

Abstract
Multidrug efflux pumps of pathogenic, Gram-negative bacteria comprise an innate resistance mechanism and are key contributors to the emerging global pandemic of antibiotic resistance. Several increasingly detailed cryo-electron microscopy maps have been resolved of an entire efflux pump complex, AcrAB-TolC, resulting in atomistic structural models. Using a recent model, we have carried out nearly 40 μs of molecular dynamics simulations to study one of the key components of the protein complex, AcrA, the membrane fusion protein that connects the inner-membrane-bound AcrB to the outer-membrane-bound TolC. We determined a three-dimensional potential of mean force (PMF) for AcrA, which displays two main conformational basins representing assembly-competent and incompetent states. Corresponding experiments show that stabilizing mutations at an inter-domain interface shift the dynamic equilibrium between these states to the incompetent one, disrupting pump assembly and function and re-sensitizing bacteria to existing antibiotics. Modulation of AcrA dynamics through pharmacological intervention therefore presents a promising route for the development of new antibiotics.
Keywords: antibiotic resistance, efflux pump, Gram-negative bacteria, molecular dynamics simulations, free-energy calculations
Graphical Abstract
The development of antibiotic resistance in pathogenic bacteria is quickly becoming one of the most dangerous global pandemics that has been observed in recent times, responsible for approximately two million illnesses and over 23,000 deaths annually in the Unites States alone according to a recent report from the CDC (https://www.cdc.gov/drugresistance/about.html). Some strains of Pseudomonas aeruginosa are now resistant to all classes of antibiotics, and Escherichia coli is not far behind, with some strains resistant to at least six classes of antibiotics.1
One of the primary sources of this resistance is the recognition and expulsion of antimicrobial drugs from the cell via large, cellular-envelope-spanning multidrug efflux pumps.1-3 The major multidrug efflux pump in E. coli is the tripartite AcrAB-TolC complex.4 Structural studies show this complex is composed of three main components, AcrB, AcrA, and TolC, in a 3:6:3 ratio.5-7 Substrate recognition and energy transduction are performed by the AcrB trimer at the inner membrane,8,9 while the TolC trimer forms a pore in the outer membrane. The membrane fusion protein (MFP), AcrA, which assembles as a trimer of dimers, completes pump assembly by binding together the inner and outer membrane components (Fig. 1A). So-called efflux pump inhibitors (EPIs) are emerging alternative therapeutics that have the potential to revive activities of existing antibiotics and to control the spread of antibiotic resistance.10 While earlier work focused on inhibiting the transport activity of AcrB,10,11 newer studies by us have begun focusing on the MFP, AcrA, in an effort to diversify the pool of potential drugs.12-14 AcrA has four main structural components: the α-hairpin, lipoyl, β-barrel, and membrane proximal (MP) domains (Fig. 1B).8 The hairpin domain interacts with the outer membrane channel, TolC, while the other domains interact with AcrB.15-18
Figure 1:

The AcrAB-TolC efflux pump complex. (A) Cryo-EM structure of the full AcrAB-TolC efflux pump complex (PDB 5O66).7 Entire structure shown in cartoon representation. One copy of the inner membrane transporter, AcrB, is colored blue, and one copy of the outer membrane channel, TolC, is colored red. Two copies of the membrane fusion protein, AcrA, in binding sites 1 and 2 are shown in green and yellow, respectively. All other copies of AcrA, AcrB, and TolC are colored gray. The approximate positions of the outer membrane (OM), peptidoglycan cell wall (PG), and inner membrane (IM) are indicated. The placement of TolC in the OM is supported by previous simulation studies.21,22 (B) Structural domains of AcrA shown in a cartoon representation, with the four domains colored and labeled. The three angles, θ, ϕ, and ψ, previously defined in Ref.20 are also labeled. (Left) Side view. θ is the angle between the α-hairpin, lipoyl, and β-barrel domains. ψ is the angle between the lipoyl, β-barrel, and membrane proximal (MP) domains. (Right) Front view. ϕ is the dihedral angle between the α-hairpin and MP domains along the axis defined by the lipoyl and β-barrel domains.
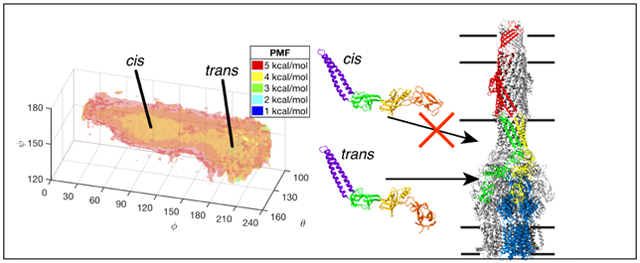
Analysis of early crystallographic structures led to the suggestion that flexible hinges in AcrA may play a role in the pump assembly and function.19 In MD of a free AcrA monomer, while the two central domains, the lipoyl and β-barrel domains, are rigid, the two terminal domains, the hairpin and MP domains, are quite flexible.20 In this work, we resolve how this flexibility affects binding to AcrB by combining mutagenesis and functional experiments with MD. MD-based free-energy calculations were used to determine multiple potentials of mean force (PMFs) for the free and bound AcrA monomer. Two main conformations, cis and trans, of free AcrA are identified from the PMFs, and mutations that shift the population into one of them compromises both the assembly and function of the efflux pump in E. coli. Differences in the AcrA free-energy landscapes for the two unique AcrB binding sites suggest distinct roles for each copy of AcrA.
Results
Conformational dynamics of a free AcrA monomer
Using self-learning umbrella sampling (SLUS, see Methods), we explored the conformational space of a free AcrA monomer in solution using the three angles, θ, ϕ, and ψ, first defined in Wang et al.20 and shown in Fig. 1B. A 3D PMF was then calculated using replica-exchange umbrella sampling (REUS, see Methods) with 1573 windows. In total, 26.7 μs of simulation time was used to calculate the full conformational free energy landscape of AcrA. The 3D PMF, as well as corresponding 2D and 1D PMFs calculated by integration of the 3D PMF along subsets of the three angles (see Methods), is shown in Figure 2.
Figure 2:

Multidimensional PMFs of free AcrA monomer in water. (A) Free energy landscape (PMF) of free AcrA in water along the θ, ϕ, and ψ angles. Isosurfaces are drawn in transparent colors, with their values labeled in the legend. The 3D PMF was calculated using replica-exchange umbrella sampling (REUS) with a total of 1573 windows simulated for 15 ns/window. Two free energy basins – cis and trans conformations – are separated by a small energy barrier. (B) Cartoon representations of the two conformations of AcrA, with side and front facing views. (C) 2D PMFs calculated by integrating out each angle of the 3D PMF. (D) 1D PMFs calculated by integrating a smoothed 3D PMF (see Fig. S1A). Block averaging of 5 ns/window blocks was used to calculate standard deviations for 1D PMFs, which are shown in gray. Individual 5 ns/window block PMFs are shown in Fig. S1B.
As predicted from previous simulation studies, free AcrA is quite flexible in the absence of AcrB and TolC, exploring a wide range of orientations of its four structural components. Our results reveal two main conformational basins: a cis-like conformation where the MP and α-helical domains point in the same direction direction and a trans-like conformation where they point in opposite directions (Fig. 2B). A small energy barrier (~1-2 kcal/mol) near ϕ = 150° separates the two conformations. The high-ϕ, trans-like conformational basin is ~1 kcal/mol deeper than the low-ϕ, cis-like conformation, but is also narrower along ϕ by ~45°. Conversely, the trans conformation exhibits a larger range in ψ, extending 20° lower than the cis conformation. The two conformations are roughly equivalent in free energy, with ΔGcis→trans = Gtrans – Gcis = −0.21 ± 0.18 kcal/mol, or about a 40/60 cis/trans equilibrium ratio. Cryo-EM structures of the full AcrAB-TolC complex7,23 show AcrA to be in the trans conformational basin in the assembled pump (ϕ > 150°; see Table 1 and Fig. 1A).
Table 1: Conformations of AcrA within the full AcrAB-TolC efflux pump complex.
Three AcrAB-TolC structures were examined: one with an open conformation of the outer membrane channel, TolC,23 and two with a closed conformation of TolC.7 Each structure has three copies of AcrA in each of two distinct binding sites on the AcrB trimer, labeled site 1 and site 2, for a total of nine copies per site. Averages and standard deviations for each site are tabulated below.
| θ (°) | ϕ (°) | ψ (°) | |
|---|---|---|---|
| Site 1 | 124.4 ± 4.3 | 181.7 ± 8.0 | 160.7 ± 1.1 |
| Site 2 | 119.6 ± 1.5 | 179.5 ± 8.9 | 168.7 ± 2.4 |
Conformation-dependent intramolecular interactions in AcrA
Our REUS simulations show that free AcrA monomers primarily exist in one of two states, either a cis- or trans-like conformation, in roughly equal proportions, with only a small energy barrier separating them. In the cis conformation (ϕ < 150°), the α-helix of the β-barrel domain forms contacts with two main regions of the MP domain: (1) the β-hairpin consisting of residues 339 to 350 and (2) the connector strands between the MP and β-barrel domains consisting of residues 48 to 53 and 299 to 307 (see Fig. 3A and B). As the protein transitions to larger ϕ dihedral angles, the contacts between these two domains are lost, with the MP and α-hairpin domains pointing in opposite directions (i.e., trans, see Figs. 2B and 3A).
Figure 3:

Interactions between β-barrel and MP domains distinguish the two main basins of the conformational landscape of free AcrA monomers. (A) Snapshots from a single equilibrium simulation of free AcrA starting from the cis conformation. Protein backbone is shown in cartoon representation, with MP domain colored reddish-orange, β-barrel domain colored yellowish-orange, and lipoyl domain colored green. Contact residues on the MP domain and the α-helix of the β-barrel domain are shown in licorice representation with hydrogen atoms omitted, colored by residue type: (blue) positively charged, (red) negatively charged, (green) polar, and (white) hydrophobic. Two residues are in contact if their side chains are within 5 Å. (B) Close up of the interfacial site between the β-barrel domain and the MP domain. Helical residues 220 to 232 are colored yellow, MP β-hairpin residues 339 to 350 are colored magenta, and connector residues 48 to 53 and 299 to 307 are colored light blue. Important contact residues are labeled. (C) Distribution of the number of MP domain contacts with the β-barrel helix over the dihedral angle, ϕ, for (top) the entire MP domain, (center) the β-hairpin, and (bottom) the connector calculated from six 250-ns equilibrium simulations. (D)-(F) Number of MP domain contacts with the β-barrel helix calculated from our REUS simulations for (D) the entire MP domain, (E) the β-hairpin, and (F) the connector. For (D)-(F), isosurfaces are drawn in transparent colors, with their values labeled in the legends.
To augment our REUS simulations, we ran an additional 1.5-μs of equilibrium simulations of free AcrA, which explore both the cis and trans ensembles, revealing an additional cis-like conformational basin for ϕ > 240° that was not observed in our REUS simulations, which was stable for up to 200 ns (see Supplemental Fig. S3). Therefore, cis-like conformations are more prevalent than our REUS simulations indicate. The free energy barrier between the two previously defined conformational basins and this second cis conformation is likely too large to be explored by the self-learning umbrella sampling algorithm, which is limited by a 3 kcal/mol free energy cutoff, i.e., the algorithm terminates if it cannot spawn any more windows that are below 3 kcal/mol. The equilibrium simulations also reveal that the primary pathway between cis conformations is via the trans conformation, with very few direct cis↔cis transitions, likely due to steric hindrance from the α-helix of the β-barrel domain (Supplemental Fig. S3). Both the REUS and equilibrium simulations reveal that contacts between the α-helix of the β-barrel domain and a β-hairpin in the MP domain define both cis conformations (see Fig. 3C and E). Additional contacts with the MP–β-barrel connector strands are also present primarily in the low-ϕ cis conformation (see Fig. 3C and F). For bound conformations of AcrA from the available cryo-EM structures of the full AcrAB-TolC complex,7,23 at most a single inter-domain contact is present when AcrA is bound to site 2 on AcrB, with no contacts present when bound to site 1, consistent with their trans-like conformations (see Table 1). Disrupting or enhancing MP–β-barrel interfacial interactions provides an avenue for assessing the importance of the cis and trans states to pump assembly and function.
Locking AcrA into the cis conformation compromises the function of the AcrAB-TolC pump
Site-directed mutagenesis was used to introduce substitutions in positions identified as involved in conformational transitions of AcrA (see Table S1 in the SI), in which the only native cysteine is lipid-modified and anchored into the inner membrane.8 AcrA variants containing Cys pairs positioned strategically at the MP and β-barrel interface were constructed to trap AcrA in the cis conformation. Blocking AcrA from accessing the trans, i.e., bound, conformation should hinder pump assembly. Three residue pairs were chosen that showed a high propensity for forming inter-domain contacts in the cis conformation, Leu50-Arg225, Ile52-Arg225, and Ile52C-Glu229, as well as two residue pairs that did not not form inter-domain contacts, Leu50-Asn232 and Ile52-Leu226. Additionally, residues Leu50, Ile52, Arg225, Leu226, Glu229, and Asn232 were substituted with cysteine one-by-one. The mutated acrA variants were co-expressed with acrB under the native promoter from plasmids in E. coli ΔAcrAB(Pore) cells. These cells lack the native acrAB operon and have controllable permeability of the outer membrane. The expression of the Pore increases the influx of antibiotics across the outer membrane and reduces the masking effect of the permeation in activities of antibiotics.24 All AcrA variants were produced at the levels comparable to the wild type protein and all AcrA variants with single substitutions complemented the antibiotic susceptible phenotype of E. coli ΔAcrAB(Pore) cells in the cells with the intact outer membrane barrier (see “No Pore” in Table 2). On the other hand, most of these AcrA mutants demonstrated reduced activities in the cells with increased influx of antibiotics across the outer membrane (see “Pore” in Table 2), the result confirming the importance of the MP domain and its interface with the β-barrel domain for AcrA-dependent antibiotic efflux, as seen in simulations.
Table 2: Antibiotic susceptibilities of E. coli ΔAcrAB(Pore) cells carrying the plasmid-borne and chromosomally produced AcrAB pump with indicated AcrA variants.
| No Pore |
Pore |
|||||
|---|---|---|---|---|---|---|
| NOV | ERY | SDS | NOV | ERY | SDS | |
| pUC18 | 1-2 | 1 | 16-32 | 0.5-1 | 0.25-0.5 | 16 |
| WT | 128-256 | 16 | >1000 | 4-8 | 1-2 | 128-256 |
| L50C R225C | 2 | 1 | 16 | <0.25 | <0.064 | 8 |
| L50C N232C | 32-64 | 8-16 | 64-128 | 0.5-1 | 0.25 | 16 |
| I52C R225C | 2-4 | 2 | 32 | ≤0.5 | 0.125 | 16 |
| I52C L226C | 64-128 | 16 | 256-512 | 2-4 | 0.5-1 | 128 |
| I52C E229C | 2 | 1-2 | 16-32 | 0.5 | ≤0.125 | 8-16 |
| L50C | 128 | 16 | >1000 | 2-4 | 0.25-0.5 | 32-64 |
| I52C | 64-128 | 16 | ≥1000 | 1-4 | 0.25-0.5 | 16-32 |
| L226C | 128 | 16 | >1000 | 4 | 0.25-0.5 | 64 |
| E229C | 128 | 8 | ≥1000 | 2-4 | 0.25-0.5 | 32-64 |
| N232C | 128 | 16 | >1000 | 4 | 0.5-1 | 64 |
The five AcrA mutants possessing the Cys-Cys pairs differed significantly in their functionalities. Three mutants, L50C-R225C, I52C-R225C, and I52C-E229C, completely lost their functions and could not complement the hypersusceptibility phenotype of ΔAcrAB(Pore) cells whether the expression of the Pore was induced or not. Furthermore, in the hyperporinated cells, the expression of these constructs further compromised the susceptibility of cells to novobiocin and erythromycin, suggesting that these AcrA variants make cells leaky to these antibiotics. Two AcrA variants, L50C-N232C and I52C-L226C, retained partial functionality with the I52C-L226C variant nearly as functional as the WT AcrA. Leu226 and Asn232 were not observed in our simulations to make contacts with the MP–β-barrel connector strands, which contain residues Leu50 and Ile52, (see Table S1). Therefore, the L50-N232C and I52C-L226C variants are far less likely to form a Cys-Cys disulfide bond and should be more flexible than the L50C-R225C, I52C-R225C, and I52C-E229C variants. The retention of partial functionality is consistent with the importance of the accessibility of the trans conformation to efflux activity.
To confirm that the observed phenotypes of the Cys-Cys AcrA variants reflect the changes in efflux activities of the pump, we next analyzed the kinetics of intracellular accumulation of a lipophilic fluorescent probe, N-phenylnaphthylamine (NPN), a known substrate of AcrAB-TolC pump.26 For this purpose, ΔAcrAB(Pore) cells with the WT and Cys-Cys AcrA variants were incubated with increasing concentrations of NPN. The fluorescence of NPN is significantly enhanced when the probe binds to membranes, reflecting the changes in the periplasmic concentration of the probe. The differences in the pump activities were compared based on the steady-state accumulation levels with the lowest levels corresponding to the most active efflux of the probe from the WT cells. In agreement with the results of functional complementation (see Table 2), the AcrA variants containing Cys pairs were deficient in the efflux of NPN, with I52C-R225C being the most affected variant (see Fig. 4).
Figure 4: Expression and function of AcrA variants integrated into the chromosome of E. coli ΔAcrAB(Pore) cells.
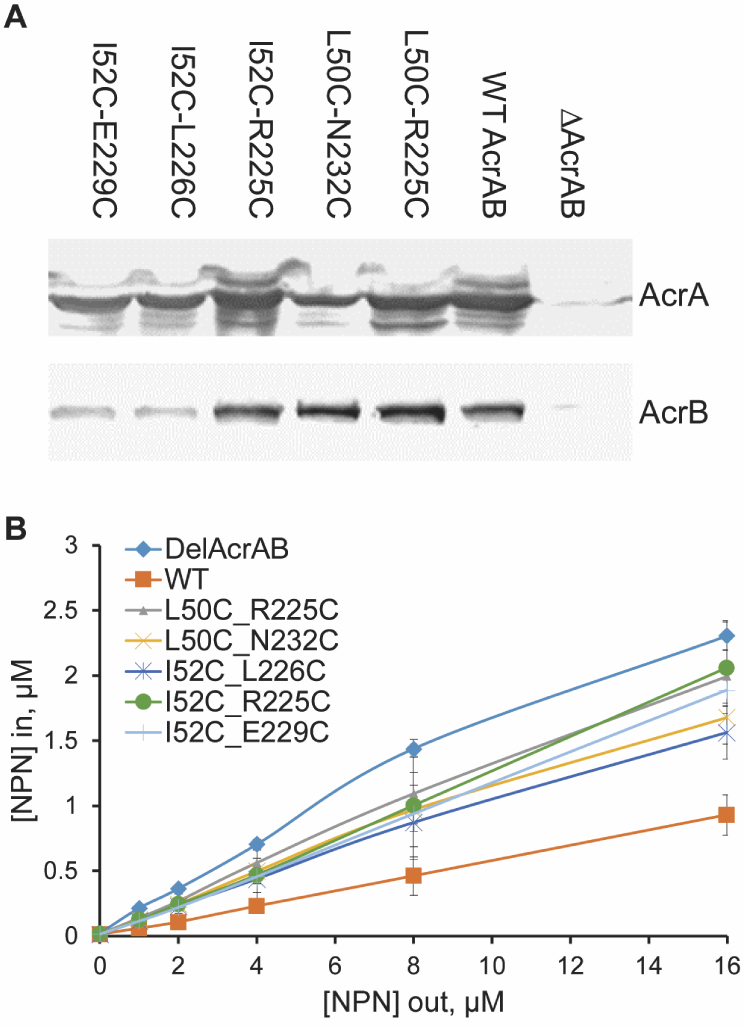
(A) Immunoblotting of whole cell extracts of ΔAcrAB(Pore) cells producing the indicated AcrA variants with anti-AcrA (top panel) and anti-AcrB (bottom panel) antibodies. (B) Concentration dependence curves of the intracellular accumulation of the fluorescent probe NPN in ΔAcrAB(Pore) cells producing the indicated AcrA variants. Cells were incubated with increasing concentrations of NPN and the changes in fluorescence intensities monitored as a function of time. The collected kinetic data were converted into concentrations and fitted into an exponential uptake model to extract the steady-state intracellular concentrations of NPN.25 The calculated intracellular concentrations ([NPN]in) are plotted as a function of the external concentration of NPN ([NPN]out). The averages of two experiments are shown.
Locking AcrA into the cis conformation compromises the stability and assembly of the AcrAB-TolC pump
In vivo proteolysis is a sensitive tool to monitor the assembly of AcrAB-TolC complex.27,28 Only if the complex is properly assembled is the T47-R315 fragment of AcrA stabilized and detectable by immunoblotting of cell extracts with anti-AcrA antibody.27,28 In these experiments, trypsin or other proteases are delivered into the periplasm by a mild osmotic shock and proteins are digested in the native environment. If the cis conformation of AcrA is stabilized by AcrA mutants with Cys-Cys pairs, the L50C-R225C, I52C-R225C, and I52C-E229C mutations, which have the strongest impact on the activity of the pump, would be expected to disrupt the formation of the complex. In contrast, the functional AcrA variants L50C-N232C and L52C-L226C are expected to assemble a complex similar to that with the WT AcrA.
We found that only the L50C-N232C and L52C-L226C variants, which are partially active in efflux (see Table 2), are assembled into the complex with AcrB and TolC, as seen from the accumulation of the characteristic Q29-K374 and T47-R315 tryptic fragments (see Fig. 5). In contrast, AcrA variants that are unable to complement the antibiotic-hypersusceptible phenotype of ΔAcrAB(Pore) cells also fail to assemble the complex and protect the T47-R315 fragment from the tryptic digest (see Fig. 5). These results demonstrate the importance of the AcrA conformation in the assembly of the complex with AcrB and TolC.
Figure 5: Trypsin digestion patterns of whole E. coli ΔAcrAB(Pore) cells producing the indicated AcrA variants.

For each reaction, 1.8 × 108 cells producing the wild type AcrA from the chromosome (WT) or the plasmid (WT(plasmid)) and the plasmid-borne AcrA with indicated double cysteine substitutions were treated with trypsin at the final concentrations of 0.0, 0.1, 1.0 and 10.0 μg/ml. Total proteins were separated by 12% SDS-PAGE and probed with polyclonal anti-AcrA antibodies. The major proteolytic fragments of AcrA assembled into the complex, Q29-K374 and T47-R315, are indicated by arrows.
Conformational selection governs binding of AcrA to one of two binding sites on AcrB
We also used MD to quantify the flexibility of AcrA after binding to AcrB. Starting from the AcrAB-TolC structure from Ref,23 we built a system with the AcrB trimer embedded in a symmetric lipid bilayer representative of the E. coli inner membrane,29,30 and with one copy of AcrA in either binding site 1 or 2 (see Fig. 1A). The AcrA MP domain was lipidated at its N-terminus, with its three acyl chains also embedded in the bilayer (see Methods). Because binding to AcrB restricts the motion of AcrA, particularly along the ψ direction, only the θ and ϕ angles were biased for the bound states.
REUS combined with the self-learning algorithm were used to determine PMFs for AcrA bound to site 1 and to site 2, requiring a total simulation time of 4.6 μs and 7.0 μs, respectively. The 2D PMFs for sites 1 and 2, as well as the θ-ϕ PMF of free AcrA, are plotted in Fig. 6. For both binding sites, we see a shift in the free energy minima from the free AcrA system towards the previously described transition region between the cis and trans states near ϕ = 150°. There is also a distinct difference in the free energy landscapes between the two sites: site 2 is more flexible than site 1. While site 1 has one main conformational basin with a range of ~90°−220° along ϕ (PMF < 3 kcal/mol), site 2 has two large basins that cover the entire range of ϕ. Site 1 reduces the conformational space of AcrA relative to the unbound state, indicative of conformational selection as the primary binding mechanism in this site.31 Interestingly, the cryo-EM conformations are not located at the bound state minima. This could be due to either the lack of additional copies of AcrA (we only have one copy in our systems), or the lack of the outer-membrane channel, TolC.
Figure 6: 2D PMFs of free and AcrB-bound AcrA.
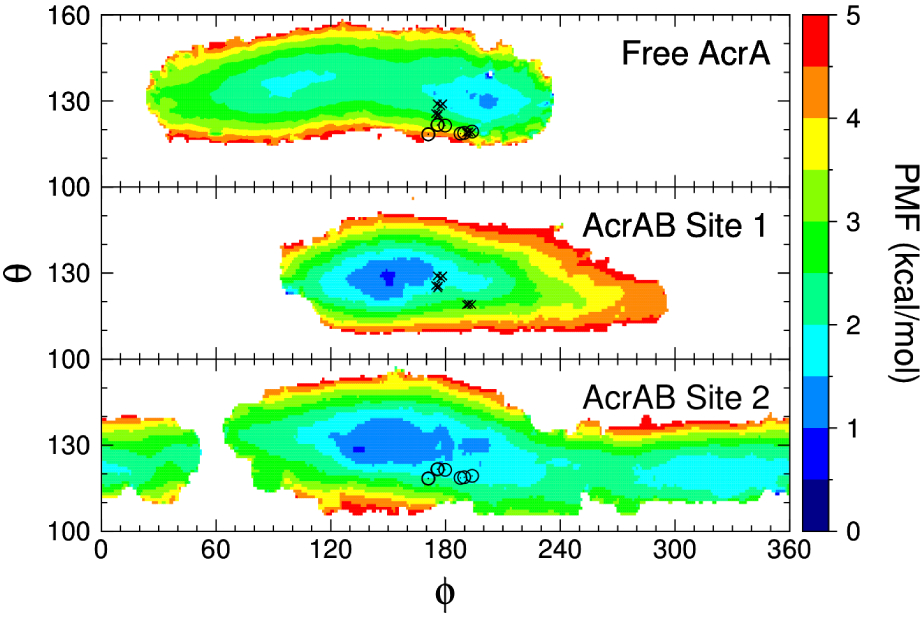
The free AcrA PMF is a 2D projection of the full 3D PMF. Cryo-EM coordinates from Refs23 and7 are also plotted with an “X” or an “O” for AcrA bound in site 1 or site 2, respectively.
Discussion
We have provided a link between the intrinsic conformational landscape of the membrane fusion protein AcrA of the tripartite AcrAB-TolC efflux pump and pump assembly and function. MD simulations show that free AcrA is very flexible, with its membrane proximal (MP) domain exhibiting the widest range of motion of all of its four structural domains. AcrA exists in two main conformations that are separated by a small energy barrier. The first is a cis conformation, in which the MP domain is in contact with the α-helix of the β-barrel domain at the interfacial site. The second is a trans conformation, in which contacts at the interfacial site are broken and the MP domain is pulled away from the β-barrel domain, pointing in the opposite direction of the α-hairpin domain; this conformation most closely resembles the bound conformations of AcrA observed in cryo-EM structures of the full efflux pump complex.7,23 Bacterial expression, fluorescent N-phenylnaphthylamine accumulation, and trypsin digestion assays for Cys-Cys mutations at the interfacial site of AcrA that lock it into the cis conformation reveal the critical role of AcrA’s conformation in AcrAB-TolC pump stability, assembly, and function.
The conformational landscape of AcrA changes in a binding-site specific manner when bound to AcrB. While one binding site exhibits a flexibility that is similar to that of the free AcrA monomer, the other site shows a sharp reduction in the accessible conformations, indicative of a conformational selection binding mechanism.31 The difference in the conformational landscapes between the two binding sites also suggests that the two copies of AcrA may play different roles in pump assembly and/or function. Efflux pumps with heterodimeric MFPs, such as the TriABC-OpmH triclosan pump in P. aeruginosa, utilize the two components at different steps of assembly, with one responsible for stimulating transport activity at the inner membrane and the other for stabilizing pore formation at the outer membrane.32,33 The reduction in flexibility of AcrA in binding site 1 suggests its role may be for stabilizing the interactions with TolC, while the second site stimulates transport with AcrB. Additionally, although a 3:6:3 AcrB:AcrA:TolC stoichiometric ratio was observed in cryo-EM structures of the full pump complex,7,23 a 3:3:3 ratio was previously suggested to be sufficient for efflux,34 with site 1, which resides between two AcrB protomers and stiffens AcrA, being the likely primary binding mode.34,35
Previous small-molecule inhibitors of AcrA designed by us were found to target the interface between the lipoyl and β-barrel domains of AcrA.12-14 The results presented here now point to the interface between the β-barrel and MP domains as a potentially fruitful region to target. Specifically, we found that pump assembly can be abrogated by restricting AcrA to its cis conformation, in which a number of inter-domain contacts are formed (Fig. 3B). Small molecules that bind here and strengthen these contacts are likely to be potent efflux inhibitors, which could be used in combination with other antibiotics, even in otherwise resistant strains. Future work will be to explore this possibility as well as determine if other pumps assemble according to the same principles laid out here.
Methods
Self learning adaptive umbrella sampling (SLUS) of free AcrA monomer
We used the model of free AcrA developed by Wang and coworkers as a starting conformation.20 This model is composed of the crystal structure for the α-hairpin, lipoyl, and β-barrel domains (PDB code: 2F1M), and a homology model of the membrane proximal (MP) domain based on the structure for MexA (PDB code: 2V4D) (see Fig. 1). The protein was solvated in water box of size 201 × 81 × 24 Å3 with 62,402 TIP3P water molecules,36 and neutralized with 0.15 M NaCl, for a total system size of 192,773 atoms. A harmonic restraint was added to align the lipoyl–β-barrel vector along the x-axis.
All simulations were performed using NAMD 2.10-1237 with the CHARMM36m protein force field.38 The temperature was fixed at 300 K using Langevin dynamics; the pressure was kept constant at 1 atm using the Langevin piston method.39 The equations of motion were integrated using the RESPA multiple time-step algorithm with a time step of 2 fs used for all bonded interactions, 2 fs for short-range non-bonded interactions, and 4 fs for long-range electrostatic interactions. Long-range electrostatic interactions were calculated using the particle-mesh Ewald method with a real-space cutoff of 12 Å.40 Short-range non-bonded Lennard-Jones interactions were cutoff at 12 Å with a potential switching function beginning at 10 Å bringing the potential energy smoothly to zero at the cut off distance. Bonds involving hydrogen atoms were constrained to their equilibrium length, employing the SETTLE algorithm41 for water molecules and the SHAKE algorithm for all others.42
We ran the self-learning adaptive biasing procedure43 starting from a set of 27 initial windows centered around (θ,ϕ,ψ) = (120°,115°,155°) generated from a 1-ns equilibrium simulation. For our collective variables, we use the three angles previously defined by Wang et al.,20 namely the angles the two terminal domains make with the two central domains (θ for the α-hairpin domain and ψ for the MP domain) and the dihedral angle between these domains about the central axis (ϕ; see Fig. 1). Umbrella sampling (US) simulations were centered on a 5° × 5° × 5° grid with a 0.2 kcal/mol force constant for all three angles. Each window was simulated for 2 ns, and PMFs were generated using the weighted histogram analysis method.44 A modified version of the WHAM code from Alan Grossfield45 that utilized the method of direct inversion in the iterative subspace (DIIS)46 was used to generate 3D PMFs.
The self-learning algorithm proceeds by first generating a PMF from the initial set of US simulations. For any windows that are below a chosen free energy cutoff – 3 kcal/mol in this study – new neighboring windows are spawned on the collective variable grid. Additional US simulations are performed for the newly created set of windows, with initial starting structures extracted from the previous set of US simulations. The PMF is then recalculated by combining all US simulations. This procedure is iterated until no new windows can be generated for the given free energy cutoff. Initially, the procedure halted near ϕ = 150° after 18 iterations resulting in a total of 743 windows, upon which we then ran 3 ns/window of replica exchange umbrella sampling (REUS)47 on all windows. After the REUS simulations, the SLUS procedure was then allowed to proceed up to ϕ = 240°, generating an additional 830 windows after another 17 iterations, for a total of 1573 windows and 35 iterations. The self-learning scheme extends the range of sampled conformations over previously reported short (20 ns) simulations of free AcrA monomers, which only covered ranges of 25°, 180°, and 60° for θ, ϕ, and ψ, respectively,20 to 45°, 210°, and 60°, respectively. We then ran a final REUS simulation on all 1573 windows for 15-ns/window, and generated a final 3D PMF. Total simulation time was 26.7 μs. Simulations were run on the petascale machine Titan at Oak Ridge National Lab.
SLUS of AcrB-bound AcrA
The AcrB trimer and one copy of bound AcrA were extracted from the cryo-EM structure of Jeong et al.23 We built two systems, one with a single copy of AcrA in binding site 1, and another with a single copy in binding site 2. All other binding sites were left unoccupied. The AcrB trimer was embedded within a symmetric bilayer composed of 924 total lipid molecules, with the following lipid composition: 409 PMPE, 110 POPE, 110 QMPE, 84 PMPG, 82 PYPG, 74 YOPE, and 54 PVCL2 lipids. AcrA was tri-acylated at Cys25 (its N-terminus), which is known to be necessary for high-affinity binding to AcrB.48 Each system was then solvated in a water box of size 190 × 190 × 256 Å3 with ~243,000 TIP3P water molecules,36 and neutralized with 0.15 M NaCl, for a total system size of ~903,000 atoms. Membrane simulations used the CHARMM36 lipid force field.49
For each system, we ran a 1-ns equilibrium simulation to seed our SLUS procedure. From this 1-ns simulation, we also extracted a set of contacts between AcrA and the AcrB trimer. We tracked when a residue on AcrA came within 5 Å of a residue on the copies of AcrB. For all such residue pairs, we measured the mean and standard deviation of their separation distance. For our system, AcrA is in contact with two of the copies of AcrB, so we picked the five residue pairs between AcrA and both of these copies of AcrB with the lowest standard deviation and restrained them to their mean distance with a 2.5 kcal/mol·Å2 force constant. With these restraints, the ψ angle between the MP and β-barrel domains is severely restricted. Therefore, for our US simulations, we only biased the θ and ϕ angles.
The SLUS procedure proceeded similarly to the free AcrA system. US simulations of 2 ns/window were performed on a 5° × 5° grid, with a 0.2 kcal/mol force constant for both angles. For the SLUS procedure, a 3 kcal/mol free energy cutoff was also used. For site 1, we started with 6 initial windows, and after 25 iterations, the SLUS procedure produced a total of 269 windows, covering a range of 90° < ϕ < 300°. High energetic barriers, particularly near ϕ > 240°, prevented the self-learning algorithm from exploring the entire conformational range of AcrA in this binding site. For site 2, we started with 28 initial windows, and after 47 iterations, the SLUS procedure produced a total of 411 windows, covering almost the entire 360°-range of ϕ. For this binding site, energetic barriers were lower than the 3 kcal/mol cutoff, which allowed the self-learning algorithm to explore the entire conformational space. For both sites, the SLUS procedure was followed by REUS simulations of 15 ns/window, for a total simulation time of 4.6 μs and 7.0 μs for site 1 and site 2, respectively. 2D PMFs were generated using WHAM.45
Mutagenesis of AcrA and complementation assays
E. coli ΔAcrAB(Pore) strain used in this study is the derivative of BW25113. A mini-Tn7T-based protocol was used to insert the Pore (fhuAΔC/Δ4L) into the E. coli chromosome using the pUC18T-R6K-mini-Tn7T suicide delivery vector along with the pTNS3 helper plasmid. The insertions were confirmed by PCR and vancomycin disc susceptibility assay.24 All substitutions in the acrA gene were constructed by QuickChange Lightning Site-Directed Mutagenesis kit (Agilent) using p151AcrABHis as the template.48 Introduced mutations and the lack of undesired mutations were verified by DNA sequencing (Oklahoma Medical Research Foundation).
Susceptibilities of E. coli cells were determined by a twofold broth dilution method.50 Cells were grown in Luria-Bertani (LB) broth (tryptone, 10 g/liter; yeast extract, 5 g/liter; NaCl, 5 g/liter) at 37°C with shaking at 200 rpm. To induce expression of the Pore, when the cells reached an optical density at 600 nm (OD600) of 0.3-0.4, L-arabinose (final concentration of 0.1%) was added and the cells were further incubated for 2-3 hours until the OD600 reached 1.0. The MICs were measured in 96-well microtiter plates. Exponentially growing cells were inoculated into wells containing LB medium in the presence of novobiocin, erythromycin, and SDS at a constant concentration of inducer. Cell growth after incubation at 37°C for 18 and 24 h was determined visually or using a Spark 10M microplate reader (TECAN).
The NPN uptake assay was performed in a temperature-controlled microplate reader (Tecan Spark 10M multimode microplate reader equipped with a sample injector) in fluorescence mode.25,26,51 Cells from frozen stocks were inoculated into LB medium and incubated for 16 h at 37°C. Cells were then subcultured into a fresh 30-ml volume of LB medium and grown at 37°C to an optical density at 600 nm (OD600) of 0.3. The cells were then induced with 0.1% arabinose and grown to an OD600 of 1.0, collected by centrifugation at 4,000 rpm for 20 min at room temperature, and washed in 25 ml HEPES-KOH buffer (50 mM; pH 7.0) containing 1 mM magnesium sulfate and 0.4 mM glucose (HMG buffer). The cells in HMG buffer were adjusted to an OD of ~1.0 and kept at room temperature during the experiment. Fluorescence intensities from NPN uptake experiments were plotted against time in Microsoft Excel and normalized to the emission before cells were added. The data were imported into MatLab (MathWorks, Inc.) to be fitted to a simple exponential equation in the form of F = A1 + A2[1 – exp(−kt)], where A1 represents the amplitude of the initial fast increase of NPN fluorescence, and A2 and k are the amplitude and rate, respectively, that are associated with the subsequent slower uptake.51
Previously, the reducing agent dithiothreitol was used to evaluate the negative impact of disulfide bonding in AcrB for the activity of the pump.52 We found however, that dithiothreitol stimulates the efflux activity of the pump even assembled with WT AcrA. Also, agar disc susceptibility assays24 were used to determine whether external dithiothreitol could restore the activity of AcrA variants. No differences in susceptibilities to novobiocin, erythromycin, or SDS were found in the absence and presence of 5 mM dithiothreitol.
Protein expression and in vivo partial proteolysis
For protein expression, whole E. coli cells were sonicated, membrane fractions isolated by ultracentrifugation, boiled in the reducing SDS-sample buffer and analyzed by SDS-PAGE followed by immunoblotting with primary polyclonal anti-AcrA (1:60,000 dilution) and anti-AcrB (1:4,000 dilution) antibodies8 followed by a secondary alkaline phosphatase-conjugated anti-rabbit immunoglobulin antibody (Sigma). 5-bromo-4-chloro-3-indolylphosphate (BCIP) and nitroblue tetrazolium (NBT) were used to visualize the bands. In vitro proteolysis experiments were carried out as described before.12 Briefly, for each reaction, 1.8 × 108 cells were washed and resuspended in buffer containing 20 mM Tris-HCl (pH 7.5), 5 mM EDTA and 20% sucrose. Trypsin was added to reactions to the final concentrations of 0.0, 0.1, 1.0 and 10.0 μg/ml. After incubation for 1 h at 37°C, proteolysis was terminated by boiling in SDS-sample buffer; total proteins were separated by 12% SDS-PAGE and probed with polyclonal anti-AcrA antibodies as described above.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01-AI052293 to H.I.Z. and R01-GM123169 to J.C.G. An award of computer time was provided by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. This research used resources of the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725. Additional computational resources were provided via the Extreme Science and Engineering Discovery Environment (XSEDE; allocation TG-MCB130173), which is supported by NSF grant number OCI-1053575.
References
- (1).Levy SB; Marshall B Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med 2004, 10, S122–S129. [DOI] [PubMed] [Google Scholar]
- (2).Routh MD; Zalucki Y; Su CC; Zhang Q; Shafer WM; Yu EW Efflux pumps of the resistance-nodulation-division family: a perspective of their structure, function, and regulation in gram-negative bacteria. Adv. Enzymol. Relat. Areas Mol. Biol 2011, 77, 109–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Delmar JA; Su CC; Yu EW Bacterial multidrug efflux transporters. Annu. Rev. Biophys 2014, 43, 93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kim J-S; Jeong H; Song S; Kim H-Y; Lee K; Hyun J; Ha N-C Structure of the tripartite multidrug efflux pump AcrAB-TolC suggests an alternative assembly mode. Mol. Cells 2015, 38, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Du D; Wang Z; James NR; Voss JE; Klimont E; Ohene-Agyei T; Venter H; Chiu W; Luisi BF Structure of the AcrAB-TolC multidrug efflux pump. Nature 2014, 509, 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Du D; van Veen HW; Luisi BF Assembly and operation of bacterial tripartite multidrug efflux pumps. Trends Microbiol. 2015, 23, 311–319. [DOI] [PubMed] [Google Scholar]
- (7).Wang Z; Fan G; Hryc CF; Blaza JN; Serysheva II; Schmid MF; Chiu W; Luisi BF; Du D An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. eLife 2017, 6, e24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zgurskaya HI; Nikaido H Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pos KM Drug transport mechanism of the AcrB efflux pump. Biochim. Biophys. Acta 2009, 1794, 782–793. [DOI] [PubMed] [Google Scholar]
- (10).Poole K Efflux pumps as antimicrobial resistance mechanisms. Ann. Med 2007, 39, 162–176. [DOI] [PubMed] [Google Scholar]
- (11).Opperman TJ; Nguyen ST Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol 2015, 6, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Abdali N; Parks JM; Haynes KM; Chaney JL; Green AT; Wolloscheck D; Walker JK; Rybenkov VV; Baudry J; Smith JC; Zgurskaya HI Reviving antibiotics: efflux pump inhibitors that interact with AcrA, a membrane fusion proein of the AcrAB-TolC multridrug efflux pump. ACS Infect. Dis 2017, 3, 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Haynes KM; Abdali N; Jhawar V; Zgurskaya HI; Parks JM; Green AT; Baudry J; Rybenkov VV; Smith JC; Walker JK Identification and structure-activity relationships of novel compounds that potentiate the activities of antibiotics in Escherichia coli. J. Med. Chem 2017, 60, 6205–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Darzynkiewicz ZM; Green AT; Abdali N; Hazel A; Fulton RL; Kimball J; Gryczynski Z; Gumbart JC; Parks JM; Smith JC; Zgurskaya HI Identification of binding sites for efflux pump inhibitors of the AcrAB-TolC component AcrA. Biophys. J 2019, 116, 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Husain F; Humbard M; Misra R Interaction between the TolC and AcrA Proteins of a Multidrug Efflux System of Escherichia coli. J. Bacteriol 2004, 186, 8533–8536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Elkins CA; Nikaido H Chimeric Analysis of AcrA Function Reveals the Importance of Its C-Terminal Domain in Its Interaction with the AcrB Multidrug Efflux Pump. J. Bacteriol 2003, 185, 5349–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tikhonova EB; Zgurskaya HI AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J. Biol. Chem 2004. , 279, 32116–32124. [DOI] [PubMed] [Google Scholar]
- (18).Zgurskaya HI; Nikaido H Cross-Linked Complex between Oligomeric Periplasmic Lipoprotein AcrA and the Inner-Membrane-Associated Multidrug Efflux Pump AcrB from Escherichia coli. J. Bacteriol 2000, 182, 4264–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mikolosko J; Bobyk K; Zgurskaya HI; Ghosh P Conformational Flexibility in the Multidrug Efflux System Protein AcrA. Structure 2006, 14, 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang B; Weng J; Fan K; Wang W Interdomain flexibility and pH-induced conformational changes of AcrA revealed by molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 3411–3420. [DOI] [PubMed] [Google Scholar]
- (21).Vaccaro L; Scott KA; Sansom MS Gating at both ends and breathing in the middle: conformational dynamics of TolC. Biophys. J 2008, 95, 5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schmidt TH; Raunest M; Fischer N; Reith D; Kandt C Computer simulations suggest direct and stable tip to tip interaction between the outer membrane channel TolC and the isolated docking domain of the multidrug RND efflux transporter AcrB. Biochim. Biophys. Acta 2016, 1858, 1419–1426. [DOI] [PubMed] [Google Scholar]
- (23).Jeong H; Kim J-S; Song S; Shigematsu H; Yokoyama T; Hyun J; Ha N-C Pseudoatomic structure of the tripartite multidrug efflux pump AcrAB-TolC reveals the intermeshing cogwheel-like interaction between AcrA and TolC. Structure 2016, 24, 272–276. [DOI] [PubMed] [Google Scholar]
- (24).Krishnamoorthy G; Wolloscheck D; Weeks JW; Croft C; Rybenkov VV; Zgurskaya HI Breaking the permeability barrier of Escherichia coli by controlled hyperporination of the outer membrane. Antimicrob. Agents Chemother 2016, 60, 7372–7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Leus IV; Weeks JW; Bonifay V; Smith L; Richardson S; Zgurskaya HI Substrate specificities and efflux efficiencies of RND efflux pumps of Acinetobacter baumannii. J. Bacteriol 2018, 200, e00049–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Krishnamoorthy G; Leus IV; Weeks JW; Wolloscheck D; Rybenkov VV; Zgurskaya HI Synergy between active efflux and outer membrane diffusion defines rules of antibiotic permeation into Gram-negative bacteria. mBio 2017, 8, e01172–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ge Q; Yamada Y; Zgurskaya HI The C-terminal domain of AcrA is essential for the assembly and function of the multidrug efflux pump AcrAB-TolC. J. Bacteriol 2009, 191, 4365–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krishnamoorthy G; Tikhonova EB; Dhamdhere G; Zgurskaya HI On the role of TolC in multidrug efflux: the function and assembly of AcrAB-TolC tolerate significant depletion of intracellular TolC protein. Mol. Microbiol 2013, 87, 982–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pandit KR; Klauda JB Membrane models of E. coli containing cyclic moieties in the aliphatic lipid chain. Biochim. Biophys. Acta Biomembr 2012, 1818, 1205–1210. [DOI] [PubMed] [Google Scholar]
- (30).Hwang H; Paracini N; Parks JM; Lakey JH; Gumbart JC Distribution of mechanical stress in the Escherichia coli cell envelope. Biochim. Biophys. Acta Biomembr 2018, 1860, 2566–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Vogt AD; Cera ED Conformational selection is a dominant mechanism of ligand binding. Biochemistry 2013, 52, 5723–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Weeks JW; Nickels LM; Ntreh AT; Zgurskaya HI Non-equilivalent roles of two periplasmic subunits in the function and assembly of triclosan pump TriABC from Pseudomonas aeruginosa. Mol. Microbiol 2015, 98, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ntreh AT; Weeks JW; Nickels LM; Zgurksaya HI Opening the channel: the two functional interfaces of Pseudomonas aeruginosa OpmH with the triclosan efflux pump TriABC. J. Bacteriol 2016, 198, 3176–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hayashi K; Nakashima R; Sakurai K; Kitagawa K; Yamasaki S; Nishino K; Yamaguchi A AcrB-AcrA Fusion Proteins That Act as Multidrug Efflux Transporters. J. Bacteriol 2016, 198, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Symmons MF; Bokma E; Koronakis E; Hughes C; Koronakis V The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. USA 2009, 106, 7173–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (37).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD Jr CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Feller SE; Zhang YH; Pastor RW; Brooks BR Constant pressure molecular dynamics simulations — The Langevin piston method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- (40).Darden TA; York DM; Pedersen LG Particle mesh Ewald: An N log N method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (41).Miyamoto S; Kollman PA Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- (42).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comp. Phys 1977, 23, 327–341. [Google Scholar]
- (43).Wojtas-Niziurski W; Meng Y; Roux B; Bernèche S Self-learning adaptive umbrella sampling method for the determination of free energy landscapes in multiple dimensions. J. Chem. Theory Comput 2013, 9, 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kumar S; Bouzida D; Swendsen RH; Kollman PA; Rosenberg JM The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem 1992, 13, 1011–1021. [Google Scholar]
- (45).Grossfield A WHAM: the weighted histogram analysis method, version 2.0.6. 2012, URL: http://membrane.urmc.rochester.edu/content/wham.
- (46).Zhang C; Lai C-L; Pettitt BM Accelerating the weighted histogram analysis method by direct inversion in the iterative subspace. Mol. Sim 2016, 42, 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sugita Y; Kitao A; Okamoto Y Multidimensional replica-exchange method for free-energy calculations. J. Chem. Phys 2000, 113, 6042. [Google Scholar]
- (48).Tikhonova EB; Yamada Y; Zgurskaya HI Sequential mechanism of assembly of multidrug efflux pump AcrAB-TolC. Chem. Biol 2011, 18, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Tikhonova EB; Wang Q; Zgurskaya HI Chimeric analysis of the multicomponent multidrug efflux transporters from gram-negative bacteria. J. Bacteriol 2002, 184, 6499–6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Westfall DA; Krishnamoorthy G; Wolloscheck D; Sarkar R; Zgurskaya HI; Rybenkov VV Bifurcation kinetics of drug uptake by Gram-negative bacteria. PLoS One 2017, 12, e0184671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Seeger MA; von Ballmoos C; Eicher T; Brandstätter L; Verrey F; Diederichs K; Pos KM Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat. Struct. Mol. Biol 2008, 15, 199–205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.