 (23) (2006) 11314–11322. [DOI] [PubMed] [Google Scholar]
- [7].Sicheri F, Kuriyan J, Structures of src-family tyrosine kinases, Curr. Opin. Struct. Biol 7 (6) (1997) 777–785. doi: 10.1016/S0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- [8].Sicheri F, Moarefi I, Kuriyan J, Crystal structure of the Src family tyrosine kinase Hck, Nature 385 (6617) (1997) 602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- [9].Xu WQ, Harrison SC, Eck MJ, Three-dimensional structure of the tyrosine kinase c-src, Nature 385 (6617) (1997) 595–602. doi:Doi 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- [10].Resh MD, Myristylation and palmitylation of src family members: The fats of the matter, Cell 76 (3) (1994) 411–413. doi: 10.1016/0092-8674(94)90104-X. [DOI] [PubMed] [Google Scholar]
- [11].Patwardhan P, Resh MD, Myristoylation and membrane binding regulate c-src stability and kinase activity, Mol. Cell. Biol 30 (17) (2010) 4094–4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Resh MD, Fatty acylation of proteins: The long and the short of it, Prog. Lipid Res 63 (2016) 120–131. doi: 10.1016/j.plipres.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lowell CA, Soriano P, Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories., Genes Dev. 10 (15) (1996) 1845–1857. doi: 10.1101/gad.10.15.1845. [DOI] [PubMed] [Google Scholar]
- [14].Sen B, Johnson FM, Regulation of Src family kinases in human cancers, J Signal Transduct 2011 (2011) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chu P-H, Tsygankov D, Berginski ME, Dagliyan O, Gomez SM, Elston TC, Karginov AV, Hahn KM, Engineered kinase activation reveals unique morphodynamic phenotypes and associated trafficking for Src family isoforms, Proc. Natl. Acad. Sci. U.S.A 111 (34) (2014) 12420–12425. doi: 10.1073/pnas.1404487111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Carrera AC, Paradis H, Borlado LR, Roberts TM, Martinez C, Lck unique domain influences lck specificity and biological function, J. Biol. Chem 270 (7) (1995) 3385–3391. [DOI] [PubMed] [Google Scholar]
- [17].Hoey JG, Summy J, Flynn DC, Chimeric constructs containing the sh4/unique domains of cyes can restrict the ability of scr(527f) to upregulate heme oxygenase-1 expression efficiently, Cell. Signal 12 (9–10) (2000) 691–701. doi:Doi 10.1016/S0898-6568(00)00116-9. [DOI] [PubMed] [Google Scholar]
- [18].Summy JM, Qian Y, Jiang BH, Guappone-Koay A, Gatesman A, Shi XL, Flynn DC, The sh4-unique-sh3-sh2 domains dictate specificity in signaling that differentiate c-yes from c-src, J. Cell. Sci 116 (12) (2003) 2585–2598. doi: 10.1242/jcs.00466. [DOI] [PubMed] [Google Scholar]
- [19].Kim PW, Sun ZY, Blacklow SC, Wagner G, Eck MJ, A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8, Science 301 (5640) (2003) 1725–8. doi: 10.1126/science.1085643. [DOI] [PubMed] [Google Scholar]
- [20].Amata I, Maffei M, Pons M, Phosphorylation of unique domains of Src family kinases, Front Genet 5 (2014) 181. doi: 10.3389/fgene.2014.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pérez Y, Maffei M, Igea A, Amata I, Gairí M, Nebreda AR, Bernadó P, Pons M, Lipid binding by the Unique and SH3 domains of c-Src suggests a new regulatory mechanism, Sci Rep 3 (2013) 1295. doi: 10.1038/srep01295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Maffei M, Arbesú M, Roux A-LL, Amata I, Roche S, Pons M, The SH3 domain acts as a scaffold for the N-terminal intrinsically disordered regions of c-Src, Structure 23 (5) (2015) 893–902. doi: 10.1016/j.str.2015.03.009. [DOI] [PubMed] [Google Scholar]
- [23].Arbesú M, Iruela G, Fuentes H, Teixeira JMC, Pons M, Intramolecular fuzzy interactions involving intrinsically disordered domains, Front Mol Biosci 5 (2018) 39. doi: 10.3389/fmolb.2018.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, Skorski T, The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells, EMBO J. 21 (21) (2002) 5766–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M, Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells, Cancer Res. 56 (15) (1996) 3589–3596. [PubMed] [Google Scholar]
- [26].Stanglmaier M, Warmuth M, Kleinlein I, Reis S, Hallek M, The interaction of the Bcr-Abl tyrosine kinase with the Src kinase Hck is mediated by multiple binding domains, Leukemia 17 (2) (2003) 283–289. [DOI] [PubMed] [Google Scholar]
- [27].Poh AR, O’Donoghue RJ, Ernst M, Hematopoietic cell kinase (HCK) as a therapeutic target in immune and cancer cells, Oncotarget 6 (18) (2015) 15752–15771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lock P, Ralph S, Stanley E, Boulet I, Ramsay R, Dunn AR, Two isoforms of murine hck, generated by utilization of alternative translational initiation codons, exhibit different patterns of subcellular localization., Molecular and Cellular Biology 11 (9) (1991) 4363–4370. doi: 10.1128/MCB.11.9.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Robbins SM, Quintrell NA, Bishop JM, Myristoylation and differential palmitoylation of the HCK protein-tyrosine kinases govern their attachment to membranes and association with caveolae., Mol. Cell. Biol 15 (7) (1995) 3507–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Möhn H, Le Cabec V, Fischer S, Maridonneau-Parini I, The src-family protein-tyrosine kinase p59hck is located on the secretory granules in human neutrophils and translocates towards the phagosome during cell activation, Biochemical Journal 309 (2) (1995) 657–665. doi: 10.1042/bj3090657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carréno S, Gouze M-E, Schaak S, Emorine LJ, Maridonneau-Parini I, Lack of palmitoylation redirects p59Hck from the plasma membrane to p61Hck-positive lysosomes, Journal of Biological Chemistry 275 (46) (2000) 36223–36229. doi: 10.1074/jbc.M003901200. [DOI] [PubMed] [Google Scholar]
- [32].Astarie-Dequeker C, Carreno S, Cougoule C, Maridonneau-Parini I, The protein tyrosine kinase Hck is located on lysosomal vesicles that are physically and functionally distinct from CD63-positive lysosomes in human macrophages, Journal of Cell Science 115 (1) (2002) 81–89. [DOI] [PubMed] [Google Scholar]
- [33].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci Signal 6 (269) (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ, COSMIC: exploring the world’s knowledge of somatic mutations in human cancer, Nucleic Acids Res. 43 (Database issue) (2015) D805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, Liang Y, Rivkin E, Wang J, Whitty B, Wong-Erasmus M, Yao L, Kasprzyk A, International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data, Database (Oxford) 2011 (2011) bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Young M, Gonfloni S, Superti-Furga G, Roux B, Kuriyan J, Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation., Cell 105 (2001) 115–126. [DOI] [PubMed] [Google Scholar]
- [37].Yang S, Banavali NK, Roux B, Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories, Proc. Natl. Acad. Sci. U.S.A 106 (2009) 3776–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yang S, Blachowicz L, Makowski L, Roux B, Multidomain assembled states of Hck tyrosine kinase in solution, Proc. Natl. Acad. Sci. U.S.A 107 (2010) 15757–15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pond MP, Blachowicz L, Roux B, 1H, 15N, 13C, resonance assignments of the intrinsically disordered SH4 and Unique domains of Hck, Biomol NMR Assign 1 (2019) 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sigal CT, Zhou W, Buser CA, McLaughlin S, Resh MD, Amino-terminal basic residues of Src mediate membrane binding through electrostatic interaction with acidic phospholipids, Proc. Natl. Acad. Sci. U.S.A 91 (25) (1994) 12253–12257. doi: 10.1073/pnas.91.25.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shenoy S, Moldovan R, Fitzpatrick J, Vanderah DJ, Deserno M, Lösche M, In-plane homogeneity and lipid dynamics in tethered bilayer lipid membranes (tBLMs), Soft Matter 6 (2010) 1263–1274. doi: 10.1039/B919988H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand É, Marcotte I, Choosing membrane mimetics for NMR structural studies of transmembrane proteins, Biochim Biophys Acta Biomembr 1808 (8) (2011) 1957–1974. doi: 10.1016/j.bbamem.2011.03.016. [DOI] [PubMed] [Google Scholar]
- [43].Piai A, Fu Q, Dev J, Chou JJ, Optimal bicelle size q for solution NMR studies of the protein transmembrane partition, Chemistry 23 (6) (2017) 1361–1367. doi: 10.1002/chem.201604206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Heinrich F, Lösche M, Zooming in on disordered systems: Neutron reflection studies of proteins associated with fluid membranes, Biochim Biophys Acta Biomembr 1838 (9) (2014) 2341–2349. doi: 10.1016/j.bbamem.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shekhar P, Nanda H, Lösche M, Heinrich F, Continuous distribution model for the investigation of complex molecular architectures near interfaces with scattering techniques, J Appl Phys 110 (10) (2011) 102216. doi: 10.1063/1.3661986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Akgun B, Satija S, Nanda H, Pirrone GF, Shi X, Engen JR, Kent MS, Conformational transition of membrane-associated terminally acylated HIV-1 Nef, Structure 21 (10) (2013) 1822–1833. doi: 10.1016/j.str.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tietjen GT, Gong Z, Chen CH, Vargas E, Crooks JE, Cao KD, Heffern CT, Henderson JM, Meron M, Lin B, Roux B, Schlossman ML, Steck TL, Lee KY, Adams EJ, Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4, Proc. Natl. Acad. Sci. U.S.A 111 (15) (2014) E1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nanda H, Heinrich F, Lösche M, Membrane association of the PTEN tumor suppressor: Neutron scattering and MD simulations reveal the structure of protein–membrane complexes, Methods 77–78 (2015) 136–146. doi: 10.1016/j.ymeth.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jaynes ET, Information Theory and Statistical Mechanics, Phys. Rev 106 (4) (1957) 620–630. [Google Scholar]
- [50].Roux B, Weare J, On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method, J Chem Phys 138 (8) (2013) 084107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Son WS, Park SH, Nothnagel HJ, Lu GJ, Wang Y, Zhang H, Cook GA, Howell SC, Opella SJ, ‘q-titration’ of long-chain and short-chain lipids differentiates between structured and mobile residues of membrane proteins studied in bicelles by solution NMR spectroscopy, J. Magn. Reson 214 (2012) 111–118. doi: 10.1016/j.jmr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zegarlinska J, Piascik M, Sikorski AF, Czogalla A, Phosphatidic acid - a simple phospholipid with multiple faces., Acta Biochim. Pol 65 (2) (2018) 163–171. doi: 10.18388/abp.2018_2592. [DOI] [PubMed] [Google Scholar]
- [53].Donepudi M, Resh MD, c-Src trafficking and co-localization with the EGF receptor promotes EGF ligand-independent EGF receptor activation and signaling, Cell. Signal 20 (7) (2008) 1359–1367. doi: 10.1016/j.cellsig.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Johnson TM, Williamson NA, Scholz G, Jaworowski A, Wettenhall RE, Dunn AR, Cheng HC, Modulation of the catalytic activity of the Src family tyrosine kinase Hck by autophosphorylation at a novel site in the unique domain., J. Biol. Chem 275 (43) (2000) 33353–33364. doi: 10.1074/jbc.M002022200. [DOI] [PubMed] [Google Scholar]
- [55].Gouri B, Swarup G, Interaction of SH3 domain of Hck tyrosine kinase with cellular proteins containing proline-rich regions: Evidence for modulation by unique domain, Indian J. Biochem. Biophys 34 (1–2) (1997) 29–39. [PubMed] [Google Scholar]
- [56].Walker F, deBlaquiere J, Burgess AW, Translocation of pp60c-src from the plasma membrane to the cytosol after stimulation by platelet-derived growth factor., J. Biol. Chem 268 (26) (1993) 19552–19558. [PubMed] [Google Scholar]
- [57].Miller WT, Determinants of substrate recognition in nonreceptor tyrosine kinases, Acc. Chem. Res 36 (6) (2003) 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].McGillivray DJ, Valincius G, Vanderah DJ, Febo-Ayala W, Woodward JT, Heinrich F, Kasianowicz JJ, Lösche M, Molecular-scale structural and functional characterization of sparsely tethered bilayer lipid membranes, Biointerphases 2 (1) (2007) 21–33. doi: 10.1116/1.2709308. [DOI] [PubMed] [Google Scholar]
- [59].Shenoy S, Shekhar P, Heinrich F, Daou M-C, Gericke A, Ross AH, Lösche M, Membrane association of the PTEN tumor suppressor: Molecular details of the protein-membrane complex from spr binding studies and neutron reflection, PLoS One 7 (4) (2012) 1–13. doi: 10.1371/journal.pone.0032591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dura JA, Pierce DJ, Majkrzak CF, Maliszewskyj NC, McGillivray DJ, Lösche M, O’Donovan KV, Mihailescu M, Perez-Salas U, Worcester DL, White SH, AND/R: Advanced neutron diffractometer/reflectometer for investigation of thin films and multilayers for the life sciences, Rev Sci Instrum 77 (7) (2006) 074301. doi: 10.1063/1.2219744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Eells R, Barros M, Scott KM, Karageorgos I, Heinrich F, Lösche M, Structural characterization of membrane-bound human immunodeficiency virus-1 Gag matrix with neutron reflectometry, Biointerphases 12 (2). doi: 10.1116/1.4983155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kirby B, Kienzle P, Maranville B, Berk N, Krycka J, Heinrich F, Majkrzak C, Phase-sensitive specular neutron reflectometry for imaging the nanometer scale composition depth profile of thin-film materials, Curr Opin Colloid Interface Sci 17 (1) (2012) 44–53. doi: 10.1016/j.cocis.2011.11.001. [DOI] [Google Scholar]
- [63].Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kale L, Schulten K, Scalable molecular dynamics with NAMD, J. Comp. Chem 26 (2005) 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jiang W, Phillips JC, Huang L, Fajer M, Meng Y, Gumbart JC, Luo Y, Schulten K, Roux B, Generalized Scalable Multiple Copy Algorithms for Molecular Dynamics Simulations in NAMD, Comput Phys Commun 185 (3) (2014) 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jo S, Kim T, Im W, Automated builder and database of protein/membrane complexes for molecular dynamics simulations, PLoS One 2 (9) (2007) 1–9. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.