

Abstract
Introduction of dynamic thiol-alkynone double addition cross-links in a polymer network enable the formation of a self-healing injectable polymer hydrogel. A four-arm polyethylene glycol (PEG) tetra-thiol star polymer is cross-linked by a small molecule alkynone via the thiol-alkynone double adduct to generate a hydrogel network under ambient aqueous conditions (buffer pH = 7.4 or 8.2, room temperature). The mechanical properties of these hydrogels can be easily tuned by varying the concentration of polymer precursors. Through the dynamic thiol-alkynone double addition cross-link, these hydrogels are self-healing and shear thinning, as demonstrated by rheological measurements, macroscopic self-healing, and injection tests. These hydrogels can be injected through a 20G syringe needle and recover after extrusion. In addition, good cytocompatibility of these hydrogels is confirmed by cytotoxicity test. This work shows the application of the thiol-alkynone double addition dynamic covalent chemistry in the straightforward preparation of self-healing injectable hydrogels, which may find future biomedical applications such as tissue engineering and drug delivery.
Polymer hydrogels are soft materials with many properties similar to those of biological tissue, leading to current and future applications as biomedical materials, for instance, in tissue engineering, wound dressing, and drug delivery.1−3 Where conventional hydrogels have a static, permanent network structure, dynamic or reversible, responsive hydrogels have recently attracted attention for application in the biomedical field, as well as in soft robotics.4 As a prime example of dynamic materials, self-healing injectable hydrogels show fascinating properties such as autonomous healing after damage and maintaining viscoelastic integrity after injection. These properties are highly important for biomedical applications such as minimally invasive implantation of cells and drug delivery vehicles.5−7 Currently, there are two general approaches to generate the dynamic interactions between hydrogel fibers or polymer chains for constructing self-healing injectable hydrogels: noncovalent bonds (e.g., hydrogen bonds, ionic bonds, host–guest interactions)8,9 and dynamic covalent bonds (e.g., boronic ester, Schiff base, and disulfide bonds).10−14 So far, a few examples of self-healing injectable hydrogels based on dynamic covalent bonds (DCBs) have been developed as well as applied in, for example, the repair of the central nervous system15 or the delivery of an antitumor drug.14 However, there are currently only a few types of dynamic covalent chemistry that meet the requirements for constructing self-healing injectable hydrogels. On one hand, some dynamic covalent bonds are only reversible under harsh conditions, impeding their application in hydrogels. For example, Diels–Alder reactions are generally only reversible at high temperatures,16 hydrazone formation and exchange requires an acidic environment.17 On the other hand, although some DCBs can be used in self-healing hydrogels, too complicated synthetic procedures for precursors or prepolymers may limit their application.11−14 Therefore, dynamic covalent chemistry that operates under ambient conditions, while allowing simple hydrogel preparation procedures, would be a valuable extension of the toolbox of reversible chemistry needed for the development of responsive hydrogel materials, as demonstrated here in the construction of a self-healing injectable hydrogel.
The reversible thiol-alkynone double conjugate addition is a recently developed dynamic covalent bond forming reaction, that now starts to find its way into some applications. Anslyn and co-workers investigated β-dithiane carbonyls (the thiol-alkynone double adduct) and β-sulfido-α,β-unsaturated carbonyls (the thiol-alkynone single adduct), demonstrating the reversibility of the thiol-alkynone double addition on small molecules.18 Based on this chemistry, applications such as dynamic combinational libraries, cleavage methods in peptide modification, and adaptable dynamic covalent polymer networks19−21 were reported in recent years. Previous studies20 found that the first addition between a thiol and a conjugated alkynone is an irreversible reaction, but the second step, the addition between the single adduct and a second thiol, is a reversible reaction (Figure 1a). Although the thiol-alkyne single addition has been applied in formation of hydrogel and polymer materials,22−24 so far there is no example of using the thiol-alkynone double addition as a reversible bond to tailor the properties of hydrogel materials. Other dynamic thiol-Michael approaches based on thiol–acrylate and thiol-benzalcyanoacetate additions have been applied in the construction of thermoresponsive polymer materials.25−28 Here, we propose the use of the thiol-alkynone double addition to construct a dynamic cross-linked hydrogel network from a four-arm polyethylene glycol (PEG) tetra-thiol star polymer and a small molecule alkynone as a cross-linker (Figure 1b). Rather than complicated synthesis of precursors, two commercially available materials are used directly to generate hydrogels at ambient conditions. The mechanical properties of the obtained hydrogels can be easily tuned by varying the concentrations of the network components. These hydrogels are self-healing, shear thinning, and can be injected through a medical syringe needle after which they spontaneously reform a gel. Importantly, such hydrogels exhibit dynamic viscoelastic behavior and biocompatibility, showing great potential in biomedical applications.
Figure 1.
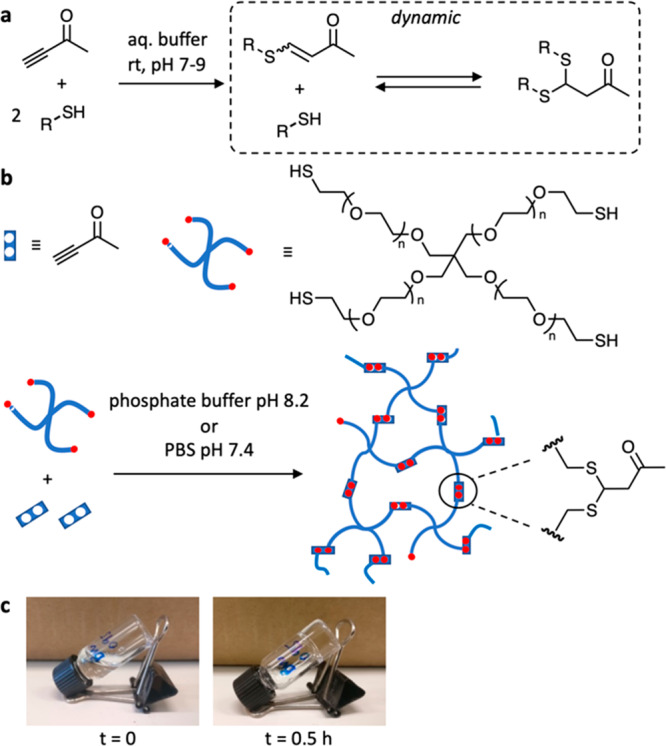
Gelation mechanism based on thiol-alkynone double conjugate addition. (a) The thiol-alkynone double conjugate addition: the first thiol addition is irreversible, generating a β-sulfido enone. The second thiol addition to the formed enone is reversible, generating a dynamic bond. (b) Schematic presentation of hydrogel formation via thiol-alkynone double addition, by cross-linking a tetra thiol star polymer with the alkynone. (c) A solution of thiol polymer and alkynone in PB8.2, at the start of the gelling process (left) and after 0.5 h (right), when a transparent gel has formed.
First, we used a reaction between low molecular weight model compounds to probe the feasibility of this reaction in mild aqueous conditions. 3-butyn-2-one (as alkynone) reacted with 2 equiv 2-mercaptoethanol to generate the mercaptoethanol-alkynone double adduct with 95% conversion after 1 h in sodium phosphate buffer (100 mM phosphate, pH = 8.2; “PB8.2”) at room temperature (Figure S1). We also tested the dynamic nature of the thiol-alkynone double addition by adding another small molecule thiol (sodium 2-mercaptoethanesulfonate) into the mercaptoethanol-alkynone double adduct PB8.2 solution. We observed a generation of the new thiol-alkynone double adduct and release of mercaptoethanol over the course of 5 h, monitored by 1H NMR, suggesting that the system was undergoing dynamic exchange in PB8.2 at room temperature (Figure S3). Then, using the same conditions, we investigated the use of a high molecular weight tetrathiol star polymer with 3-butyn-2-one as a low molecular weight cross-linker. Upon simply mixing 3-butyn-2-one PB8.2 solution (0.39 μL in 100 μL, 50 mM) and 4-arm PEG thiol PB8.2 solution (25 mg polymer (Mw = 10 kDa, Đ ≤ 1.05) in 150 μL, 16.7 wt %) in a 1:2 molar ratio of alkynone and thiol groups at room temperature, the storage modulus (G′) surpassed the loss modulus (G′′) ∼ 30 min after mixing the two solutions, indicating the formation of a hydrogel (Figure 2a). This process resulted in the formation of a transparent, colorless hydrogel with a G′ of 3.9 × 103 Pa and tan δ (G′′/G′) of 4.0 × 10–3 (10 wt % network content). We also determined the progress of single and double addition product formation in the hydrogel, in relation to gel formation, using 1H NMR spectroscopy and the tube-inversion method. 1H NMR showed the disappearance of alkynone together with the appearance and decrease of single adduct followed by the appearance and increase of double adduct on a time scale of minutes (single adduct) to hours (double adduct) (Figures S4 and S5). Gelation coincided with conversion to the double adduct (the cross-link) surpassing ∼60%.
Figure 2.
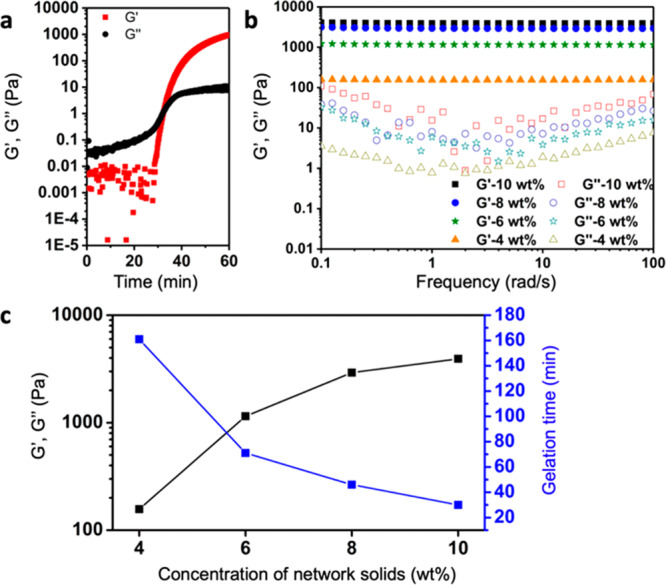
Rheological properties of hydrogels. (a) Time sweep measurement of the gelation process of a 10 wt % hydrogel (γ = 1%, ω = 1 Hz, 25 °C). (b) Frequency sweeps of the hydrogels with 4, 6, 8, and 10 wt % solid concentration (γ = 1%, ω = 0.1–100 rad/s, 25 °C). (c) The storage moduli (G′) and gelation time of hydrogels with solid concentration 4, 6, 8, and 10 wt %.
To investigate the influence of alkynone cross-linker on hydrogel formation we performed experiments varying the molar ratio of alkynone and thiol group (2:1, 1:1, 1:4, 1:8; Table S1). The vial-inversion method and rheological time sweep measurements were used for checking hydrogel formation and gelation time. At 2:1 and 1:1 alkynone/thiol ratios no hydrogel formation is observed, the mixtures remain liquid. Under these conditions, the excess of alkynone limits the reaction to the formation of the single addition product, which does not function as a cross-link in the material. At 1:4 and 1:8 alkynone/thiol ratios hydrogels did form, albeit on slightly longer time scales (∼50 min and ∼90 min, respectively) and lower final storage modulus (2.1 × 103 Pa and 1.2 × 103 Pa, respectively) than the 1:2 hydrogel (Figure S6). The low alkynone content likely leads to slower cross-linking and fewer cross-links formed, causing longer gelation times and gels with a lower final G′. In the following experiments, we set the ratio of alkynone and thiol groups to 1:2 to ensure efficient cross-linking in these hydrogels.
The influence of polymer content on the gelation and mechanical properties of these hydrogels was studied by rheological time and frequency sweep experiments on hydrogels with varying solid concentration (4–10 wt %; Figures 2b and S7). From Figure 2b it can be seen that for all hydrogels, G′ remained constant between 0.1 and 100 rad/s oscillatory frequency and was always larger than G′′, which indicates elastic behavior and a gel-like state of the samples. The gelation time and mechanical properties of these hydrogels show a dependence on polymer concentration. G′ increases from 1.6 × 102 Pa for the 4 wt % hydrogel to 3.9 × 103 Pa for the 10 wt % hydrogel and gelation times decrease from ∼160 min for the 4 wt % hydrogel to ∼30 min for the 10 wt % hydrogel (Figure 2c). Facile control over mechanical properties of hydrogels is desired for various tissue engineering and in vivo applications, where a match in mechanical strength between hydrogel and tissue is required.29
As phosphate buffered saline (PBS) is more relevant to physiological conditions, we also tested a 10 wt % hydrogel formed in PBS (100 mM, pH = 7.4, “PBS7.4”) for rheological behavior (Figure S8). Hydrogel formation in PBS (Gel-PBS7.4) takes 6.5 h, which is much longer than the 0.5 h gelation time in PB8.2 (Gel-PB8.2). The long gelation time in PBS7.4 may hinder application for cell encapsulation. However, the final storage modulus observed for the 10 wt % hydrogel formed in PB8.2 (G′ = 3.9 × 103 Pa) is similar to that of the hydrogel formed in PBS (G′ = 3.8 × 103 Pa), indicating a similar cross-link density. The origin of the difference in gelation rate in different buffer conditions was studied using small molecule model tests. The rate of double addition between small molecules 3-butyn-2-one (1 equiv) and 2-mercaptoethanol (2 equiv) in PB8.2 and PBS7.4 were monitored by 1H NMR (Scheme S4 and Figures S9–S11), which showed that the equilibration time of double addition in PBS7.4 (∼4 h) is much slower than in PB8.2 (∼1 h). Still, in both cases, a similar final conversion to double adduct was observed (∼95%). PB8.2 and PBS7.4 differ in pH and salt concentration. According to a previous report, the rate of thiol-alkynone double addition increases with pH,20 which explains the increased rate of gelation in PB8.2.
We investigated the response of these hydrogels to mechanical failure. Initially, a rheological strain sweep was measured on the 10 wt % Gel-PB8.2 to determine the critical strain value to disrupt the hydrogel network and induce a gel–sol transition. As can be seen in Figure 3a, G′ starts to decrease substantially when a strain over 80% is applied, showing the beginning of the nonlinear viscoelastic region. There is a crossover point of G′ and G′′ at the critical strain value of 212%. Next, a step strain measurement was performed on PB8.2 hydrogels, starting at 1% strain, then going to 300% strain, and back to 1% strain, with for each value a 1 min interval of constant strain. As illustrated by Figure 3b, when the hydrogel was subjected to 300% strain, G′ dropped from 3.9 × 103 Pa to 8 × 102 Pa. Tan δ (G′′/G′) became >1, suggesting the collapse of the hydrogel network and conversion to a viscous fluid state. Return to 1% strain led to a quick recovery of the initial G′ value and a tan δ < 1, which means the hydrogel network recovered immediately. Next, we performed a macroscopic self-healing test by reconnecting two pieces of hydrogel (Figures 3c and S14). Two disk shape hydrogels were stained with either fluorescein (yellow) or rhodamine B (red). Both were cut into equal halves using a scalpel. Two different color pieces were then pressed together at the side of the cut. After 15 min, the two pieces had reconnected and the integrated hydrogel could be lifted by a tweezer, bearing its own weight. After ∼10 h, the dyes diffused into the opposite piece, indicating the formation of a continuous gel structure. The crack visually disappeared over the course of 2 days. The self-healing properties of a 10 wt % Gel-PBS7.4 were also investigated by step strain measurement and macroscopic self-healing experiments (Figures S12 and S15). We observed macroscopic self-healing of cut gels as well as quick recovery after applying a 300% strain. Two notable differences were that, even at 300% strain, tan δ remained <1 and the gel recovered to only 88% of the initial G′ after removing the strain.
Figure 3.
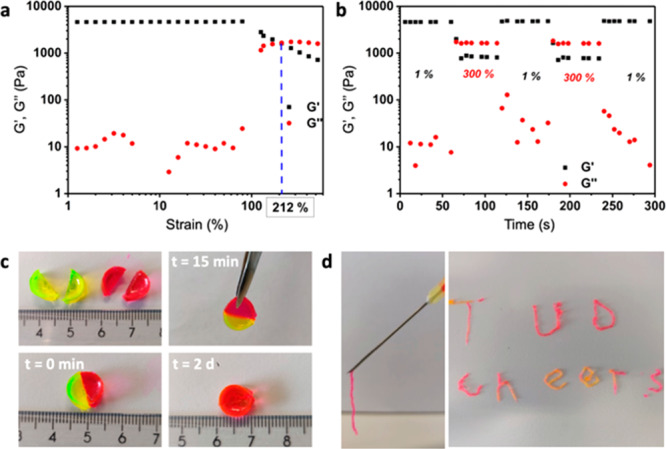
Self-healing and injectable properties of hydrogels. (a) Strain sweep of 10 wt % Gel-PB8.2. (b) Step strain measurement of 10 wt % Gel-PB8.2, strain is switched from 1% to 300% to 1%, for two cycles. (c) Macroscopic self-healing of 10 wt % Gel-PB8.2 (thickness: 4 mm; diameter: 9 mm). Two gel cylinders were made, each containing a dye (the yellow dye is fluorescein, the red dye is rhodamine B, and both are incorporated in the gel by mixing them into the gelling solution). Both were cut in half using a scalpel, and then one-half of each was pressed together with the other color gel. After 15 min, the two parts had adhered and could hold their own weight. After 2 days, the dyes diffused into the opposite piece and the crack visually disappeared. (d) Gel injection: immediate gel formation upon hand-pressing an 8 wt % Gel-PB8.2 through a 20G needle, leading to a 0.6 ± 0.2 mm diameter strip-shaped hydrogel. Fluorescein and rhodamine B dyes are added to the gel for visualization. The extruded Gel-PB8.2 allowed printing stable structures, in this case words with feature sizes on the mm scale.
Rheological viscosity-shear rate flow step measurements showed that these hydrogels are shear-thinning (Figure S13). The viscosity of a 10 wt % hydrogel (Gel-PB8.2) decreased with increasing shear rate, from 5.0 × 103 Pa·s at 0.1 s–1 to 0.13 × 103 Pa·s at 80 s–1. When a large shear stress is applied, the viscosity of a hydrogel will decrease and the hydrogel should show viscous flow through a needle. A 10 wt % hydrogel formed in PB8.2 was unable to pass through a 20G needle (0.9 mm diameter, sufficiently narrow for subcutaneous injections) probably due to its highly dense, cross-linked network. However, an 8 wt % hydrogel formed in PB8.2 and a 10 wt % hydrogel formed in PBS7.4 can both be successfully injected through a 20G syringe needle (Figures 3d and S15). The extruded hydrogel instantly recovers upon exiting the needle, as at that point the shear stress is removed. We demonstrated gel injection and recovery by writing letters made from 8 wt % Gel-PB8.2 passed through a 20G needle. This property of these hydrogels should also allow future 3D-printing to construct structured gel materials.30
The cytotoxicity of 10 wt % Gel-PB8.2 and Gel-PBS7.4 was evaluated using the live/dead staining assay with NIH/3T3 cells (mouse fibroblast cells). The cells were loaded on a piece of hydrogel and cocultured in the cell culture media at 37 °C in 5%/95% CO2/air atmosphere. After incubating for 48 h, the cell viability was checked. As shown in Figure 4a,b, the cell viability in the control experiment was around 98%, and only slightly lower viability was observed in both Gel-PB8.2 (93%) and Gel-PBS7.4 (94%). It indicates that the thiol-alkynone double addition hydrogels exhibit a good biocompatibility.
Figure 4.
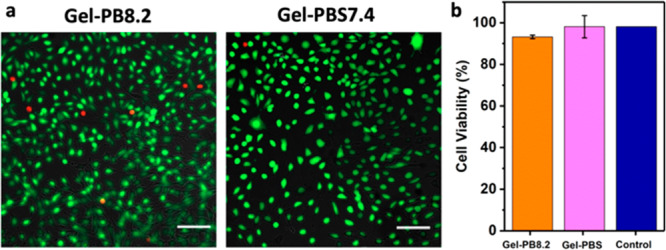
Biocompatibility of hydrogels. (a) Fluorescence microscopy images of NIH/3T3 cells after live/dead assay with calcein AM (green, live cells) and propidium iodide (red, dead cells). The cells were incubated together with Gel-PB8.2 (left) and Gel-PBS7.4 (right) around 48 h. Scale bar = 100 μm. (b) Cell viability of NIH/3T3 cells after incubating with Gel-PB8.2 and Gel-PBS7.4 and without hydrogel (control experiment). Error bars indicate the s.d. of three independent experiments.
In conclusion, we have developed a novel self-healing injectable hydrogel based on dynamic thiol-alkynone double addition chemistry. The thiol-alkynone double addition reaction enables facile synthesis of dynamic polymer hydrogels. The mechanical properties and gelation times are easily tuned by changing the concentration of polymer precursors during hydrogel preparation. Furthermore, the dynamic thiol-alkynone double addition endows shear-thinning and self-healing properties to these hydrogels, confirmed by rheological measurements and macroscopic self-healing and injection tests. As a result, these gels can be injected through a 20G needle, to afford stable gel objects upon extrusion. A 48 h cytotoxicity test confirmed a good biocompatibility of these hydrogels. In all, these self-healing, injectable hydrogels show promising potential in biomedical applications such as tissue engineering and drug delivery.
Acknowledgments
Financial support by the Chinese Scholarship Council (B.F.) and the European Research Council (R.E., ERC Consolidator Grant 726381) is acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmacrolett.0c00241.
Experimental details and supporting figures and tables (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Y. S.; Khademhosseini A. Advances in Engineering Hydrogels. Science 2017, 356, 3627. 10.1126/science.aaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talebian S.; Mehrali M.; Taebnia N.; Pennisi C. P.; Kadumudi F. B.; Foroughi J.; Hasany M.; Nikkhah M.; Akbari M.; Orive G.; Dolatshahi-Pirouz A. Self-Healing Hydrogels: The Next Paradigm Shift in Tissue Engineering?. Adv. Sci. 2019, 6, 1801664. 10.1002/advs.201801664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Mooney D. J. Designing Hydrogels for Controlled Drug Delivery. Nat. Rev. Mater. 2016, 1, 1–17. 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eelkema R.; Pich A. Pros and Cons: Supramolecular or Macromolecular: What Is Best for Functional Hydrogels with Advanced Properties?. Adv. Mater. 2020, 1906012. 10.1002/adma.201906012. [DOI] [PubMed] [Google Scholar]
- Tu Y.; Chen N.; Li C.; Liu H.; Zhu R.; Chen S.; Xiao Q.; Liu J.; Ramakrishna S.; He L. Advances in Injectable Self-Healing Biomedical Hydrogels. Acta Biomater. 2019, 90, 1–20. 10.1016/j.actbio.2019.03.057. [DOI] [PubMed] [Google Scholar]
- Lu H. D.; Charati M. B.; Kim I. L.; Burdick J. A. Injectable Shear-Thinning Hydrogels Engineered with a Self-Assembling Dock-and-Lock Mechanism. Biomaterials 2012, 33, 2145–2153. 10.1016/j.biomaterials.2011.11.076. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Yang J. H.; Zhou J.; Xu F.; Zrínyi M.; Dussault P. H.; Osada Y.; Chen Y. M. Self-Healing Gels Based on Constitutional Dynamic Chemistry and Their Potential Applications. Chem. Soc. Rev. 2014, 43, 8114–8131. 10.1039/C4CS00219A. [DOI] [PubMed] [Google Scholar]
- Phadke A.; Zhang C.; Arman B.; Hsu C.-C.; Mashelkar R. A.; Lele A. K.; Tauber M. J.; Arya G.; Varghese S. Rapid Self-Healing Hydrogels. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 4383–4388. 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. L.; in het Panhuis M. Self-Healing Hydrogels. Adv. Mater. 2016, 28, 9060–9093. 10.1002/adma.201601613. [DOI] [PubMed] [Google Scholar]
- Scheutz G. M.; Lessard J. J.; Sims M. B.; Sumerlin B. S. Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets. J. Am. Chem. Soc. 2019, 141, 16181–16196. 10.1021/jacs.9b07922. [DOI] [PubMed] [Google Scholar]
- Yesilyurt V.; Webber M. J.; Appel E. A.; Godwin C.; Langer R.; Anderson D. G. Injectable Self-Healing Glucose-Responsive Hydrogels with PH-Regulated Mechanical Properties. Adv. Mater. 2016, 28, 86–91. 10.1002/adma.201502902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Wang W.; Diaz-Dussan D.; Peng Y. Y.; Chen Y.; Narain R.; Hall D. G. In Situ Forming, Dual-Crosslink Network, Self-Healing Hydrogel Enabled by a Bioorthogonal Nopoldiol-Benzoxaborolate Click Reaction with a Wide PH Range. Chem. Mater. 2019, 31, 4092–4102. 10.1021/acs.chemmater.9b00769. [DOI] [Google Scholar]
- Wang W.; Xiang L.; Gong L.; Hu W.; Huang W.; Chen Y.; Asha A. B.; Srinivas S.; Chen L.; Narain R.; Zeng H. Injectable, Self-Healing Hydrogel with Tunable Optical, Mechanical, and Antimicrobial Properties. Chem. Mater. 2019, 31, 2366–2376. 10.1021/acs.chemmater.8b04803. [DOI] [Google Scholar]
- Li Y.; Yang L.; Zeng Y.; Wu Y.; Wei Y.; Tao L. Self-Healing Hydrogel with a Double Dynamic Network Comprising Imine and Borate Ester Linkages. Chem. Mater. 2019, 31, 5576–5583. 10.1021/acs.chemmater.9b01301. [DOI] [Google Scholar]
- Tseng T. C.; Tao L.; Hsieh F. Y.; Wei Y.; Chiu I. M.; Hsu S. H. An Injectable, Self-Healing Hydrogel to Repair the Central Nervous System. Adv. Mater. 2015, 27, 3518–3524. 10.1002/adma.201500762. [DOI] [PubMed] [Google Scholar]
- Adzima B. J.; Kloxin C. J.; Bowman C. N. Externally Triggered Healing of a Thermoreversible Covalent Network via Self-Limited Hysteresis Heating. Adv. Mater. 2010, 22, 2784–2787. 10.1002/adma.200904138. [DOI] [PubMed] [Google Scholar]
- Tan K. L.; Jacobsen E. N. Indium-Mediated Asymmetric Allylation of Acylhydrazones Using a Chiral Urea Catalyst. Angew. Chem., Int. Ed. 2007, 46, 1315–1317. 10.1002/anie.200603354. [DOI] [PubMed] [Google Scholar]
- Joshi G.; Anslyn E. V. Dynamic Thiol Exchange with β-Sulfido-α,β-Unsaturated Carbonyl Compounds and Dithianes. Org. Lett. 2012, 14, 4714–4717. 10.1021/ol301781u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matysiak B. M.; Nowak P.; Cvrtila I.; Pappas C.; Liu B.; Komaromy D.; Otto S. Antiparallel Dynamic Covalent Chemistries. J. Am. Chem. Soc. 2017, 139, 6744–6751. 10.1021/jacs.7b02575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu H. Y.; Chan T. C.; Ho C. M.; Liu Y.; Wong M. K.; Che C. M. Electron-Deficient Alkynes as Cleavable Reagents for the Modification of Cysteine-Containing Peptides in Aqueous Medium. Chem. - Eur. J. 2009, 15, 3839–3850. 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- Van Herck N.; Maes D.; Unal K.; Guerre M.; Winne J. M.; Du Prez F. E. Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol-Yne Cross-Linking. Angew. Chem., Int. Ed. 2020, 59, 3609–3617. 10.1002/anie.201912902. [DOI] [PubMed] [Google Scholar]
- Truong V. X.; Dove A. P. Organocatalytic, Regioselective Nucleophilic “Click” Addition of Thiols to Propiolic Acid Esters for Polymer-Polymer Coupling. Angew. Chem., Int. Ed. 2013, 52, 4132–4136. 10.1002/anie.201209239. [DOI] [PubMed] [Google Scholar]
- Macdougall L. J.; Pérez-Madrigal M. M.; Arno M. C.; Dove A. P. Nonswelling Thiol-Yne Cross-Linked Hydrogel Materials as Cytocompatible Soft Tissue Scaffolds. Biomacromolecules 2018, 19, 1378–1388. 10.1021/acs.biomac.7b01204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall L. J.; Truong V. X.; Dove A. P. Efficient in Situ Nucleophilic Thiol-Yne Click Chemistry for the Synthesis of Strong Hydrogel Materials with Tunable Properties. ACS Macro Lett. 2017, 6, 93–97. 10.1021/acsmacrolett.6b00857. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Digby Z. A.; Flum J. A.; Chakma P.; Saul J. M.; Sparks J. L.; Konkolewicz D. Dynamic Thiol-Michael Chemistry for Thermoresponsive Rehealable and Malleable Networks. Macromolecules 2016, 49, 6871–6878. 10.1021/acs.macromol.6b01061. [DOI] [Google Scholar]
- Kuhl N.; Geitner R.; Bose R. K.; Bode S.; Dietzek B.; Schmitt M.; Popp J.; Garcia S. J.; van der Zwaag S.; Schubert U. S.; Hager M. D. Self-Healing Polymer Networks Based on Reversible Michael Addition Reactions. Macromol. Chem. Phys. 2016, 217, 2541–2550. 10.1002/macp.201600353. [DOI] [Google Scholar]
- El-Zaatari B. M.; Ishibashi J. S.; Kalow J. A. Cross-Linker Control of Vitrimer Flow. Polym. Chem. 2020, 10.1039/D0PY00233J. [DOI] [Google Scholar]
- Herbert K. M.; Getty P. T.; Dolinski N. D.; Hertzog J. E.; de Jong D.; Lettow J. H.; Romulus J.; Onorato J. W.; Foster E. M.; Rowan S. J. Dynamic Reaction-Induced Phase Separation in Tunable, Adaptive Covalent Networks. Chem. Sci. 2020, 10.1039/D0SC00605J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher D. E.; Mooney D. J.; Zandstra P. W. Growth Factors, Matrices, and Forces Combine and Control Stem Cells. Science 2009, 324, 1673–1677. 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truby R. L.; Lewis J. A. Printing Soft Matter in Three Dimensions. Nature 2016, 540, 371–378. 10.1038/nature21003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.