

CONSPECTUS:
The characterization of catalytic materials under working conditions is of paramount importance for a realistic depiction and comprehensive understanding of the system. Under such relevant environments, catalysts often exhibit properties or reactivity not observed under standard spectroscopic conditions. Fulfilling such harsh environments as high temperature and pressure is a particular challenge for solid-state NMR where samples spin several thousand times a second within a strong magnetic field. To address concerns about the disparities between spectroscopic environments and operando conditions, novel MAS NMR technology has been developed that enables the probing of catalytic systems over a wide range of pressures, temperatures, and chemical environments. In this Account, new efforts to overcome the technical challenges in the development of operando and in situ MAS NMR will be briefly outlined. Emphasis will be placed on exploring the unique chemical regimes that take advantage of the new developments. With the progress achieved, it is possible to interrogate both structure and dynamics of the environments surrounding various nuclear constituents (1H, 13C, 23Na, 27Al, etc.), as well as assess time-resolved interactions and transformations. Operando and in situ NMR enables the direct observation of chemical components and their interactions with active sites (such as Brønsted acid sites on zeolites) to reveal the nature of the active center under catalytic conditions. Further, mixtures of such constituents can also be assessed to reveal the transformation of the active site when side products, such as water, are generated. These interactions are observed across a range of temperatures (−10 to 230 °C) and pressures (vacuum to 100 bar) for both vapor and condensed phase analysis. When coupled with 2D NMR, computational modeling, or both, specific binding modes are identified where the adsorbed state provides distinct signatures. In addition to vapor phase chemical environments, gaseous environments can be introduced and controlled over a wide range of pressures to support catalytic studies that require H2, CO, CO2, etc. Mixtures of three phases may also be employed. Such reactions can be monitored in situ to reveal the transformation of the substrates, active sites, intermediates, and products over the course of the study. Further, coupling of operando NMR with isotopic labeling schemes reveals specific mechanistic insights otherwise unavailable. Examples of these strategies will be outlined to reveal important fundamental insights on working catalyst systems possible only under operando conditions. Extension of operando MAS NMR to study the solid–electrolyte interface and solvation structures associated with energy storage systems and biomedical systems will also be presented to highlight the versatility of this powerful technique.
Graphical Abstract
INTRODUCTION AND MODERN APPROACH
Modern chemical transformations extensively rely on catalytic chemistry to provide the desired product. Catalysts are involved in the production of an estimated 30% of all global products and 90% of chemicals,1 covering a diverse set of industries such as petrochemicals,2 emissions controls,3 and agriculture,4 to name a few. A solid understanding of how these systems function, through either structural characterization or mechanistic insight, is imperative for the thoughtful design of new materials or optimization of chemical processes. To address such questions, advanced characterization techniques have been developed to elucidate the underlying structures and reaction mechanisms of chemical systems. In situ and operando characterization is of particular interest due to the relevance of revealing the state of the catalyst under operating conditions, often at significantly elevated temperatures or pressures. Magic-angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy is an excellent tool for such investigations on heterogeneous catalysts due to the nondestructive nature of the characterization and the sensitivity to small changes in the chemical environment of the nucleus. As such, the development of in situ and operando solid-state NMR technologies for high temperature and pressure has been a subject of great interest to catalysis and other fields.5,6 Though a powerful technique used to probe chemical systems, overcoming the technical difficulties of a nonmagnetic vessel capable of withstanding high temperatures and pressures while spinning several thousand times per second has been a challenging endeavor.
The early history of operando NMR has been extensively detailed elsewhere.5 Briefly, early adaptations took the form of flame-sealed tubes7–9 or polymer inserts,10,11 which exhibited undesirable operational limitations. Newer efforts for improved rotor performance have centered on rotor designs that can handle high temperatures and pressures under conditions of fast spinning rates, which were extensively used to reveal the nature of stored CO2 for carbon sequestration.12,13 Modern rotor schemes have suffered from limitations in the maximum operating temperature or sample volume due to the use of epoxy or ceramic inserts.12–14 The present state of modern rotor design is depicted in Figure 1. The all-zirconia cavern-style rotor sleeve is milled with a threaded top to allow for a secure seal. Reverse threading prevents sample rotation from loosening the zirconia cap and an O-ring (or two) constitutes the sealing surface, enabling a mechanical strength, chemical resistance, and temperature tolerance that is maximized for flexibility of performance. These designs are suitable for temperatures and pressures up to at least 250 °C and 100 bar, limited by readily available NMR probe technology. A complementary technology was recently developed that substantially reduces unit production costs by employing simple snap-in features in a commercial rotor sleeve.15 Termed WHiMS, this rotor avoids the need for specialized loading equipment by employing a check valve for gas loading. This design has been demonstrated to withstand 275 bar of pressure (20 °C) or 325 °C at significantly decreased pressure. Both of these rotor technologies are quite similar in sample volumes, ~400 μL for the 7.5 mm OD versions. The preparation of solids and liquids in the rotors is nearly identical, and both are dependent on a force applied to an O-ring for sealing. The key differences in these designs stem from the required production and operation resources as well as the flexibility of gas atmosphere introduction. The WHiMS is more restrictive to a loading pressure higher than 5 bar, and the rotor size (outer diameter) is limited to 5 mm and above due to the difficulties of engineering high mechanical strength plastic valves. The all-zirconia design is more scalable to smaller rotor sizes to accommodate limited sample applications and the requirement of faster sample spinning at high field for studying quadrupolar nuclei (e.g., smaller than 3.2 mm rotor size and spinning rates exceeding 15 kHz).
Figure 1.
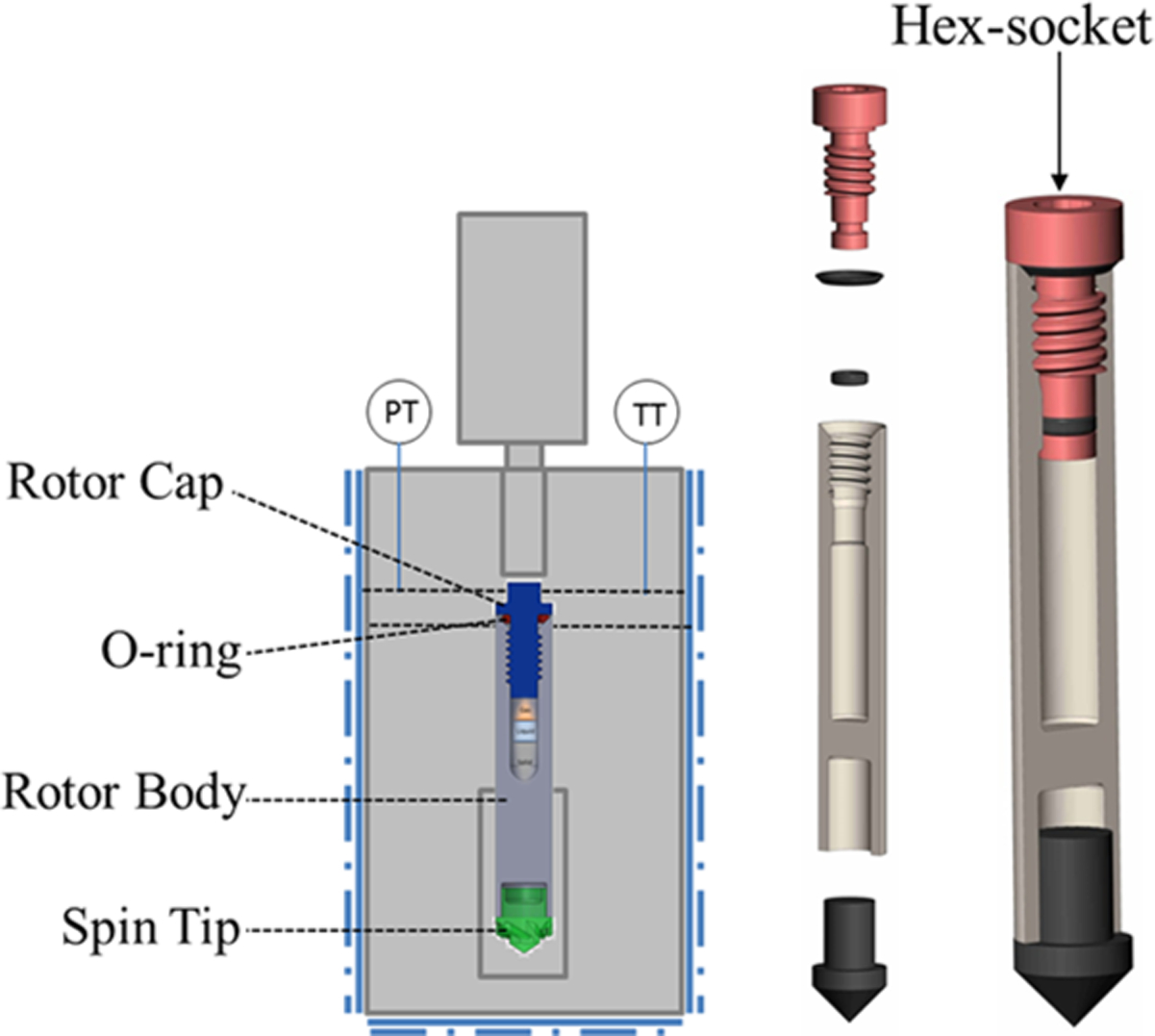
Schematic of high-temperature, high-pressure operando NMR rotors loaded within the specially designed loading chamber and an internal component view. Reprinted with permission from ref 16. Copyright 2015 Royal Society of Chemistry.
The WHiMS rotor takes advantage of a check valve to maintain internal pressure and offers the ease of gas introduction by simple exposure to a high-pressure external atmosphere. Gases may be sequentially added to the desired makeup, provided the external gas pressure exceeds both the internal pressure and the pressure required to open the check valve (>5 bar gauge for a 5 mm OD rotor). To charge the all-zirconia rotors with a gas atmosphere, a specially designed rotor loading chamber must be utilized. The all-zirconia rotor would be fixed within the loading chamber, and gases of varying pressures (from vacuum up to >2000 psi) and temperatures (<0 to >100 °C) could be introduced to the chamber volume around the rotor by use of gas manifolds. When the conditions are satisfactory, the rotor cap is threaded closed using a bit piece that matches the cap style (exploded hex or socket of the appropriate size) and is magnetically coupled to a rotating rod on the exterior of the chamber, enabling tight environmental control and eliminating the need for a rotating high pressure sealing surface. Viewing windows on the chamber enable the observation of the cap seating while maintaining the desired environment. The modular nature of the interior enables rotors from 9.5 mm to 2.5 mm to be used in the same system with a small change in the rotor stage components. As such, the complementary technologies offer options for either preparation convenience and fewer resource requirements or a more expansive range of control of the internal environment of the rotor. Due to the range of convenience and control options, these two technologies have been used for an array of applications to strengthen scientific understanding of catalytic systems.
In addition to batch-style in situ NMR investigations, continuous flow NMR probes have been employed to better mimic the conditions of a fixed-bed reactor while taking advantage of the benefits afforded by MAS.17–21 In these designs, an axial tube delivers gases to the bottom of the catalyst bed, which were then allowed to flow up and leave through the top of the rotor. Alternatively, the gas profile enters through the top and exits via the bottom with the aid of a slight vacuum on the exit channel to encourage flow.22 While such efforts certainly have brought unique insight and represent advancement in technical capabilities, the technique suffers from channeling of the catalyst bed and other issues of flow induced by back pressure or mixing of gases with the bearing and drive gas of the MAS technique.22 Nevertheless, such technologies continue to provide unique insight and offer new opportunities to couple characterization techniques. The inclusion of a quartz window on the bottom of the NMR rotor, for example, has been shown to be an effective method for coupling NMR measurements with UV–vis spectroscopy.23
MATERIALS STRUCTURE AND TRANSFORMATION
The scalable nature of rotor design and modular loading chamber components make it an excellent tool for multinuclear investigations on the structure of catalyst materials since an array of probes, and thus spinning rates, and magnetic fields may be employed to provide a thorough illustration of the material. A recent publication has reviewed the characterization of active metal sites in zeolites by NMR,24 but the evolution of the materials synthesis process, which generates an active catalyst structure, is uniquely possible with specialized NMR capabilities that enable high pressure and temperature resilience. Such transformations have been reported for the crystallization of AlPO4-5 where operando 1H, 13C, 27Al, and 31P MAS NMR results were combined to reveal a semicrystalline phase that served as an intermediate during the hydrolysis and condensation reactions that constitute the crystallization mechanism.16,25,26 In this example, NMR was able to clearly show the expulsion of excess water, phosphate, and aluminum to yield a crystalline catalytic solid. Similar principles can be applied to understand the formation of catalytic materials in zeolites.
23Na and 27Al are attractive nuclei to observe such operando transitions due to their sensitivity and presence in a variety of synthesis gels.27 For example, the formation of the faujasite (FAU) zeolite crystal structure was previously described by operando 23Na and 27Al MAS NMR, revealing the kinetics of crystallization of the material structure at elevated temperature and pressure.28 The time-resolved spectroscopic data presented in Figure 2 illustrate not only the importance of temporal resolution for observing the evolution of center band features but also how spinning sidebands can offer unique insight into the evolution of the structure. Figure 2A–C reveals the presence and evolution of two 27Al species: solid tetrahedral aluminum from the framework (AlFt at 62 ppm), which shifted from 60 ppm as the extended framework formed, and a narrow, liquid 27Al species (Al(OH) X− at ~76 ppm), which shifts slightly depending on hydroxyl replacement with siloxy groups. The intensity of AlFt increased and the line width decreased during the crystallization process, indicating the improved crystallinity over the course of the experiment. The Al(OH) X− species decreased in intensity as it was incorporated into the framework, while the line width was relatively invariant.
Figure 2.
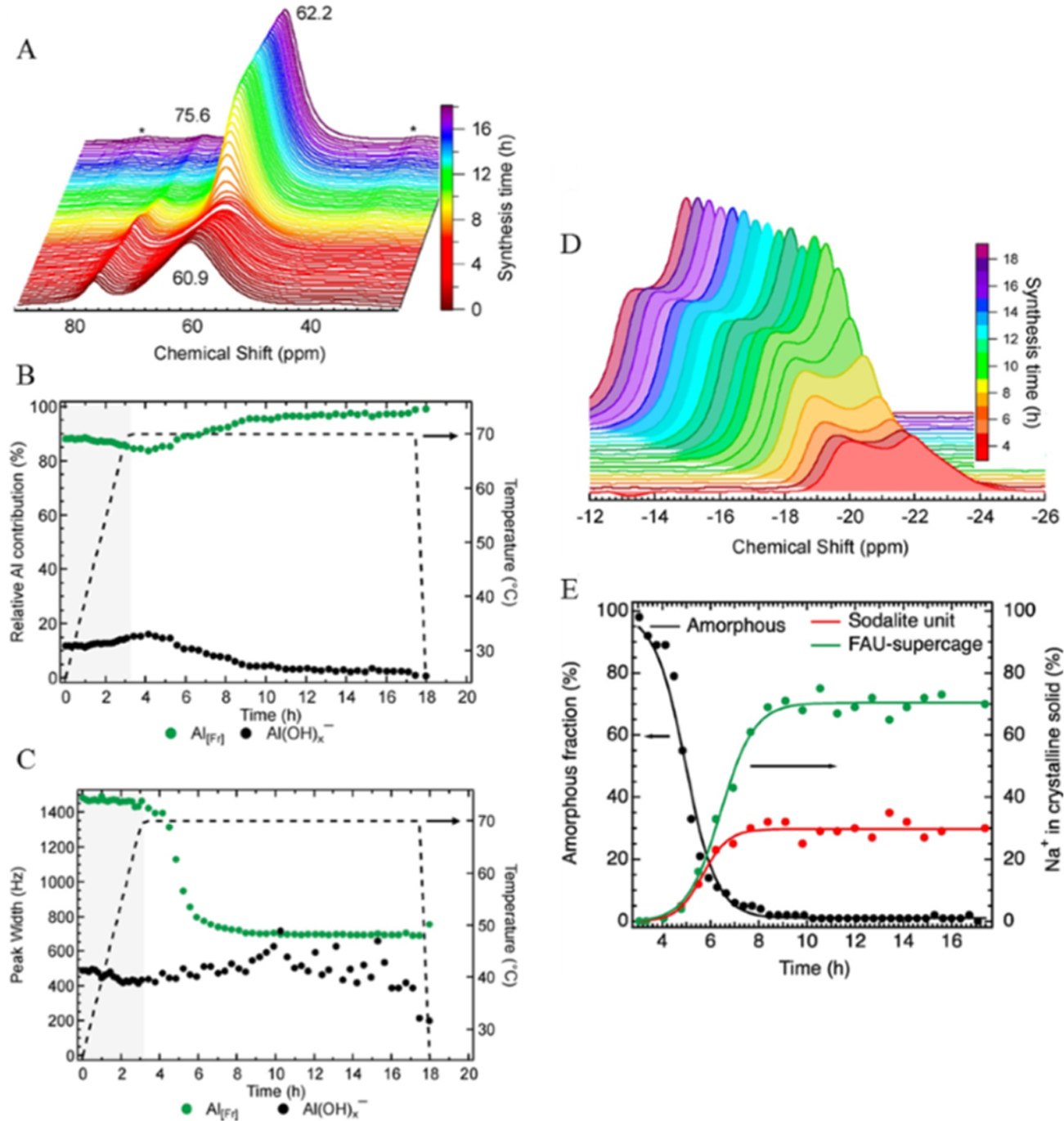
(A) In situ27Al MAS NMR spectra showing the changes during the synthesis of FAU. Deconvolution of the spectra (3 h heating period shaded in gray) led to modulation of the peak area (B) and line width (C) observed for liquid Al(OH)x− and solid tetrahedral Al (Al[Fr]). Changes in the spinning sideband associated with solid Na+ material as a function of synthesis time (D). A high-field peak and a low-field spinning sideband peak were identified at −22 and −20 ppm, respectively. (E) Kinetic transformation of amorphous material into crystalline FAU as directed by the speciation of Na+ ions [plotted as formed fraction of the final concentration of sodalite (−20 ppm) and the supercage (−22 ppm)] with lines included to guide the eyes. Reprinted with permission from ref 28. Copyright 2018 American Chemical Society.
The 23Na species were also monitored during framework formation. While the asymmetric center band feature narrowed and slightly moved downfield, this was shown to constitute both the solid and aqueous sodium that were superimposed and complicated a detailed analysis of the formation of FAU. Second-order quadrupolar interactions were sufficiently suppressed under these conditions; however, the spinning sidebands (Figure 2D,E) exhibited two distinct framework cationic sites at −20 ppm (sodalite positions) and −22 ppm (supercage positions) from the corresponding isotropic chemical shifts at 2.8 and 0.8 ppm, respectively. Both species grew in abundance as the crystallization process completed, and the sodalite population reached saturation prior to the supercage. Further, it is shown that cationic sites populate supercage and sodalite sites at a ratio of 70:30, providing direct evidence as to the types of confined environments experienced by substrate molecules and their reactive intermediate states. It should be noted that a quantitative comparison of the quadrupolar species abundance based on the sideband patterns alone, without simulation, is only possible if the quadrupolar coupling constants are known.
Not only is the formation of an extended crystal structure of a material of great importance, but so is the behavior of the catalytically active site. Identification of the active center of a catalyst is appropriately regarded as an integral step in evaluating the performance of a catalyst material and discerning the underlying reaction mechanisms. Material surfaces have dynamic structures that are significantly impacted by the local environments around the active centers.29,30 As such, it is necessary to conduct spectroscopic measurements under conditions relevant to the reaction of interest to gain a firm understanding of the true nature of the active centers. Acidic zeolite sites, for example, are well-known to catalyze an abundance of reactions. Zeolitic acid sites are highly sensitive to the chemical environment and readily adapt to changes in the surrounding atmosphere.
Recently, the impact of water on zeolite HZSM-5 was detailed by employing a combination of in situ 1H single-pulse and 1H–29Si cross-polarization (CP) NMR, depicted in Figure 3.29 It was shown that under conditions of hydration, the Brønsted acid site (BAS) proton would progressively detach from the framework oxygen atom. At low water loadings, the detachment took the form of an elongated HBA–OF bond due to hydrogen bonding of the acid proton with one adsorbed water molecule. Within this regime, the 1H–29Si CP NMR illustrated an increase in silicon signal due to the more efficient polarization of silicon atoms from the additional proximal protons. With the subsequent addition of water at hydration levels exceeding two water molecules per BAS, the in situ 1H–29Si CP NMR signals were suppressed. Concurrently, selective excitation spin–echo experiments revealed a 1H peak at 9 ppm, which represents a hydronium ion (H3O+), that is difficult to observe otherwise. Experimental and computational evidence confirmed the presence and assignment of hydrated hydronium ions that solvate the acid proton from the framework sites and undergo extensive molecular motion and exchange with surrounding water molecules. Both the longer 1H–29Si distances and the increased mobility led to smaller signal in the CP experiment. This exchange was found to be extensive at elevated temperatures, even at low water concentrations. The employment of Hahn-echo and cross-polarization pulse sequences have also been used to discriminate between molecules reacting in the pores and at the pore mouths of zeolites to identify the active centers. It was found that the etherification of citronellene with ethanol primarily occurs on the pore mouth rather than within the zeolite pore due to the preferential adsorption of ethanol in the pore and the easily accessible acid sites at the pore opening.31
Figure 3.
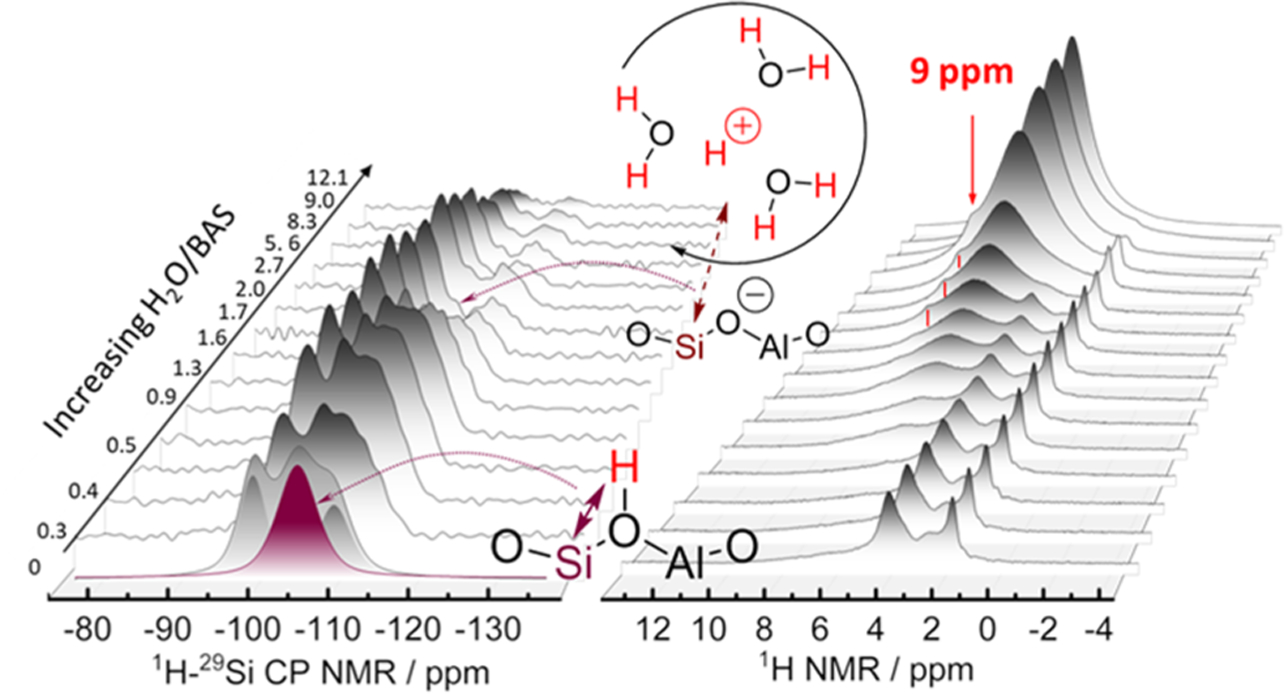
Schematic representation of the genesis and stability of hydronium ions in HZSM-5 as a function of water content in the framework. Adapted with permission from ref 29. Copyright 2019 American Chemical Society.
REACTION INTERMEDIATES AND MECHANISMS
A firm mechanistic description is also of great interest for understanding the chemistry operating during a reaction on the defined active centers. In situ NMR offers extensive opportunities to highlight the underlying chemical pathways of reactivity for heterogeneous catalysts. This may come from isotopic labeling to trace out a pathway or identification of the presence and dynamics of surface intermediates.32,33 Such efforts can be extended to simultaneously measure product formation rates and selectivity to provide a truly operando spectroscopic technique.16 In particular, extensive work has been conducted to trace out the mechanisms of reactions within zeolite frameworks, owing to the relatively well-defined structures they offer.34–36 Propane activation over Zn/H-MFI, for example, was explored by in situ NMR to highlight the formation of a zinc-propyl species in concert with Zn–OH groups prior to propane conversion. This dissociative adsorption initiates reactivity on the zinc center and contrasts the protonation mechanism observed on the Brønsted acid sites of unmodified zeolites.37–39 Zinc modification was also studied for the carbonylation of ethane, which reveals zinc-ethyl and methoxy species as the key intermediates.40
Due to the elevated temperatures and pressures required for condensed phase reactions, sealed rotors are uniquely suited for in situ analyses of the evolution of species during such processes. The dehydration of cyclohexanol, for example, has also been of interest and explored in detail by in situ NMR in conjunction with kinetic studies, which revealed no apparent mass transport limitations and enabled measurable reactivity, similar to the well-matching activation barriers between NMR and batch reactor studies.14,16 It was demonstrated that the aqueous-phase dehydration of 1-13C-cyclohexanol on H-BEA zeolite occurs through a cyclohexyl cation, which undergoes a 1,2-hydride shift via an E1 mechanism. Such insights were extended to more complex reaction networks involving cyclohexanol and water. For example, phenol alkylation in H-BEA zeolite has been the focus of multiple studies due to the strong interest in catalytic conversion of lignin-derived phenolic compounds to improve the value of the chemicals and identify the key reaction pathways for the transformation thereof.41,42
Important chemical transformations of phenol include alkylation and dealkylation transitions that adjust the carbon number of substrate species and pose interesting mechanistic questions, which can be addressed by in situ NMR.43 The alkylation of phenol is an electrophilic aromatic substitution, which can take place with either alcohols (protonated alcohol) or alkenes (carbenium) as alkylation agents. The alkylation of phenol with cyclohexanol and cyclohexene in decalin was studied by in situ 13C MAS NMR, revealing a detailed pathway whereby alkylation products were minimal until a majority of the cyclohexanol co-reactant was dehydrated to cyclohexene on the acid sites.41 This behavior was caused by the absence of reactive electrophile, which was present as a cyclohexyl carbenium when the cyclohexanol concentration was suppressed. This carbenium is directly formed by the protonation of cyclohexene by the Brønsted acid site and not during the dehydration of cyclohexanol. Subsequent work greatly expanded upon these observations in high detail only possible with in situ NMR to provide a thorough view of how alkylating reagents and solvents alter the reaction pathways of H-BEA-catalyzed phenol alkylation.42
Figure 4 reveals the time-resolved development of phenol alkylation with cyclohexanol and cyclohexene with decalin as the solvent. As previously reported, dehydration of cyclohexanol (red squares) to cyclohexene (blue diamonds) and dicyclohexyl ether was the primary transformation during the first period of time of the reaction at 160 °C. Phenol (green triangles) remained relatively unperturbed during this induction period. Not until ~70% of the cyclohexanol was dehydrated did C–C and C–O bond coupling alkylated phenol products form. The C–C alkylation products steadily increased with time, while the C–O alkylation products exhibited a transient behavior characterized by an enhancement in signal intensity after the induction period, followed by a decrease in concertation at longer reactions times. In contrast, when cyclohexene was used as an alkylation agent, the reaction initiated immediately. The concentration of cyclohexanol in the aqueous phases did not play a role in the observed reaction rate, but this was an influencing factor when decalin was used as a solvent. Based upon this observation and the detailed results of the reaction with cyclohexanol as the alkylating agent, a scheme was proposed based on cyclohexanol monomer and dimer pathways.
Figure 4.
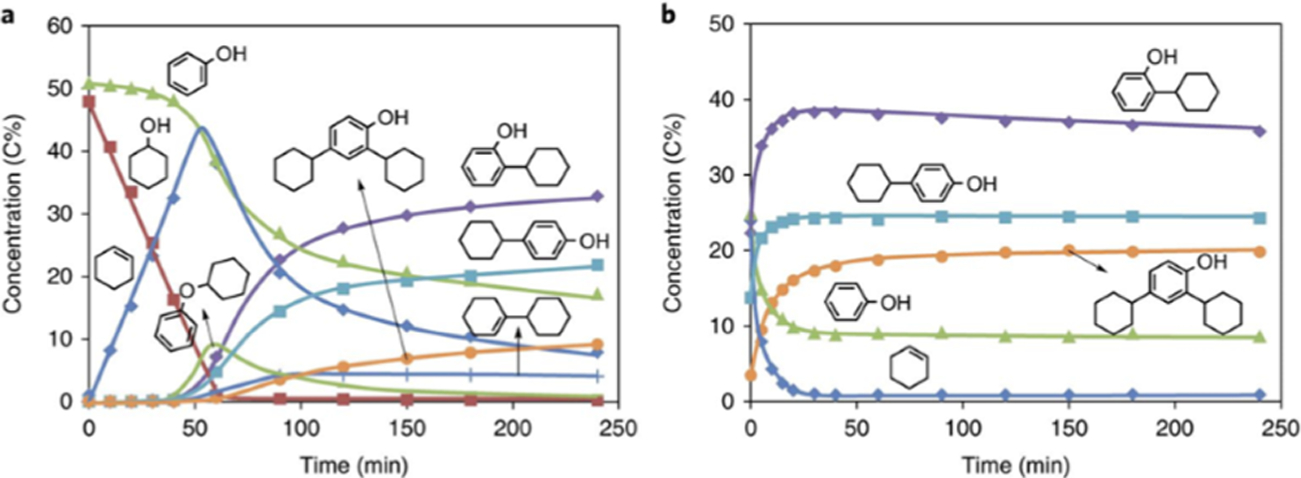
Species concentrations determined by 13C NMR for phenol alkylation in decalin as a function of reaction time: reaction with (a) cyclohexanol or (b) cyclohexene. Green triangles, phenol; red squares, cyclohexanol; blue diamonds, cyclohexene; purple diamonds, 2-cyclohexylphenol (2-CHP); blue squares, 4-cyclohexylphenol (4-CHP); orange circles, 2,4-dicyclohexylphenol (2,4-DCHP); green line, cyclohexyl phenyl ether (CHPE); blue crosses, 1-cyclohexylcyclohexene (1-CC). Reaction conditions: 5.0 g of phenol, 5.0 g of cyclohexanol (a) or cyclohexene (b), 0.2 g (a) or 1.0 g (b) of H-BEA-150, 100 mL ofdecalin, 5 MPa (ambient temperature) H2, stirred at 700 rpm, 160 °C. Reprinted with permission from ref 42. Copyright 2018 Springer.
Isotopic-labeling schemes may also be coupled with in situ studies to unravel more complex mechanistic puzzles (Figure 5). 13C scrambling of cyclohexanol dehydration in decalin solvent was shown to occur between 3- and 4-13C-cyclohexenes after an induction period, in direct contrast to aqueous-phase observations where 1-, 3-, and 4-13C-cyclohexenes scramble from the initial phase of the reaction. It was suggested that cyclohexanol does not form the lower-activity alcohol–alcohol dimer in water-filled pores, which are prevalent in aprotic solvents such as decalin. In the initial stage of the reaction (0 to 200 min), the dimer-mediated pathway facilitates dehydration to 1-13C-cyclohexene, according to the scheme and evidenced by the NMR-derived concentrations. Readsorption of cyclohexene was hindered due to the notable abundance of cyclohexanol dimers on the active sites during this induction period. The monomer/dimer ratio is proposed to slowly increase as cyclohexanol was consumed over the 200 to 500 min time span but not substantially, which resulted in a lower concentration of carbenium ions compared to the aqueous phase during the induction period. The scrambling is understood to directly result from the readsorption of cyclohexene onto the active center at the later stages of the reaction (500 to 780 min) when cyclohexanol dimers are sufficiently suppressed. Such cyclohexanol dimer species are proposed to inhibit the phenol–cyclohexanol alkylation reaction in decalin. It was also revealed that hydrated hydronium ions exhibited less-favorable carbenium ion formation than the nonhydrated acid sites present in apolar solvents, despite the necessity of cyclohexanol to dehydrate prior to carbenium ion formation from cyclohexene. Such insights were made possible by the contributions of in situ NMR observing the fine details of dynamic development of chemical species.
Figure 5.
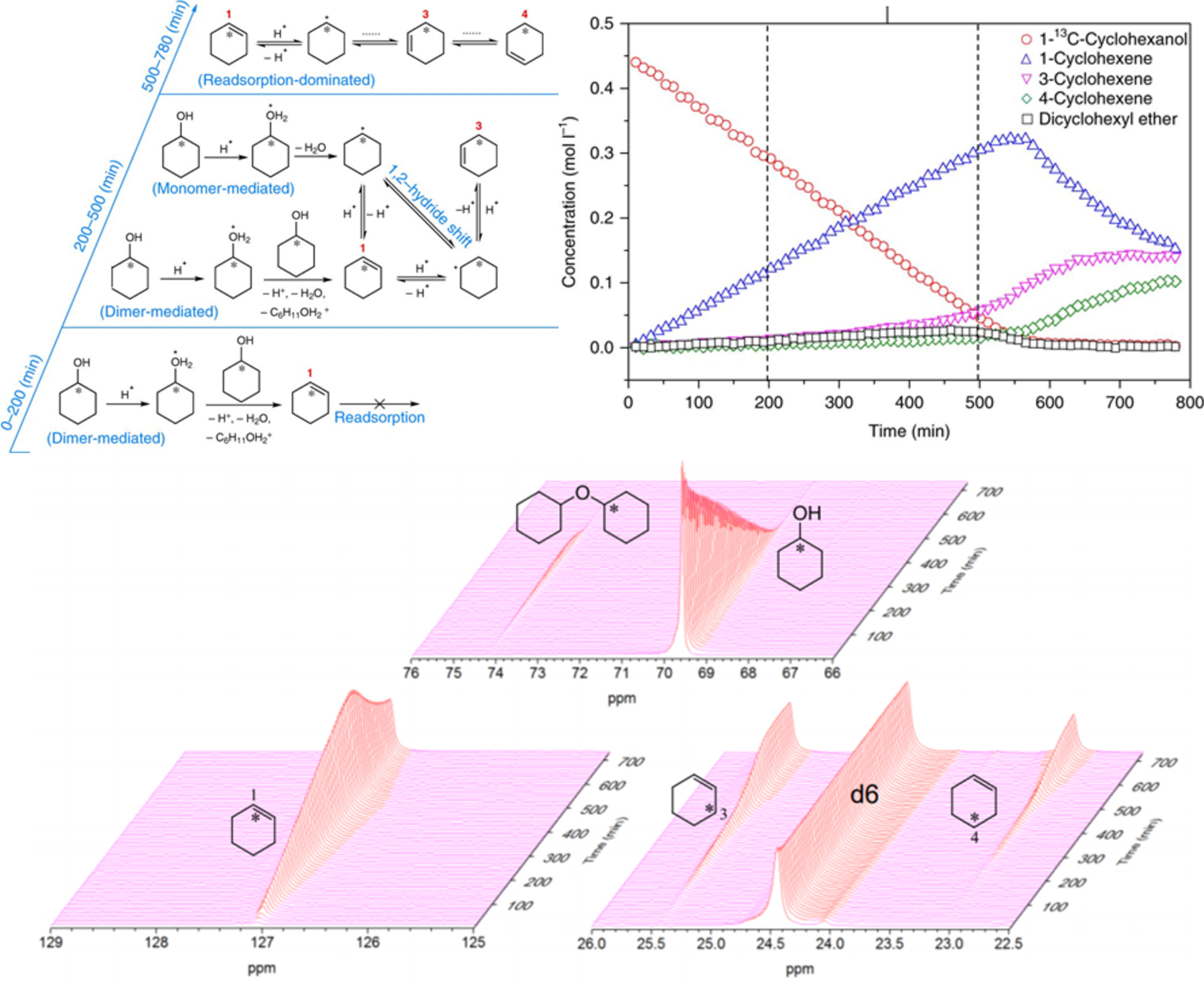
Reaction pathways proposed on the basis of in situ 13C NMR measurements of 1-13C-cyclohexanol dehydration on HBEA in decalin at 126 °C. Within 0–200 min, a significant fraction of the reaction occurs via elimination from alcohol dimer species (the monomer path not shown), while cyclohexene readsorption is severely hindered; within 200–500 min, olefin formation occurs via monomeric (increased contribution) and dimeric cyclohexanol (reduced contribution), and cyclohexene readsorption becomes less hindered with decreasing surface abundance of dimer species; within 500–780 min, cyclohexene readsorption becomes more pronounced after more than 70% of cyclohexanol is converted, and the distribution of labels becomes fully randomized at the end. The corresponding stacked plots for these 13C MAS NMR-derived concentration data are presented on the bottom. Adapted with permission from ref 42. Copyright 2018 Springer.
In addition to the consumption of phenol, its generation from the hydrogenolysis of benzyl phenyl ether, catalyzed by Ni/γ-Al2O3 has been investigated by MAS NMR employing the WHiMS rotor system.44 This systems reveals the unique role a co-reactant can play in the chemical transformation of interest. The key finding enabled by the use of in situ NMR showed that 2-propanol, the solvent employed in the study, served as the source of H2 during the reaction, ultimately forming acetone as the result of dehydrogenation.
ADAPTABILITY OF SEALED MAS ROTORS FOR ADDITIONAL SCIENTIFIC DISCOVERY
In addition to monitoring reactions for heterogeneous catalysis, sealed MAS rotors offer distinct advantages in monitoring the transformations occurring in biological systems as well as the interphase and solvation structures in electrochemical energy storage systems. This rotor technology has demonstrated suitability for use in biological tissue examination, as shown in Figure 6.16 An early example of in situ MAS NMR analysis of an in-tact tissue from a mouse liver enabled the observation of a variety of chemical constituents such as glucose, glycogen, and an array of functional groups. Modulation of the temperature without a loss in signal indicates that such sealing capabilities will eliminate the concern for biofluid leaking, advantageous for both safety and experimental accuracy. Since the rotor may easily accommodate low temperatures, it enables extended analysis at conditions that prevent biological tissue degradation, which is an area of concern for the accurate determination of chemical species in biological samples. Such a sealed rotor simplifies the metabolic profiling of intact tissues by MAS NMR.
Figure 6.
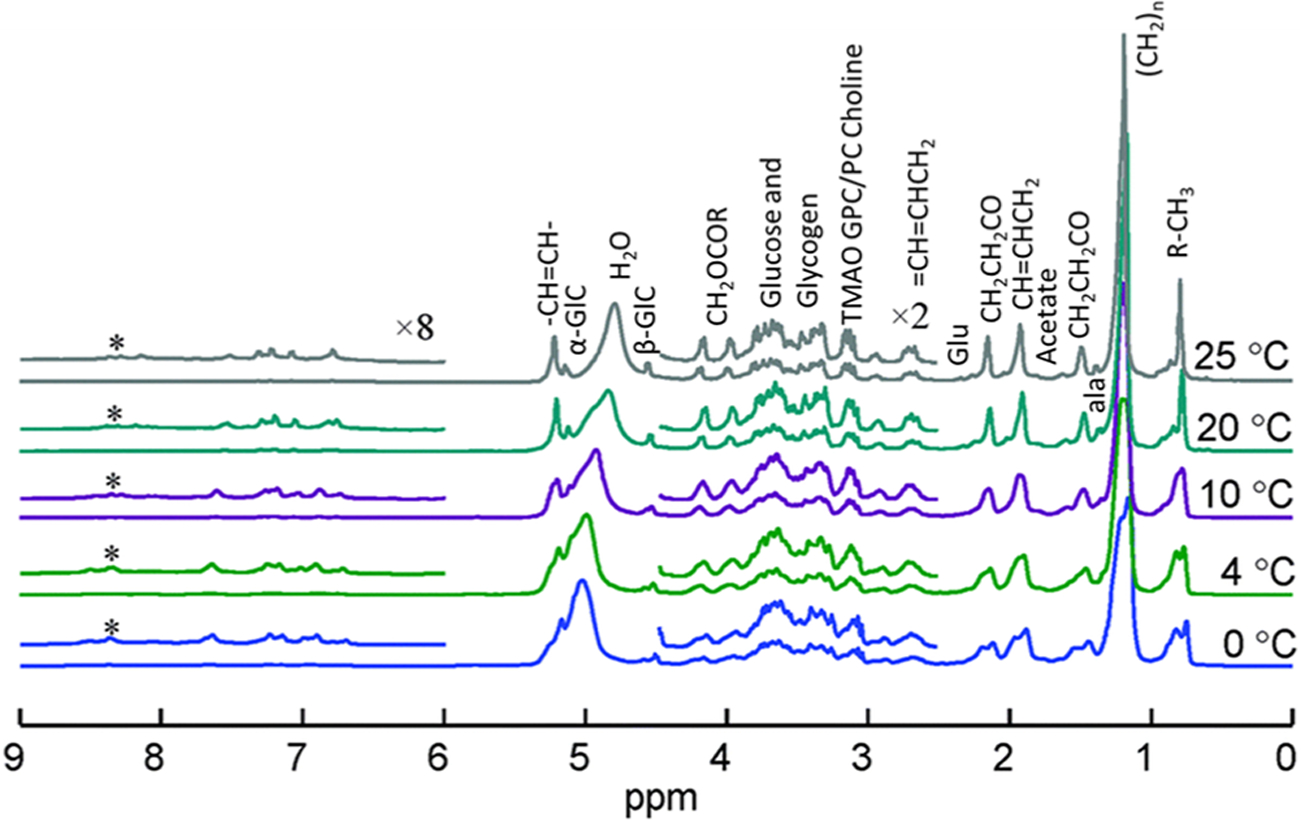
Stacked plot of the variable-temperature 1H MAS NMR spectrum of 280 mg of mouse liver from 0 to 25 °C. Adapted with permission from ref 16. Copyright 2015 Royal Society of Chemistry.
Another related subject includes that of understanding the interfacial interactions and solvation structures of materials for energy storage. Similar to catalysis applications, much of the critical chemistry for electrochemical cells occurs at the surface. Figure 7 highlights this concept for electrolytes 0.1 M Mg(TFSI)2 and 1.0 M Mg(TFSI)2 in diglyme (G2) interacting with a simulated MgO passivation layer for a Mg electrode.45 Employing sealed rotors enabled the investigation of the thermal adsorption and decomposition of the electrolytes without a loss of solution. At elevated temperatures, diglyme solvent was shown to decompose on the MgO layer to form surface methoxy species (with Mg-methoxy at 50 ppm and Mg-(O-CH2CH2)2-OCH3 at 56.96, 69, and 72.43 ppm). With low Mg(TFSI)2 concentrations, solvent-separated ion pairs were the dominating feature of the spectrum, evidenced by the large amount of solution G2 (~63%). G2 also adsorbs onto the MgO surface without decomposition (58.45, 70.65, and 72.24 ppm). At high salt concentrations, however, contact ion pairs form between Mg and TFSI in the solution and the MgTFSI(G2)2(G2)6+ cation adsorbs onto the MgO surface (59.28, 69.74, and 71.49 ppm). Elevated temperatures stimulate desolvation converting the surface-interacting solvation structure to MgTFSI(G2)2+ (60.94, 68.85, and 70.63 ppm) and releasing G2 molecules from the second solvation shell of the original cluster into the solution (58.75 and 72.6 ppm). Employing such a technique for battery applications demonstrates that the composition of the electrolyte has a directing role in the species present on the electrode surface, which may impact the structures and constituents of the solid–electrolyte interface on working electrodes. Such detailed observations of surface-interacting and solutions species at elevated temperatures are greatly benefitted by sealed in situ rotor technologies.
Figure 7.
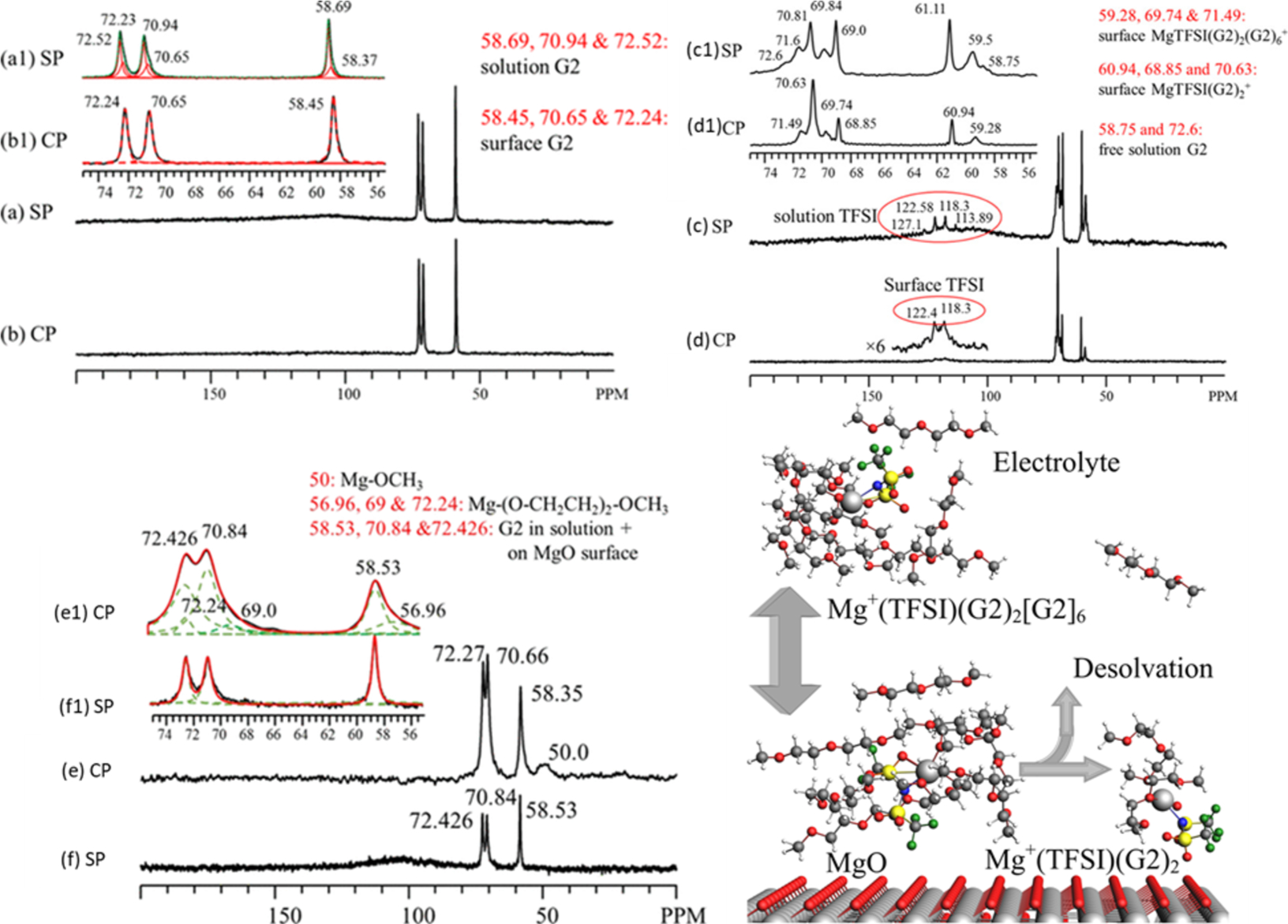
(a) 13C SP/MAS (a) and CP/MAS (b) spectra obtained on MgO550 (clean surface) + 0.1 M Mg(TFSI)2 in G2 after in situ heat treatment at 180 °C for 1 h. Traces a1 and b1 are horizontally expanded regions (55 to 75 ppm) of traces a and b, highlighting MgO surface-adsorbed G2 with peaks located at approximately 58.4, 70.65, and 72.23 ppm. No electrolyte decomposition is observed for this sample. 13C SP/MAS (c) and CP/MAS (d) spectra obtained on MgO and 1.0 M Mg(TFSI)2 in G2 after in situ heat treatment at 180 °C for 1 h. Traces c1 and d1 are horizontally expanded regions of traces c and d, highlighting MgO surface-mediated adsorption products with peaks at approximately 60.94, 68.85, and 70.63 ppm, surface adsorbed G2 with peaks located at approximately 59.28, 69.74, and 71.49 ppm, and surface adsorbed TFSI– at 118.3 and 122.4 ppm. No electrolyte decomposition is observed for this sample. (e) 13C CP/MAS and SP/MAS (f) spectra obtained on MgO550 + G2 after reacting at 180 °C for 1 h. Traces e1 and f1 are horizontally expanded regions of traces e and f. The bottom right schematic depicts the desolvation process. Reprinted with permission from ref 45. Copyright 2019 American Chemical Society.
SUMMARY AND OUTLOOK
The studies described here represent the capabilities of the current state of in situ NMR technology whereby the formation and identification of the nature of the catalytic active sites can be described in detail and a robust analysis of reaction mechanisms at relevant conditions is possible. Reactions occurring in vacuum atmospheres up to several hundred bar and from low temperatures (well below 0 °C) to 250 °C are realizable and reproducible in systems containing mixtures of solids, liquids, and gases. Such a diverse and inclusive operating range sets broad limits for the types of systems that can be analyzed by in situ and operando NMR methods. Future work will be based on employing the current and evolving technologies to explore the challenging mixed-phase systems at finely controlled pressures, not previously possible, while addressing key molecular-level questions posed in catalysis and related fields. Such efforts will involve extending operations to smaller rotor sizes for faster spinning at higher magnetic fields, which will enable well-resolved in situ NMR of quadrupolar nuclei including studies of 27Al MAS NMR at 850 MHz. These efforts are already underway and represent a dramatic improvement over current capabilities. Our current experiments indicate that such an approach is possible with all-zirconia rotors as small as at least 3.2 mm. This may enable detection of NMR-active species at spinning rates of up to 25 kHz, which would dramatically increase the quality of detection and resolution for some quadrupolar nuclei at high fields under conditions of high temperature and pressure. The use of even smaller rotor sizes (2.5 or 1.6 mm) may become possible in the future, which would facilitate faster spinning rates (35 and 45 kHz, respectively). The adaptability of such methods to a wider array of rotor sizes and fields of study positions in situ NMR as an attractive option for a variety of applications. In fact, not only is it relevant to catalysis, but in situ and operando NMR is gaining popularity within fields such as geochemistry,46–48 energy storage,6,45 and biology.16,49
ACKNOWLEDGMENTS
The review of catalyst applications was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Biosciences, and Geosciences. The review of biomedical applications was supported by the National Institute of Health, National Institute of Environmental Health Sciences, Grant R21ES029778. The review of energy storage applications was supported by the Joint Center for Energy Storage Research, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. Pacific Northwest National Laboratory is a multiprogram national laboratory operated by Battelle for the US Department of Energy under Contract DE-AC05-76RL0 1830.
Biographies
Nicholas Jaegers earned his B.S. in Chemical Engineering from Iowa State University in 2014. He is a Ph.D. candidate in Chemical Engineering at Washington State University within Dr. Yong Wang’s research group and started working four years ago at Pacific Northwest National Laboratory as a Ph.D. intern, focusing on applications of nuclear magnetic resonance in catalysis under the mentorship of Dr. Jian Zhi Hu.
Karl Mueller received his Ph.D. from the University of California, Berkeley, and is the Chief Science and Technology Officer for Physical and Computational Sciences at Pacific Northwest National Laboratory. His research focuses on the use of NMR methods to address structural and dynamic questions in complex systems, including batteries and catalyst materials.
Yong Wang obtained his Ph.D. in Chemical Engineering from Washington State University in 1993. He holds joint appointments at Pacific Northwest National Laboratory and Washington State University where he serves as the Voiland Distinguished Professor in Chemical Engineering.
Jian Zhi Hu obtained his Ph.D. in Applied Physics in 1994 from a Joint-Training Program between Wuhan Institute of Physics, the Chinese Academy of Sciences, and the Department of Chemistry, University of Utah. Currently, he is a senior staff scientist at Pacific Northwest National Laboratory.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.9b00557
The authors declare the following competing financial interest(s): J.Z.H. and his colleagues hold patents on rotor technology. J.Z.H., N.R.J., et al. have filed a provisional patent application on the exposure device.
Contributor Information
Nicholas R. Jaegers, Pacific Northwest National Laboratory, Richland, Washington, and Washington State University, Pullman, Washington;.
Karl T. Mueller, Pacific Northwest National Laboratory, Richland, Washington;.
Yong Wang, Pacific Northwest National Laboratory, Richland, Washington, and Washington State University, Pullman, Washington;.
Jian Zhi Hu, Pacific Northwest National Laboratory, Richland, Washington;.
REFERENCES
- (1).Toulhoat H Heterogeneous Catalysis: Use of Density Functional Theory In Encyclopedia of Materials: Science and Technology; Buschow, Cahn KHJ, Flemings RW, Ilschner MC, Kramer B, Mahajan EJ, Veyssiére S, P., Eds.; Elsevier: Oxford, 2010; pp 1–7. [Google Scholar]
- (2).Primo A; Garcia H Zeolites as catalysts in oil refining. Chem. Soc. Rev 2014, 43, 7548–7561. [DOI] [PubMed] [Google Scholar]
- (3).Jaegers NR; Lai JK; He Y; Walter E; Dixon DA; Vasiliu M; Chen Y; Wang CM; Hu MY; Mueller KT; Wachs IE; Wang Y; Hu JZ Mechanism by which Tungsten Oxide Promotes the Activity of Supported V2O5/TiO2 Catalysts for NOX Abatement: Structural Effects Revealed by V-51 MAS NMR Spectroscopy. Angew. Chem 2019, 131, 12739–12746. [DOI] [PubMed] [Google Scholar]
- (4).Catalytic Ammonia Synthesis: Fundamentals and Practice; Springer; US: New York, 1991. [Google Scholar]
- (5).Jaegers NR; Hu MY; Hoyt DW; Wang Y; Hu JZ Development and Application of In Situ High-Temperature, High-Pressure Magic Angle Spinning NMR In Modern Magnetic Resonance; Webb GA, Ed., Springer International Publishing: Cham, 2017; pp 1–19. [Google Scholar]
- (6).Hu JZ; Jaegers NR; Hu MY; Mueller KT In situ and ex situ NMR for battery research. J. Phys.: Condens. Matter 2018, 30, 463001. [DOI] [PubMed] [Google Scholar]
- (7).Miyoshi T; Takegoshi K; Terao T 13C High-Pressure CPMAS NMR Characterization of the Molecular Motion of Polystyrene Plasticized by CO2 Gas. Macromolecules 1997, 30, 6582–6585. [Google Scholar]
- (8).Miyoshi T; Takegoshi K; Terao T 129Xe n.m.r. study of free volume and phase separation of the polystyrene/poly(vinyl methyl ether) blend. Polymer 1997, 38, 5475–5480. [Google Scholar]
- (9).Miyoshi T; Takegoshi K; Terao T Effects of Xe Gas on Segmental Motion in a Polymer Blend As Studied by 13C and 129Xe High-Pressure MAS NMR. Macromolecules 2002, 35, 151–154. [Google Scholar]
- (10).Yonker CR; Linehan JC The use of supercritical fluids as solvents for NMR spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc 2005, 47, 95–109. [Google Scholar]
- (11).Deuchande T; Breton O; Haedelt J; Hughes E Design and performance of a high pressure insert for use in a standard magic angle spinning NMR probe. J. Magn. Reson 2006, 183, 178–182. [DOI] [PubMed] [Google Scholar]
- (12).Turcu RVF; Hoyt DW; Rosso KM; Sears JA; Loring JS; Felmy AR; Hu JZ Rotor design for high pressure magic angle spinning nuclear magnetic resonance. J. Magn. Reson 2013, 226, 64–69. [DOI] [PubMed] [Google Scholar]
- (13).Hoyt DW; Turcu RVF; Sears JA; Rosso KM; Burton SD; Felmy AR; Hu JZ High-pressure magic angle spinning nuclear magnetic resonance. J. Magn. Reson 2011, 212, 378–385. [DOI] [PubMed] [Google Scholar]
- (14).Vjunov A; Hu MY; Feng J; Camaioni DM; Mei DH; Hu JZ; Zhao C; Lercher JA Following Solid-Acid-Catalyzed Reactions by MAS NMR Spectroscopy in Liquid Phase-Zeolite-Catalyzed Conversion of Cyclohexanol in Water. Angew. Chem., Int. Ed 2014, 53, 479–482. [DOI] [PubMed] [Google Scholar]
- (15).Chamas A; Qi L; Mehta HS; Sears JA; Scott SL; Walter ED; Hoyt DW High temperature/pressure MAS-NMR for the study of dynamic processes in mixed phase systems. Magn. Reson. Imaging 2019, 56, 37–44. [DOI] [PubMed] [Google Scholar]
- (16).Hu JZ; Hu MY; Zhao ZC; Xu SC; Vjunov A; Shi H; Camaioni DM; Peden CHF; Lercher JA Sealed rotors for in situ high temperature high pressure MAS NMR. Chem. Commun 2015, 51, 13458–13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hunger M; Horvath T A New Mas Nmr Probe for in-Situ Investigations of Hydrocarbon Conversion on Solid Catalysts under Continuous-Flow Conditions. J. Chem. Soc., Chem. Commun 1995, 1423–1424. [Google Scholar]
- (18).Hunger M; Horvath T Conversion of propan-2-ol on zeolites LaNaY and HY investigated by gas chromatography and in situ MAS NMR spectroscopy under continuous-flow conditions. J. Catal 1997, 167, 187–197. [Google Scholar]
- (19).Hunger M; Horvath T; Weitkamp J Methyl tertiary-butyl ether synthesis on zeolite HBeta investigated by in situ MAS NMR spectroscopy under continuous-flow conditions. Microporous Mesoporous Mater. 1998, 22, 357–367. [Google Scholar]
- (20).Hunger M; Seiler M; Buchholz A In situ MAS NMR spectroscopic investigation of the conversion of methanol to olefins on silicoaluminophosphates SAPO-34 and SAPO-18 under continuous flow conditions. Catal. Lett 2001, 74, 61–68. [Google Scholar]
- (21).Jiang YJ; Huang J; Marthala VRR; Ooi YS; Weitkamp J; Hunger M In situ MAS NMR-UV/Vis investigation of H-SAPO-34 catalysts partially coked in the methanol-to-olefin conversion under continuous-flow conditions and of their regeneration. Microporous Mesoporous Mater. 2007, 105, 132–139. [Google Scholar]
- (22).Hu JZ; Sears JA; Mehta HS; Ford JJ; Kwak JH; Zhu KK; Wang Y; Liu J; Hoyt DW; Peden CHF A large sample volume magic angle spinning nuclear magnetic resonance probe for in situ investigations with constant flow of reactants. Phys. Chem. Chem. Phys 2012, 14, 2137–2143. [DOI] [PubMed] [Google Scholar]
- (23).Wang W; Jiang YJ; Hunger M Mechanistic investigations of the methanol-to-olefin (MTO) process on acidic zeolite catalysts by in situ solid-state NMR spectroscopy. Catal. Today 2006, 113, 102–114. [Google Scholar]
- (24).Xu J; Wang Q; Deng F Metal Active Sites and Their Catalytic Functions in Zeolites: Insights from Solid-State NMR Spectroscopy. Acc. Chem. Res 2019, 52, 2179–2189. [DOI] [PubMed] [Google Scholar]
- (25).Zhao ZC; Xu SC; Hu MY; Bao XH; Hu JZ In Situ High Temperature High Pressure MAS NMR Study on the Crystallization of AlPO4–5. J. Phys. Chem . C 2016, 120, 1701–1708. [Google Scholar]
- (26).Xu S; Zhao Z; Hu MY; Han X; Hu JZ; Bao X Investigation of water assisted phase transformation process from AlPO4–5 to AlPO4-tridymite. Microporous Mesoporous Mater. 2016, 223, 241. [Google Scholar]
- (27).Hu JZ; Zhang X; Jaegers NR; Wan C; Graham TR; Hu M; Pearce CI; Felmy AR; Clark SB; Rosso KM Transitions in Al Coordination during Gibbsite Crystallization Using High-Field Al-27 and Na-23 MAS NMR Spectroscopy. J. Phys. Chem . C 2017, 121, 27555–27562. [Google Scholar]
- (28).Prodinger S; Vjunov A; Hu JZ; Fulton JL; Camaioni DM; Derewinski MA; Lercher JA Elementary Steps of Faujasite Formation Followed by in Situ Spectroscopy. Chem. Mater 2018, 30, 888–897. [Google Scholar]
- (29).Wang M; Jaegers NR; Lee MS; Wan C; Hu JZ; Shi H; Mei DH; Burton SD; Camaioni DM; Gutierrez OY; Glezakou VA; Rousseau R; Wang Y; Lercher JA Genesis and Stability of Hydronium Ions in Zeolite Channels. J. Am. Chem. Soc 2019, 141, 3444–3455. [DOI] [PubMed] [Google Scholar]
- (30).Jaegers NR; Wan C; Hu MY; Vasiliu M; Dixon DA; Walter E; Wachs IE; Wang Y; Hu JZ Investigation of Silica-Supported Vanadium Oxide Catalysts by High Field V-51 Magic-Angle Spinning NMR. J. Phys. Chem . C 2017, 121, 6246–6254. [Google Scholar]
- (31).Radhakrishnan S; Goossens PJ; Magusin PCMM; Sree SP; Detavernier C; Breynaert E; Martineau C; Taulelle F; Martens JA In Situ Solid-State C-13 NMR Observation of Pore Mouth Catalysis in Etherification of beta-Citronellene with Ethanol on Zeolite Beta. J. Am. Chem. Soc 2016, 138, 2802–2808. [DOI] [PubMed] [Google Scholar]
- (32).Qi L; Alamillo R; Elliott WA; Andersen A; Hoyt DW; Walter ED; Han KS; Washton NM; Rioux RM; Dumesic JA; Scott SL Operando Solid-State NMR Observation of Solvent-Mediated Adsorption-Reaction of Carbohydrates in Zeolites. ACS Catal 2017, 7, 3489–3500. [Google Scholar]
- (33).Jaegers NR; Khivantsev K; Kovarik L; Klas DW; Hu JZ; Wang Y; Szanyi J Catalytic activation of ethylene C-H bonds on uniform d8 Ir(i) and Ni(ii) cations in zeolites: toward molecular level understanding of ethylene polymerization on heterogeneous catalysts. Catal. Sci. Technol 2019, 9, 6570–6576. [Google Scholar]
- (34).Haw JF; Richardson BR; Oshiro IS; Lazo ND; Speed JA Reactions of Propene on Zeolite Hy Catalyst Studied by Insitu Variable-Temperature Solid-State Nuclear Magnetic-Resonance Spectroscopy. J. Am. Chem. Soc 1989, 111, 2052–2058. [Google Scholar]
- (35).Moreno-Gonzalez M; Hueso B; Boronat M; Blasco T; Corma A Ammonia-Containing Species Formed in Cu-Chabazite As Per In Situ EPR, Solid-State NMR, and DFT Calculations. J. Phys. Chem. Lett 2015, 6, 1011–1017. [DOI] [PubMed] [Google Scholar]
- (36).Xu J; Wang Q; Li S; Deng F In Situ Solid-State NMR Investigation of Catalytic Reactions on Zeolites In Solid-State NMR in Zeolite Catalysis; Xu J, Wang Q, Li S, Deng F, Eds.; Springer Singapore: Singapore, 2019; pp 199–254. [Google Scholar]
- (37).Kolyagin YG; Ordomsky VV; Khimyak YZ; Rebrov AI; Fajula F; Ivanova II Initial stages of propane activation over Zn/MFI catalyst studied by in situ NMR and IR spectroscopic techniques. J. Catal 2006, 238, 122–133. [Google Scholar]
- (38).Ivanova II; Pomakhina EB; Rebrov AI; Derouane EG C-13 MAS NMR mechanistic study of the initial stages of propane activation over H-ZSM-5 zeolite. Top. Catal 1998, 6, 49–59. [Google Scholar]
- (39).Ivanova II; Blom N; Derouane EG Controlled-atmosphere C-13 MAS NMR study of the initial stages of propane activation over H-ZSM-5. Stud. Surf. Sci. Catal 1995, 94, 419–426. [Google Scholar]
- (40).Wang XM; Xu J; Qi GD; Wang C; Wang WY; Gao P; Wang Q; Liu XL; Feng ND; Deng F Carbonylation of ethane with carbon monoxide over Zn-modified ZSM-5 zeolites studied by in situ solid-state NMR spectroscopy. J. Catal 2017, 345, 228–235. [Google Scholar]
- (41).Zhao ZC; Shi H; Wan C; Hu MY; Liu YS; Mei DH; Camaioni DM; Hu JZ; Lercher JA Mechanism of Phenol Alkylation in Zeolite H-BEA Using In Situ Solid-State NMR Spectroscopy. J. Am. Chem. Soc 2017, 139, 9178–9185. [DOI] [PubMed] [Google Scholar]
- (42).Liu YS; Barath E; Shi H; Hu JZ; Camaioni DM; Lercher JA Solvent-determined mechanistic pathways in zeolite-H-BEA-catalysed phenol alkylation. Nat. Catal 2018, 1, 141–147. [Google Scholar]
- (43).Ilias S; Bhan A The mechanism of aromatic dealkylation in methanol-to-hydrocarbons conversion on H-ZSM-5: What are the aromatic precursors to light olefins? J. Catal 2014, 311, 6–16. [Google Scholar]
- (44).Walter ED; Qi L; Chamas A; Mehta HS; Sears JA; Scott SL; Hoyt DW Operando MAS NMR Reaction Studies at High Temperatures and Pressures. J. Phys. Chem . C 2018, 122, 8209–8215. [Google Scholar]
- (45).Hu JZ; Jaegers NR; Chen Y; Han KS; Wang H; Murugesan V; Mueller KT Adsorption and Thermal Decomposition of Electrolytes on Nanometer Magnesium Oxide: An in Situ C-13 MAS NMR Study. ACS Appl. Mater. Interfaces 2019, 11, 38689–38696. [DOI] [PubMed] [Google Scholar]
- (46).Graham TR; Dembowski M; Martinez-Baez E; Zhang X; Jaegers NR; Hu JZ; Gruszkiewicz MS; Wang HW; Stack AG; Bowden ME; Delegard CH; Schenter GK; Clark AE; Clark SB; Felmy AR; Rosso KM; Pearce CI In Situ Al-27 NMR Spectroscopy of Aluminate in Sodium Hydroxide Solutions above and below Saturation with Respect to Gibbsite. Inorg. Chem 2018, 57, 11864–11873. [DOI] [PubMed] [Google Scholar]
- (47).Zhang X; Huestis PL; Pearce CI; Hu JZ; Page K; Anovitz LM; Aleksandrov AB; Prange MP; Kerisit S; Bowden ME; Cui W; Wang Z; Jaegers NR; Graham TR; Dembowski M; Wang H-W; Liu J; N’Diaye AT; Bleuel M; Mildner DFR; Orlando TM; Kimmel GA; La Verne JA; Clark SB; Rosso KM Boehmite and Gibbsite Nanoplates for the Synthesis of Advanced Alumina Products. ACS Applied Nano Materials 2018, 1, 7115–7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhang X; Cui W; Hu JZ; Wang H-W; Prange MP; Wan C; Jaegers NR; Zong M; Zhang H; Pearce CI; Li P; Wang Z; Clark SB; Rosso KM Transformation of Gibbsite to Boehmite in Caustic Aqueous Solution at Hydrothermal Conditions. Cryst. Growth Des 2019, 19, 5557–5567. [Google Scholar]
- (49).Tfaily M; Hu JZ; Heyman H; Toyoda J; Jaegers NR; Wilson R; Chanton J Tracking the Fate of new C in Northern Peatlands by a Compound-Specific Stable Isotope-Labeling Approach coupled with multiple analytical techniques and gas fluxes analysis. AGU Fall Meeting Abstracts, 2017. [Google Scholar]