

An efficient CRISPR-Cas9 editing tool based on a previous two-plasmid system was developed for Clostridium acetobutylicum and used to investigate the contribution of chromosomal butanol dehydrogenase genes during solventogenesis. Thanks to the control of cas9 expression by inducible promoters and of Cas9-guide RNA (gRNA) complex activity by an anti-CRISPR protein, this genetic tool allows relatively fast, precise, markerless, and iterative modifications in the genome of this bacterium and potentially of other bacterial species. As an example, scarless mutants in which up to three genes coding for alcohol dehydrogenases are inactivated were then constructed and characterized through fermentation assays. The results obtained show that in C. acetobutylicum, other enzymes than the well-known AdhE1 are crucial for the synthesis of alcohol and, more globally, to perform efficient solventogenesis.
KEYWORDS: anti-CRISPR, CRISPR-Cas9, Clostridium acetobutylicum, alcohol dehydrogenases, metabolic engineering
ABSTRACT
Although Clostridium acetobutylicum is the model organism for the study of acetone-butanol-ethanol (ABE) fermentation, its characterization has long been impeded by the lack of efficient genome editing tools. In particular, the contribution of alcohol dehydrogenases to solventogenesis in this bacterium has mostly been studied with the generation of single-gene deletion strains. In this study, the three butanol dehydrogenase-encoding genes located on the chromosome of the DSM 792 reference strain were deleted iteratively by using a recently developed CRISPR-Cas9 tool improved by using an anti-CRISPR protein-encoding gene, acrIIA4. Although the literature has previously shown that inactivation of either bdhA, bdhB, or bdhC had only moderate effects on the strain, this study shows that clean deletion of both bdhA and bdhB strongly impaired solvent production and that a triple mutant ΔbdhA ΔbdhB ΔbdhC was even more affected. Complementation experiments confirmed the key role of these enzymes and the capacity of each bdh copy to fully restore efficient ABE fermentation in the triple deletion strain.
IMPORTANCE An efficient CRISPR-Cas9 editing tool based on a previous two-plasmid system was developed for Clostridium acetobutylicum and used to investigate the contribution of chromosomal butanol dehydrogenase genes during solventogenesis. Thanks to the control of cas9 expression by inducible promoters and of Cas9-guide RNA (gRNA) complex activity by an anti-CRISPR protein, this genetic tool allows relatively fast, precise, markerless, and iterative modifications in the genome of this bacterium and potentially of other bacterial species. As an example, scarless mutants in which up to three genes coding for alcohol dehydrogenases are inactivated were then constructed and characterized through fermentation assays. The results obtained show that in C. acetobutylicum, other enzymes than the well-known AdhE1 are crucial for the synthesis of alcohol and, more globally, to perform efficient solventogenesis.
INTRODUCTION
Clostridium acetobutylicum has been established as the model organism for the study of acetone-butanol-ethanol (ABE) fermentation. The need for sustainable alternatives to fossil-based ways of producing these chemicals has renewed interest for this microorganism in the last few decades. Since C. acetobutylicum mainly produces alcohols, enzymes implicated in their production have been thoroughly studied.
In total, seven genes encoding enzymes with predicted alcohol dehydrogenase activity have been identified in the genome of C. acetobutylicum DSM 792 (also known as ATCC 824) (1). Genes CA_P0162 and CA_P0035 code for AdhE1 and AdhE2, respectively. Those enzymes contain both aldehyde dehydrogenase and alcohol dehydrogenase domains, although AdhE1 only retains aldehyde dehydrogenase activity (2). The five other identified genes (CA_C3298, CA_C3299, CA_C3375, CA_C3392, and CA_P0059) code for alcohol dehydrogenases (1). The contributions of the different enzymes involved in butanol flux have been recently characterized in C. acetobutylicum (2). During acidogenesis, the small amount of butyraldehyde produced by AdhE2 is reduced to butanol mainly by the BdhB dehydrogenase but also by AdhE2 and BdhA. During solventogenesis, most of the butanol flux depends on AdhE1, which converts butyryl coenzyme A (butyryl-CoA) to butyraldehyde, and on BdhB, which converts butyraldehyde to butanol (2).
Of the six enzymes with alcohol dehydrogenase activity identified, three (BdhA, BdhB, and BdhC, encoded by CA_C3299, CA_C3298, and CA_C3392, respectively) have been shown to be NADPH dependent, unlike AdhE2, which is NADH dependent (2). Mutants of strains ATCC 824 and DSM 1731 were constructed previously in two different studies (3, 4) in which the three corresponding genes were inactivated separately with ClosTron-based approaches (5); these studies showed that those genes have little or no contribution to solvent production. However, to date, we are not aware of any studies about the concomitant deletion of several copies of bdh genes.
In fact, difficulties in genetically modifying this bacterium, mainly because of poor transformability and low homologous recombination (HR) efficiencies (6), still limit a full understanding of its metabolism and physiology. To overcome this issue, genetic tools dedicated to the Clostridium genus in general and to C. acetobutylicum in particular have been developed in the recent years (7). Lately, several research groups working on Clostridium have developed tools based on CRISPR-Cas (8–14). Briefly, the Cas nuclease is guided to a target site by a so-called guide RNA (gRNA), where it generates a double-strand break (DSB). For most bacteria, including those belonging to the Clostridium genus, those DSBs are lethal when occurring in an essential nucleic acid, such as in the chromosome, and are repaired through HR when a template is available. Providing a template altering the target site will allow the bacteria to repair its genome and to prevent another DSB. The strength of the strategy is that the bacteria can survive only if the target site is modified through the allele exchange event using the provided editing template.
In some cases (11), endogenous CRISPR-Cas systems have been exploited, but since none of them has been identified in C. acetobutylicum strains so far, the heterologous Cas9 from Streptococcus pyogenes is, to date, the most commonly used nuclease in this bacterium. Given the high toxicity of Cas9 and the low transformation and HR frequencies in Clostridium, the use of constitutive promoters for the expression of cas9 and gRNA is generally not possible. In fact, only a few publications reported on the successful generation of mutants when using constitutively expressed nucleases and gRNA in this genus (8, 14). To alleviate this issue, several groups used Cas9n, a variant of Cas9 that catalyzes a single-strand break (SSB) instead of a DSB, which can be repaired by the cell and is therefore less toxic (9, 15). Ultimately, the cell will be protected against the nickase action only if the target site is modified through HR with the provided editing template. On one hand, it is therefore easier to obtain transformants but, on the other hand, mutants are generally isolated at lower frequencies and after subculturing, because of the less selective action of the nickase (7). Another strategy relies on the use of inducible promoters to drive the expression of cas9 and/or the gRNA (10, 12, 13). Once transformants in which cas9 expression is repressed have been obtained, it is possible to trigger the synthesis of the ribonucleoprotein complex through the addition of the inducer and to select edited cells.
Our group recently developed an inducible CRISPR-Cas9 tool for Clostridium based on the use of two plasmids (16). In this system, the cas9 gene is located on a first plasmid, pCas9ind, under the control of Pcm-tetO2/1, together with the tetR gene, whose product represses the transcription of cas9 in the absence of anhydrotetracycline (aTc) (17). While this first plasmid is unique and modification independent, the second plasmid, generically designated pGRNA, is target-specific and contains a gRNA expression cassette and the appropriate editing template. Both pCas9ind and pGRNA plasmids are introduced into the cell sequentially, and cas9 and gRNA expression is induced by aTc in transformants to select genome-edited mutants.
Although this system has been successfully used to target several genes in the genome of C. acetobutylicum, we report here on the difficulties encountered in obtaining transformants containing both pCas9 and pGRNA in some cases, most likely because of a basal leaky expression in the absence of the inducer of both the nuclease and the gRNA. To overcome this problem, we optimized pCas9ind by cloning a gene encoding the AcrIIA4 anti-Cas9 protein (18) under the control of a lactose-inducible promoter (19). Anti-Cas proteins have been discovered recently in CRISPR-Cas antagonists that have evolved inhibitors of those bacterial defense mechanisms (20, 21). The ability to regulate at a posttranslational level the action of Cas nucleases opens applications for these proteins in CRISPR-based genetic tools, in particular, for organisms with low transformation efficiencies (22). In this work, the use of AcrIIA4 allowed us to limit the undesired activity of the CRISPR-Cas system at the electroporation step, resulting in an increased transformation efficiency of pGRNA plasmids.
This refined tool allowed us to investigate the role of the NADPH-dependent alcohol dehydrogenase-encoding genes bdhA, bdhB, and bdhC in the industrially relevant C. acetobutylicum DSM 792. Mutants were generated in which one, two, or all of the corresponding genes were deleted and were further characterized in batch fermentation assays. Subsequently, mutants displaying stronger effects were complemented with plasmids carrying each bdh copy to confirm the phenotypes obtained and investigate the contribution of each gene to alcohol production.
RESULTS
Improvement of a CRISPR-Cas9 genetic tool.
To determine the contribution of BdhA, BdhB, and BdhC to butanol production in C. acetobutylicum DSM 792, we sought to create mutant strains using our CRISPR-Cas9 genetic tool (16). First, we implemented a protocol for the rapid replacement of the gRNA sequence in the targeting plasmids. A cassette containing the aTc-inducible promoter Pcm-2tetO1 (17) and the gRNA chimeric sequence (23), separated by a 38-nucleotide (nt) cassette containing two BsaI sites, was cloned into pEC750C, yielding pGRNAind. Upon restriction with BsaI, overhang extremities were generated, allowing the insertion of two hybridized single-strand oligonucleotides with extremities compatible with the linearized pGRNAind in order to generate a new target-specific plasmid (Fig. 1A). This simple modification allowed us to generate new pGRNA plasmids faster than through the cloning of a synthesized complete gRNA expression cassette, as previously done in our laboratory (16). We used this strategy to construct plasmids targeting bdhA, bdhB, and bdhC, designated pGRNA-bdhA, pGRNA-bdhB and pGRNA-bdhC, respectively.
FIG 1.

Easy reprogramming of the two-plasmid CRISPR-Cas9 genetic tool for multiple genome editing. (A) Retargeting of the pGRNA plasmid by cloning two hybridized primers between the aTc-inducible promoter Pcm-2tetO1 and the gRNA scaffold. (B) Insertion of the editing template within the pGRNA plasmid by restriction enzyme-based cloning. (C) Iterative genome modification. Step 1, the strain is transformed with pCas9acr and transformants are selected on erythromycin-containing medium. Step 2, the resulting strain is transformed with a pGRNA editing plasmid, and transformants are selected on medium containing erythromycin, thiamphenicol, and lactose to activate the transcription of acrIIA4. Step 3, expression of cas9 is triggered on medium containing erythromycin and thiamphenicol and devoid of lactose so that transcription of acrIIA4 is no longer induced. The medium contains aTc to trigger the transcription of cas9 to select mutants in which the editing event occurred. Step 4, once aTc-resistant colonies have been confirmed, pGRNA is curated on medium containing erythromycin and no thiamphenicol, yielding cells ready for a new round of modification. ori, replication origin for E. coli; pIP404 and repH, compatible replication origins for C. acetobutylicum; catP, thiamphenicol-chloramphenicol resistance gene; ermB, erythromycin resistance gene; RS, restriction site; RE, restriction enzyme; DSB, double-strand break; HR, homologous recombination.
The next step consisted of cloning of splicing by overhang extension (SOE) PCR products that can be used as an editing template for the reparation of the DSB created through the action of the Cas9-gRNA complex (Fig. 1B). Editing templates were constructed in order to delete most of the targeted genes so that only a few nucleotides at the extremities of the coding sequence were still present, leading to the synthesis of small peptides. These templates were cloned into the appropriate vectors, yielding pGRNA-ΔbdhA, pGRNA-ΔbdhB and pGRNA-ΔbdhC. Since bdhA and bdhB are located next to each other in the DSM 792 chromosome, one single editing template was designed for the deletion of both genes at once and cloned into pGRNA-bdhB to obtain pGRNA-ΔbdhA ΔbdhB.
Those plasmids were then introduced in C. acetobutylicum DSM 792 containing pCas9ind (further referred to as DSM 792 [pCas9ind]). Notably, most of the assays performed to introduce plasmids pGRNA-ΔbdhB or pGRNA-ΔbdhA ΔbdhB into this recombinant strain failed, as no transformants were obtained. We hypothesized that this result might be due to a toxic leaky expression of both the cas9 gene and the gRNA, despite their control by inducible promoters Pcm-tetO2/1 and Pcm-2tetO1, respectively, which would prevent the generation of transformants. We therefore modified the pCas9ind plasmid by inserting the acrIIA4 gene from Listeria monocytogenes under the control of Pbgal and the associated transcriptional repressor-encoding gene bgaR (19, 24), yielding pCas9acr. This modification was aimed at controlling the action of the Cas9-gRNA complex during the transformation step by the addition of lactose to the medium. Once transformants were obtained, the expression of the Cas9-gRNA complex was triggered with aTc on lactose-free medium. The procedure by which pCas9acr can be used in combination with pGRNA plasmids to create multiple mutations in the genome of DSM 792 is shown in Fig. 1C.
Transformation of pGRNA plasmids into DSM 792 (pCas9ind) and DSM 792 (pCas9acr) was further assessed using lactose-supplemented plates for the selection of transformants. Transformation efficiencies are shown in Fig. 2. Although plasmids pGRNA-ΔbdhA and pGRNA-ΔbdhC were introduced at similar rates in DSM 792 (pCas9ind) or DSM 792 (pCas9acr), the generation of transformants containing pGRNA-ΔbdhB and pGRNA-ΔbdhA ΔbdhB was greatly improved when using a strain containing pCas9acr instead of pCas9ind. Therefore, all further experiments were performed using DSM 792 (pCas9acr).
FIG 2.
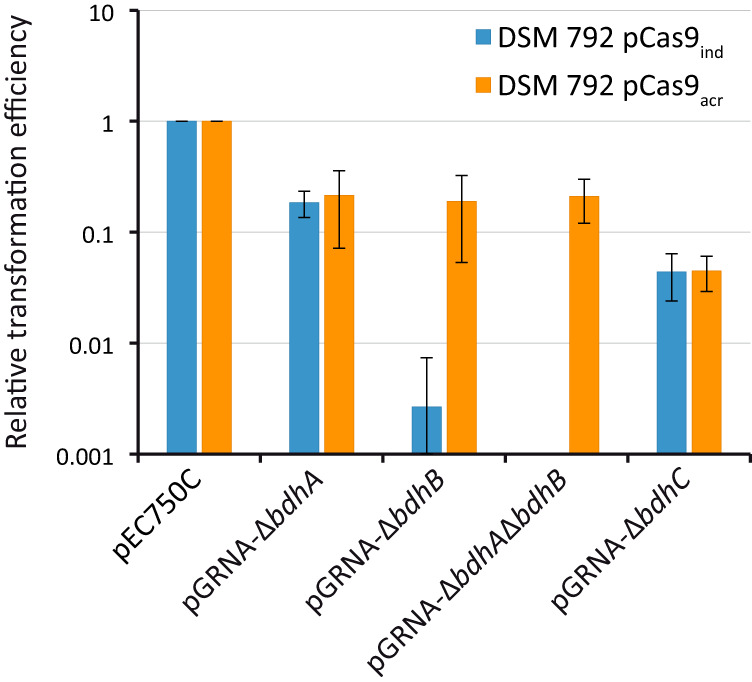
Relative transformation efficiencies of pGRNA template plasmids in DSM 792 containing pCas9ind or pCas9acr. Results shown are average values ± standard deviations from at least four independent replicates for each transformation. Absolute transformation efficiencies for pEC750C in DSM 792 (pCas9ind) and DSM 792 (pCas9acr) were 148 ± 37 CFU μg−1 and 121 ± 92 CFU μg−1, respectively.
Bdh mutant construction.
Once transformants containing pCas9acr and either pGRNA-ΔbdhA, pGRNA-ΔbdhB, pGRNA-ΔbdhC, or pGRNA-ΔbdhA ΔbdhB were obtained, expression of cas9 was induced as previously described (16). For each modification, at least three aTc-resistant colonies obtained from independent transformants were analyzed and confirmed by PCR, yielding the corresponding mutants that we stored for further experiments. As for the early version of our tool (16), the editing efficiency for every single modification was 100% (data not shown). To generate DSM 792 ΔbdhA ΔbdhC, DSM 792 ΔbdhB ΔbdhC, and DSM 792 ΔbdhA ΔbdhB ΔbdhC, one DSM 792 ΔbdhC mutant was randomly chosen (i.e., the first mutant obtained), streaked on yeast-tryptone-glucose (2YTG) plates devoid of thiamphenicol until pGRNA-ΔbdhC was cured, and further transformed with plasmids pGRNA-ΔbdhA, pGRNA-ΔbdhB, and pGRNA-ΔbdhA ΔbdhB, respectively. Once transformants were obtained, expression of cas9 was induced, and aTc-resistant colonies obtained from independent transformants were analyzed by PCR, confirmed, and saved for further experiments. For each type of deletion, six independent colonies were tested, and the editing efficiency for this new round of modification was again 100% (data not shown). PCR analyses of bdhA-bdhB and bdhC loci in each different mutant are shown in Fig. 3.
FIG 3.

CRISPR-Cas9 strategy and PCR analyses of representative deletion mutants constructed in this study. (A and B) bdh loci from DSM 792. Genes are shown in green, gRNA target sites are indicated in violet, and homology sequenced between gDNA and the editing templates is in blue. (A) bdhA-bdhB locus. (B) bdhC locus. (C) Amplification of the bdhA-bdhB and bdhC loci. Amplification with primer pair P30-P31 yields a PCR product of 5,173, 4,027, 4,024, or 2,578 bp in wild-type (WT), ΔbdhA, ΔbdhB, or ΔbdhA ΔbdhB strains, respectively. Amplification with primer pair P32-P33 yields a PCR product of 3,124 bp or 1,993 bp in WT or ΔbdhC strains, respectively. Lane M, 2-log DNA ladder (0.1 to 10 kb; NEB); lane H2O, amplification on water.
Phenotypic analysis.
Those mutants were then characterized by batch fermentation performed in flasks on Gapes medium enriched in ammonium acetate. For each type of mutation (i.e., deletion of one, two, or the three butanol dehydrogenase-encoding genes), the three randomly chosen independent mutants selected were assessed. Final fermentation products are shown in Table 1. After 72 h, the wild-type strain yielded a classical ABE product pattern composed of 22 to 23 g/liter of solvents and less than 5 g/liter of acids. No significant difference was observed for the single deletion mutants compared to the wild-type strain or for the ΔbdhA ΔbdhC and ΔbdhB ΔbdhC double mutants. On the contrary, final fermentation product concentrations were strongly affected in ΔbdhA ΔbdhB and ΔbdhA ΔbdhB ΔbdhC mutants. Lower solvent titers (7.4 and 4.2 g/liter, respectively) and higher acid concentrations (9.7 and 11.4 g/liter, respectively) were detected, resulting in lower final pH and consistent with a so-called “acid crash” event. Both ethanol and butanol productions were strongly decreased in those mutants, and final titers were lower than that of acetone, which became the main fermentation product in terms of solvents.
TABLE 1.
Fermentation products of the mutants analyzed in this studya
| Strain or genotype | Product (g/liter) |
Glucose consumption (g/liter) | Final pH | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solvents |
Acids |
|||||||||
| Ethanol | Acetone | Butanol | Total | Acetate | Butyrate | Lactate | Total | |||
| WT | 1.3 ± 0.1 | 9.0 ± 0.5 | 12.4 ± 1.4 | 22.8 ± 2.0 | 2.6 ± 0.1 | 2.4 ± 0.1 | NDb | 5.0 ± 0.2 | 69.2 ± 0.2 | 5.1 ± 0.0 |
| ΔbdhA | 1.2 ± 0.1 | 9.1 ± 0.4 | 11.6 ± 0.4 | 22.0 ± 0.1 | 2.4 ± 0.1 | 2.3 ± 0.1 | ND | 4.7 ± 0.0 | 69.4 ± 0.2 | 5.2 ± 0.0 |
| ΔbdhB | 0.9 ± 0.0 | 9.2 ± 0.2 | 12.1 ± 0.6 | 22.2 ± 0.6 | 2.2 ± 0.1 | 2.7 ± 0.1 | ND | 4.9 ± 0.2 | 71.0 ± 2.4 | 5.3 ± 0.0 |
| ΔbdhC | 1.3 ± 0.2 | 9.3 ± 0.3 | 12.8 ± 1.0 | 23.4 ± 1.4 | 2.6 ± 0.4 | 2.1 ± 0.5 | ND | 4.7 ± 0.9 | 71.4 ± 2.6 | 5.3 ± 0.1 |
| ΔbdhA ΔbdhB | 0.5 ± 0.0 | 3.8 ± 0.6 | 3.2 ± 0.7 | 7.4 ± 1.4 | 4.5 ± 0.4 | 5.0 ± 0.2 | 0.2 ± 0.1 | 9.7 ± 0.4 | 35.9 ± 2.2 | 4.8 ± 0.0 |
| ΔbdhA ΔbdhC | 1.1 ± 0.2 | 9.5 ± 0.5 | 13.3 ± 0.1 | 23.9 ± 0.4 | 2.2 ± 0.2 | 2.8 ± 0.4 | ND | 5.0 ± 0.2 | 72.0 ± 0.6 | 5.2 ± 0.0 |
| ΔbdhB ΔbdhC | 1.0 ± 0.1 | 9.3 ± 1.1 | 11.6 ± 0.7 | 21.9 ± 1.6 | 2.0 ± 0.2 | 2.9 ± 0.5 | ND | 4.9 ± 0.2 | 73.5 ± 1.8 | 5.3 ± 0.0 |
| ΔbdhA ΔbdhB ΔbdhC | 0.4 ± 0.0 | 2.5 ± 0.2 | 1.3 ± 0.2 | 4.2 ± 0.3 | 5.3 ± 0.3 | 5.9 ± 0.2 | 0.2 ± 0.1 | 11.4 ± 0.5 | 26.7 ± 1.8 | 4.7 ± 0.0 |
Results shown are average values ± standard deviations from at least three independent technical replicates. For mutant assays, values are from biological replicates.
ND, not detected.
Hypothesizing that the inability of the mutants to efficiently assimilate acids and convert them into solvents led to an irreversible pH decrease, we performed pH-controlled batch fermentations of the wild-type strain as well as ΔbdhA ΔbdhB and ΔbdhA ΔbdhB ΔbdhC mutants in a bioreactor (Table 2). Although solvent production of the mutants was slightly increased compared to that of flask fermentations, the final solvent concentrations were still strongly reduced compared to those of the wild-type strain. Unlike what was observed for flask fermentations, butanol was still the main solvent detected at the end of the fermentation. Interestingly, the major difference observed compared to flask fermentation was a stronger accumulation of lactate, reaching up to 2 g/liter in the pH-controlled bioreactor instead of 0.2 g/liter in flask assays.
TABLE 2.
Fermentation products in pH-controlled bioreactors
| Strain or genotype | Product (g/liter) |
Glucose consumption (g/liter) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Solvents |
Acids |
||||||||
| Ethanol | Acetone | Butanol | Total | Acetate | Butyrate | Lactate | Total | ||
| WT | 1.4 | 8.0 | 15.7 | 25.2 | 2.0 | 1.8 | NDa | 3.8 | 72.5 |
| ΔbdhA ΔbdhB | 0.3 | 3.1 | 5.0 | 8.4 | 4.3 | 4.3 | 1.8 | 10.4 | 41.9 |
| ΔbdhA ΔbdhB ΔbdhC | 0.2 | 1.9 | 2.8 | 4.9 | 4.9 | 4.9 | 2.0 | 11.8 | 36.8 |
ND, not detected.
Complementation assays.
To confirm that the deletion of bdh genes was responsible for the phenotypes observed, we complemented ΔbdhA ΔbdhB and ΔbdhA ΔbdhB ΔbdhC mutants with plasmids allowing the expression of each gene separately under the control of its native promoter. As a control, the wild-type strain was also transformed with those plasmids. For each complementation (i.e., each plasmid introduced in each strain), three randomly chosen transformants were analyzed through batch fermentation assays performed in flasks as performed previously, and final fermentation products are presented in Table 3. In the wild-type strain, overexpression of each gene had no significant effect compared to that in the presence of the empty control vector. However, the ΔbdhA ΔbdhB and ΔbdhA ΔbdhB ΔbdhC mutants assessed were successfully complemented with any of the bdh copies, without a significant difference in terms of the product pattern of selectivity. The two mutants assessed still underwent acid crash when transformed with an empty vector. Interestingly, even the phenotype of the ΔbdhA ΔbdhB mutant was reversed by the overexpression of bdhC, which led to fermentation performances in the range of those of the wild-type strain.
TABLE 3.
Complementation assaysa
| Strain or geneotype | Plasmid | Products (g/liter) |
Glucose consumption (g/liter) | Final pH | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solvents |
Acids |
||||||||||
| Ethanol | Acetone | Butanol | Total | Acetate | Butyrate | Lactate | Total | ||||
| WT | pFW01 | 1.8 ± 0.1 | 8.9 ± 0.4 | 13.3 ± 0.7 | 24.0 ± 1.1 | 2.2 ± 0.2 | 2.5 ± 0.1 | NDb | 4.7 ± 0.2 | 64.0 ± 1.3 | 5.3 ± 0.1 |
| pFW01-bdhA | 2.2 ± 0.1 | 9.9 ± 0.2 | 11.9 ± 0.6 | 24.0 ± 0.9 | 1.7 ± 0.3 | 1.4 ± 0.4 | ND | 3.2 ± 0.7 | 64.8 ± 1.5 | 5.3 ± 0.1 | |
| pFW01-bdhB | 1.6 ± 0.3 | 8.4 ± 1.1 | 12.1 ± 1.3 | 22.1 ± 2.6 | 2.3 ± 0.3 | 2.7 ± 0.2 | ND | 5.0 ± 0.5 | 61.5 ± 2.5 | 5.1 ± 0.0 | |
| pFW01-bdhC | 1.8 ± 0.4 | 9.6 ± 0.5 | 13.4 ± 0.7 | 24.8 ± 1.5 | 2.4 ± 0.1 | 2.3 ± 0.1 | ND | 4.7 ± 0.2 | 64.2 ± 0.6 | 5.2 ± 0.0 | |
| ΔbdhA ΔbdhB | pFW01 | 0.7 ± 0.0 | 2.6 ± 0.7 | 2.7 ± 0.8 | 6.1 ± 1.5 | 5.4 ± 0.4 | 4.1 ± 0.6 | 0.6 ± 0.1 | 10.1 ± 1.0 | 21.4 ± 3.4 | 4.7 ± 0.1 |
| pFW01-bdhA | 2.0 ± 0.1 | 10.6 ± 1.1 | 13.7 ± 0.8 | 26.3 ± 0.9 | 1.7 ± 0.5 | 1.3 ± 0.1 | ND | 3.0 ± 0.5 | 70.9 ± 2.2 | 5.5 ± 0.1 | |
| pFW01-bdhB | 1.8 ± 0.1 | 8.8 ± 0.5 | 13.6 ± 0.1 | 24.2 ± 0.3 | 2.1 ± 0.4 | 1.6 ± 0.5 | ND | 3.7 ± 0.9 | 68.7 ± 0.6 | 5.4 ± 0.3 | |
| pFW01-bdhC | 1.4 ± 0.1 | 10.2 ± 0.4 | 13.2 ± 0.7 | 24.8 ± 1.2 | 1.9 ± 0.1 | 2.5 ± 0.1 | ND | 4.4 ± 0.2 | 67.7 ± 1.0 | 5.4 ± 0.0 | |
| ΔbdhA ΔbdhB ΔbdhC | pFW01 | 0.5 ± 0.1 | 2.3 ± 0.3 | 2.1 ± 0.3 | 4.9 ± 0.6 | 5.7 ± 0.5 | 4.6 ± 0.4 | 0.6 ± 0.1 | 10.9 ± 1.0 | 18.8 ± 3.4 | 4.6 ± 0.0 |
| pFW01-bdhA | 1.4 ± 0.1 | 9.6 ± 0.3 | 13.2 ± 0.3 | 24.2 ± 0.6 | 1.9 ± 0.0 | 1.8 ± 0.1 | ND | 3.7 ± 0.2 | 68.6 ± 1.1 | 5.4 ± 0.1 | |
| pFW01-bdhB | 1.7 ± 0.2 | 8.7 ± 0.8 | 13.8 ± 0.7 | 24.2 ± 1.7 | 1.9 ± 0.0 | 1.7 ± 0.1 | ND | 3.6 ± 0.1 | 68.6 ± 1.2 | 5.5 ± 0.1 | |
| pFW01-bdhC | 1.2 ± 0.3 | 10.4 ± 0.8 | 12.3 ± 0.5 | 23.9 ± 0.7 | 1.8 ± 0.4 | 2.1 ± 1.1 | ND | 3.9 ± 1.5 | 67.4 ± 0.4 | 5.4 ± 0.1 | |
Results shown are average values ± standard deviations from at least three independent biological replicates.
ND, not detected.
DISCUSSION
Although studied for more than one century, the ABE metabolism of C. acetobutylicum is still only partially understood. At the molecular level, the role of the main solventogenic actors, the alcohol dehydrogenase genes, has only been investigated using single-gene deletion approaches due to a lack of efficient genome-editing tools. To overcome this issue, several dedicated tools have been developed in recent years, including those based on CRISPR-Cas approaches which proved their efficiency in other organisms. Although powerful, CRISPR-Cas-based genetic tools also have drawbacks. In Clostridium, the main one is the toxicity of the system, which makes it difficult to use because of the low transformation and HR frequencies observed in these bacteria. In some cases, Cas9n has been used instead of Cas9 because the toxicity of the latter is too high, even when placing the corresponding gene under the control of an inducible promoter (9, 15). However, the use of nickases can be laborious for the very same reason, i.e., because of a lowered toxicity that does not allow the elimination of all unmodified cells. In other words, introduction of the CRISPR-Cas machinery is facilitated when Cas9n is employed, but mutant selection is more efficient with the use of a native Cas9.
In this study, we intended to use a two-plasmid CRISPR-Cas tool recently developed to inactivate bdhA, bdhB, and bdhC, the three genes identified in the genome of C. acetobutylicum DSM 792 that code for NADPH-dependent alcohol dehydrogenases. We first failed in trying to generate DSM 792 ΔbdhB and DSM 792 ΔbdhA ΔbdhB mutants because we did not manage to obtain transformants containing both pCas9ind and the corresponding pGRNA plasmids. Although it cannot be ruled out that the transformation efficiency of pGRNA plasmids is affected by the editing template, we believe that it is most likely linked to the gRNA strength. In the example reported in this study, only one assay allowed us to obtain three transformants of DSM 792 (pCas9ind) containing pGRNA-ΔbdhB, after dozens of attempts. Similarly, no transformant was ever obtained when introducing pGRNA-ΔbdhA ΔbdhB in DSM 792 (pCas9ind). Obviously, an alternative to circumvent this problem would have been to change the gRNA sequence targeting bdhB, especially since we facilitated the retargeting of pGRNA plasmids compared to that with the early version of the tool (Fig. 1A). However, we also faced this toxicity problem with a few other targeting plasmids in our laboratory (data not reported), and we therefore wanted to modify our tool so that it would be possible to generate transformants with any pGRNA plasmid used. Therefore, we added extra control of the Cas9-gRNA complex nuclease activity through the use of a recently described anti-CRISPR protein (18). Interestingly, the transformation efficiency of pGRNA-ΔbdhA and pGRNA-ΔbdhC was not improved, suggesting that placing cas9 under the control of an inducible promoter was sufficient when using those gRNA sequences. However, transformation efficiencies with pGRNA-ΔbdhB and pGRNA-ΔbdhA ΔbdhB were greatly increased when DSM 792 (pCas9acr) was used, demonstrating that we effectively improved our tool. To the best of our knowledge, the use of anti-CRISPR proteins to realize CRISPR-based genome editing has been documented only in human cells (25) and in the archaeon Sulfolobus islandicus (26). Combining two antagonist activities controlled by inducible promoters, we constructed a fast, reliable, and efficient genome-editing tool. Although only deletions were performed in this study, the CRISPR/anti-CRISPR strategy can also be employed to perform insertion and single nucleotide modification as shown with the first version of the tool (16), without the presence of any unwanted scar or antibiotic resistance marker in the genome, and can therefore be used to perform several rounds of modification.
Two studies previously analyzed the role of the genes investigated in this study through the construction of single-gene deletion mutants obtained with ClosTron (5), yielding slightly different outcomes (3, 4). In the first of these studies (3), bdhA and bdhB were successfully inactivated but the corresponding strains Cac-bdhA459s::CT and Cac-bdhB475a::CT were not significantly different from the wild-type strain, in accordance with our results. The deletion of bdhC was not investigated in this work (3). In a more recent study (4), BdhB was shown to be relevant for alcohol production, since the corresponding mutant had lower titers of butanol and ethanol. Moreover, BdhC (referred to as YqhD) was shown to be involved in ethanol production following the observation that a ΔyqhD mutant had a lower final ethanol titer but was not affected in terms of butanol production (4). Those results contrast with ours, since we did not observe any significant effect of either bdhB or bdhC deletion on ethanol and butanol production. Similarly, the deletion of bdhA had no significant effect, as reported in both previous studies (3, 4).
Despite the fact that the ClosTron has revolutionized the genetics of Clostridium by allowing fast inactivation of genes in bacteria which were difficult to modify, it suffers from some limitations. First, it has been shown that the disruption of a gene rather than its clean deletion can trigger polar effects, which could explain the differences observed between those two studies concerning the importance of BdhB. Moreover, another limitation of the ClosTron is that the inactivation of a gene requires its disruption with an intron that contains an antibiotic resistance marker, which therefore limits the number of modifications than can be performed in the recombinant strain. For this last reason, the two studies cited above (3, 4) focused on the creation of single-gene deletion strains. Using our improved CRISPR-Cas9 genetic tool, single, double, and triple marker-free insertion-free mutants in which targeted genes were cleanly deleted were obtained. The high editing efficiency of the tool allowed us to generate numerous independent mutants, providing a higher confidence in the fermentation results obtained.
Results obtained in this study suggest that none of the bdh genes are essential for solvent production. Indeed, every single deletion mutant retained the ability to efficiently produce solvents under the conditions tested, without clear differences in the final product patterns. Therefore, it can be hypothesized that the two remaining bdh copies allow the synthesis of a sufficient Bdh pool in the cell for the efficient production of solvents. This result is in accordance with most of the conclusions drawn from previous studies, although the lower ethanol production in the ΔbdhC mutant and the diminished production of both ethanol and butanol in the ΔbdhB mutant were not observed, unlike what was shown in one of those studies (4). Interestingly, we observed that the deletion of several bdh copies had strong effects on the solvent production capacities of C. acetobutylicum. In particular, bdhA and bdhB apparently have a greater role in solventogenesis than bdhC, since ΔbdhA ΔbdhB is the only double mutant with a significantly affected fermentation product pattern compared to that of the wild-type strain.
These results are in accordance with a recent study in which transcriptomic and proteomic data were obtained from phosphate-limited chemostat cultures of C. acetobutylicum maintained in different metabolic states (2). Interestingly, the data collected indicate that all three genes are expressed throughout fermentation, unlike genes from the sol operon that are mainly expressed during solventogenesis. Transcription of bdhA does not vary between acidogenic and solventogenic phases, unlike the transcription of bdhC, which slightly increases, and transcription of bdhB, which is 73% higher. During solventogenesis, transcripts of bdhA, bdhB, and bdhC represent 17%, 59%, and 24% of the bdh mRNA pool, respectively. Protein quantification indicates that BdhB represents 62% of the Bdh pool in acidogenesis, followed by BdhA (27%) and BdhC (11%). Increased transcription of bdhB in solventogenesis results in an increase of BdhB in the pool of Bdh enzymes (83%) compared to BdhA (11%) and BdhC (6%). Activities of the purified enzymes were also reported in the same publication and show comparable butanol dehydrogenase activities for all enzymes (2).
Taken together, these data suggest that BdhB is the main butanol dehydrogenase. A smaller quantity of enzymes is transcribed from the bdhA gene, although it represents one quarter of the Bdh enzyme pool during acetogenesis. Our results are in accordance with those data, since the fact that a ΔbdhA ΔbdhB mutant cannot perform an efficient solventogenesis while other double mutants can indicates that BdhA and BdhB have a more important role than BdhC. Interestingly, a ΔbdhB mutant is still capable of performing efficient fermentation, suggesting that the two remaining genes can compensate the loss of this copy, providing an explanation why so many copies of the same gene can be found in the genome. As intuited, the ΔbdhA ΔbdhB ΔbdhC triple mutant was even more heavily affected than the ΔbdhA ΔbdhB mutant, although neither butanol nor ethanol production was abolished, suggesting that other enzymes are still capable of compensating for the lower alcohol dehydrogenase activity in the mutant, most likely AdhE2 and/or the products of CA_C3375 and CA_P0059.
Notably, fermentations performed in pH-controlled bioreactors did not prevent acid accumulation, showing the mutant's incapacity to reduce acids. Instead, the triple mutant produced a significant amount of lactate under these conditions, most likely to regenerate its NAD(P)+ pool, which was no longer produced by Bdh enzymes. Interestingly, although final solvent titers were lower than for the wild-type strain, a classical ratio of ethanol-acetone-butanol was obtained, unlike what was observed in flask fermentation assays.
Complementation assays showed that any bdh copy is capable of reverting the acid crash phenotype of ΔbdhA ΔbdhB and ΔbdhA ΔbdhB ΔbdhC mutants in non-pH-controlled flask fermentations. Although those genes have been introduced in the cells under the control of their native promoter, the plasmid copy number most probably allows their overexpression. In fact, previous complementation assays performed with the same plasmid in Clostridium beijerinckii DSM 6423 resulted in an 80- to 90-fold overexpression compared to expression from the gene located on the chromosome of the bacterium (27). Interestingly, while a chromosomal bdhC is not sufficient to prevent acid crash, its overexpression in ΔbdhA ΔbdhB and ΔbdhA ΔbdhB ΔbdhC mutants results in efficient fermentation and highlights the crucial influence of the expression level of a particular gene for the whole metabolism of the microorganism. It is also interesting to point out that expression of either bdhA, bdhB, or bdhC in the ΔbdhA ΔbdhB ΔbdhC mutant had no strong impact on the ethanol/butanol ratio, suggesting that the corresponding enzymes are all capable of efficiently reducing acetaldehyde in ethanol and butyraldehyde in butanol. A better strategy to optimize the ethanol/butanol ratio could be to modify the selectivity of AdhE1 and AdhE2 for acetaldehyde and butyraldehyde, as recently attempted (28).
In summary, the improvement of our CRISPR-Cas9 genetic tool dedicated to C. acetobutylicum through the control of the Cas9/gRNA activity using the anti-CRISPR protein AcrIIA4 will allow us to step up the construction of clean mutants in order to enhance our understanding of the molecular physiology of this industrially relevant microorganism and other microorganisms belonging to the Clostridium genus. As an example, it allowed us to investigate the importance of the main butanol dehydrogenases of C. acetobutylicum DSM 792. In fact, while previous studies only focused on single gene deletions that did not allow the respective authors to figure out the major role of these enzymes, our tool allowed us to construct multiple deletion mutants and to highlight the major role of BdhA and BdhB in solvent production as well as the capacity of BdhC to compensate for the absence of the other Bdhs in a triple mutant strain. The results obtained indicate that efficient alcohol production in C. acetobutylicum DSM 792 requires a pool of enzymes with butanol dehydrogenase activity, which can be synthesized from any of the three genes investigated in this study. Butanol dehydrogenase activity is still observed in the triple deletion mutant, suggesting that the products of other genes not investigated in this study, most probably AdhE2, CA_C3375, and CA_P0059, are also part of this pool, although not in quantities sufficient to avoid an acid crash. This observation might explain why solventogenic strains carry so many alcohol dehydrogenase-encoding genes on their chromosome. In conclusion, efficient metabolic engineering in Clostridium in order to modify the fermentation product pattern requires editing of numerous genes that can be achieved with iterative and precise genetic tools, such as the one described in this study.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Relevant characteristics of the bacterial strains and plasmids used in this study are listed in Table 4. C. acetobutylicum was grown anaerobically at 34°C in liquid 2YTG medium (16 g liter−1 tryptone, 10 g liter−1 yeast extract, 4 g liter−1 NaCl, and 5 g liter−1 glucose) or on solid 2YTG with 1.5% agar, supplemented with 40 μg ml−1 erythromycin and/or 15 μg ml−1 thiamphenicol, if necessary. Escherichia coli was grown aerobically at 37°C and 200 rpm in liquid LB medium or solid LB with 1.5% agar supplemented with erythromycin (500 μg ml−1 for solid medium and 100 μg ml−1 for liquid medium), chloramphenicol (25 μg ml−1 for solid medium and 12.5 μg ml−1 for liquid medium), or tetracycline (20 μg ml−1) if necessary.
TABLE 4.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| C. acetobutylicum DSM 792 | Wild type | DSMZ |
| E. coli NEB 10-beta | Cloning strain | NEB |
| Plasmids | ||
| pAN2 | tetA, Φ3T I gene, p15A origin | 5, 30 |
| pFW01 | ermB, ColE1 origin, pCB102 origin | 16 |
| pCas9ind | ermB, ColE1 origin, pCB102 origin, cas9 (Pcm-tetO2/1 promoter), tetR | 16 |
| pCas9acr | pCas9ind derivative with acrIIA4 (Pbgal promoter) and bgaR insertions | This study |
| pEC750C | catP, ColE1 origin, pIP404 origin | 16 |
| pGRNAind | pEC750C derivative with gRNA expression cassette (Pcm-2tetO1 promoter) insertion | This study |
| pGRNA-bdhA | pGRNAind derivative targeting bdhA | This study |
| pGRNA-ΔbdhA | pGRNA-bdhA derivative with ΔbdhA editing template insertion | This study |
| pGRNA-bdhB | pGRNAind derivative targeting bdhB | This study |
| pGRNA-ΔbdhB | pGRNA-bdhB derivative with ΔbdhB editing template insertion | This study |
| pGRNA-ΔbdhA ΔbdhB | pGRNA-bdhB derivative with ΔbdhA ΔbdhB editing template insertion | This study |
| pGRNA-bdhC | pGRNAind derivative targeting bdhC | This study |
| pGRNA-ΔbdhC | pGRNA-bdhC derivative with ΔbdhC editing template insertion | This study |
| pFW01-bdhA | pFW01 derivative with bdhA insertion | This study |
| pFW01-bdhB | pFW01 derivative with bdhB insertion | This study |
| pFW01-bdhC | pFW01 derivative with bdhC insertion | This study |
DNA manipulation and plasmid construction.
All enzymes used for DNA modification were purchased from New England BioLabs (NEB). The Q5 high-fidelity polymerase (NEB) was used for all PCR amplifications, except for colony PCRs, which were performed using OneTaq DNA polymerase (NEB). Genomic DNA (gDNA) was extracted from C. acetobutylicum using the ChargeSwitch gDNA Mini bacteria kit (Thermo Fisher Scientific). Plasmids and PCR product purification kits were purchased from Qiagen. All primers used in this study are listed in Table 5.
TABLE 5.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′→3′) |
|---|---|
| P1 | CAGATTGTACTGAGAGTGCACC |
| P2 | GTGAGCGGATAACAATTTCACAC |
| P3 | TCATGCACTTAACTCGTGTTCCAT |
| P4 | AAACATGGAACACGAGTTAAGTGC |
| P5 | TCATGCTTATTACGACATAACACA |
| P6 | AAACTGTGTTATGTCGTAATAAGC |
| P7 | TCATGCTCTTGTATCATAGTCCGT |
| P8 | AAACACGGACTATGATACAAGAGC |
| P9 | AAAAAAGGATCCTTAGGAGCCATATCTGGATG |
| P10 | TATGCTAAGTTTTAAATCTTATTAATAGAAACTGTAGAGG |
| P11 | TTAATAAGATTTAAAACTTAGCATACTTCTTACC |
| P12 | AAAAAAGTCGACCTTCTAATCTCCTCTACTATTTTAG |
| P13 | ATGCATGGATCCAAACGAACCCAAAAAGAAAGTTTC |
| P14 | GGTTGATTTCAAATCTGTGTAAACCTACCG |
| P15 | ACACAGATTTGAAATCAACCACTTTAACCC |
| P16 | ATGCATGTCGACTCTTAAGAACATGTATAAAGTATGG |
| P17 | GCTAAGTTTTAAATCTGTGTAAACCTACCG |
| P18 | ACACAGATTTAAAACTTAGCATACTTCTTACC |
| P19 | ATGCATGTCGACCTTCTAATCTCCTCTACTATTTTAG |
| P20 | AAAAAACTCGAGCTATAAATAATATTACCCCCATAACTG |
| P21 | ATAATTTTGATGAGGGTGCAATGTAAGTTTG |
| P22 | TTGCACCCTCATCAAAATTATACATAAGATTATCCC |
| P23 | AAAAAATCTAGAGCAAAGGAATTAGTAGAAATTAC |
| P24 | AAAAAACTCGAGTTTAACCCCTCCTGTTTAGATTAT |
| P25 | AAAAAAGTCGACCGAAAAATTCACCCCCTCAA |
| P26 | AAAAAACTCGAGAAAATACTCCCCAAGATATTAATGCT |
| P27 | AAAAAAGTCGACTAGAAACTGTAGAGGTATTTTTATAATTTAAAAG |
| P28 | AAAAAACTCGAGTTTCTCCACCTTTAATACTAAAATATTTTTCA |
| P29 | AAAAAAGTCGACAAAAATACCCCACTATTCTAATTTTATTCA |
| P30 | AGTACCTCCTAAGCCTTTTTATGA |
| P31 | TATGGATTGCCCTACAGCCC |
| P32 | CCACTGCCACCATTTTCTAGC |
| P33 | CAAGGACAGATAGGTGGGGC |
A fragment containing bgaR and a codon-optimized acrIIA4 gene from L. monocytogenes under the control of Pbgal was synthesized and cloned into the pEX-K4 vector (Eurofins Genomics). The whole cassette was amplified with primers P01 and P02, digested with SacI, and cloned into pCas9ind linearized with the same restriction enzyme, yielding pCas9acr. A fragment containing the Pcm-2tetO1 promoter fused with the chimeric gRNA sequence, separated by a 38-nt spacer containing two BsaI restriction sites, was synthesized and cloned into the pEX-K4 vector (Eurofins Genomics). This cassette was amplified with primers P01 and P02, digested with SacI, and cloned into pEC750C, yielding pGRNAind. Sequences of the synthesized fragments are listed in the supplemental material.
The gRNA target sites were chosen using Geneious R10.2 (Biomatters, Ltd., Aukland, New Zealand), which provides for each CRISPR-Cas9 potential target site activity scores (on-target activity, ranging from 0 to 1) predicted with an algorithm (29), and specificity scoring, i.e., the probability that the gRNA will not target another location in the genome (off-target activity, ranging from 0% to 100%, with a higher score meaning better specificity and less off-target activity). Target sites were chosen so that the on-target score was superior to 0.2 and the off-target score was superior to 98% when up to five mismatches, including one insertion or deletion, were allowed to detect off-targets. The pGRNA-bdhA, pGRNA-bdhB, and pGRNA-bdhC plasmids were constructed by ligating the hybridization product of 5′ P-modified oligonucleotide couples P3/P4, P5/P6, and P7/P8, respectively, into the dephosphorylated BsaI-digested pGRNAind. Briefly, hybridization was performed in a thermocycler according to the following protocol: 500 nmol of each primer was mixed in a 10-μl reaction mixture containing 1 mM Tris-HCl, denatured at 95°C during 30 s, and further hybridized lowering the temperature 1°C every 5 s 70 times.
The pGRNA-ΔbdhA plasmid was constructed by cloning the amplicon, obtained using a splicing by overhang extension (SOE) PCR with primer pairs P9/P10 and P11/P12 with DSM 792 gDNA, into pGRNA-bdhA digested with BamHI and SalI. The pGRNA-ΔbdhB plasmid was constructed by cloning the SOE PCR product obtained using primer pairs P13/P14 and P15/P16 with DSM 792 gDNA into pGRNA-bdhB digested with BamHI and SalI. Similarly, the pGRNA-ΔbdhA ΔbdhB plasmid was constructed by cloning the SOE PCR product obtained using primer pairs P13/P17 and P18/P19 with DSM 792 gDNA into the same BamHI-SalI doubly digested pGRNA-bdhB. The pGRNA-ΔbdhC plasmid was constructed by cloning the SOE PCR product obtained using primer pairs P20/P21 and P22/P23 with DSM 792 gDNA into pGRNA-bdhC digested with XhoI and XbaI.
For complementation assays, plasmids pFW01-bdhA, pFW01-bdhB, and pFW01-bdhC were constructed by cloning the PCR products obtained with primer pairs P24/P25, P26/P27, and P28/P29, respectively, into pFW01 using XhoI and SalI restriction enzymes.
Transformation, induction of cas9 expression, and mutant confirmation.
Plasmids were transformed into chemically competent NEB 10-beta competent E. coli cells containing pAN2 (5) for DNA methylation. The vectors were then isolated, and 1 μg was used for electroporation of C. acetobutylicum DSM 792 as previously described (30). Cells of DSM 792 containing either pCas9ind or pCas9acr were further transformed with pEC750C and derivative plasmids (i.e., plasmids containing a gRNA expression cassette with or without editing template) and selected on 2YTG solid medium supplemented with erythromycin, thiamphenicol, and 40 mM lactose. Expression of cas9 was induced in transformants by exposure to aTc as described previously (16), except that the inducer concentration was 1 μg ml−1. Mutants were confirmed by amplification of the bdhA-bdhB and bdhC loci performed on gDNA using primer pairs P30/P31 and P32/P33, respectively.
Fermentation assays.
Flask batch fermentations were performed in modified Gapes medium (31) containing 2.5 g liter−1 yeast extract, 1 g liter−1 KH2PO4, 0.6 g liter−1 K2HPO4, 1 g liter−1 MgSO4·7H2O, 6.6 mg liter−1 FeSO4·7H2O, 0.1 g liter−1 4-aminobenzoic acid, 5.8 g liter−1 CH3COONH4, and 80 g liter−1 glucose. Erythromycin (40 μg ml−1) was used for complementation assays. Fresh colonies were used to inoculate 5 ml Gapes medium. After 72 h of growth in an anaerobic chamber at 34°C without agitation, 1 ml of these precultures was inoculated into 100-ml flasks containing 19 ml of the same liquid medium. The flasks were sealed with rubber stoppers, and a pressure relief valve system was punctured through the rubber stoppers to prevent overpressure. Flasks were further incubated outside the anaerobic chamber for 72 h at 34°C with agitation at 120 rpm.
For bioreactor batch fermentations, fresh colonies were used to inoculate 5 ml modified Gapes medium (31). After 72 h of growth in an anaerobic chamber at 34°C without agitation, 1 ml of these precultures was inoculated into a 100-ml Erlenmeyer flask containing 40 ml of the same liquid medium and incubated for another 24 h in the anaerobic chamber. This second preculture was then used to inoculate a Biostat Q bioreactor (Sartorius) containing 360 ml of the same medium. Throughout the fermentation, the pH was controlled so as not to decrease below 5.0 by the addition of 5 M KOH.
The fermentation samples were centrifuged at 5,000 × g for 5 min, and the supernatant was diluted with an internal standard (0.5 g liter−1 propanol). The concentrations of the solvents produced were determined by gas chromatography on a PoraBOND-Q column (25-m length, 0.32-mm internal diameter, 0.5-μm film thickness; Agilent Technologies) equipped with a flame ionization detector. Helium was used as carrier gas at a flow rate of 1.6 ml min−1. The column was gradually heated from 50°C to 250°C in a 30-min run.
Acid concentrations were quantified by high-performance liquid chromatography (HPLC) (Aminex HPX-87H from Bio-Rad coupled to a Spectra System RI 150 refractometer and a Waters 2487 dual λ UV detector set at 210 nm); 0.01 M sulfuric acid mobile phase was used at a flow rate of 0.6 ml min−1. The column temperature was set to 60°C.
Residual sugar quantities were determined by HPLC using an Aminex HXP-87P (Bio-Rad) coupled to a Varian 350 RI refractometer for detection. Water was used as a mobile phase at a flow rate of 0.4 ml/min.
Supplementary Material
ACKNOWLEDGMENTS
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
We thank Hélène Velly and Sandra Menir for technical assistance during bioreactor assays.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Nölling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, Dubois J, Qiu D, Hitti J, Wolf YI, Tatusov RL, Sabathe F, Doucette-Stamm L, Soucaille P, Daly MJ, Bennett GN, Koonin EV, Smith DR. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol 183:4823–4838. doi: 10.1128/JB.183.16.4823-4838.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoo M, Bestel-Corre G, Croux C, Riviere A, Meynial-Salles I, Soucaille P. 2015. A quantitative system-scale characterization of the metabolism of Clostridium acetobutylicum. mBio 6:e01808-15. doi: 10.1128/mBio.01808-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooksley CM, Zhang Y, Wang H, Redl S, Winzer K, Minton NP. 2012. Targeted mutagenesis of the Clostridium acetobutylicum acetone-butanol-ethanol fermentation pathway. Metab Eng 14:630–641. doi: 10.1016/j.ymben.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Dai Z, Dong H, Zhang Y, Li Y. 2016. Elucidating the contributions of multiple aldehyde/alcohol dehydrogenases to butanol and ethanol production in Clostridium acetobutylicum. Sci Rep 6:28189. doi: 10.1038/srep28189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron. A universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 6.McAllister KN, Sorg JA, Margolin W. 2019. CRISPR genome editing systems in the genus Clostridium: a timely advancement. J Bacteriol 201:e00219-19. doi: 10.1128/JB.00219-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joseph RC, Kim NM, Sandoval NR. 2018. Recent developments of the synthetic biology toolkit for Clostridium. Front Microbiol 9:154. doi: 10.3389/fmicb.2018.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Zhang Z-T, Seo S-O, Choi K, Lu T, Jin Y-S, Blaschek HP. 2015. Markerless chromosomal gene deletion in Clostridium beijerinckii using CRISPR/Cas9 system. J Biotechnol 200:1–5. doi: 10.1016/j.jbiotec.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 9.Xu T, Li Y, Shi Z, Hemme CL, Li Y, Zhu Y, van Nostrand JD, He Z, Zhou J, Spormann AM. 2015. Efficient Genome Editing in Clostridium cellulolyticum via CRISPR-Cas9 Nickase. Appl Environ Microbiol 81:4423–4431. doi: 10.1128/AEM.00873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagaraju S, Davies NK, Walker DJF, Köpke M, Simpson SD. 2016. Genome editing of Clostridium autoethanogenum using CRISPR/Cas9. Biotechnol Biofuels 9:219. doi: 10.1186/s13068-016-0638-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyne ME, Bruder MR, Moo-Young M, Chung DA, Chou CP. 2016. Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in Clostridium. Sci Rep 6:25666. doi: 10.1038/srep25666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Zhang Z-T, Seo S-O, Lynn P, Lu T, Jin Y-S, Blaschek HP. 2016. Bacterial genome editing with CRISPR-Cas9. Deletion, integration, single nucleotide modification, and desirable “clean” mutant selection in Clostridium beijerinckii as an example. ACS Synth Biol 5:721–732. doi: 10.1021/acssynbio.6b00060. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Dong S, Wang P, Tao Y, Wang Y, Kelly RM. 2017. Genome Editing in Clostridium saccharoperbutylacetonicum N1-4 with the CRISPR-Cas9 System. Appl Environ Microbiol 83:e00233-17. doi: 10.1128/AEM.00233-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang H, Chai C, Li N, Rowe P, Minton NP, Yang S, Jiang W, Gu Y. 2016. CRISPR/Cas9-based efficient genome editing in Clostridium ljungdahlii an autotrophic gas-fermenting bacterium. ACS Synth Biol 5:1355–1361. doi: 10.1021/acssynbio.6b00044. [DOI] [PubMed] [Google Scholar]
- 15.Li Q, Chen J, Minton NP, Zhang Y, Wen Z, Liu J, Yang H, Zeng Z, Ren X, Yang J, Gu Y, Jiang W, Jiang Y, Yang S. 2016. CRISPR-based genome editing and expression control systems in Clostridium acetobutylicum and Clostridium beijerinckii. Biotechnol J 11:961–972. doi: 10.1002/biot.201600053. [DOI] [PubMed] [Google Scholar]
- 16.Wasels F, Jean-Marie J, Collas F, López-Contreras AM, Lopes Ferreira N. 2017. A two-plasmid inducible CRISPR/Cas9 genome editing tool for Clostridium acetobutylicum. J Microbiol Methods 140:5–11. doi: 10.1016/j.mimet.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Dong H, Tao W, Zhang Y, Li Y. 2012. Development of an anhydrotetracycline-inducible gene expression system for solvent-producing Clostridium acetobutylicum: a useful tool for strain engineering. Metab Eng 14:59–67. doi: 10.1016/j.ymben.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 18.Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, Bondy-Denomy J. 2017. Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell 168:150.e10–158.e10. doi: 10.1016/j.cell.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartman AH, Liu H, Melville SB. 2011. Construction and characterization of a lactose-inducible promoter system for controlled gene expression in Clostridium perfringens. Appl Environ Microbiol 77:471–478. doi: 10.1128/AEM.01536-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borges AL, Davidson AR, Bondy-Denomy J. 2017. The discovery, mechanisms, and evolutionary impact of anti-CRISPRs. Annu Rev Virol 4:37–59. doi: 10.1146/annurev-virology-101416-041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanley SY, Maxwell KL. 2018. Phage-encoded anti-CRISPR defenses. Annu Rev Genet 52:445–464. doi: 10.1146/annurev-genet-120417-031321. [DOI] [PubMed] [Google Scholar]
- 22.Marino ND, Pinilla-Redondo R, Csörgő B, Bondy-Denomy J. 16 March 2020. Anti-CRISPR protein applications. Natural brakes for CRISPR-Cas technologies. Nat Methods doi: 10.1038/s41592-020-0771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Al-Hinai MA, Fast AG, Papoutsakis ET. 2012. Novel system for efficient isolation of Clostridium double-crossover allelic exchange mutants enabling markerless chromosomal gene deletions and DNA integration. Appl Environ Microbiol 78:8112–8121. doi: 10.1128/AEM.02214-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin J, Jiang F, Liu J-J, Bray NL, Rauch BJ, Baik SH, Nogales E, Bondy-Denomy J, Corn JE, Doudna JA. 2017. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv 3:e1701620. doi: 10.1126/sciadv.1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayo-Muñoz D, He F, Jørgensen J, Madsen P, Bhoobalan-Chitty Y, Peng X. 2018. Anti-CRISPR-based and CRISPR-based genome editing of Sulfolobus islandicus rod-shaped virus 2. Viruses 10:695. doi: 10.3390/v10120695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hocq R, Bouilloux-Lafont M, Lopes Ferreira N, Wasels F. 2019. σ54 (σL) plays a central role in carbon metabolism in the industrially relevant Clostridium beijerinckii. Sci Rep 9:578. doi: 10.1038/s41598-019-43822-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho C, Hong S, Moon HG, Jang Y-S, Kim D, Lee SY, Lovley DR. 2019. Engineering clostridial aldehyde/alcohol dehydrogenase for selective butanol production. mBio 10:e02683-18. doi: 10.1128/mBio.02683-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. 2014. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol 32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mermelstein LD, Papoutsakis ET. 1993. In vivo methylation in Escherichia coli by the Bacillus subtilis phage phi 3T I methyltransferase to protect plasmids from restriction upon transformation of Clostridium acetobutylicum ATCC 824. Appl Environ Microbiol 59:1077–1081. doi: 10.1128/AEM.59.4.1077-1081.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gapes JR, Nimcevic D, Friedl A. 1996. Long-term continuous cultivation of Clostridium beijerinckii in a two-stage chemostat with on-line solvent removal. Appl Environ Microbiol 62:3210–3219. doi: 10.1128/AEM.62.9.3210-3219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.