

Abstract

Selenium (Se) is an essential dietary element for humans and animals, and the atmosphere is an important source of Se to soils. However, estimates of global atmospheric Se fluxes are highly uncertain. To constrain these uncertainties, we use a global model of atmospheric Se cycling and a database of more than 600 sites where Se in aerosol has been measured. Applying Bayesian inference techniques, we determine the probability distributions of global Se emissions from the four major sources: anthropogenic activities, volcanoes, marine biosphere, and terrestrial biosphere. Between 29 and 36 Gg of Se are emitted to the atmosphere every year, doubling previous estimates of emissions. Using emission parameters optimized by aerosol network measurements, our model shows good agreement with the aerosol Se observations (R2 = 0.66), as well as with independent aerosol (0.59) and wet deposition measurements (0.57). Both model and measurements show a decline in Se over North America in the last two decades because of changes in technology and energy policy. Our results highlight the role of the ocean as a net atmospheric Se sink, with around 7 Gg yr–1 of Se transferred from land through the atmosphere. The constrained Se emissions represent a substantial step forward in understanding the global Se cycle.
Introduction
The environmental cycle of selenium (Se) impacts human and animal health, as Se is an essential dietary element. Although Se toxicity can also lead to deleterious health effects, Se deficiency is a more widespread problem in humans.1,2 The Se content of plant-based foods is controlled by the amount of bioavailable Se in agricultural soils. Several studies have hypothesized that Se in soils is largely supplied through atmospheric deposition,2−7 meaning that atmospheric Se cycling can influence soil Se concentrations and thus Se levels in plant-based food. Therefore, it is essential to produce accurate Se deposition maps, in order to identify agricultural areas that may be at risk for Se deficiency. A recent study has also suggested that atmospheric deposition is a driver of marine Se concentrations.8 In the ocean, Se acts as an essential element for phytoplankton growth, yet at higher concentrations it can be toxic.9−11
A few studies, dating back to the 1980s, have attempted to calculate the global atmospheric Se budget by assessing the available information about Se sources.12−14 According to these studies, approximately 13–19 Gg of Se cycle annually through the atmosphere. Volatile Se is emitted by natural sources — including volcanic outgassing (∼5% of total emissions), marine biota (35%), and terrestrial biota (15%)—and anthropogenic sources (40%)—including coal combustion, metal smelting, and biomass burning.15 Wind-blown dust and sea salt are also natural sources of Se, but in general these particles have a low Se content and shorter atmospheric lifetimes than those of the volatile forms of Se. The previous global Se emission estimates were mainly made with a bottom-up approach, which rely on measured or estimated emission factors and source activities. The alternative to the bottom-up approach is a top-down approach, which uses atmospheric models to derive emission fluxes that are compatible with observations. Mosher and Duce12 used a simple top-down approach, based on available aerosol Se observations at the time, to estimate the global fluxes from the marine and terrestrial biosphere.
Since this time, new studies have been published that investigate Se emission processes. Selenium emission fluxes are often reported as ratios with the corresponding sulfur (S) species because S and Se are in the same group in the periodic table and have analogous atmospheric cycles. For example, Amouroux et al.16 measured volatile Se species in marine environments and found a correlation with the analogous S species. Volcanic emissions of Se, and the Se/S ratio in these plumes, have been monitored using gas and particulate trapping,17 as well as with moss sampling.18 In addition to the new information about Se sources, hundreds of publications since the previous Se budgets have analyzed the concentration of Se in atmospheric samples, especially in aerosol and precipitation. Several networks in North America19−21 and Europe22,23 measure elemental concentrations, including Se, in particulate samples using standardized methods. These studies potentially contain an abundance of information about atmospheric Se cycling, though they have not yet been exploited to produce top-down estimates of Se emissions because of the lack of a suitable global Se model to invert.
Recently, we implemented the atmospheric Se cycle in the global chemistry-climate model SOCOL-AER.24 The model tracks the emission of volatile Se species, chemical transformations, uptake in the aerosol phase, transport, and deposition. A global sensitivity analysis showed that although there are large uncertainties associated with the chemical reaction rates of Se, the uncertainty in Se deposition maps mainly arises from uncertainties in global emission fluxes. For example, the uncertainties in global Se emissions from the marine biosphere and volcanoes span multiple orders of magnitude because of the high variability of past flux measurements.24 We aim to reduce the uncertainty in global Se emission fluxes by comparing available measurements with the global Se model, using Bayesian inversion methods. Similar approaches have been used to constrain emissions of greenhouse gases,25,26 as well as trace elements such as mercury.27,28 In the present study, we compile a large database of aerosol Se concentrations measured by networks and publications in the literature. We use this database to produce constrained emission fluxes of Se by applying Bayesian inversion techniques to the SOCOL-AER Se model.
Materials and Methods
SOCOL-AER Model
SOCOL-AER is an extended version of the SOCOLv3 chemistry-climate model,29 including an additional module for sulfate aerosol microphysics.30 The chemical scheme in the model is based on the MEZON submodel,31 which represents atmospheric chemistry processes with a suite of around 90 chemical species and 300 reactions. The dynamical core of the model is the middle atmosphere version of ECHAM5.32 The chemically coupled aerosol module, based on the AER model,33 tracks the sulfate particle size distribution in 40 size bins between 0.39 nm and 3.2 μm. The model calculates changes in the particle size distribution because of microphysical processes, for example, nucleation, condensation, evaporation, coagulation, and sedimentation. For this study, we use the newest version of SOCOL-AER,34 which includes interactive wet and dry deposition schemes and improved aerosol number and mass conservation. The model is run in T42 resolution (∼2.8° × 2.8°) with 39 vertical levels from the surface to 80 km. To balance computational constraints and accuracy, different time steps are employed depending on the model routine: a 2 h time step is used for chemistry and radiation, a 15 min time step for dynamics and deposition, and a 6 min time step for aerosol microphysics routines.
The atmospheric Se cycle was implemented in SOCOL-AER based on the existing information in the literature and by analogy with the S cycle.24 The model includes seven gas phase species of Se: dimethylselenide (DMSe), carbonyl selenide (OCSe), carbon diselenide (CSe2), thiocarbonyl selenide (CSSe), hydrogen selenide (H2Se), oxidized organic Se, oxidized inorganic Se, and 40 tracers of condensed Se, one for each sulfate aerosol size bin from 0.39 nm and 3.2 μm. We calculate the changes in the Se particulate size distribution during sulfate aerosol evaporation, condensational growth, and coagulation. The rate constants of gas phase Se reactions are taken from the literature or are estimated from the analogous S reaction rates (Table 2 in Feinberg et al.24). Photolysis of Se species is calculated using the measured absorption cross sections of H2Se and OCSe35,36 and assuming that CSe2 and CSSe have the same cross section as CS2.37 The oxidized Se species can condense onto sulfate aerosol particles, taking into account their gas phase diffusion and assuming a mass accommodation coefficient of 1. We assign the oxidized inorganic and organic Se tracers a high Henry’s law constant in the deposition schemes, namely ∼1012 mol m–3 atm–1, which effectively takes into account the high solubilities of oxidized Se species.38,39 Deposition rates of particulate Se depend on the host aerosol radius and grid cell meteorology.
In this study, we constrain the global Se emissions from each main emission source (anthropogenic, volcanic, marine biogenic, and terrestrial biogenic). We assume base maps for each Se source and apply Bayesian inference to calculate scaling factors for each map, that is total Se emissions from each source. For the first three sources, the spatial distribution of Se emissions is assumed to vary identically to S. We use sulfur dioxide (SO2) emissions from the Community Emissions Data Systems (CEDS),40 which were developed for phase 6 of the Coupled Model Intercomparison Project (CMIP6), as a base map for the anthropogenic emissions of Se. A known issue with this dataset is that the SO2 emissions in the Western US are overestimated and Eastern US are underestimated compared to the US Environmental Protection Agency (EPA) state-level data, and, therefore, we apply a corrective grid over the US for Se emissions that reduces Western US emissions by 50–70% and increases Eastern US emissions by 15%.41 The SO2 emission dataset is extended from 2015 to 2017 by extrapolating continental trends in SO2 emissions between 2012 and 2014. For the speciation of anthropogenic emissions, we assume that 3% of emissions occur as OCSe, 3% as CSe2, 3% as CSSe, 3% as H2Se, and 88% as oxidized inorganic Se, using mean values from our previous uncertainty analysis.24 Marine DMSe concentrations are scaled from the dimethylsulfide (DMS) climatology of Lana et al.42 and subsequent emissions are calculated online with a wind-driven parameterization.43 Volcanic emissions of Se are distributed according to SO2 emissions from the GEIA inventory, representing background degassing emissions.44,45 Volcanic Se is emitted as 6.5% H2Se and 93.5% oxidized inorganic Se. Although DMSe emissions have been measured from terrestrial biogenic sources in several field and lab studies,2 very little is known about the spatial distribution of terrestrial Se emissions. We, therefore, tested two different maps in our analysis. The first assumes that emission of Se from land is constant over all nonglaciated areas.24 For the second map, terrestrial DMSe emissions follow the spatial distribution of volatile organic compounds (VOCs) in the MEGAN-MACC inventory46 (Supporting Information, Section S1). The MEGAN-MACC Se emission map is likely more realistic because it represents the emission of DMSe from the terrestrial biosphere as a function of leaf area, light, and temperature. For brevity, we refer to these different emission assumptions as CLAND and MEGAN, respectively.
To conduct the Bayesian inversion, the aerosol Se field must be calculated under many different combinations of emission estimates. We conducted a sensitivity analysis of the aerosol Se field to all relevant uncertainties, similar to the approach in Feinberg et al.24 for deposition, and found that in most areas only uncertainties in emissions affect aerosol Se concentrations (Section S2). As Se is not expected to have any effect on climate or atmospheric chemistry because of its low concentration in the atmosphere, the aerosol Se field responds linearly to changes in emissions. Therefore, we calculate the aerosol Se concentration under an array of emission scaling assumptions using a linear model
| 1 |
θ’s refer to scaling factors that will be derived by Bayesian inversion and the subscripts A, V, M, and T refer to anthropogenic, volcanic, marine, and terrestrial sources, respectively. The R’s are response functions of the aerosol Se field as a function of space and time to a fixed amount of global emissions from each source. These response functions are calculated from four simulations of SOCOL-AER for 1970–2017, each with only one Se source turned on and all others turned off. For each of the four simulations, we choose emission scaling factors so that global Se emissions are close to the median of previously estimated ranges for each source; however, because we assume linear responses to changes in Se emissions, their specific values are not very important. For the volcanic and terrestrial simulations, the Se emissions are fixed at 1.9 and 2.7 Gg Se yr–1, respectively. Marine emissions of Se are calculated online from marine DMSe maps, which are scaled by a factor of 10–4 from marine DMS molar concentrations. For the transient anthropogenic Se emissions, SO2 emissions are scaled by 9.81 × 10–5 g Se (g S)−1. Using the four simulations and eq 1, we can calculate aerosol Se for all model grid cells between 1970 and 2017 under different emissions assumptions. We run the model for 1979–2017 in nudged mode, that is, model dynamics are forced toward ERA-Interim data,47 so that the modeled meteorology is similar to observed meteorology during this time period.
Compilation of Se Observations
We compiled the Se data from several aerosol networks: Interagency Monitoring of Protected Visual Environments (IMPROVE),19 National Air Pollution Surveillance Program (NAPS),20 Environment and Climate Change Canada’s Monitoring and Surveillance in the Great Lakes Basin (GLBM),21 European Monitoring and Evaluation Programme (EMEP),22 and the UK Heavy Metals Monitoring Network (UKHMMN).23 Further information about the measurement methods of these networks is available in Table S1. For three of the networks, IMPROVE,48 NAPS,49 and GLBM,50 we downloaded individual Se measurements and calculated annual means for each measurement station, replacing all undetected values with half of the detection limit. We compiled annual mean values for EMEP from annual reports51 and for UKHMMN from annual reports52 and an online database.53 Each annual mean at a certain site is treated as a separate data point in our Bayesian inversion, to be compared with the model annual mean at the site coordinates. For our analysis, we remove site annual means if more than 30% of the data are below the detection limit or less than nine months of the year were measured. Stations that are close to point sources or traffic in the UKHMMN and NAPS databases are excluded (19 out of 57 stations), to avoid biasing the comparison with the coarse resolution global model. Model grid cells represent an area of ∼300 × 300 km, which can include both urban and rural areas, while the aerosol measurements are made in point locations. We include all stations from the GLBM, IMPROVE, and EMEP networks because they are generally located away from urban areas. Although the aerosol networks are spread throughout North America and Europe, they lack coverage in Asia and the Southern Hemisphere. In order to supplement the aerosol networks, we also reviewed the literature for studies measuring Se in aerosol.
We conducted the systematic review in Web of Science (Clarivate Analytics), searching for the terms “aerosol selenium”, “aerosol metal* Se”, and “aerosol element* Se”. The final search was made in November 2019, for a total of 590 results. We screened these papers for studies that measured Se in aerosol for longer than a week in a stationary location. We combine the particulate data from different size cutoffs—particulate matter less than 2.5 μm in diameter (PM2.5), PM10, and total suspended particulate—because many papers have suggested that Se is concentrated in the fine aerosol fraction.15,54−56 The compiled dataset includes a total of 232 papers covering 397 measurement sites. We distinguish between urban and nonurban sites in the database in our analysis, basing these categories on site descriptions in the original publications and a gridded population density dataset.57 In addition to the mean, median, and standard deviation of Se concentrations, we extracted the metadata from the compiled publications: geographic coordinates, altitude, sampling and analysis methods, the number of samples, and references (Supporting Information Spreadsheet).
We separate the compiled database into two subsets: a training dataset, which is used in the Bayesian framework to constrain Se emission estimates, and an independent validation dataset, which is not included in the Bayesian framework (Figure 1). The validation dataset will be used to verify whether the inversion procedure is successful and confirm that the derived emission estimates are robust. In the training dataset, we include all network measurements except for UKHMMN, totaling 1840 means at 205 sites in North America and Europe for the period 1980–2017. The UKHMMN data are included in the validation dataset as they may bias the overall European results because of their high spatial coverage in a region with only several model grid cells. The validation dataset also includes all of the compiled literature measurements, for a total of 421 site means between 1970 and 2017. To compare model and measurements, the model is sampled for the same time period as the observations. The model is interpolated horizontally and vertically to the coordinates and altitudes of the observations.
Figure 1.

Map showing aerosol Se measurement sites. Circle markers show sites in the training dataset and square markers show sites in the validation dataset.
Bayesian Framework
We will constrain the global emission magnitudes from the four major volatile Se sources: anthropogenic, volcanic, marine biogenic, and terrestrial biogenic. To test whether the Se/S ratio for anthropogenic emissions varies by the region, we separately constrain the North American and European anthropogenic emission total by separating the EMEP data from the IMPROVE, NAPS, and GLBM data. To conduct the Bayesian inference, we assume that the aerosol Se measurements (Y) can be described as
| 2 |
Because the Se concentrations vary
by orders
of magnitude in the observations and model, we first log transform
both quantities (Y and 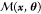 ) before comparison. The
random error term
(ε) should be constructed in a way that adequately
captures the differences between the simulated and observed values.
We consider the error between model and observations to follow a Gaussian
distribution, that is ε = N(0,σ2). Several error types are included in the error term, including
variability error, the mismatch error, and a residual error, expressed
here by their standard deviations (σ)
) before comparison. The
random error term
(ε) should be constructed in a way that adequately
captures the differences between the simulated and observed values.
We consider the error between model and observations to follow a Gaussian
distribution, that is ε = N(0,σ2). Several error types are included in the error term, including
variability error, the mismatch error, and a residual error, expressed
here by their standard deviations (σ)
| 3 |
The variability in the annual mean is calculated from the observations by taking the standard deviation of individual network measurements in logarithmic space (σvariability). The mismatch error considers how representative each model grid cell is for a point measurement, and should be larger in areas with higher regional variability in Se concentrations. We determine this error following Chen and Prinn,25 by calculating the standard deviation of aerosol Se (in log space) in the nine model grid cells surrounding the measurement station in the horizontal plane (σmismatch). Both the variability error and the mismatch error are multiplied by coefficients that are inferred in the Bayesian inversion (θϵ1 and θϵ2). A residual error term, represented by its standard deviation θϵ3, is also inferred in the Bayesian inversion.
Bayes’ theorem relates posterior the probability distributions of parameters (fpost(θ, y)) to the likelihood function (f(y | θ)) and prior knowledge of the parameter distributions (fprior(θ))
| 4 |
In our case, θ represents the four Se emission parameters (eq 1) and three error coefficients (eq 3), and y is the vector of 1840 aerosol Se annual means from European and North American networks. The likelihood function represents the probability of observing the set of measurements y given a certain set of parameters θ. Assuming that the residual error for different observations is independent and normally distributed, the likelihood function becomes the product of probability densities from the Gaussian distributions:58
| 5 |
where i is the index of a single annual mean observation, Ny is the total number of observations in the training dataset (1840), and fN(y; μ, σ2) refers to the probability density at y for a Gaussian function with mean μ and variance σ2. We assume uniform or loguniform prior probability distributions for the emission input parameters.24 The prior probability distributions were purposefully kept broad to allow the measurement inference to control the shape of the posterior probability distribution.
Using Markov Chain Monte Carlo (MCMC) sampling with the Metropolis algorithm,59 we can sample from the posterior distribution of the model parameters. At each stage of MCMC sampling, a new parameter set (θk+1) is drawn in the vicinity of the previous parameter set (θk), that is from a normal distribution with θk as the mean. The posterior probability of the new parameter set is calculated (eq 4) and compared to the previous set (r = fk+1/fk). If r is greater than 1, the sample set is accepted and if r is less than 1 the sample set is accepted with probability r (and rejected with probability 1 – r). If the new parameter set is rejected, we retain the previous parameter set (θk+1 = θk). This procedure is repeated for 106 samples. We use these samples to compute summary statistics on the posterior distributions of the parameters. We can calculate aerosol Se concentrations by inputting the posterior parameter distributions in eq 1. The posterior modeled Se values are then compared to measurements in the training and validation sets, to evaluate the quality of the SOCOL-AER model and the Bayesian inversion procedure.
Results and discussion
Constraining Se Emissions
Using the training aerosol Se dataset, we constrain the scaling factors for the Se emission sources (Table 1). The derived Se/S anthropogenic emission ratio is similar for both North American and European regions, with overlapping probability distributions. The European anthropogenic emissions are more uncertain, likely because of the smaller amount of observations compared to North America (83 vs 1757). To estimate anthropogenic Se emissions outside North America and Europe, we create an overall scaling factor that is the composite of the two probability distributions from North America and Europe. The derived marine biogenic scaling factor of 5.2 × 10–4, representing the mass ratio of Se to S emissions, is similar in magnitude to past ratios measured in North Atlantic (2.5 × 10–4) blooms.16 The derived volcanic Se/S scaling factor (3 × 10–4) is close to the median value of the reviewed volcanic fluxes in Floor and Román-Ross17 (3.5 × 10–4). The derived global terrestrial Se emissions depend on the choice of emission map: MEGAN emissions lead to higher derived global emissions (5.0 Gg Se yr–1) than in CLAND (2.1 Gg Se yr–1). This is mainly because of the emission distribution outside of the regions covered by the training dataset. MEGAN VOC emissions are higher in tropical areas, where there are no network measurements of Se. Therefore, more field campaigns measuring terrestrial Se emissions, especially in tropical regions, would be useful for constraining the global magnitude of this source. We also ran several sensitivity runs with other assumptions for the marine and anthropogenic emission maps, which did not have a large effect on the derived parameter distributions (Section S4).
Table 1. Constrained Emission Parameter Distributions from the Bayesian Analysisa.
| source | units | MEGAN | CLAND |
|---|---|---|---|
| anthropogenic North America | g Se/104 g S | 1.8 (1.7–1.9) | 2.0 (1.9–2.1) |
| anthropogenic Europe | g Se/104 g S | 2.1 (1.7–2.5) | 2.0 (1.7–2.4) |
| anthropogenic overall | g Se/104 g S | 1.9 (1.7–2.4) | 2.0 (1.7–2.3) |
| marine biogenic | g Se/104 g S | 5.2 (4.1–6.4) | 5.5 (4.3–6.8) |
| volcanic | g Se/104 g S | 3.0 (1.5–5.0) | 3.6 (1.7–6.0) |
| terrestrial biogenic | Gg Se yr–1 | 5.0 (4.3–5.7) | 2.1 (1.8–2.4) |
Median values are listed, with 2nd to 98th percentile values in parentheses. Parameters are shown for two terrestrial emission assumptions (MEGAN and CLAND).
We calculate global emission totals from each source (Figure 2) using the scaling factors from Table 1. Using our top-down approach, we have constrained the uncertainty in global Se emissions compared to prior ranges given in the literature, especially for the marine and volcanic sources. Selenium budget estimates in the 1980s12,14 estimated global marine emissions to be between 0.4 and 9 Gg Se yr–1. Later on, volatile Se flux measurements from marine cruises were extrapolated to global scales, leading to larger estimates of 28.5 and 35 Gg Se yr–1.16,60 Our median marine emission estimate (12.6 Gg Se yr–1), constrained by network observations, is within the range of these previous estimates. Measured volcanic emission fluxes show high variability in Se/S ratios,17 leading to extrapolated global emissions between 0.076 and 49.1 Gg Se yr–1.24 Given the available measurements, we find global volcanic emissions totaling 3.8 Gg Se yr–1. This flux only represents background volcanic degassing. Sporadic volcanic eruptions likely also emit significant amounts of Se, leading to regional perturbations in the atmospheric Se measurements. However, there are not enough measurements available to constrain Se emissions from these events. Median anthropogenic emissions, 10.9 Gg Se yr–1 for the years 2000–2015, are almost double the estimate of past Se budgets.12,13 Our anthropogenic emissions are still around double the budget estimates even if we use the same base year as Nriagu and Pacyna,13 1983 (12.3 vs 6.3 Gg Se yr–1), or as Pacyna and Pacyna,61 1995 (11.3 vs 4.6 Gg Se yr–1). The terrestrial Se emissions that we derive (2–5 Gg Se yr–1) are similar in range to previous assessments. Overall, we find global Se emissions are estimated between 29 and 36 Gg Se yr–1, around double the most recent estimate from Wen and Carignan15 of 13–19 Gg Se yr–1. Our assessment is based on a large dataset of Se measurements and a novel atmospheric Se model, which were not available in previous budget estimates. The modeled source fractions are similar to those reported in past studies: anthropogenic sources contribute 30–42% of global Se emissions, marine sources 32–50%, volcanic sources 6–22%, and terrestrial sources 5–18%.
Figure 2.

Probability distributions of global Se emissions from the major sources averaged for 2000–2015. For the values from this study, markers represent the median value and error bars are the 2nd to 98th percentile limits. For the literature values, markers represent the mean values from prior budget studies12−14 and error bars show the range in previous estimates, as reviewed in Feinberg et al.24 Derived values for terrestrial emissions under different assumptions (MEGAN and CLAND) are shown, with the error bars smaller than the markers; for other sources, only the MEGAN assumption is shown.
Measurement–Model Comparison
The optimized emission parameters are used to compute aerosol Se concentrations (eq 1), which can then be compared with observations. We tested the agreement of the model with observations under several different emission map assumptions (Section S4); however, for the main paper we will focus on the MEGAN emission set. We first compare the simulated values with the training dataset, for which we would expect good agreement because the model parameters are calibrated to these data (Figure 3a). Indeed, we find a very good log–log fit between model and measurements (R2 = 0.66). Overall, 85% of the modeled concentrations are within a factor of 2 of the Se measurements. The model fit is also reasonable when the results are separated into individual networks, with all networks showing more than 71% of sites matched within a factor of 2 by the model (Table S3). The fit of modeled Se with measurements is on par with the agreement our model achieves for S deposition measurements.34 This is a substantial achievement for modeling a trace element (Se), compared to an element for which much more atmospheric data are available (S).
Figure 3.

(a) Comparison of modeled aerosol Se with observed values in the training dataset between 1980 and 2017, separated by the network. Error bars show the 2nd to 98th percentile values, arising because of parametric uncertainties and random error terms (eq 3). (b) Comparison of modeled aerosol Se with observed values in the validation dataset (1970–2017). Measurement sites are categorized into urban and nonurban sites. Only nonurban sites are used to calculate R2 and the fit. The total number of sites is listed in parentheses. (c) Comparison of modeled Se wet deposition fluxes with observed fluxes (1976–2017) compiled by Feinberg et al.24 Again, only nonurban sites are used to calculate R2 and the fit. Error bars are not shown because the variability of the measured fluxes is not adequately known.
The Se emission scaling parameters are fixed over time in our Bayesian framework (eq 1). To test whether this assumption is appropriate, in Figure 4 we compare the modeled and observed trend in aerosol Se in several regions between 1988 and 2017. The Western US region shows low concentrations of aerosol Se with similar interannual variability to the model. The increase in aerosol Se in the Western US after 2011 is likely artificial. The IMPROVE network changed its analytical system from a custom energy-dispersive X-ray fluorescence (EDXRF) device to a PANalytical Epsilon 5 EDXRF in January 2011,62 which raised the detection limit for Se by around a factor of 10 to around 0.2 ng m–3. Because only sites with >70% of measurements above this new detection limit are included in the model and observation means, aerosol Se shows an increase after 2011 in the Western US. The Appalachian region in eastern US is characterized by high atmospheric Se concentrations in the 1990s because of the prevalence of coal-fired power plants, a major source of anthropogenic Se.63 Both measured and modeled Se in Appalachia decrease by around half between the periods 1995–1999 and 2011–2015. In Ontario, Canada, the model shows higher Se concentrations than the GLBM measurements between 1997 and 2001. However, the modeled decline in Se matches the measurements after 2005. In 2003, the Ontario government launched its plan of shutting down coal-fired power plants in the province, which was accomplished by 2014.64 The elimination of coal in Ontario, along with other policy and technology changes, led to a 55–70% reduction in both modeled and observed aerosol Se during this period. Overall, the model captures the trends in aerosol Se over time, even though a constant Se/S ratio is used for anthropogenic emissions. Therefore, our current assumption that Se can be directly scaled to SO2 emissions seems to be reasonable, at least in North America where continuous long-term measurement records are available.
Figure 4.

Aerosol Se trend in observations (IMPROVE and GLBM networks) and the model. Annual means are calculated for three regions in both observations and the model. The interquartile range (IQR) is shown for observation sites as shading; if the IQR disappears, it means that only one site measured in that region for that year. The regions shown are US Appalachia, including the states AL, DC, GA, KY, MD, MS, NY, NC, OH, PA, SC, TN, VA, and WV; Western US, including AZ, CA, CO, ID, MT, NM, NV, OR, UT, WA, and WY; and Ontario, Canada.
The validation dataset acts as an independent test of our model because the compiled measurements include sites outside of North America and Europe (Figure 1). We would expect the model to show worse agreement with the validation dataset than the training dataset, for two reasons. First, the model parameters were calibrated specifically to the measurements in the training dataset. Second, the compiled literature measurements of Se represent a much more heterogeneous dataset. The compiled studies have used a diverse range of sampling and analytical techniques to measure Se in aerosol. In addition, many measurements in the compiled dataset were made in urban locations that would be difficult for our global model to capture. To treat this issue, we have differentiated measurement sites into urban and nonurban locations in the comparison plot (Figure 3b). Despite our previous concerns, the model shows excellent agreement with the nonurban sites in the validation database (R2 = 0.59). The model agrees with measurements within a factor of 2 at 65% of nonurban sites (Table S4). Even in nonurban sites (n = 56) outside of North America and Europe, the model shows a good fit to the measurements (R2 = 0.67) and the fraction of sites within a factor of 2 (52%). Therefore, our assumption of applying the derived anthropogenic Se/S scaling factors globally is supported by our current dataset. Measurements of Se and S in coal have suggested that mean Se/S ratios in Chinese and US coal are similar,65−68 which supports our extrapolation of emission parameters to other regions. Nevertheless, increased measurements of atmospheric Se and Se emission factors in other regions would be highly beneficial for testing this assumption. While some sites classified as urban in the validation database do fall close to the 1–1 line, measurements in urban areas are generally much higher than the corresponding model values. For example, several studies have measured Se concentrations above 100 ng m–3 in urban regions (outside of the plot axes in Figure 3b), up to 580 ng m–3 in a metal smelting area in South Africa.69 On the other hand, the maximum modeled concentrations are around 10 ng m–3. Regional atmospheric models would be necessary to resolve Se concentrations in urban environments. Overall, the presently available aerosol Se data in nonurban areas lend confidence to the optimized SOCOL-AER model.
In addition to using independent aerosol Se measurements to validate our model, we also compare our model to measured Se wet deposition fluxes in Figure 3c. The wet deposition measurements were compiled in Feinberg et al.24 and compared in that study to the model in the year 2000. We can now compare observations with transient wet deposition fluxes calculated with optimized parameters. The model shows similar agreement to that with the independent aerosol dataset: R2 = 0.57 and 72% of modeled fluxes are within a factor of 2 of nonurban measurements (Table S5). The fact that our model matches two different Se observables (aerosol and wet deposition) is a further confirmation of our derived emission fluxes.
Environmental Significance and Outlook
The atmospheric Se cycle in the constrained SOCOL-AER model is summarized in Figure 5 for 2000–2015. Between 29 and 36 Gg of Se cycle through the atmosphere every year. Around 65% of the emissions flux is from natural sources and 35% of emissions are anthropogenic. A portion of current natural emissions may actually be historical anthropogenic Se emissions that continue to be recycled in the surface environment, which is the case for the global mercury cycle70 but is not taken into account here. The sensitivity analysis in Feinberg et al.24 suggested that the global atmospheric Se lifetime ranges between 2.9 and 6.4 d for the year 2000, with median 4.4 d. For 2000–2015, we find a median atmospheric Se lifetime of around 5.0 d, slightly shifted from the previous median because of different meteorological settings and the use of fixed values for the Se rate constants. The total atmospheric burden of Se is around 0.45 Gg, with 96% being in the aerosol phase. Selenium is mainly removed from the atmosphere by wet deposition (81%), rather than dry deposition (19%). Wet deposition is relatively more important (87%) over the ocean compared to over land (74%).
Figure 5.

Diagram of the model’s global atmospheric Se cycle for the period 2000–2015. All values are in units Gg Se yr–1, unless otherwise stated. Median values are listed with 2nd to 98th percentile values in parentheses. Fluxes are calculated using MEGAN terrestrial emissions. The 2nd to 98th percentile values for the global Se atmospheric lifetime are taken from the sensitivity analysis in Feinberg et al.,24 including parametric uncertainties in the reaction rate constants, aerosol partitioning, and speciation.
A striking feature of the budget in Figure 5 is that there is a 7 Gg Se yr–1 imbalance between marine emission and deposition of Se. Our model indicates that the ocean is a net sink for atmospheric Se and that significant transfer of Se from the land to the ocean occurs through atmospheric transport. The riverine flux of Se has been estimated by previous studies to be 2.6–8.9 Gg Se yr–1.8,71,72 Therefore, the atmospheric transport pathway is of similar magnitude, if not larger, than riverine Se transport from the land to ocean. The atmospheric Se cycle could, therefore, play a significant role in marine Se cycling. Indeed, measurements suggest that subsurface Se concentrations have increased by around 10 to 20% in the North Pacific between 1977 and 2011, likely because of the increased anthropogenic inputs from the atmosphere.8 The increase in Se concentrations is important because Se can be both an essential and toxic element for marine phytoplankton, depending on the concentration.8−11 In addition, some studies hypothesize that Se is involved in the detoxification of mercury in marine biota.73−75 Although there is already significant interest in the effect of atmospheric Se deposition on terrestrial soil systems, the link between atmospheric Se deposition and marine biogeochemistry should also be further explored.
A main conclusion from our analysis is that higher amounts of Se cycle through the atmosphere (29–36 Gg Se yr–1) than previously expected from budget estimates (13–19 Gg Se yr–1).15 In addition, current anthropogenic Se emissions (10.9 Gg Se yr–1) are almost double previous assessments.12,13,61 Our new estimates, derived by integrating a global Se model and a large measurement database, represent a significant advance in our understanding of the atmospheric Se cycle. One clear implication of our results is that the atmosphere may be a more important transport medium for Se than previously thought. Our estimate for the mean atmospheric deposition over continents, 870 mg ha–1 yr–1, can be compared to other sources of Se to soils. Combining the mean Se content in the Earth’s crust, 0.05 mg kg–1,38 with an estimate for the mean soil formation rate, 700 kg ha–1 yr–1,76 the mean bedrock source of Se to soils is 35 mg ha–1 yr–1, much lower than mean atmospheric inputs. Apart from soil formation and atmospheric deposition, other sources of Se to soils include irrigation and fertilization,2,7,77 which are expected to be minor sources in most locations but more data are required to quantify their global importance in the soil mass balance. A global biogeochemical Se cycle model developed by Mason et al.8 indicates small increases in the concentration of Se in soils and vegetation (1%) and the surface and subsurface ocean (16%) from 1850 to 2010 because of anthropogenic emissions. However, their estimates for atmospheric Se fluxes and burden are significantly different from the constrained results in our study. Our new atmospheric information can improve biogeochemical Se models and quantification of the amount of historical anthropogenic Se in different environmental compartments.
This study is the first data-driven approach investigating the atmospheric Se cycle. On the global scale, our model agrees with available atmospheric Se measurements. Implementation of the Se cycle in high-resolution regional models may enable a more in-depth comparison between measurements and models, specifically in urban areas. Increased high-quality measurements of atmospheric Se, especially in areas that currently have low measurement coverage, would be necessary to further validate SOCOL-AER. Field and laboratory measurements of Se fluxes from different sources2,17,18,78 can be used to develop new hypotheses about emission processes. Combined experimental and modeling approaches will be essential to evaluate the anthropogenic perturbation to the biogeochemical Se cycle and its consequences for ecosystem and human health.
Acknowledgments
This work was supported by a grant from ETH Zurich under the project ETH-39 15-2. We thank all individuals involved in the collection, analysis, and publication of the aerosol Se data as part of networks or individual scientific studies. IMPROVE is a collaborative association of state, tribal, and federal agencies, and international partners. US Environmental Protection Agency is the primary funding source of IMPROVE, with contracting and research support from the National Park Service. The Air Quality Group at the University of California, Davis is the central analytical laboratory of IMPROVE, with ion analysis provided by Research Triangle Institute, and carbon analysis provided by Desert Research Institute. For the Canadian aerosol data, we acknowledge researchers involved in the regional, provincial, and territorial partners of the NAPS network and Environment and Climate Change Canada’s Monitoring and Surveillance in the Great Lakes Basin (GLB) under the Chemicals Management Plan (CMP). We thank all researchers who contributed to the EMEP monitoring program. We acknowledge the Department for Environment, Food & Rural Affairs (Defra) for providing UK aerosol Se measurements. Thanks to Anthony Prenni (National Park Service), Bret Schichtel (National Park Service), Helena Dryfhout-Clark (Environment and Climate Change Canada), and Sharon Goddard (National Physical Laboratory) for providing additional information about aerosol network measurements. Thanks to Steven Smith (Pacific Northwest National Laboratory) for providing information about the CMIP6 SO2 emissions. We thank Elian Minoggio (ETH Zurich) and Franziska Genter (University of Technology Sydney), who contributed to the compilation and preliminary analysis of the aerosol Se dataset.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.0c01408.
The authors declare no competing financial interest.
Supplementary Material
References
- Fordyce F. M.Essentials of Medical Geology; Springer, 2013; pp 375–416. [Google Scholar]
- Winkel L.; Vriens B.; Jones G.; Schneider L.; Pilon-Smits E.; Bañuelos G. Selenium cycling across soil-plant-atmosphere interfaces: a critical review. Nutrients 2015, 7, 4199–4239. 10.3390/nu7064199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Låg J.; Steinnes E. Regional distribution of selenium and arsenic in humus layers of Norwegian forest soils. Geoderma 1978, 20, 3–14. 10.1016/0016-7061(78)90045-9. [DOI] [Google Scholar]
- Haygarth P. M.; Cooke A. I.; Jones K. C.; Harrison A. F.; Johnston A. E. Long-term change in the biogeochemical cycling of atmospheric selenium: Deposition to plants and soil. J. Geophys. Res. Atmos. 1993, 98, 16769–16776. 10.1029/93JD01023. [DOI] [Google Scholar]
- Fan M.-S.; Zhao F.-J.; Poulton P. R.; McGrath S. P. Historical changes in the concentrations of selenium in soil and wheat grain from the Broadbalk experiment over the last 160 years. Sci. Total Environ. 2008, 389, 532–538. 10.1016/j.scitotenv.2007.08.024. [DOI] [PubMed] [Google Scholar]
- Blazina T.; Sun Y.; Voegelin A.; Lenz M.; Berg M.; Winkel L. H. E. Terrestrial selenium distribution in China is potentially linked to monsoonal climate. Nat. Commun. 2014, 5, 4717. 10.1038/ncomms5717. [DOI] [PubMed] [Google Scholar]
- Song T.; Cui G.; Su X.; He J.; Tong S.; Liu Y. The origin of soil selenium in a typical agricultural area in Hamatong River Basin, Sanjiang Plain, China. Catena 2020, 185, 104355. 10.1016/j.catena.2019.104355. [DOI] [Google Scholar]
- Mason R. P.; Soerensen A. L.; DiMento B. P.; Balcom P. H. The Global Marine Selenium Cycle: Insights From Measurements and Modeling. Global Biogeochem. Cycles 2018, 32, 1720–1737. 10.1029/2018GB006029. [DOI] [Google Scholar]
- Harrison P.; Yu P.; Thompson P.; Price N.; Phillips D. Survey of selenium requirements in marine phytoplankton. Mar. Ecol.: Prog. Ser. 1988, 47, 89–96. 10.3354/meps047089. [DOI] [Google Scholar]
- Doblin M.; Blackburn S.; Hallegraeff G. Comparative study of selenium requirements of three phytoplankton species: Gymnodinium catenatum, Alexandrium minutum (Dinophyta) and Chaetoceros cf. tenuissimus (Bacillariophyta). J. Plankton Res. 1999, 21, 1153–1169. 10.1093/plankt/21.6.1153. [DOI] [Google Scholar]
- Cutter G. A.; Cutter L. S. Sources and cycling of selenium in the western and equatorial Atlantic Ocean. Deep Sea Res., Part II 2001, 48, 2917–2931. 10.1016/S0967-0645(01)00024-8. [DOI] [Google Scholar]
- Mosher B. W.; Duce R. A. A global atmospheric selenium budget. J. Geophys. Res. Atmos. 1987, 92, 13289–13298. 10.1029/JD092iD11p13289. [DOI] [Google Scholar]
- Nriagu J. O.; Pacyna J. M. Quantitative assessment of worldwide contamination of air, water and soils by trace metals. Nature 1988, 333, 134. 10.1038/333134a0. [DOI] [PubMed] [Google Scholar]
- Nriagu J. O. A global assessment of natural sources of atmospheric trace metals. Nature 1989, 338, 47. 10.1038/338047a0. [DOI] [Google Scholar]
- Wen H.; Carignan J. Reviews on atmospheric selenium: emissions, speciation and fate. Atmos. Environ. 2007, 41, 7151–7165. 10.1016/j.atmosenv.2007.07.035. [DOI] [Google Scholar]
- Amouroux D.; Liss P. S.; Tessier E.; Hamren-Larsson M.; Donard O. F. X. Role of oceans as biogenic sources of selenium. Earth Planet. Sci. Lett. 2001, 189, 277–283. 10.1016/S0012-821X(01)00370-3. [DOI] [Google Scholar]
- Floor G. H.; Román-Ross G. Selenium in volcanic environments: a review. Appl. Geochem. 2012, 27, 517–531. 10.1016/j.apgeochem.2011.11.010. [DOI] [Google Scholar]
- Arndt J.; Calabrese S.; D’Alessandro W.; Planer-Friedrich B. Using mosses as biomonitors to study trace element emissions and their distribution in six different volcanic areas. J. Volcanol. Geoth. Res. 2017, 343, 220–232. 10.1016/j.jvolgeores.2017.07.004. [DOI] [Google Scholar]
- Malm W. C.; Sisler J. F.; Huffman D.; Eldred R. A.; Cahill T. A. Spatial and seasonal trends in particle concentration and optical extinction in the United States. J. Geophys. Res. Atmos. 1994, 99, 1347–1370. 10.1029/93JD02916. [DOI] [Google Scholar]
- Galarneau E.; Wang D.; Dabek-Zlotorzynska E.; Siu M.; Celo V.; Tardif M.; Harnish D.; Jiang Y. Air toxics in Canada measured by the National Air Pollution Surveillance (NAPS) program and their relation to ambient air quality guidelines. J. Air Waste Manag. Assoc. 2016, 66, 184–200. 10.1080/10962247.2015.1096863. [DOI] [PubMed] [Google Scholar]
- Blanchard P.; Audette C.; Hulting M.; Basu I.; Brice K.; Backus S.; Dryfhout-Clark H.; Froude F.; Hites R.; Neilson M.; Wu R.. Atmospheric Deposition of Toxic Substances to the Great Lakes: IADN Results Through 2005. 2005, https://nepis.epa.gov/Exe/ZyPDF.cgi?Dockey=P100A9SI.PDF. Last access: March 5, 2020.
- Tørseth K.; Aas W.; Breivik K.; Fjæraa A. M.; Fiebig M.; Hjellbrekke A. G.; Lund Myhre C.; Solberg S.; Yttri K. E. Introduction to the European Monitoring and Evaluation Programme (EMEP) and observed atmospheric composition change during 1972–2009. Atmos. Chem. Phys. 2012, 12, 5447–5481. 10.5194/acp-12-5447-2012. [DOI] [Google Scholar]
- Goddard S. L.; Williams K. R.; Robins C.; Butterfield D. M.; Brown R. J. C. Concentration trends of metals in ambient air in the UK: a review. Environ. Monit. Assess. 2019, 191, 683. 10.1007/s10661-019-7824-2. [DOI] [PubMed] [Google Scholar]
- Feinberg A.; Maliki M.; Stenke A.; Sudret B.; Peter T.; Winkel L. H. E. Mapping the drivers of uncertainty in atmospheric selenium deposition with global sensitivity analysis. Atmos. Chem. Phys. 2020, 20, 1363–1390. 10.5194/acp-20-1363-2020. [DOI] [Google Scholar]
- Chen Y.-H.; Prinn R. G. Estimation of atmospheric methane emissions between 1996 and 2001 using a three-dimensional global chemical transport model. J. Geophys. Res. Atmos. 2006, 111, D10307. 10.1029/2005JD006058. [DOI] [Google Scholar]
- Stohl A.; et al. An analytical inversion method for determining regional and global emissions of greenhouse gases: Sensitivity studies and application to halocarbons. Atmos. Chem. Phys. 2009, 9, 1597–1620. 10.5194/acp-9-1597-2009. [DOI] [Google Scholar]
- Song S.; et al. Top-down constraints on atmospheric mercury emissions and implications for global biogeochemical cycling. Atmos. Chem. Phys. 2015, 15, 7103–7125. 10.5194/acp-15-7103-2015. [DOI] [Google Scholar]
- Denzler B.; Bogdal C.; Henne S.; Obrist D.; Steinbacher M.; Hungerbühler K. Inversion Approach to Validate Mercury Emissions Based on Background Air Monitoring at the High Altitude Research Station Jungfraujoch (3580 m). Environ. Sci. Technol. 2017, 51, 2846–2853. 10.1021/acs.est.6b05630. [DOI] [PubMed] [Google Scholar]
- Stenke A.; Schraner M.; Rozanov E.; Egorova T.; Luo B.; Peter T. The SOCOL version 3.0 chemistry–climate model: description, evaluation, and implications from an advanced transport algorithm. Geosci. Model Dev. 2013, 6, 1407–1427. 10.5194/gmd-6-1407-2013. [DOI] [Google Scholar]
- Sheng J.-X.; Weisenstein D. K.; Luo B.-P.; Rozanov E.; Stenke A.; Anet J.; Bingemer H.; Peter T. Global atmospheric sulfur budget under volcanically quiescent conditions: Aerosol-chemistry-climate model predictions and validation. J. Geophys. Res. Atmos. 2015, 120, 256–276. 10.1002/2014JD021985. [DOI] [Google Scholar]
- Egorova T.; Rozanov E.; Zubov V.; Karol I. Model for investigating ozone trends (MEZON). Izv. Atmos. Ocean. Phys. 2003, 39, 277–292. [Google Scholar]
- Roeckner E.; Bäuml G.; Bonaventura L.; Brokopf R.; Esch M.; Giorgetta M.; Hagemann S.; Kirchner I.; Kornblueh L.; Manzini E.; Rhodin A.; Schlese U.; Schulzweida U.; Tompkins A.. The atmospheric general circulation model ECHAM 5. PART I: Model description. 2003, http://www.mpimet.mpg.de/fileadmin/publikationen/Reports/max_349.pdf. Last access: March 5, 2020.
- Weisenstein D. K.; Yue G. K.; Ko M. K. W.; Sze N.-D.; Rodriguez J. M.; Scott C. J. A two-dimensional model of sulfur species and aerosols. J. Geophys. Res. Atmos. 1997, 102, 13019–13035. 10.1029/97JD00901. [DOI] [Google Scholar]
- Feinberg A.; Sukhodolov T.; Luo B.-P.; Rozanov E.; Winkel L. H. E.; Peter T.; Stenke A. Improved tropospheric and stratospheric sulfur cycle in the aerosol–chemistry–climate model SOCOL-AERv2. Geosci. Model Dev. 2019, 12, 3863–3887. 10.5194/gmd-12-3863-2019. [DOI] [Google Scholar]
- Goodeve C. F.; Stein N. O. The absorption spectra and the optical dissociation of the hydrides of the oxygen group. Trans. Faraday Soc. 1931, 27, 393–402. 10.1039/TF9312700393. [DOI] [Google Scholar]
- Finn E. J.; King G. W. The absorption spectrum of carbonyl selenide. J. Mol. Spectrosc. 1975, 56, 39–51. 10.1016/0022-2852(75)90201-5. [DOI] [Google Scholar]
- Burkholder J.; Sander S.; Abbatt J.; Barker J.; Huie R.; Kolb C.; Kurylo M.; Orkin V.; Wilmouth D.; Wine P.. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18. 2015, https://jpldataeval.jpl.nasa.gov/pdf/JPL_15-10.pdf. Last access: March 5, 2020.
- Rumble J. R.Handbook of Chemistry and Physics, 100th ed. (Internet Version); CRC Press/Taylor & Francis: Boca Raton, FL, 2019; http://hbcponline.com. Last access: March 3, 2020. [Google Scholar]
- Rael R. M.; Tuzaon E. C.; Frankenberger W. T. Jr Gas-phase reactions of dimethyl selenide with ozone and the hydroxyl and nitrate radicals. Atmos. Environ. 1996, 30, 1221–1232. 10.1016/1352-2310(95)00447-5. [DOI] [Google Scholar]
- Hoesly R. M.; Smith S. J.; Feng L.; Klimont Z.; Janssens-Maenhout G.; Pitkanen T.; Seibert J. J.; Vu L.; Andres R. J.; Bolt R. M.; Bond T. C.; Dawidowski L.; Kholod N.; Kurokawa J.-i.; Li M.; Liu L.; Lu Z.; Moura M. C. P.; ÓRourke P. R.; Zhang Q. Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS). Geosci. Model Dev. 2018, 11, 369–408. 10.5194/gmd-11-369-2018. [DOI] [Google Scholar]
- Smith S.Western USA SO2 emissions too high. CEDS Github Repository. https://github.com/JGCRI/CEDS/issues/2. Last access: March 5, 2020.
- Lana A.; Bell T. G.; Simó R.; Vallina S. M.; Ballabrera-Poy J.; Kettle A. J.; Dachs J.; Bopp L.; Saltzman E. S.; Stefels J.; Johnson J. E.; Liss P. S. An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean. Global Biogeochem. Cycles 2011, 25, GB1004. 10.1029/2010GB003850. [DOI] [Google Scholar]
- Nightingale P. D.; Malin G.; Law C. S.; Watson A. J.; Liss P. S.; Liddicoat M. I.; Boutin J.; Upstill-Goddard R. C. In situ evaluation of air-sea gas exchange parameterizations using novel conservative and volatile tracers. Global Biogeochem. Cycles 2000, 14, 373–387. 10.1029/1999GB900091. [DOI] [Google Scholar]
- Andres R. J.; Kasgnoc A. D. A time-averaged inventory of subaerial volcanic sulfur emissions. J. Geophys. Res. Atmos. 1998, 103, 25251–25261. 10.1029/98JD02091. [DOI] [Google Scholar]
- Dentener F.; Kinne S.; Bond T.; Boucher O.; Cofala J.; Generoso S.; Ginoux P.; Gong S.; Hoelzemann J. J.; Ito A.; Marelli L.; Penner J. E.; Putaud J.-P.; Textor C.; Schulz M.; van der Werf G. R.; Wilson J. Emissions of primary aerosol and precursor gases in the years 2000 and 1750 prescribed data-sets for AeroCom. Atmos. Chem. Phys. 2006, 6, 4321–4344. 10.5194/acp-6-4321-2006. [DOI] [Google Scholar]
- Sindelarova K.; Granier C.; Bouarar I.; Guenther A.; Tilmes S.; Stavrakou T.; Müller J.-F.; Kuhn U.; Stefani P.; Knorr W. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years. Atmos. Chem. Phys. 2014, 14, 9317–9341. 10.5194/acp-14-9317-2014. [DOI] [Google Scholar]
- Dee D. P.; et al. The ERA-Interim reanalysis: Configuration and performance of the data assimilation system. Q. J. R. Meteorol. Soc. 2011, 137, 553–597. 10.1002/qj.828. [DOI] [Google Scholar]
- IMPROVE Data. http://vista.cira.colostate.edu/Improve/. Last access: March 5, 2020.
- NAPS Program. http://data.ec.gc.ca/data/air/monitor/national-air-pollution-surveillance-naps-program/. Last access: March 5, 2020.
- Environment and Climate Change Canada Data. http://donnees.ec.gc.ca/data/air/monitor/monitoring-of-atmospheric-particles/. Last access: March 5, 2020.
- EMEP/CCC Reports. https://projects.nilu.no//ccc/reports.html. Last access: March 5, 2020.
- UK AIR Search Library. https://uk-air.defra.gov.uk/library/search. Last access: March 5, 2020.
- UK AIR Annual and Exceedence Statistics. https://uk-air.defra.gov.uk/data/exceedence?. Last access: March 5, 2020.
- Rahn K. A.; Winchester J. W.. Sources of Trace Elements in Aerosols: an Approach to Clean Air; ORA Project, 1971; p 089030.
- Querol X.; et al. Spatial and temporal variations in airborne particulate matter (PM10 and PM2.5) across Spain 1999–2005. Atmos. Environ. 2008, 42, 3964–3979. 10.1016/j.atmosenv.2006.10.071. [DOI] [Google Scholar]
- Qie G.; Wang Y.; Wu C.; Mao H.; Zhang P.; Li T.; Li Y.; Talbot R.; Hou C.; Yue T. Distribution and sources of particulate mercury and other trace elements in PM2.5 and PM10 atop Mount Tai, China. J. Environ. Manage. 2018, 215, 195–205. 10.1016/j.jenvman.2018.03.050. [DOI] [PubMed] [Google Scholar]
- Center for International Earth Science Information Network (CIESIN) Columbia University . Gridded Population of the World, Version 4 (GPWv4): Population Density, Revision 11, 2018; last access: March 5, 2020. [Google Scholar]
- Kavetski D. In Handbook of Hydrometeorological Ensemble Forecasting; Duan Q., Pappenberger F., Wood A., Cloke H. L., Schaake J. C., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2019; pp 481–522. [Google Scholar]
- Ramachandran K. M.; Tsokos C. P.. Mathematical Statistics with Applications; Elsevier Academic Press, 2009. [Google Scholar]
- Amouroux D.; Donard O. F. X. Maritime emission of selenium to the atmosphere in eastern Mediterranean seas. Geophys. Res. Lett. 1996, 23, 1777–1780. 10.1029/96GL01271. [DOI] [Google Scholar]
- Pacyna J. M.; Pacyna E. G. An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide. Environ. Rev. 2001, 9, 269–298. 10.1139/a01-012. [DOI] [Google Scholar]
- Hyslop N. P.; Trzepla K.; White W. H. Assessing the Suitability of Historical PM2.5 Element Measurements for Trend Analysis. Environ. Sci. Technol. 2015, 49, 9247–9255. 10.1021/acs.est.5b01572. [DOI] [PubMed] [Google Scholar]
- Eldred R. A. Comparison of Selenium and Sulfur at Remote Sites. J. Air Waste Manag. Assoc. 1997, 47, 204–211. 10.1080/10473289.1997.10464423. [DOI] [Google Scholar]
- Government of Ontario . The End of Coal. https://www.ontario.ca/page/end-coal. Last access: March 5, 2020.
- Chiou K. Y.; Manuel O. K. Tellurium and selenium in aerosols. Environ. Sci. Technol. 1986, 20, 987–991. 10.1021/es00152a003. [DOI] [PubMed] [Google Scholar]
- Hong Y.; Zhang H.; Zhu Y. Sulfur isotopic characteristics of coal in China and sulfur isotopic fractionation during coal-burning process. Chin. J. Geochem. 1993, 12, 51–59. 10.1007/BF02869045. [DOI] [Google Scholar]
- Ren D.; Zhao F.; Wang Y.; Yang S. Distributions of minor and trace elements in Chinese coals. Int. J. Coal Geol. 1999, 40, 109–118. 10.1016/S0166-5162(98)00063-9. [DOI] [Google Scholar]
- Lee K.; Hong S.-B.; Lee J.; Chung J.; Hur S.-D.; Hong S. Seasonal variation in the input of atmospheric selenium to northwestern Greenland snow. Sci. Total Environ. 2015, 526, 49–57. 10.1016/j.scitotenv.2015.04.082. [DOI] [PubMed] [Google Scholar]
- Van Zyl P. G.; Beukes J. P.; Du Toit G.; Mabaso D.; Hendriks J.; Vakkari V.; Tiitta P.; Pienaar J. J.; Kulmala M.; Laakso L. Assessment of atmospheric trace metals in the western Bushveld Igneous Complex, South Africa. S. Afr. J. Sci. 2014, 110, 1–10. 10.1590/sajs.2014/20130280. [DOI] [Google Scholar]
- Amos H. M.; Jacob D. J.; Streets D. G.; Sunderland E. M. Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Global Biogeochem. Cycles 2013, 27, 410–421. 10.1002/gbc.20040. [DOI] [Google Scholar]
- Gaillardet J.; Viers J.; Dupré B. In Treatise on Geochemistry; Holland H. D., Turekian K. K., Eds.; Pergamon: Oxford, 2003; pp 225–272. [Google Scholar]
- Stüeken E. E. Selenium Isotopes as a Biogeochemical Proxy in Deep Time. Rev. Mineral. Geochem. 2017, 82, 657–682. 10.2138/rmg.2017.82.15. [DOI] [Google Scholar]
- Li M.; Juang C. A.; Ewald J. D.; Yin R.; Mikkelsen B.; Krabbenhoft D. P.; Balcom P. H.; Dassuncao C.; Sunderland E. M. Selenium and stable mercury isotopes provide new insights into mercury toxicokinetics in pilot whales. Sci. Total Environ. 2020, 710, 136325. 10.1016/j.scitotenv.2019.136325. [DOI] [PubMed] [Google Scholar]
- Nakazawa E.; Ikemoto T.; Hokura A.; Terada Y.; Kunito T.; Tanabe S.; Nakai I. The presence of mercury selenide in various tissues of the striped dolphin: evidence from μ-XRF-XRD and XAFS analyses. Metallomics 2011, 3, 719–725. 10.1039/C0MT00106F. [DOI] [PubMed] [Google Scholar]
- Gajdosechova Z.; Lawan M. M.; Urgast D. S.; Raab A.; Scheckel K. G.; Lombi E.; Kopittke P. M.; Loeschner K.; Larsen E. H.; Woods G.; Brownlow A.; Read F. L.; Feldmann J.; Krupp E. M. In vivo formation of natural HgSe nanoparticles in the liver and brain of pilot whales. Sci. Rep. 2016, 6, 1–11. 10.1038/srep34361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki T.; Rasyidin A. Rates of weathering and soil formation. Geoderma 1992, 52, 251–263. 10.1016/0016-7061(92)90040-E. [DOI] [Google Scholar]
- Yu T.; Yang Z.; Lv Y.; Hou Q.; Xia X.; Feng H.; Zhang M.; Jin L.; Kan Z. The origin and geochemical cycle of soil selenium in a Se-rich area of China. J. Geochem. Explor. 2014, 139, 97–108. 10.1016/j.gexplo.2013.09.006. [DOI] [Google Scholar]
- Lanceleur L.; Tessier E.; Bueno M.; Pienitz R.; Bouchard F.; Cloquet C.; Amouroux D. Cycling and atmospheric exchanges of selenium in Canadian subarctic thermokarst ponds. Biogeochemistry 2019, 145, 193–211. 10.1007/s10533-019-00599-w. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.