

ABSTRACT
Prostate cancer (PrCa) cells crosstalk with the tumour microenvironment by releasing small extracellular vesicles (sEVs). sEVs, as well as large extracellular vesicles (LEVs), isolated via iodixanol density gradients from PrCa cell culture media, express the epithelial-specific αvβ6 integrin, which is known to be induced in cancer. In this study, we show sEV-mediated protein transfer of αvβ6 integrin to microvascular endothelial cells (human microvascular endothelial cells 1 – HMEC1); we demonstrate that de novo αvβ6 integrin expression is not caused by increased mRNA levels. Incubation of HMEC1 with sEVs isolated from PrCa PC3 cells that express the αvβ6 integrin results in a highly significant increase in the number of nodes, junctions and tubules. In contrast, incubation of HMEC1 with sEVs isolated from β6 negative PC3 cells, generated by shRNA against β6, results in a reduction in the number of nodes, junctions and tubules, a decrease in survivin levels and an increase in a negative regulator of angiogenesis, pSTAT1. Furthermore, treatment of HMEC1 with sEVs generated by CRISPR/Cas9-mediated down-regulation of β6, causes up-regulation of pSTAT1. Overall, our findings suggest that αvβ6 integrin in cancer sEVs regulates angiogenesis during PrCa progression.
KEYWORDS: Angiogenesis, endothelial cell, extracellular vesicle, integrin, prostate cancer, survivin
Introduction
Among US men, prostate cancer (PrCa) is the most common malignancy and the second leading cause of cancer death [1]. To reduce mortality from PrCa, it is necessary to understand the underlying biochemical events and molecular mechanisms involved in PrCa progression. In particular, tumour angiogenesis plays a role in the progression of PrCa [2] based on findings that microvessel density in PrCa strongly correlates with Gleason grade and may predict disease progression [3]. Recent studies have focused on small extracellular vesicles (sEVs) as crucial mediators of tumour angiogenesis [4,5] and as modulators of the tumour microenvironment (TME), thereby supporting aggressive cancer [6–8].
While large extracellular vesicles (LEVs) are plasma membrane-derived extracellular vesicles (EVs) 100-1000 nm in size, recovered by a 10,000 g centrifugation step [9], the sEVs are a population of EVs recovered by a 100,000 g high-speed ultracentrifugation step, < 200 nm in size, of endosomal or non-endosomal in origin and secreted upon fusion with the plasma membrane [9–12]. The sEV subtype sediments in the light fractions of the high-speed density gradient ultracentrifugation, and it is enriched in tetra-spanins (CD9, CD63 and CD81) [11]. The sEVs carry proteins, mRNAs and miRNAs as cargo to mediate intercellular communication and modify the functional state of the recipient cells that interact with these secreted sEVs [13–15].
Integrins are transmembrane receptors that are expressed on PrCa cell-derived sEVs [6,16–19]. During tumour angiogenesis, integrins appear to play an important role in endothelial cell migration and survival [20,21]. However, the impact of PrCa cell-derived sEV-associated integrins on endothelial cells has not been explored so far. In particular, researchers have identified αvβ6 integrin as an epithelial-specific integrin that is not expressed in endothelial cells under normal conditions but can be induced [22–25]. The αvβ6 integrin is known to be up-regulated in many cancers [25] and correlates with poor survival in breast cancer [26–28], non-small cell lung cancer [29] and colon cancer [30,31] patients. It is not expressed in healthy prostate but is highly expressed in primary and metastatic PrCa [32,33]. Our previous studies have shown that the PrCa cell-derived sEV-associated αvβ6 integrin functionally modulates cells of the prostate TME [17,19]. The αvβ6 integrin is actively packaged into sEVs isolated from PrCa cell lines, and is efficiently transferred via these sEVs to β6-negative PrCa cells or monocytes, thus resulting in increased migration of recipient PrCa cells [17] and M2 polarisation of recipient monocytes, respectively [19]. These previous studies led us to hypothesise that PrCa cell-derived sEVs that express αvβ6 integrin (αvβ6-positive sEVs) may functionally impact endothelial cells.
In this study, we demonstrate for the first time that PrCa cell-derived αvβ6 integrin is transferred via sEVs as a functionally active molecule to β6-negative endothelial cells and significantly impact the angiogenic potential of endothelial cells. Despite the important role of angiogenesis in PrCa progression, clinical trials with anti-angiogenic therapy in this disease have not been effective [34–36]. Owing to our novel findings, targeting αvβ6 integrin in combination with current anti-angiogenic therapies may provide a novel approach to develop effective therapies against PrCa.
Materials and methods
Cell lines
Bovine aortic endothelial cells (BAECs) were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 100 μg/mL streptomycin and 100 U/mL penicillin (Corning Cellgro, USA) in a humidified atmosphere of 5% CO2 at 37°C [37].
Human microvascular endothelial cells 1 (HMEC1) were cultured in endothelial cell growth media supplemented with endothelial cell growth supplement (R&D Systems, Cat. # CCM027), 100 μg/mL streptomycin and 100 U/mL penicillin (Corning Cellgro, USA) in a humidified atmosphere of 5% CO2 at 37°C.
C4-2B cell lines were maintained in Roswell park memorial institute (RPMI) media with L-glutamine (Corning, USA) supplemented with 5% FBS, 1 mM sodium pyruvate (Corning Cellgro, USA), non-essential amino acids (Corning Cellgro, USA), 100 μg/mL streptomycin and 100 U/mL penicillin (Corning Cellgro, USA) in a humidified atmosphere of 5% CO2 at 37°C. The C4-2B PrCa cells stably transfected with either empty vector (C4-2B-Mock) or β6 cDNA-expression vector (C4-2B-αvβ6) were maintained as previously described [32].
PC3 cell lines were maintained in RPMI media with L-glutamine (Corning, USA) supplemented with 10% FBS, 100 μg/mL streptomycin and 100 U/mL penicillin (Corning Cellgro, USA) in a humidified atmosphere of 5% CO2 at 37°C. PC3 cells stably transfected with control shRNA (PC3-shCtrl) or shRNA specifically targeting β5 integrin subunit (PC3-shβ5) or β6 integrin subunit (PC3-shβ6) were maintained as previously described [32,38].
For genomic depletion of the β6 integrin subunit, PC3 cells were transfected with pX458 (Addgene plasmid #48,138), a plasmid expressing eGFP, spCas9 and a sgRNA targeting the fifth coding exon of β6 integrin (seed sequence: 5ʹ-GCTAATATTGACACACCCGA-3ʹ) using Lipofectamine LTX with Plus Reagent (ThermoFisher Scientific, Waltham, Massachusetts). At 72 h after transfection, eGFP-positive cells were single-cell sorted by a FACSAria II flow cytometer (BD Biosciences, San Jose, California). Clonally expanded cell populations were screened for frame-shifting indels by amplifying the target locus by polymerase chain reaction (PCR) (forward primer: 5ʹ-CAGTGAGATTCATAGCTGAGTTGCAG-3ʹ; reverse primer: 5ʹ-GTAGAGACAGCAAACTTCCGAAGC-3ʹ) and Sanger sequenced using both forward and reverse primers above. Complete knockout was confirmed in PC3-WT-clone 1, PC3-β6 KO-clone 5, PC3-β6 KO-clone 7 cells by immunoblotting (IB) using an antibody (Ab) to the αvβ6 integrin.
Antibodies for immunoblotting
The following primary Abs were used for IB analyses: mouse monoclonal Abs against: ALIX (Abcam, ab117600), αvβ6 integrin (6.2A1) [39], CD9 (Santa Cruz, sc18869), CD63 (Abcam, ab8219), CD81 (Abcam, ab23505); rabbit polyclonal Abs against: actin (Sigma Aldrich, A2066), CANX (Santa Cruz, sc-11,397), STAT1 (Santa Cruz, sc-346), TSG101 (Abcam, ab30871); and rabbit monoclonal Abs against integrin β5 subunit (Cell Signaling, 3629), pSTAT1(Y701) (Cell Signaling, 7649 S) and survivin (Cell Signaling, 2808). The following secondary Abs were used for IB analyses: HRP-linked anti-mouse IgG (Cell Signaling, 7076 S) and HRP-linked anti-rabbit IgG (Cell Signaling, 7074 S).
LEV and sEV isolation and analysis
LEVs include large and intermediate EVs and were isolated as described previously [5,11]. Briefly, PrCa cells (PC3-parental) were plated in 150 mm cell culture dishes (ThermoScientific) in their respective cell line complete media. After 48 h of incubation at 37°C, cells were transferred to starvation media (complete media devoid of FBS) for the next 48 h. LEVs were isolated from culture supernatant (SN) collected after 48 h of serum starvation by differential centrifugation. Briefly, the dead cells and cell debris were spun down from SN at 2000 g, 4°C for 20 min. The SN collected was spun at 10,000 g, 4°C for 35 min in a Beckman Type 45 Ti rotor using a Beckman L8-70 M Ultracentrifuge. The 10,000 g pellet was washed in phosphate buffer saline (PBS) followed by a second spin at 16,000 g, 4°C for 40 min in a tabletop centrifuge. The final LEV pellet (PC3 LEVs) was resuspended in PBS.
For sEV isolation, PrCa cells (PC3-parental, -shCtrl, -shβ6, -shβ5, -WT, -β6 KO-5, -β6 KO-7 and C4-2B-Mock, -αvβ6) were plated in 150 mm cell culture dishes (ThermoScientific) in their respective complete media. After 48 h of incubation at 37°C, cells were transferred to starvation media (complete media devoid of FBS) for the next 48 h. sEVs were isolated from SN collected after 48 h of serum starvation by high-speed differential ultracentrifugation. Briefly, the dead cells and cell debris were spun down from SN at 2000 g, 4°C for 20 min. The SN collected was spun at 10,000 g, 4°C for 35 min in a Beckman Type 45 Ti rotor using a Beckman L8-70 M Ultracentrifuge. The SN collected without disturbing the 10,000 g pellet was spun at 100,000 g, 4°C for 70 min in a Beckman Type 45 Ti rotor using a Beckman L8-70 M Ultracentrifuge; the pellet was washed in PBS followed by a second spin at 100,000 g, 4°C for 70 min in a Beckman Type 45 Ti rotor using a Beckman L8-70 M Ultracentrifuge. The final sEV pellet from each cell type mentioned above was resuspended in PBS to get PC3 sEVs, PC3-shCtrl sEVs, PC3-shβ6 sEVs, PC3-shβ5 sEVs, PC3-WT sEVs, PC3-β6 KO-5 sEVs, PC3-β6 KO-7 sEVs, C4-2B-Mock sEVs and C4-2B-αvβ6 sEVs. The total cell lysates (TCLs; 10–40 μg), LEV or sEV lysates were prepared using radio immuno precipitation assay (RIPA) buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100 and 1% sodium deoxycholate) supplemented with protease inhibitors (calpain, aprotinin, leupeptin, pepstatin, sodium fluoride and sodium orthovanadate). The total protein concentration of sEVs was determined using BioRad DCTM protein assay kit as per the manufacturer’s protocol. Equal amounts of proteins in non-reducing (heated without 2-mercaptoethanol) and reducing conditions (heated with 2-mercaptoethanol) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membranes (immobilon-E PVDF membrane, pore size 0.45 µm, Millipore), blocked with blocking buffers [5% non-fat dry milk in Tris Buffer Saline with 0.1% Tween 20 (TBST) or 5% bovine serum albumin (BSA) in TBST] for 1 h at room temperature, incubated overnight with primary Abs as described above, followed by TBST washes (4 × 10 min) at room temperature, incubation with horseradish peroxidase (HRP)-conjugated anti-mouse or -rabbit secondary Abs for 1 h at room temperature, followed by TBST washes (4 × 10 min) at room temperature. For visualisation, WesternBrightTM ECL HRP substrate kits (Advansta Inc., CA, USA) were used. The size distribution (mean and mode) and concentration of LEVs or sEVs were determined by nanoparticle tracking analyses (NTAs) detailed below.
Iodixanol density gradient
For iodixanol density gradient separation, a previously described procedure was used [19]. Briefly, the LEVs obtained from PC3-parental cells or sEVs obtained from PC3 cells (PC3-parental, -shCtrl, -shβ5 and -shβ6) or C4-2B cells (C4-2B-Mock, -αvβ6) were suspended in 1.636 mL of 30% iodixanol solution [made by mixing 1:1 of 60% (wt/vol) stock solution of iodixanol (OptiPrep™, Sigma # 1556) with a buffer (0.25 M sucrose, 10 mM Tris pH 8.0, 1 mM EDTA, pH 7.4)] and layered at the bottom of an ultracentrifugation tube. Next, 0.709 mL of 20% (wt/vol) iodixanol and 0.654 mL of 10% (wt/vol) iodixanol solutions were successively layered on top of the 30% iodixanol-vesicle suspension to create a discontinuous gradient. Gradient samples were centrifuged for 1 h at 350,000 g, 4°C in a SW55Ti rotor using a Beckman L8-70 M Ultracentrifuge. Ten consecutive fractions of 0.267 mL were collected from top to bottom of the gradient. The refractive index of each fraction was assessed with an ABBE-3 L refractometer (Fisher Scientific) and density was calculated. All 10 fractions were diluted with 1 mL PBS and centrifuged for 70 min at 100,000 g, 4°C in a TLA-100.2 rotor using a Beckman, Optima TL Ultracentrifuge. The pellets thus obtained in 10 respective fractions were again washed in 1 mL PBS and centrifuged for 70 min at 100,000 g, 4°C in a TLA-100.2 rotor using a Beckman, Optima TL Ultracentrifuge. The final pellet in each fraction was resuspended in 30 μL of PBS and stored at −80°C or utilised for analysis by NTA or IB in non-reducing and reducing conditions or functional assays.
Nanoparticle tracking analysis
The size distribution and concentration of sEVs isolated from the PrCa cells (PC3-parental, -shCtrl, -shβ5, -shβ6, -WT, -β6 KO-5, -β6 KO-7, C4-2B-Mock and -αvβ6 cells) were analysed using NanoSight NS300 instrument (Malvern Panalytical, UK). Briefly, sEV suspensions were diluted 1:1000 and/or 1:200 (for iodixanol density gradient separated fractions) in PBS, and the analysis was performed using camera levels ranging from 11 to 13 to see the EV particles clearly in a way that they do not appear saturated (coloured pixels). Using the script SOP standard measurement, video files of 30- or 60-s duration (repeated three times or five times) were captured with a frame rate of 25 frames per second of particles moving under Brownian motion at a temperature ranging from 22°C to 25°C. The analysis of videos was performed at a detection threshold ranging from 3 to 5 using NTA software version 3.1 (build 3.1.54).
Quantitative real-time PCR
HMEC1 or BAEC (2 × 105) were seeded on six-well cell culture dishes. Next day, cells were washed with PBS, incubated with endothelial cell basal media and either PBS or sEVs (40 μg/mL) isolated from PC3 cells (4, 8 and 16 h for BAEC or 2, 4, 8, 16 and 24 h for HMEC1). After incubation, cells were washed with PBS, trypsinised and total RNA from HMEC1, BAEC or PC3 PrCa cells (positive control for β6 mRNA expression) was isolated using the Qiagen RNeasy Kits (Qiagen, Valencia, CA, USA) as per manufacturer’s protocol. RNA (1 μg) was reverse transcribed with random hexamer oligos (Invitrogen) and SuperScript II RNase H-reverse transcriptase enzyme (Invitrogen). Subsequently, for real-time PCR analysis, complementary DNA (cDNA) was amplified using the QuantStudio 12 K Flex Real-Time PCR system. The gene expression of β6, BIRC5 and GAPDH were profiled using primers for β6 (forward primer, 5ʹ-GGTCTCATCTGGAAGCTACTGGTGTCA-3ʹ; reverse primer, 5ʹ-GGTCTCCCAGATGCACAGTAGGACAACC-3ʹ), BIRC5 (forward primer, 5ʹ-GACTTGGCTCGATGCTGTGG-3ʹ; reverse primer, 5ʹ-TACGCCAGACTTCAGCCCTG-3ʹ) and GAPDH (forward primer, 5ʹ-GGGAAGGTGAAGGTCGGAGT-3ʹ; reverse primer, 5ʹ-GTTCTCAGCCTTGACGGTGC-3ʹ). The relative mRNA expression was calculated using the 2ΔΔCT method. Each reaction was carried out in triplicate; mean and standard error of mean were calculated using Excel (Microsoft) software.
Analysis of sEV-mediated αvβ6 integrin transfer and impact on angiogenic signalling via immunoblotting
To evaluate the PC3-sEV-mediated internalisation of αvβ6 integrin, the HMEC1 or BAEC (2 × 105) cultured in serum- and growth factor-starved conditions were incubated with the PBS as vehicle control or the same dose of sEVs (20 µg/mL) at different time lengths (6, 16 and 24 h for HMEC1 and 4, 8 and 16 h for BAEC). The efficiency of PC3 sEV-mediated αvβ6 integrin internalisation in HMEC1 was also evaluated after acid wash [sodium acetate buffer (0.2 M acetic acid/0.5 M NaCl, pH 2.8]. The sEV-mediated internalisation of αvβ6 integrin in HMEC1 was also tested upon incubation of HMEC1 (2 × 105) cultured in serum- and growth factor-starved conditions with 40 µg/mL of sEVs derived from PrCa cells (PC3-WT, -β6 KO-5 and -β6 KO-7) for 18 h. After incubation with sEVs, HMEC1 or BAEC were washed with PBS, trypsinised, collected, lysed in RIPA buffer containing protease inhibitors and TCL were subjected to IB analysis to measure β6 levels.
The effect of PrCa sEVs on angiogenic signalling in HMEC1 was also evaluated after incubation of HMEC1 (2 × 105) with PBS or 40 µg/mL sEVs derived from PrCa cells (PC3-WT, -β6 KO-5, -β6 KO-7, -shCtrl, -shβ6, -shβ5 and C4-2B-Mock or -αvβ6) for 18 h. After incubation with sEVs, HMEC1 were washed with PBS, trypsinised, collected, lysed in RIPA buffer containing protease inhibitors and TCL were subjected to IB analysis to measure levels of angiogenic signalling molecules.
Analysis of sEV-mediated αvβ6 integrin transfer and cell surface expression via FACS
Fluorescence-activated cell sorting (FACS) analysis was employed to detect sEV-mediated αvβ6 integrin transfer and expression on the cell surface of HMEC1. HMEC1 (2 × 106) plated in 100 mm cell culture dishes were serum- and growth factor-starved and incubated with PBS or 40 µg/mL PC3 sEVs for 18 h. After incubation with PBS or sEVs, HMEC1 were trypsinised, washed with PBS and 3 × 105 cells were incubated with the Ab specific to αvβ6 integrin (mAb 6.4B4, 10 µg/mL in HMEC1 media) or mouse IgG as isotype control (10 µg/mL in HMEC1 media) for 45 min at 4°C. Samples were washed three times with complete media (RPMI with 10% FBS), pelleted and incubated with Alexa FluorTM 488 F(ab’)2 fragment of rabbit anti-mouse-IgG (H + L) (Invitrogen) in HMEC1 complete media for 30 min at 4°C, washed three times with complete media (RPMI with 10% FBS), pelleted and resuspended in 500 µL of PBS and analysed. The data were acquired using BD Celesta flow cytometer (BD Biosciences) and analysed by FlowJo software.
Trypan blue dye exclusion assay
HMEC1 (2 × 105) were seeded on six-well cell culture dishes (replicates n = 3). Next day, cells were washed with PBS, incubated with endothelial cell basal media and either PBS or iodixanol density gradient separated sEVs (40 μg/mL) isolated from PrCa cells (PC3-shCtrl, -shβ6, -shβ5, C4-2B-Mock and -αvβ6) at 37ºC for 18 h. After 18 h of incubation with PBS or respective sEVs, HMEC1 form each condition were washed with PBS, trypsinised, collected and resuspended in endothelial cell basal media. One part of the cell suspension was mixed with one part of 0.4% trypan blue. The mixture was allowed to incubate ~3 min at room temperature and 10 μL of the trypan blue/cell mixture was applied to a haemocytometer and the unstained cells (viable) were counted using a hand tally counter in 4 separate grids (each having 16 squares) of haemocytometer using the inverted microscope under 10× objective. The total number of viable cells per mL = (the total number of viable cells counted in 4 grids/4) × 10,000 × dilution factor. For PC3-shCtrl, -shβ6 and -shβ5 cells (1 × 106) were seeded on 100 mm cell culture dishes (replicates n = 3) in complete media, incubated for 72 h at 37ºC and after incubation, the viable cells were counted by trypan blue dye exclusion method as described above.
Boyden chamber assay
HMEC1 (5 × 104) were seeded on Transwell chambers (replicates n = 3) and incubated with PBS or iodixanol density gradient separated sEVs (0.3 × 109 vesicles) isolated from the PrCa cells (PC3-parental, -shCtrl, -shβ5, -shβ6, C4-2B-Mock and -αvβ6). To acquire the 0.3 × 109 sEVs utilised for the functional analyses, ~12 × 104 PC3 cells and ~2 × 105 C4-2B cells are required. HMEC1 complete media were placed in the bottom chamber as a chemoattractant for sEV-incubated HMEC1 in Transwell chambers and incubated for 24 h at 37ºC. In another set of experiments, HMEC1 basal media were placed in the bottom chamber for sEV-incubated HMEC1 in Transwell chambers and incubated for 24 h at 37ºC. After 24 h, Transwell inserts were placed in 100% methanol for 10 min to allow the fixation of migrated HMEC1. A cotton-tipped applicator was used to remove the remaining methanol from the top of the membranes and they were allowed to dry. For staining the fixed HMEC1, the membranes were positioned in a 0.5% crystal violet solution and incubated at room temperature for 10 min. Excess crystal violet was removed from the top of the membranes with a cotton-tipped applicator. Membranes were gently rinsed in distilled water to remove the excess crystal violet. Pictures were captured underneath an inverted microscope and the number of migrated cells was counted manually in different fields of view (n = 6 or 9) to obtain an average total number of cells that have migrated through the membranes towards the chemo-attractant and attached to the underside of the membranes.
Tube formation assay
For tube formation assays, 96-well plates were coated with 70 µL of BD Matrigel™ Basement Membrane Matrix (Cat. #354,230). Plates were incubated for 3 h at 37ºC to allow the Matrigel to form a gel. A single-cell suspension of 1.5 × 104 HMEC1 or BAEC/well (replicates n = 3) were plated on to the solidified Matrigel using 100 µL media/well and incubated with PBS, or iodixanol density gradient separated sEVs (0.3 × 109 vesicles) isolated from the PrCa cells (PC3-shCtrl, -shβ5, -shβ6, C4-2B-Mock and -αvβ6) and incubated for 5 h for HMEC1 or 8 h for BAEC at 37ºC. The endothelial tubes formed were examined after 5 h for HMEC1 or 8 h for BAEC using a light microscope and images were captured in different fields of view (n = 6). Using the ImageJ Angiogenesis Analyser Plugin, the photomicrographs were analysed and quantified for nodes, junctions and tubules formed.
Human tissue specimens
All formalin-fixed and paraffin-embedded human tissue specimens used in this study were de-identified and processed in accordance with IRB approved protocols. Seven metastatic prostate adenocarcinoma tissue samples (Gleason Score [GS] 8 [n = 1], GS 9 [n = 2], GS 10 [n = 4]) were obtained from the Department of Pathology at Thomas Jefferson University (Philadelphia, PA). Additionally, nine human malignant prostate adenocarcinomas tissue samples (GS 7 [n = 5], GS 8 [n = 2], GS 9 [n = 1] and GS 10 [n = 1]) were obtained from the Cooperative Human Tissue Network (CHTN) western division at Vanderbilt University Medical Centre, TN, or Mid-Atlantic division at University of Virginia, VA. The CHTN is funded by the National Cancer Institute and other investigators may have received specimens from the same subjects.
Immunohistochemistry (IHC)
Following a standardised protocol, immunohistochemistry (IHC) was performed on the PrCa tissue sections. The tissue sections were baked at 60°C for 1 h, followed by deparaffinisation with xylene (3 min × 2), rehydration with graded alcohols (100%, 90%, 70%, 50% and 30% for 3 min each) followed by deionised water (3 min × 2). The sections were incubated with 3% H2O2 solution for quenching endogenous peroxidase activity, followed by antigen retrieval by pepsin (0.5% in 5 mM HCl) digestion for αvβ6 integrin for 15 min at 37°C or proteinase K (20 μg/mL in Tris-EDTA buffer, pH 8.0) digestion for von Willebrand factor (vWF) for 15 min at 37°C or sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) for CD31 for 15 min at 95°C. Sections were washed once with deionised water for 5 min, followed by PBS wash for 5 min and blocked with 5% goat serum or horse serum made in PBS with 0.1% Tween 20 (PBST) for 2 h. The tissue sections were incubated overnight at 4°C with an Ab to αvβ6 integrin (6.2A1, 2 μg/mL) or isotype control mouse-IgG (mIgG, 2 μg/mL), vWF (Dako A0082, 3 μg/mL) or isotype control rabbit-IgG (rbIgG, 3 μg/mL) and CD31 (ab28364, 1 μg/mL) or isotype control rabbit-IgG (rbIgG, 1 μg/mL). The following day, the tissue sections were washed with PBST (5 min × 2) followed by PBS (5 min) and incubated with secondary Ab in PBST (biotinylated horse anti-mouse-IgG [BA-2000, Vector Laboratories, Burlingame, CA] or biotinylated goat anti-rabbit-IgG [BA-1000, Vector Laboratories, Burlingame, CA], 10 μg/mL) for 30 min at room temperature. The unbound secondary Ab was washed with PBST (5 min × 2), followed by PBS (5 min). The tissue sections were incubated with streptavidin horseradish peroxidase (SAP) in PBS (SA-5004, Vector Laboratories, Burlingame, CA, 5 μg/mL) for 30 min at room temperature. The unbound SAP was washed with PBST (5 min × 2), followed by PBS (5 min). The colour was developed by adding substrate chromogen, 3,3′-diaminobenzidine solution (DAB peroxidase substrate kit, SK-4100, Vector Laboratories, Burlingame, CA). A brown precipitate indicated positive expression. The DAB reaction was stopped by rinsing the tissue sections in deionised water. The sections were counterstained with Harris haematoxylin, dehydrated in graded ethanol (30%, 50%, 70%, 90% and 100% for 5 min each) followed by xylene (5 min × 2), dried and finally mounted with Permount (Vector Laboratories, Burlingame, CA).
At least two members of the team reviewed each tumour section. The immunostaining intensity of αvβ6 integrin in PrCa epithelial cells or vWF, CD31 in endothelial cells within each specimen was evaluated by the pathologist and given a score. The scoring of immunostaining intensity is summarised and grouped as follows: negative, 0; negligible, 1+; weak, 2+ and strong, 3+. For a given intensity, the percentage of PrCa cells positive for αvβ6 integrin within a given specimen was also scored on a scale of 0–100: 0%, no cell staining; 100%, all cells positively stained.
Statistical analysis
For statistical analysis, Student’s t-test is used for comparing two group means. One-way ANOVA with post hoc Fisher’s LSD test are applied to compare the means of three or more independent groups. A two-sided P value of ≤ 0.05 is considered statistically significant. Software GraphPad Prism 7 is used for data analysis.
Results
Characterisation of prostate cancer cell-derived αvβ6-positive LEVs and αvβ6-positive sEVs
EV subtypes include LEVs that are 100–1000 nm in size and sEVs that are < 200 nm size [11]. These EV subtypes can be isolated by flotation into iodixanol density gradients on the basis of different buoyant densities and sizes [11]. We have previously shown that αvβ6 integrin is enriched in sEVs derived from PrCa cells. We now also show the expression of αvβ6 integrin in LEVs. We utilised serum-starved conditioned media from PrCa cells (PC3 cells) endogenously expressing αvβ6 integrin for isolation of LEVs by differential centrifugation (10,000 g). LEVs were then floated on an iodixanol density gradient and 10 fractions were collected from top to bottom. Our IB analysis shows that β6 and tetraspanins (CD63 and CD81) are expressed in iodixanol density gradient fractions (density range: 1.099–1.169 g/mL) from PC3-derived LEV samples (Figure 1a). A previous study has also shown expression of tetraspanins like CD81 in LEVs at varying levels [40], although in one study, it was shown that expression of CD81 in LEVs is lower than sEVs [41].
Figure 1.
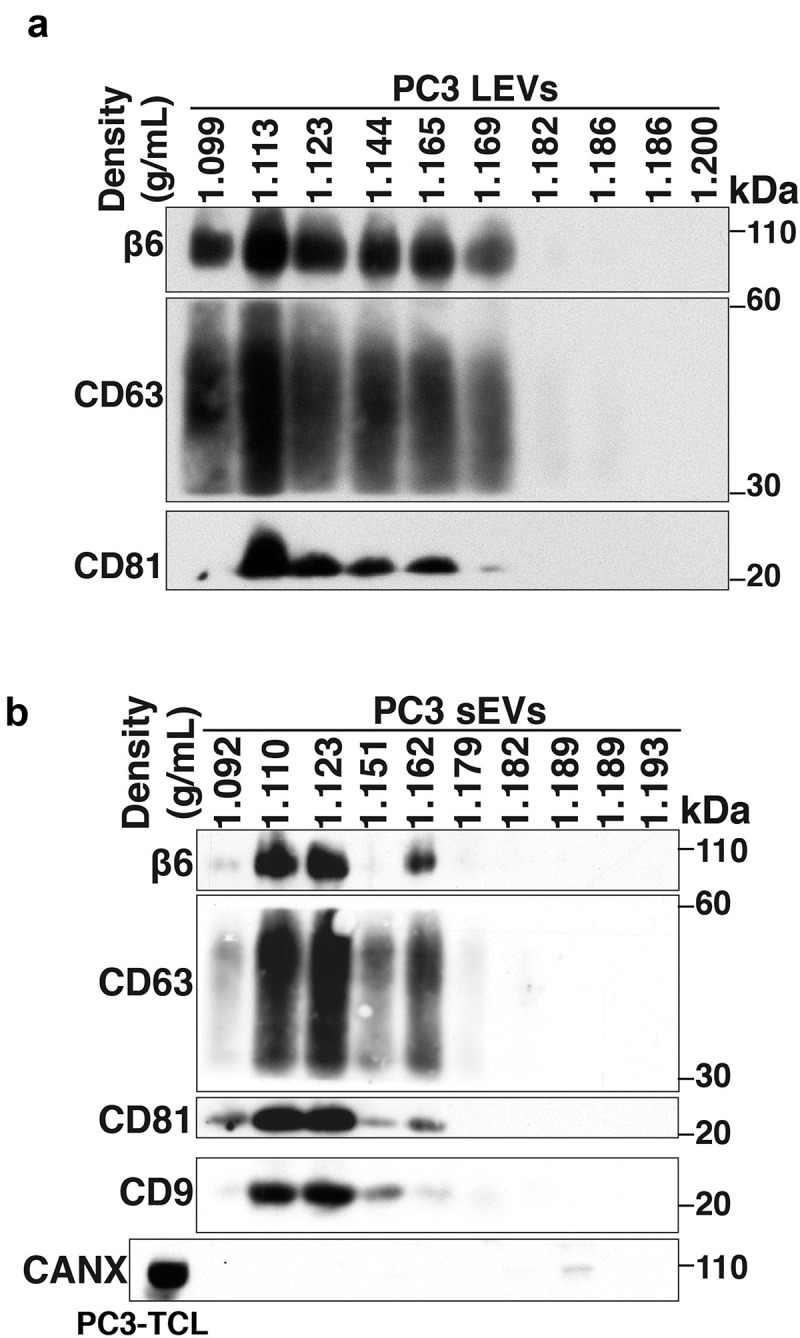
Characterisation of prostate cancer cell-derived αvβ6-positive LEVs and αvβ6-positive sEVs.
After removal of LEVs, sEVs were isolated by high-speed differential ultracentrifugation (100,000 g) followed by flotation on an iodixanol density gradient. We analyse the levels of the β6 and sEV markers (CD63, CD81 and CD9) in each iodixanol density gradient fraction from the PC3-derived sEV samples and observed that their levels are the highest in the 1.123 g/mL density fraction (Figure 1b). None of the 10 iodixanol density gradient fractions shows the expression of Calnexin (CANX), an endoplasmic reticulum (ER) marker known to be absent in sEVs which is instead detected in PC3-total cell lysates (PC3-TCL) (Figure 1b). Our proteomic analysis of PC3 sEVs [19] had shown that several aberrations follow down-regulation of αvβ6 integrin and indicated that these aberrant sEVs might impact endothelial cell behaviour [19]. Given our results showing that the levels of αvβ6 integrin and sEV markers are the highest in the 1.123 g/mL fraction, we used this fraction [11] to narrow down the subset that carries the functional αvβ6 integrin and its downstream effectors.
Prostate cancer cell-derived small extracellular vesicular-αvβ6 integrin is transferred to endothelial cells
Endothelial cells are an important component of the prostate TME and their role in increased angiogenesis has been associated with prostate tumour progression [2,3]. We have previously shown that the αvβ6 integrin is packaged in sEVs shed by PrCa cells, transferred via sEVs to recipient prostate cells and monocytes, and is able to functionally modulate these cells [17,19]. We hypothesised that transfer of PrCa cell-derived sEVs that express αvβ6 integrin (αvβ6-positive sEVs) to β6-negative endothelial cells might promote the angiogenic potential of these recipient endothelial cells.
To study PrCa cell-derived sEV-mediated transfer of αvβ6 integrin to endothelial cells, we selected human microvascular endothelial cells (HMEC1) and bovine aortic endothelial cells (BAEC) that do not express β6. To investigate whether αvβ6 integrin is transferred to endothelial cells via iodixanol density gradient isolated sEVs, we first collected the 100,000 g pellet and characterised it by NTA and IB (Figure 2a,b). Then we isolated them using iodixanol density gradient as described in the previous section. Our NTA shows that the sEVs from PC3 cells have a diameter of < 150 nm, confirming their vesicular subtype identification as sEVs (Figure 2a). The 100,000 g pellet is also characterised for expression of sEV-specific markers by IB; sEVs from PC3 cells show enrichment of β6 and sEV markers CD63, CD81, ALIX and TSG101 compared to TCL (Figure 2b). Furthermore, the sEVs do not express CANX, an ER marker known to be present in TCL (Figure 2b). Incubation of BAEC and HMEC1 with PBS or αvβ6-positive sEVs from PC3 cells for different time points (4, 8 and 16 h for BAEC and 2, 4, 8, 16 and 24 h for HMEC1) shows that β6 mRNA expression is not induced in endothelial cells (Figure 2c,d). To test whether αvβ6 integrin is transferred as protein via sEVs to endothelial cells, we incubated HMEC1 and BAEC with PBS or 100,000 g isolated αvβ6-positive sEVs from PC3 cells for different time periods [6, 24 h for HMEC1 (Figure 2e,f) and 4, 8, 16 h for BAEC (Fig. S1)]. We observe that a low level of β6 integrin subunit is transferred in 6 h while a more robust amount is transferred in 24 h in HMEC1 (Figure 2e). While in BAEC, a low level of β6 integrin subunit is transferred in as few as 4 h, a more robust amount is transferred in 8 h (Fig. S1). Furthermore, the results show that after sEV incubation, acid wash (which removes non-specifically trapped proteins from the cell surface) of recipient HMEC1 does not reduce β6 levels transferred to HMEC1, indicating an efficient microvascular endothelial cell internalisation of the transferred β6 integrin subunit (Figure 2e). These results were confirmed using iodixanol density gradient isolated αvβ6-positive sEVs and show that incubation of HMEC1 with the β6-positive sEV fraction, corresponding to 1.123 g/mL density, exhibits a very efficient transfer of β6 integrin subunit to HMEC1 in as little as 6 h (Figure 2f). Since the αvβ6 integrin is a cell-surface receptor, we further evaluated de novo cell-surface expression of αvβ6 integrin after transfer via sEVs into HMEC1. Using FACS analyses, we show that upon incubation with PC3 sEVs, the αvβ6 integrin is transferred to HMEC1 and detected on the cell surface (Figure 2g). Overall, our data show that de novo expression of αvβ6 integrin on the plasma membrane of endothelial cells can be attributed to efficient protein transfer via PC3 sEVs.
Figure 2.
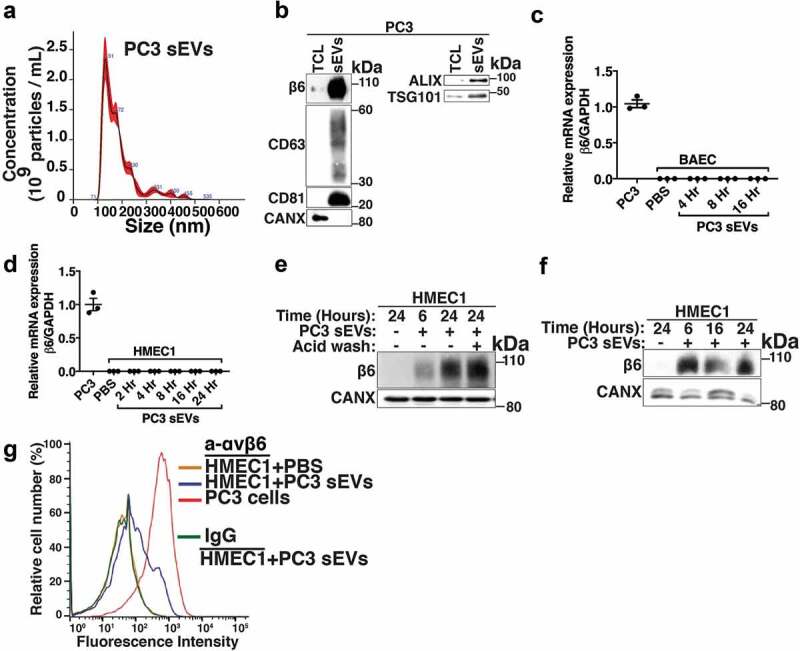
Transfer of prostate cancer cell-derived αvβ6-positive sEVs to microvascular and aortic endothelial cells.
αvβ6 integrin in sEVs does not affect expression of sEV markers
To more specifically evaluate the functional implications of αvβ6-positive sEVs on endothelial cells, we utilised PC3 cells stably transfected with control shRNA (PC3-shCtrl), shRNA to β6 integrin subunit (PC3-shβ6) or shRNA to β5 integrin subunit (PC3-shβ5) and C4-2B PrCa cells transfected with either empty vector (C4-2B-Mock) or β6 cDNA-expression vector (C4-2B-αvβ6). We isolated sEVs from PC3-shCtrl, -shβ6 and -shβ5 cells through high-speed differential ultracentrifugation (100,000 g) and further removed contaminants by iodixanol density gradient centrifugation. The sEV fractions corresponding to a density of 1.12 g/mL were characterised for size distribution by NTA. The majority of the PC3-shCtrl sEVs, -shβ6 sEVs and -shβ5 sEVs are < 150 nm in size (Figure 3a). The average yield of sEVs from PC3 cells is ~2.5 × 103 sEVs/cell/48 h. Furthermore, NTA data from C4-2B-Mock sEVs and -αvβ6 sEVs also show that the majority of the sEVs exhibit a particle size of < 150 nm (Figure 3b). The average yield of sEVs from C4-2B cells is ~1.5 × 103 sEVs/cell/48 h. IB analysis of the TCL and the sEVs from PC3-shβ5, -shCtrl and -shβ6 cells shows similar levels of β6 integrin subunit in -shβ5 sEVs and -shCtrl sEVs whereas its expression is significantly reduced in the sEVs from -shβ6 cells (Figure 3c). Expression of sEV markers (CD63, CD81 and TSG101) is enriched for sEVs in comparison to respective TCL (Figure 3c). IB analysis of TCL and sEVs from C4-2B-Mock and -αvβ6 cells shows the expression of β6 only in the TCL and sEVs from C4-2B-αvβ6 cells. Expression of sEV markers (ALIX, TSG101 and CD9) is enriched for both C4-2B-Mock sEVs and -αvβ6 sEVs in comparison to their respective TCL (Figure 3d). Overall, our data show that the αvβ6 integrin does not alter expression of sEV-specific markers in sEVs.
Figure 3.
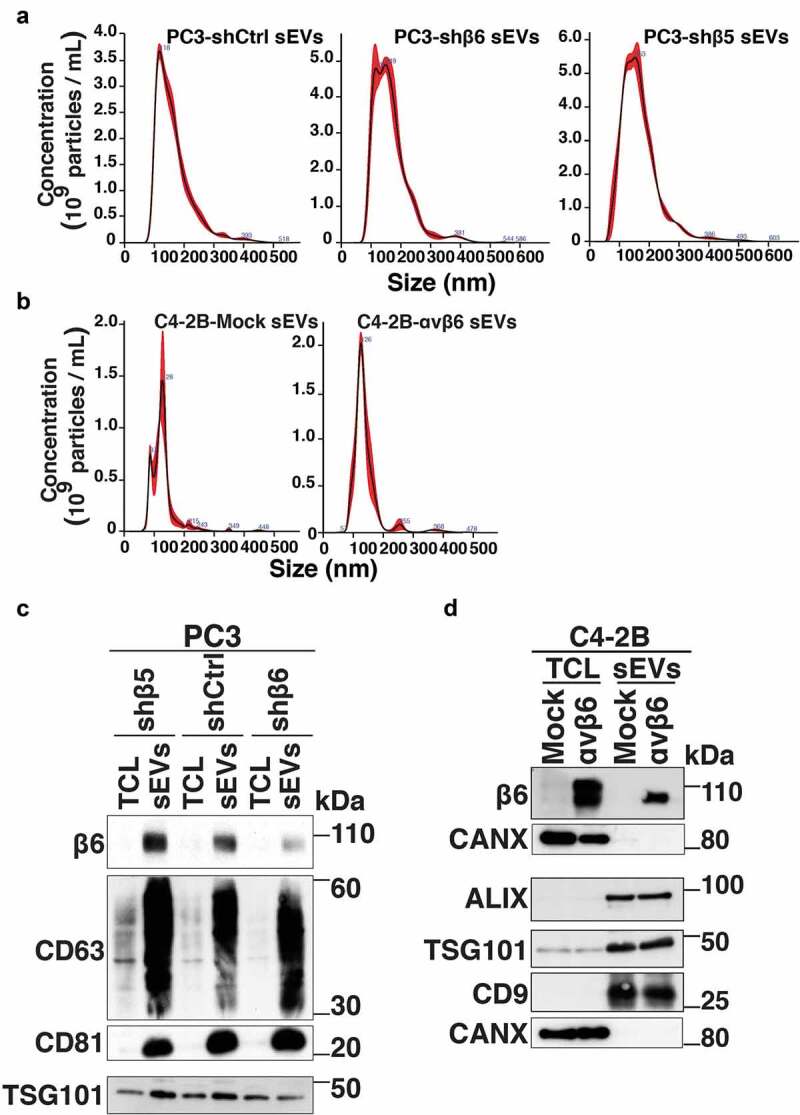
Characterisation of prostate cancer cell-derived sEVs upon knockdown or expression of β6 integrin subunit in prostate cancer cells.
αvβ6 integrin in prostate cancer sEVs increases motility and tube forming potential of endothelial cells
Increased proliferation, motility and tube formation by endothelial cells are considered to be hallmarks of angiogenesis [42]. To test the impact of αvβ6-positive sEVs on HMEC1 viability, we incubated HMEC1 with PBS or sEVs from PC3-shCtrl, -shβ6, -shβ5 (data not shown) and C4-2B-Mock, -αvβ6 cells and performed trypan blue dye exclusion assays (Fig. S2). Our analysis shows that relative to incubation with PBS, there is no significant change in the viability of HMEC1 upon incubation with respective sEVs (Fig. S2). We also investigated the impact of β6 or β5 integrin subunit knockdown on the viability of PrCa cells. Our analysis shows that there is no significant change in the viability of PC3-shβ6 or PC3-shβ5 cells compared to PC3-shCtrl cells (data not shown).
To further evaluate the role of αvβ6-positive sEVs on the motility of HMEC1, we performed Boyden chamber assays. In comparison to incubation with PBS, there is a significant increase (P < 0.05) in the motility of HMEC1 upon incubation with PC3-shCtrl sEVs (Fig. S3A). Compared to incubation with PC3-shCtrl sEVs, motility of HMEC1 is significantly decreased (P < 0.0005) upon incubation with PC3-shβ6 sEVs (Fig. S3A) and not significantly altered upon incubation with PC3-shβ5 sEVs (Fig. S3A). Furthermore, there is a significant increase in motility of HMEC1 upon incubation with C4-2B-αvβ6 sEVs compared to both incubation with PBS (P < 0.005) or C4-2B-Mock sEVs (P < 0.05) (Fig. S3B). Finally, we investigated the impact of PC3 sEVs on the motility of HMEC1 in the absence of chemotaxis. We demonstrate that even in the absence of chemotaxis, there is a significant increase in motility of HMEC1 upon incubation with PC3 sEVs compared to incubation with PBS (P < 0.005) (Fig. S3C). Overall, the results from our study show that αvβ6-positive sEVs significantly increase the motility of HMEC1.
Angiogenesis is known to be a vital process associated with PrCa progression [2]; however, the key molecular mechanisms that regulate PrCa cell-mediated angiogenesis have been elusive. Owing to the significant impact that αvβ6-positive sEVs have on the motility of microvascular endothelial cells, we further evaluated the implications of αvβ6-positive sEVs on tube forming potential of endothelial cells. We performed tube formation assays which provided a rapid and quantitative method for assessing the angiogenic potential of HMEC1 and BAEC plated on Matrigel and incubated with PBS or sEVs from PC3-shCtrl, -shβ6, -shβ5 and C4-2B-Mock, -αvβ6 cells. The phase-contrast microscopy images from these tube formation assays show extensive branching and tube formation of HMEC1 upon incubation with PC3-shCtrl sEVs (Figure 4, upper panel) compared to incubation with the PBS-only control. On the other hand, branching and tube formation is abrogated upon incubation with PC3-shβ6 sEVs (Figure 4, upper panel) and mildly reduced upon incubation with PC3-shβ5 sEVs (Figure 4, upper panel). The phase-contrast microscopy images were further quantified for the number of nodes, junctions and tubules formed in each sEV incubation group. Compared to incubation with PBS, there is a significant increase in the number of nodes (P < 0.05), junctions (P < 0.05) and tubules (P < 0.05) formed upon incubation with PC3-shCtrl sEVs (Figure 4, lower panel). Compared to incubation with PC3-shCtrl sEVs, there is a highly significant reduction in the number of nodes (P < 0.0005), junctions (P < 0.0005) and tubules (P < 0.0005) formed upon incubation with PC3-shβ6 sEVs (Figure 4, lower panel). In comparison to incubation with PBS, there is no impact of PC3-shβ5 sEVs on formation of nodes, junctions and tubules (Figure 4, lower panel). A significant reduction in the number of nodes (P < 0.005), junctions (P < 0.005) and tubules (P < 0.05) formed by HMEC1 is observed upon incubation with PC3-shβ5 sEVs compared to incubation with PC3-shCtrl sEVs (Figure 4, lower panel). A significant decrease in formation of nodes (P < 0.05), junctions (P < 0.05) and tubules (P < 0.005) formed by HMEC1 is observed upon incubation with PC3-shβ6 sEVs compared to incubation with PC3-shβ5 sEVs (Figure 4, lower panel). Furthermore, the phase-contrast microscopy images show extensive branching and tube formation of HMEC1 upon incubation with C42B-αvβ6 sEVs compared to incubation with PBS controls (Figure 5a, upper panel). The quantification of phase-contrast microscopy images for number of nodes, junctions and tubules formed in each incubation group also shows that there is a highly significant increase in nodes (P < 0.0005), junctions (P < 0.0005) and tubules (P < 0.005) formation by HMEC1 upon incubation with C4-2B-αvβ6 sEVs compared to incubation with PBS (Figure 5a, lower panel). In comparison to incubation with C4-2B-Mock sEVs, there is a significant increase in nodes (P < 0.005), junctions (P < 0.05) and tubules (P < 0.05) formation by HMEC1 upon incubation with C4-2B-αvβ6 sEVs (Figure 5a, lower panel).
Figure 4.

Down-regulation of αvβ6 integrin in prostate cancer sEVs modulates the angiogenic potential of microvascular endothelial cells.
Figure 5.
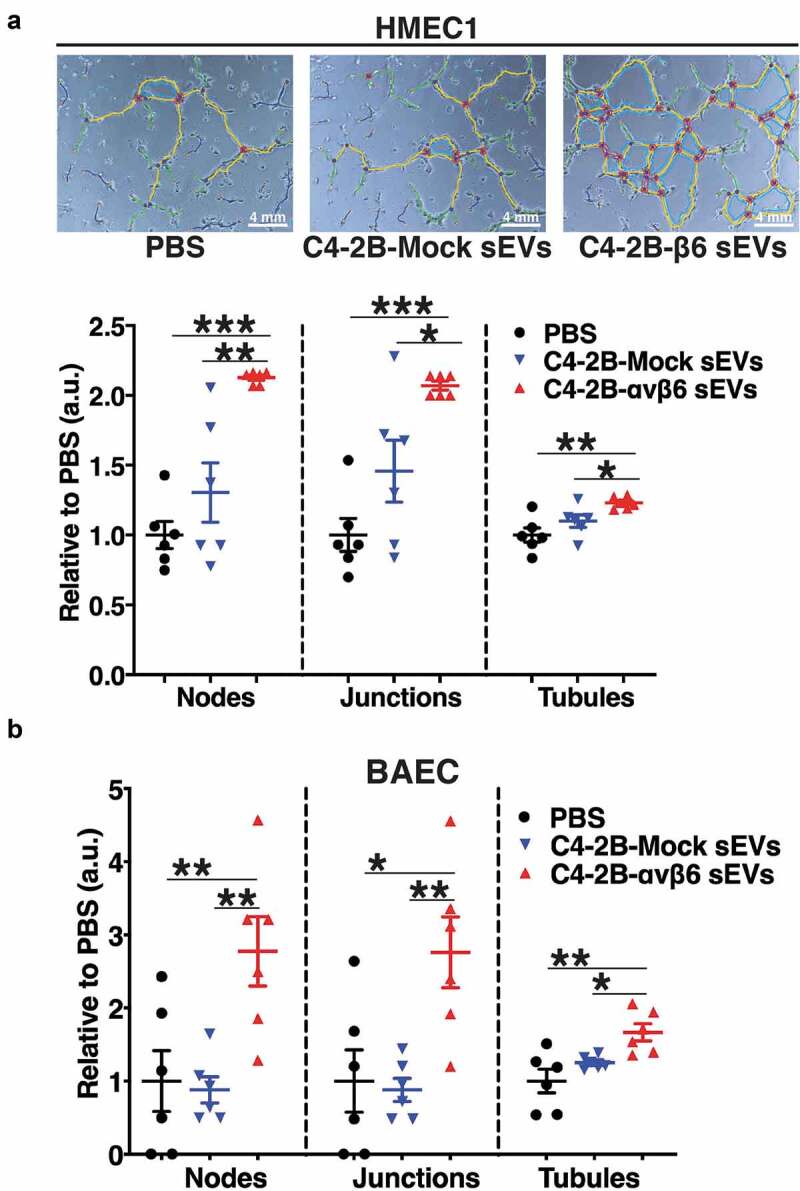
Expression of αvβ6 integrin in prostate cancer sEVs modulates the tube forming potential of endothelial cells.
To test whether the impact of αvβ6-positive sEVs is not limited only to microvascular endothelial cells, we included another endothelial cell type, bovine aortic endothelial cells (BAEC). Compared to incubation with PBS, there is a significant increase in nodes (P < 0.005), junctions (P < 0.05) and tubules (P < 0.005) formed by BAEC upon incubation with C4-2B-αvβ6 sEVs (Figure 5b). In addition, compared to incubation with C4-2B-Mock sEVs, there is a significant increase in nodes (P < 0.005), junctions (P < 0.005) and tubules (P < 0.05) formed by BAEC upon incubation with C4-2B-αvβ6 sEVs (Figure 5b). Overall, the results from our study show that αvβ6-positive sEVs support the tube formation capability of endothelial cells.
Uptake of prostate cancer cell-derived αvβ6-positive sEVs regulate angiogenic signalling in microvascular endothelial cells
We have previously demonstrated that the αvβ6 integrin negatively regulates protein levels of the signalling molecule STAT1 both in PC3 cells and in the sEVs derived from them [19]. Since elevated STAT1 is known to be a negative regulator of angiogenesis [43,44], we hypothesised that the transfer of αvβ6-positive sEVs to HMEC1 might negatively regulate expression and activation of STAT1 protein in HMEC1. To test this hypothesis, we utilised PrCa cells harbouring CRISPR/Cas9-mediated down-regulation of β6 integrin subunit (PC3-β6 KO-5 and PC3-β6 KO-7) or CRISPR/Cas9 construct transfected cells that endogenously express β6 integrin subunit (PC3-WT). We isolated sEVs from PC3-WT, -β6 KO-5 and -β6 KO-7 cells through high-speed differential ultracentrifugation (100,000 g) and characterised them for size distribution by NTA. The majority of PC3-WT, -β6 KO-5 and -β6 KO-7 sEVs are < 150 nm in size (Figure 6a). IB analysis of the sEVs and respective TCL from PC3-WT, -β6 KO-5 and -β6 KO-7 cells shows expression of β6 integrin subunit only in TCL and sEVs from PC3-WT cells (Figure 6b). CANX is used as the loading control for TCL and known to be absent in sEVs (Figure 6b). As expected, the levels of CD63, CD81, CD9 and TSG101 are highly enriched in the sEV preparations as compared to TCL (Figure 6b). The absence of CANX in the sEV preparations confirms the removal of contaminants in our isolated sEVs (Figure 6b). Upon incubation with sEVs from αvβ6 integrin expressing cells (PC3-WT), by IB analysis we demonstrate the efficient uptake of β6 in HMEC1, while β6 is not present in HMEC1 incubated with sEVs derived from β6-negative cells (PC3-β6 KO-5 and PC3-β6 KO-7) (Figure 6c). Further evaluation of αvβ6-positive sEV-mediated impact on HMEC1 signalling shows that incubation of HMEC1 with sEVs from PC3-β6 KO-5 or -β6 KO-7 cells results in increased expression of pSTAT1(Y701) in comparison with HMEC1 incubated with sEVs from PC3-WT cells or PBS (Figure 6d).
Figure 6.
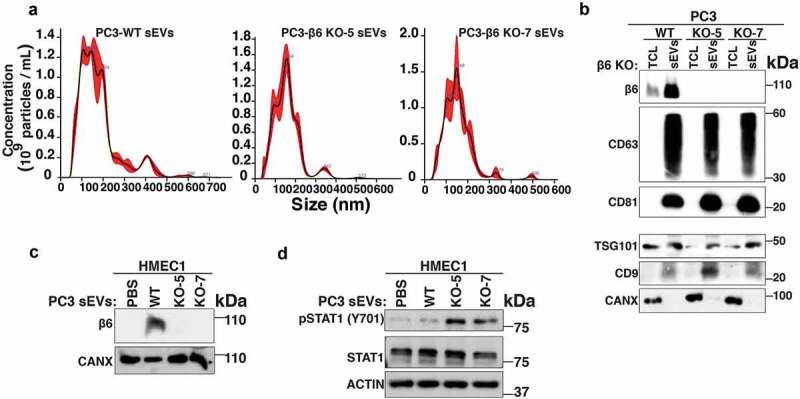
Transfer of αvβ6-positive sEVs derived from CRISPR/Cas9 genetically modified prostate cancer cells to microvascular endothelial cells regulates STAT1 signalling.
We further corroborated our findings with PC3-shCtrl, -shβ6 and -shβ5 sEVs by testing their impact on angiogenic pathways in recipient HMEC1. Incubation of HMEC1 with PC3-shβ6 sEVs results in the up-regulation of pSTAT1(Y701) levels compared to incubation with PBS or PC3-shCtrl and -shβ5 sEVs (Figure 7a). Similar to the results with PC3 sEVs, incubation of HMEC1 with C4-2B-αvβ6 sEVs results in lower pSTAT1(Y701) levels compared to incubation with PBS or C4-2B-Mock sEVs (Fig. S4).
Figure 7.
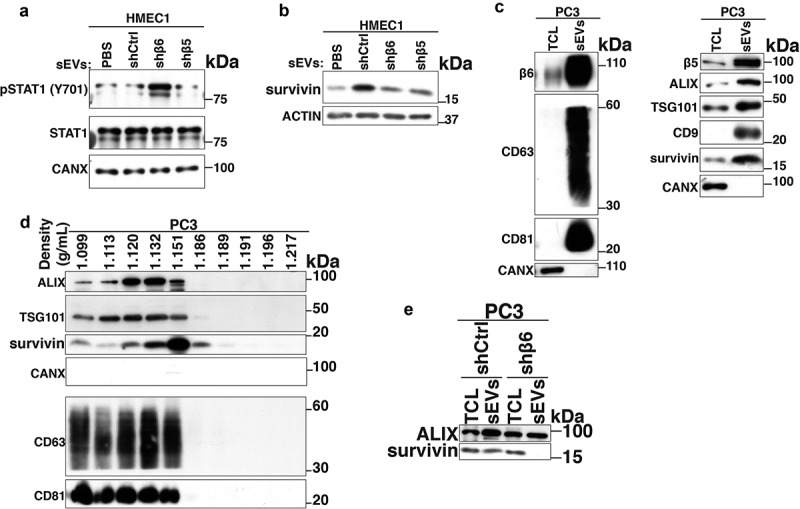
Transfer of prostate cancer cell-derived αvβ6-positive sEVs to microvascular endothelial cells regulates survivin levels.
Since αvβ6 integrin is known to regulate survivin expression in PrCa cells [33] and survivin is known to promote angiogenesis [45–47], we also investigated changes in the levels of survivin in HMEC1 upon incubation with αvβ6-positive sEVs. Our data demonstrate that the expression of survivin protein is reduced in HMEC1 upon incubation with PC3-shβ6 sEVs compared to incubation with PC3-shCtrl sEVs (Figure 7b). This led us to investigate whether the BIRC5 (survivin) mRNA expression is altered in HMEC1 upon treatment with PC3 sEVs. The qPCR data show no significant differences in survivin mRNA levels in HMEC1 upon incubation with sEVs from PC3-shCtrl, -shβ5 or -shβ6 (data not shown). We also characterised PC3 sEVs for expression of survivin by IB. In IB, the sEVs from PC3 cells show enrichment of survivin, β5 integrin subunit (Figure 7c, right panel) along with β6 integrin subunit, sEV markers CD63, CD81 (Figure 7c, left panel), ALIX, TSG101 and CD9 (Figure 7c, right panel) compared to TCL (Figure 7c). sEVs did not express CANX (Figure 7c). We also observe the expression of survivin along with sEV markers (ALIX, TSG101, CD9, CD63 and CD81) in iodixanol density gradient fractions from PC3-derived sEV samples (Figure 7d). The ER marker CANX known to be absent in sEVs is not expressed in any of the 10 fractions (Figure 7d). Interestingly, the expression of survivin remains unaltered in PC3-shβ6 TCL compared to PC3-shCtrl TCL, whereas it is significantly reduced in sEVs from PC3-shβ6 cells compared to sEVs from PC3-shCtrl cells (Figure 7e).
These findings suggest that incubation of HMEC1 with PrCa cell-derived sEVs harbouring down-regulation of β6 integrin subunit and associated cargo may increase STAT1 signalling or decrease survivin expression to modulate the angiogenic potential of HMEC1.
αvβ6 integrin is expressed in blood vessels found in prostate cancer patient tissues
Based on our observation that αvβ6 integrin is transferred via PC3 sEVs to endothelial cells, we hypothesised that αvβ6 integrin might be expressed in blood vessels in human PrCa tissue specimens. To test this hypothesis, we performed IHC on serial sections from PrCa tissue specimens to evaluate the expression of αvβ6 integrin in blood vessels. The αvβ6 integrin is expressed in the epithelial compartment in 11 of 16 PrCa cases evaluated. Among the αvβ6 integrin-positive PrCa cases, the αvβ6 integrin is expressed in up to 75% of PrCa cells (3+ intensity). vWF or CD31, a marker of blood vessels, is expressed (3+ intensity) in blood vessels. Using vWF and CD31, we show that in three of 16 PrCa cases, αvβ6 integrin is detected in endothelial cells (Figure 8). All these three cases have a Gleason score 10 diagnosis. The cases diagnosed as Gleason scores 7, 8, 9 do not show expression of αvβ6 integrin in blood vessels. In 5 of 16 PrCa cases, both epithelial and endothelial cells are negative for expression of αvβ6 integrin (Fig. S5A). In 8 of 16 PrCa cases, αvβ6 integrin is expressed only in epithelial cells but not in endothelial cells (Fig. S5B). In conclusion, this finding that the αvβ6 integrin is in endothelial cells in PrCa patient samples is novel since αvβ6 integrin was previously known to be either expressed by cells of epithelial origin during conditions of injury, wound healing and cancer [48,49] or de novo induced in endothelial cells under conditions of infection and injury [22–24].
Figure 8.
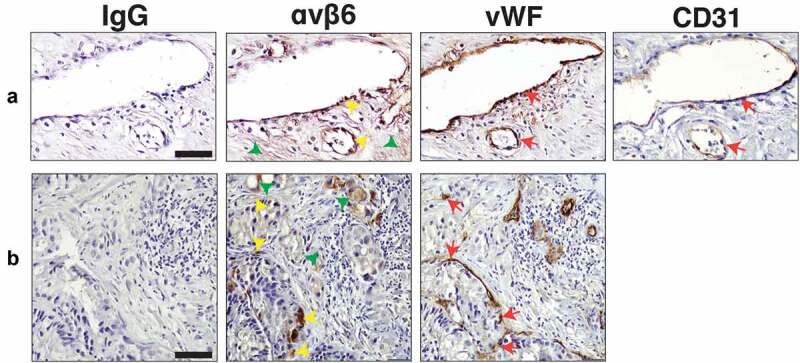
Expression of αvβ6 integrin, vWF and CD31 in blood vessels of human prostate cancer tissues.
Discussion
This is the first study that explores the functional impact of PrCa cell-derived sEVs that express αvβ6 integrin (αvβ6-positive sEVs) on endothelial cells in the TME. The present study demonstrates for the first time that the αvβ6 integrin protein is transferred by αvβ6-positive sEVs to endothelial cells and is functionally active in the recipient endothelial cells. Furthermore, this study also demonstrates that the αvβ6-positive sEVs promote microvascular endothelial cell motility and significantly increase tube formation of endothelial cells. Overall, the αvβ6-positive sEVs impact mechanistically endothelial cell functions.
We show for the first time that the αvβ6 integrin is also expressed in PC3 LEVs; however, in this study, our focus is on sEVs since a prior proteomic analysis of PC3 sEVs had shown that several aberrations follow down-regulation of αvβ6 integrin and had indicated that these aberrant sEVs might influence endothelial cell behaviour [19].
De novo induction of αvβ6 integrin expression in endothelial cells under conditions of infection and injury has been reported [22–24]. In human dermal microvascular endothelial cells and in tumour-associated endothelial cells purified from human breast carcinomas (B-TEC), oxytocin induces αvβ6 integrin expression [22]. Among other examples, exposure to lipopolysaccharide (LPS) enhances expression of αvβ6 integrin in a toll like receptor 4 (TLR)4-dependent manner in cardiac endothelial cells (CEC) [23]. Similarly, cytomegalovirus (CMV) infection induces αvβ6 integrin expression in both epithelial and endothelial cells of pulmonary, uterine and placental blood vessels at the sites of CMV infection-mediated injury [24]. Furthermore, αvβ6 integrin expressed on small vascular tufts during later stages of a healing skin wound has been assumed to impact remodelling of the vasculature [50]. In view of these previous studies, and on the fact that sEVs are known to carry mRNA as their cargo, we investigated the possibility of PrCa cell-derived αvβ6-sEVs-mediated β6 mRNA transfer or induction in HMEC1. We do not detect an increase of β6 mRNA in endothelial cells upon incubation with αvβ6-positive sEVs (Figure 2c,d), suggesting that β6 mRNA is neither transferred nor induced in endothelial cells. This finding also excludes the possibility that cytokines or growth factors in αvβ6-positive sEVs could be responsible for the induction of β6 integrin in endothelial cells.
Tumour angiogenesis involves multiple cellular processes, including endothelial cell proliferation, migration, extracellular matrix reorganisation and tube formation [42]. It is known that inactivation of the gene encoding for the β6 integrin subunit results in reduced keratinocyte migration [51]. The αvβ6 integrin transferred by sEVs could be crucial for the adhesion and migration of PrCa cells [17]. Similar to these previous studies, results from our motility assays suggest that αvβ6 and/or αvβ6-regulated cargo in αvβ6-positive sEVs drives migration of microvascular endothelial cells. Here, using in vitro tube formation assays that mimic angiogenesis [52], we present strong evidence that sEVs, derived from PrCa cells with down-regulated β6 integrin subunit, cause a highly significant reduction of capillary-like tube formation by endothelial cells (Figs. 4 & 5). In contrast, incubation of endothelial cells with sEVs from PC3 cells transfected with shRNA to another αv-binding subunit, β5, only slightly reduces tube formation, possibly by using the αvβ6 integrin that remains expressed in PC3-shβ5 sEVs.
We have demonstrated that siRNA-mediated down-regulation of β6 integrin subunit in PrCa cells results in increased STAT1 protein levels both in cells and sEVs derived from them [19]. STAT1 may act as a tumour suppressor in PrCa [19,53] and has been shown to be a negative regulator of angiogenesis [43,44]. We observe that uptake of αvβ6-positive sEVs led to down-regulation of pSTAT1(Y701) levels in microvascular endothelial cells (Figs. 6d & 7a). However, the total STAT1 levels remain unchanged in microvascular endothelial cells; we, therefore, propose a novel pathway that differed from the pathway identified in PrCa cells and their sEVs via our proteomic data [19]. The findings from this study are suggestive of different αvβ6-mediated regulatory mechanisms of STAT1 signalling in cancer cells versus microvascular endothelial cells. With respect to these STAT1 findings, the αvβ6 integrin is also known to be a major activator of TGF-β1 [24,54–56]. Also, the αvβ6 integrin interacts with TGFβ receptor II (TβRII) through the β6 cytoplasmic domain [38]. TGF-β1 plays a critical role in tumour angiogenesis [57–59] and the antagonistic effect of TGF-β1 on STAT1 signalling has previously been shown [60]. Therefore, we speculate that upon transfer from PrCa cell-derived sEVs to microvascular endothelial cells, αvβ6 integrin activates TGF-β1 leading to inhibition of STAT1 signalling and thus increased angiogenesis.
We have previously demonstrated that expression of αvβ6 integrin in C4-2B and LNCaP PrCa cells results in an androgen receptor-mediated increased expression of survivin [33]. We found that survivin is expressed in PC3 sEVs and its levels are significantly reduced in PC3-shβ6 sEVs; however, survivin levels are unaltered in PC3-shβ6 cells compared to PC3-shCtrl cells. This discrepancy of αvβ6 integrin not regulating survivin in PC3 cells might be attributed to the absence of androgen receptor in these cells. Survivin has been detected in sEVs derived from plasma of PrCa patients [61] and is known to be a critical mediator of angiogenesis [45–47]. This led us to hypothesise that the uptake of αvβ6-positive sEVs by microvascular endothelial cells may increase survivin levels in endothelial cells. We observe that mRNA levels of survivin are unaltered in microvascular endothelial cells upon incubation with αvβ6-positive sEVs, whereas survivin protein levels are increased. We conclude that survivin protein is transferred via αvβ6-positive sEVs to microvascular endothelial cells. We speculate that the lack of αvβ6 integrin in PC3 cells may inhibit the sorting of survivin in sEVs; this may result in a reduced transfer of survivin via PC3-shβ6 sEVs to microvascular endothelial cells. Furthermore, the antagonistic effect between STAT1 and survivin shown in gastric cancer cells and tissues [62] supports our evidence of an angiogenic signalling pathway mediated by the uptake of αvβ6-positive sEVs in microvascular endothelial cells.
In summary, our study demonstrates that by transferring αvβ6 integrin and/or potential angiogenic cargoes via sEVs from αvβ6-positive PrCa cells to recipient endothelial cells, angiogenic programmes are potently modulated in the recipient cells.
Supplementary Material
Acknowledgments
We would like to thank Dr. Eric B. Kmiec and Dr. Pawel A. Bialk from the Gene Editing Institute, Christiana Health Care System, for generating the cell lines with CRISPR/Cas9-mediated β6 integrin subunit down-regulation; Dr. Joseph A. Madri, Department of Pathology, Yale University School of Medicine, New Haven, CT, USA for BAEC; Dr. Michael Root at Thomas Jefferson University for refractometer; Dr. James Keen and Yolanda Covarrubias, Sidney Kimmel Cancer Center (SKCC) Bio-imaging facility at Thomas Jefferson University for support with confocal imaging; Dr. Lei Yu and Amir Yarmahmoodi, SKCC Flow Cytometry facility at Thomas Jefferson University for technical support with NTA and FACS experiments; Dr. Zhijiu Zhong, Translational Research/Pathology facility at Thomas Jefferson University for technical support with immunohistochemistry experiments; Cancer Genomics & Bioinformatics Core facility, led by Dr. Paolo Fortina at Thomas Jefferson University for technical support with q-PCR experiments; Dr. Mark Fortini and Jennifer Wilson at Thomas Jefferson University for editing the manuscript; Veronica Robles for administrative assistance with the preparation of the manuscript.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Author contributions
SRK and LRL conceptualised the study and designed experiments. SRK performed the experiments. IS generated Figure 1b and reviewed the manuscript. FQ assisted in performing an experiment and reviewed the manuscript. NN generated Figure 1a and reviewed the manuscript. EA assisted in performing an experiment and reviewed the manuscript. QL performed statistical analysis on data. SS and JK assisted in performing an experiment and reviewed the manuscript. PAM provided PrCa tissue sections, reviewed immunohistochemical staining and the manuscript. PHW and SV provided anti-αvβ6 integrin Abs (6.2A1 and 6.4B4) and reviewed the manuscript. SRK, DCA and LRL analysed results and wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s). Paul H. Weinreb is an employee and shareholder of Biogen. Shelia M. Violette is currently an employee of Admirx and, during this study, was an employee and shareholder of Biogen.
References
- [1].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2019. C.A. Cancer J. Clin. 2019;69(1):7–19. [DOI] [PubMed] [Google Scholar]
- [2].Hwang C, Heath EI. Angiogenesis inhibitors in the treatment of prostate cancer. J Hematol Oncol. 2010;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Weidner N, Carroll PR, Flax J, et al. Tumor angiogenesis correlates with metastasis in invasive prostate carcinoma. Am J Pathol. 1993;143(2):401–409. [PMC free article] [PubMed] [Google Scholar]
- [4].Salem KZ, Moschetta M, Sacco A, et al Exosomes in tumor angiogenesis. Methods Mol Biol. 2016;1464:25–34. [DOI] [PubMed] [Google Scholar]
- [5].Sato S, Vasaikar S, Eskaros A, et al EPHB2 carried on small extracellular vesicles induces tumor angiogenesis via activation of ephrin reverse signaling. JCI Insight. 2019;4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hoshino A, Costa-Silva B, Shen TL, et al Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kosaka N, Iguchi H, Hagiwara K, et al. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem. 2013;288(15):10849–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ludwig N, Whiteside TL. Potential roles of tumor-derived exosomes in angiogenesis. Expert Opin Ther Targets. 2018;22(5):409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Thery C, Witwer KW, Aikawa E, et al Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jimenez L, Yu H, McKenzie AJ, et al Quantitative proteomic analysis of small and large extracellular vesicles (EVs) Reveals Enrichment of Adhesion Proteins in Small EVs. J Proteome Res. 2019;18(3):947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kowal J, Arras G, Colombo M, et al Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mathieu M, Martin-Jaular L, Lavieu G, et al. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21(1):9–17. [DOI] [PubMed] [Google Scholar]
- [13].Bruno S, Chiabotto G, Favaro E, et al. Role of extracellular vesicles in stem cell biology. Am J Physiol Cell Physiol. 2019;317(2):C303–C13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lasser C, Jang SC, Lotvall J. Subpopulations of extracellular vesicles and their therapeutic potential. Mol Aspects Med. 2018;60:1–14. [DOI] [PubMed] [Google Scholar]
- [15].Spinelli C, Adnani L, Choi D, et al. Extracellular vesicles as conduits of non-coding RNA emission and intercellular transfer in brain tumors. Noncoding RNA. 2018;5:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].DeRita RM, Sayeed A, Garcia V, et al Tumor-derived extracellular vesicles require beta1 integrins to promote anchorage-independent growth. iScience. 2019;14:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fedele C, Singh A, Zerlanko BJ, et al. The alphavbeta6 integrin is transferred intercellularly via exosomes. J Biol Chem. 2015;290(8):4545–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Krishn SR, Singh A, Bowler N, et al Prostate cancer sheds the alphavbeta3 integrin in vivo through exosomes. Matrix Biol. 2019;77:41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu H, Bowler N, Harshyne LA, et al Exosomal alphavbeta6 integrin is required for monocyte M2 polarization in prostate cancer. Matrix Biol. 2018;70:20–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8(8):604–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Weis SM, Cheresh DA. alphaV integrins in angiogenesis and cancer. Cold Spring Harb Perspect Med. 2011;1(1):a006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cassoni P, Marrocco T, Bussolati B, et al Oxytocin induces proliferation and migration in immortalized human dermal microvascular endothelial cells and human breast tumor-derived endothelial cells. Mol Cancer Res. 2006;4(6):351–359. [DOI] [PubMed] [Google Scholar]
- [23].Song J, Chen X, Wang M, et al. Cardiac endothelial cell-derived exosomes induce specific regulatory B cells. Sci Rep. 2014;4:7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tabata T, Kawakatsu H, Maidji E, et al Induction of an epithelial integrin alphavbeta6 in human cytomegalovirus-infected endothelial cells leads to activation of transforming growth factor-beta1 and increased collagen production. Am J Pathol. 2008;172(4):1127–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Niu J, Li Z. The roles of integrin alphavbeta6 in cancer. Cancer Lett. 2017;403:128–137. [DOI] [PubMed] [Google Scholar]
- [26].Allen MD, Marshall JF, Jones JL. alphavbeta6 expression in myoepithelial cells: a novel marker for predicting DCIS progression with therapeutic potential. Cancer Res. 2014;74(21):5942–5947. [DOI] [PubMed] [Google Scholar]
- [27].Allen MD, Thomas GJ, Clark S, et al Altered microenvironment promotes progression of preinvasive breast cancer: myoepithelial expression of alphavbeta6 integrin in DCIS identifies high-risk patients and predicts recurrence. Clin Cancer Res. 2014;20(2):344–357. [DOI] [PubMed] [Google Scholar]
- [28].Moore KM, Thomas GJ, Duffy SW, et al Therapeutic targeting of integrin alphavbeta6 in breast cancer. J Natl Cancer Inst. 2014;106:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Elayadi AN, Samli KN, Prudkin L, et al A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007;67(12):5889–5895. [DOI] [PubMed] [Google Scholar]
- [30].Bates RC, Bellovin DI, Brown C, et al Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 2005;115(2):339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cantor DI, Cheruku HR, Nice EC, et al. Integrin alphavbeta6 sets the stage for colorectal cancer metastasis. Cancer Metastasis Rev. 2015;34(4):715–734. [DOI] [PubMed] [Google Scholar]
- [32].Dutta A, Li J, Lu H, et al Integrin alphavbeta6 promotes an osteolytic program in cancer cells by upregulating MMP2. Cancer Res. 2014;74(5):1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lu H, Wang T, Li J, et al alphavbeta6 integrin promotes castrate-resistant prostate cancer through JNK1-mediated activation of androgen receptor. Cancer Res. 2016;76(17):5163–5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Antonarakis ES, Carducci MA. Targeting angiogenesis for the treatment of prostate cancer. Expert Opin Ther Targets. 2012;16(4):365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bilusic M, Wong YN. Anti-angiogenesis in prostate cancer: knocked down but not out. Asian. J. Androl. 2014;16(3):372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Melegh Z, Oltean S. Targeting angiogenesis in prostate cancer. Int J Mol Sci. 2019;20:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sumpio BE, Yun S, Cordova AC, et al MAPKs (ERK1/2, p38) and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J Biol Chem. 2005;280(12):11185–11191. [DOI] [PubMed] [Google Scholar]
- [38].Dutta A, Li J, Fedele C, et al alphavbeta6 integrin is required for TGFbeta1-mediated matrix metalloproteinase2 expression. Biochem J. 2015;466(3):525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Weinreb PH, Simon KJ, Rayhorn P, et al Function-blocking integrin alphavbeta6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem. 2004;279(17):17875–17887. [DOI] [PubMed] [Google Scholar]
- [40].Crescitelli R, Lasser C, Jang SC, et al Subpopulations of extracellular vesicles from human metastatic melanoma tissue identified by quantitative proteomics after optimized isolation. J Extracell Vesicles. 2020;9(1):1722433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Minciacchi VR, You S, Spinelli C, et al Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget. 2015;6(13):11327–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol. 2002;282(5):C947–70. [DOI] [PubMed] [Google Scholar]
- [43].Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006;66(7):3649–3657. [DOI] [PubMed] [Google Scholar]
- [44].Huang S, Bucana CD, Van Arsdall M, et al. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 2002;21(16):2504–2512. [DOI] [PubMed] [Google Scholar]
- [45].Li Z, Ren W, Zeng Q, et al Effects of survivin on angiogenesis in vivo and in vitro. Am J Transl Res. 2016;8(2):270–283. [PMC free article] [PubMed] [Google Scholar]
- [46].O’Connor DS, Schechner JS, Adida C, et al Control of apoptosis during angiogenesis by survivin expression in endothelial cells. Am J Pathol. 2000;156(2):393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sanhueza C, Wehinger S, Castillo Bennett J, et al. The twisted survivin connection to angiogenesis. Mol Cancer. 2015;14:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Breuss JM, Gallo J, DeLisser HM, et al Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci. 1995;108(Pt 6):2241–2251. [DOI] [PubMed] [Google Scholar]
- [49].Koivisto L, Bi J, Hakkinen L, et al. Integrin alphavbeta6: structure, function and role in health and disease. Int. J. Biochem. Cell. Biol. 2018;99: 186–196. [DOI] [PubMed] [Google Scholar]
- [50].Christofidou-Solomidou M, Bridges M, Murphy GF, et al. Expression and function of endothelial cell alpha v integrin receptors in wound-induced human angiogenesis in human skin/SCID mice chimeras. Am J Pathol. 1997;151(4):975–983. [PMC free article] [PubMed] [Google Scholar]
- [51].Huang X, Wu J, Spong S, et al. The integrin alphavbeta6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J Cell Sci. 1998;111(Pt 15):2189–2195. [DOI] [PubMed] [Google Scholar]
- [52].DeCicco-Skinner KL, Henry GH, Cataisson C, et al Endothelial cell tube formation assay for the in vitro study of angiogenesis. J Vis Exp. 2014;91:e51312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hatziieremia S, Mohammed Z, McCall P, et al Loss of signal transducer and activator of transcription 1 is associated with prostate cancer recurrence. Mol Carcinog. 2016;55(11):1667–1677. [DOI] [PubMed] [Google Scholar]
- [54].Koth LL, Alex B, Hawgood S, et al Integrin beta6 mediates phospholipid and collectin homeostasis by activation of latent TGF-beta1. Am J Respir Cell Mol Biol. 2007;37(6):651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Munger JS, Huang X, Kawakatsu H, et al The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–328. [DOI] [PubMed] [Google Scholar]
- [56].Puthawala K, Hadjiangelis N, Jacoby SC, et al Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177(1):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ferrari G, Cook BD, Terushkin V, et al. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol. 2009;219(2):449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Iruela-Arispe ML, Sage EH. Endothelial cells exhibiting angiogenesis in vitro proliferate in response to TGF-beta 1. J Cell Biochem. 1993;52(4):414–430. [DOI] [PubMed] [Google Scholar]
- [59].Vinals F, Pouyssegur J. Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-alpha signaling. Mol Cell Biol. 2001;21(21):7218–7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Penafuerte C, Bautista-Lopez N, Bouchentouf M, et al. Novel TGF-beta antagonist inhibits tumor growth and angiogenesis by inducing IL-2 receptor-driven STAT1 activation. J Immunol. 2011;186(12):6933–6944. [DOI] [PubMed] [Google Scholar]
- [61].Stobiecka M, Ratajczak K, Jakiela S. Toward early cancer detection: focus on biosensing systems and biosensors for an anti-apoptotic protein survivin and survivin mRNA. Biosens Bioelectron. 2019;137:58–71. [DOI] [PubMed] [Google Scholar]
- [62].Deng H, Zhen H, Fu Z, et al. The antagonistic effect between STAT1 and survivin and its clinical significance in gastric cancer. Oncol Lett. 2012;3(1):193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.