

Estuarine salt marshes are highly productive ecosystems subjected to regular disturbances by hydrodynamic exchange. However, little is known about how distinct assembly processes and co-occurrence of taxa influence the structure of the abundant and rare bacterial biospheres in these soil systems. This study aims at unravelling these intricacies by studying a typical estuarine salt marsh located in Hangzhou Bay, China. Our study provides important pieces of evidence on the diverse distribution of rare and abundant bacterial biospheres. We show that a few abundant taxa are central nodes in species co-occurrence, potentially playing important roles as keystone species in the system. In addition, we highlight a dynamic interplay of assembly processes structuring these two subcommunities.
KEYWORDS: assembly processes, co-occurrence network, keystone taxa, rare biosphere, salt marsh
ABSTRACT
Understanding how species interaction and assembly processes structure the abundant and rare bacterial biospheres in soils is crucial for predicting how biodiversity influences ecosystem functioning. Here, we profiled the bacterial communities across a salt marsh ecosystem gradient to investigate the co-occurrence patterns across taxa and the relative influence of ecological processes mediating the assembly of the abundant and rare biospheres in soil. Our results revealed abundant taxa to be ubiquitous across all sites, whereas the distributions of the rare taxa were relatively more site specific. The α-diversity indices and β-diversity of rare subcommunities were significantly higher than those of the abundant subcommunities. Besides, both the taxonomic and functional composition of soil bacterial communities differed significantly between the two biospheres. Furthermore, the influence of stochasticity differed in each subcommunity. In particular, stochastic processes were relatively more important in constraining the assembly of rare taxa. Co-occurrence network analysis revealed that a few abundant taxa occupy central nodes within the networks, possibly indicating crucial roles as keystone taxa. Collectively, these findings suggest that abundant and rare bacterial biospheres have distinct distributions underpinned by a dynamic interplay of ecological processes and taxon co-occurrence patterns.
IMPORTANCE Estuarine salt marshes are highly productive ecosystems subjected to regular disturbances by hydrodynamic exchange. However, little is known about how distinct assembly processes and co-occurrence of taxa influence the structure of the abundant and rare bacterial biospheres in these soil systems. This study aims at unravelling these intricacies by studying a typical estuarine salt marsh located in Hangzhou Bay, China. Our study provides important pieces of evidence on the diverse distribution of rare and abundant bacterial biospheres. We show that a few abundant taxa are central nodes in species co-occurrence, potentially playing important roles as keystone species in the system. In addition, we highlight a dynamic interplay of assembly processes structuring these two subcommunities.
INTRODUCTION
Estuarine salt marshes rank among the most productive ecosystems on Earth. Sediment bacterial communities in these systems are fundamental for ecosystem functioning and often display a diverse set of taxa and metabolic functions (1, 2). Understanding the processes and mechanisms structuring bacterial diversity, taxon interaction, and distributional patterns is of key importance for ecosystem biogeochemistry and stabilization in salt marshes.
The distribution of soil bacterial communities often comprises a few abundant taxa with ubiquitous distributions and numerous rare taxa with restricted distributions (3). In recent years, the ecological importance of the rare taxa has been increasingly emphasized. The rare biosphere encompasses a diverse pool of genes and functions (4) and substantial metabolically active lineages (5, 6). Furthermore, some rare bacterial taxa are known to perform essential functions related to element cycling (7). However, existing studies on the distribution of bacterial taxa in estuarine marshes have often been hampered by the massive numbers of species in most communities and have thus neglected differences between abundant and rare taxa (5, 8). Determining the influence of the ecological processes governing community assemblies has been a central effort of researchers in microbial ecology (9, 10). The structure of abundant and rare bacterial biospheres is likely subject to different controlling factors or ecological processes (6, 11). For instance, in marine ecosystems, rare subcommunities were shown to be mostly governed by local factors (environmental filtering and biotic interactions), whereas abundant taxa were mainly influenced by regional factors (dispersal-related processes) (11).
Microbe-microbe interactions also play an important role in structuring bacterial communities (12). Mounting examples have supported the facets of interactions within abundant taxa (8, 12–14), while a few studies have investigated the complex relationships between both biospheres (e.g., abundant and rare taxa). Co-occurrence network analysis is a powerful tool to determine potential interaction among bacterial taxa (15, 16). The different roles of abundant and rare taxa in the co-occurrence network were revealed in recent studies (6, 17–20). For instance, Jiao et al. (17) reported a more important role for abundant taxa than for rare taxa in the network of oil-contaminated soils.
The salt marsh located in south Hangzhou Bay is one of eight salt marshes in China and is characterized by multiple environmental gradients and high biodiversity (21). In our previous study, we reported the overall distribution of bacterial communities in this system (21). Therefore, future studies should also pay fine attention to the rare bacterial subcommunity, which is critical for ecosystem restoration and environmental management. Here, sediments were collected from a distance gradient in this ecosystem. Specifically, we aim at investigating (i) the distributional patterns of abundant and rare taxa, (ii) the co-occurrence patterns of these two biospheres, and (iii) the relative influence of the assembly processes structuring them.
RESULTS
Alpha diversity and composition of the abundant and rare biospheres.
In our total data set, 258 operational taxonomic units (OTUs) (0.62%) encompassing 30.12% of all sequences were abundant and present across all samples, while 26,250 OTUs (62.98%) encompassing 5.26% of all sequences were classified as rare taxa. The Venn diagram showed that most of the abundant OTUs were shared among the transects, accounting for a substantial fraction of the sequences (40.0 to 68.4%), whereas most of the transect-specific OTUs belonged to rare taxa (Fig. 1a). The rare subcommunity had a higher α-diversity than its abundant counterpart, both in terms of OTU richness (richness) and the Shannon-Wiener index (Shannon) (see Fig. S1 in the supplemental material). The α-diversity of the whole community and that of the rare subcommunity exhibited a similar pattern (Fig. S1). Gammaproteobacteria, Anaerolineae, Deltaproteobacteria, and Betaproteobacteria were the most abundant taxa, whereas Deltaproteobacteria, Gammaproteobacteria, Alphaproteobacteria, and Planctomycetes were the most common taxa present in the rare biosphere (see Fig. S2 in the supplemental material). Last, we found functions related to bacterial secretion systems, such as ABC transporters, methane metabolism, and amino sugar metabolism, to be more prevalent in the rare subcommunity than in the abundant one (P < 0.05) (see Fig. S3 in the supplemental material).
FIG 1.

Community structure of bacterial communities across the three transects. See Table 1 for transect details. (a) Venn diagram displaying the numbers of unique and shared OTUs between the three transects. (b) Principal-coordinate analysis (PCoA) of bacterial communities based on Bray-Curtis distances. (c) The pairwise Bray-Curtis dissimilarity of bacterial communities across different points. Different letters above bars indicate significant differences (P < 0.05) according to the nonparametric Mann-Whitney U test.
Beta diversity of the abundant and rare biospheres.
The distribution of both abundant and rare subcommunities showed significant separation following a clear trend with the sampling transects (Fig. 1b and c), which was confirmed by the results of analysis of similarity (ANOSIM) between sampling transects (Table 1). The distance-decline regression analyses of physicochemical properties and bacterial communities had significant positive and negative slopes, respectively (Fig. 2). This indicates that they both were undergoing a directional change. The slope of the rare subcommunity was lower than that of the overall and abundant community slopes (Fig. 2b), and the rare subcommunity exhibited a significantly higher β-diversity than the abundant subcommunity. The whole community showed the lowest β-diversity (Fig. 3). However, across the three sampling transects, the abundant subcommunity exhibited greater niche breadth values than the rare subcommunity (see Fig. S4 in the supplemental material). Besides, the partition of β-diversity demonstrated that species richness rather than species replacement (turnover) accounted for the majority of the β-diversity and promoted the shift in community composition (Fig. 3). Mantel tests showed a more similar structure between the abundant subcommunity and the whole community than between than the rare biosphere and the whole community (abundant versus whole: R2 = 0.9859, P < 0.01; rare versus whole: R2 = 0.9201, P < 0.01). Furthermore, the ANOSIM showed that the rare subcommunity holds greater community dissimilarity across the transects than those of the abundant subcommunity and whole community (Fig. 4).
TABLE 1.
Analysis of similarities (ANOSIM) of bacterial community groups at different spatial scalesa
| Grouping | Whole community |
Abundant |
Rare |
|||
|---|---|---|---|---|---|---|
| R | P value | R | P value | R | P value | |
| Global | 0.895 | 0.001 | 0.861 | 0.001 | 0.909 | 0.001 |
| S-M vs S-N | 0.971 | 0.001 | 0.945 | 0.001 | 0.986 | 0.001 |
| S-M vs S-F | 0.811 | 0.001 | 0.769 | 0.001 | 0.818 | 0.001 |
| S-N vs S-F | 0.946 | 0.001 | 0.929 | 0.001 | 0.938 | 0.001 |
The S-N sampling transect was located about 300 m away from the water-land junction and was without vegetation; the S-M sampling transect was located about 1,000 m away from the water-land junction and was mainly covered with Scirpus mariqueter; the S-F sampling transect was located about 2,500 m away from the water-land junction and was mainly covered with Phragmites australis. All P values are significant.
FIG 2.

Distance-decay regression analysis of changes in environmental parameters (a) and bacterial β-diversity (b) of the entire community and of the abundant and the rare biosphere. The physicochemical properties used in the regression analysis include electrical conductivity, pH, phosphorus, potassium, nitrogen, organic carbon, Fe2O3, ammonium, and clay, silt, and sand content.
FIG 3.
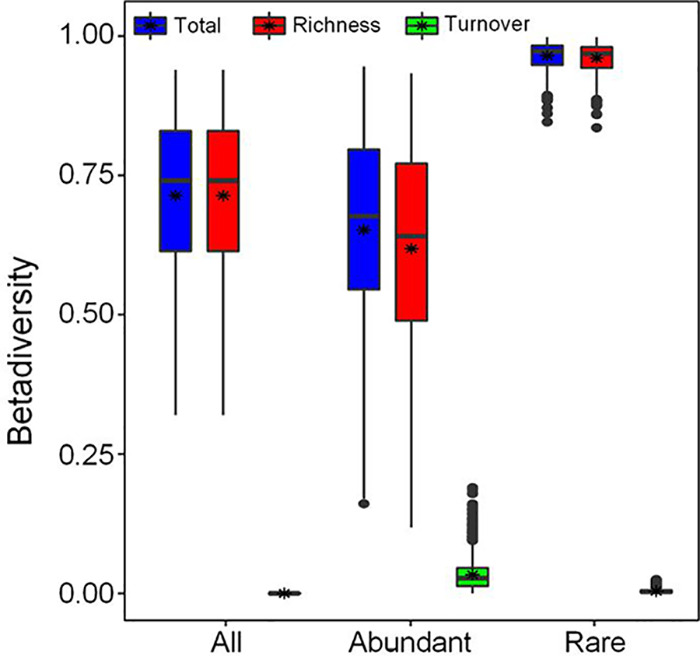
The contribution of community turnover and OTU richness to the β-diversity patterns of the entire community, the abundant biosphere, and the rare biosphere, respectively. The top and bottom boundaries of each box indicate the 75th and 25th quartile values, and lines within each box represent the median values; asterisks (*) indicate mean values.
FIG 4.

Analysis of similarity (ANOSIM) and dissimilarity of the entire community and of the abundant and the rare biospheres across the three transects. See Table 1 for transect details. Significant values were determined at a P value of <0.05. The ANOSIM R and P values were calculated based on the Bray-Curtis dissimilarity matrices. Data are shown as means ± standard deviation (SD).
Co-occurrence of bacterial taxa.
Co-occurrence analysis displayed scale-free characteristics (Table 2), which indicates that the network was nonrandomly generated. The resulting network was composed of 2,590 nodes associated with 20,661 edges (Table 2). There were 3,290 edges between the abundant and other nodes and 71 edges between the rare and other nodes. No edge was detected between the abundant and rare nodes. These suggested that the rare taxa seldom co-occurred with the abundant taxa, whereas the other taxa frequently co-occurred with abundant taxa (Fig. 5a). Besides, no negative correlation was found within the rare taxa. We further compared the specific node-level topological characteristics among three subcommunities. The degree, betweenness, centrality, and eigencentrality showed significantly higher (P < 0.05) values in the abundant taxa than in the rare taxa. Significantly higher closeness centrality values were obtained for the rare taxa than for other taxa (Fig. 5b). Keystone species, which have an integral part in maintaining the structure and function of bacterial communities, were identified as having a high degree (>70) of connection and low betweenness centrality values (<5,000). In total, 17 OTUs were identified as keystone taxa, including Anaerolineaceae (3 OTUs), Cytophagaceae (2 OTUs), and Sphingomonadaceae (2 OTUs) (see Table S1 in the supplemental material), and two of these belong to the abundant taxa.
TABLE 2.
Topological properties of co-occurrence networks of soil bacterial communities and their associated random networksa
| Statistic or network type | Value |
|---|---|
| Real network | |
| No. of nodesb | 2,590 |
| No. of edgesc | 20,661 |
| Modularityd | 0.624 |
| Avg clustering coefficiente | 0.149 |
| Network diamf | 17 |
| Avg path lengthg | 5.348 |
| Avg degreeh | 12.722 |
| Random network | |
| Modularity (SD)i | 0.157 (0.003) |
| Avg clustering coefficient (SD)i | 0.006 (0.000) |
| Avg path length (SD)i | 3.113 (0.000) |
Random networks were generated by rewiring all links with the same numbers of nodes and edges to the real networks.
Number of OTUs with a correlation ρ > 0.8 or ρ < –0.8 and statistical significance (P < 0.01).
Number of strong and significant correlations between nodes.
A modularity value of >0.4 suggests that the network has a modular structure. It indicates that there are nodes in the network that are more densely connected between each other than with the rest of the network and that their density is noticeably higher than the graph’s average.
Average clustering coefficient indicates how nodes are embedded in their neighborhood and the degree to which nodes tend to cluster together.
The maximum distance between all possible pairs of nodes.
The average number of steps along the shortest paths for all possible pairs of network nodes.
Node connectivity showing how many connections (on average) each node has to the other nodes in the network.
Numbers in parentheses indicate the standard deviations of topological properties of the 1,000 Erdös-Rényi random networks.
FIG 5.

Properties of the correlation-based network. (a) Networks analysis showing the intra-associations within each subcommunity and the interassociations between different subcommunities. A connection stands for a strong (Spearman’s ρ > 0.8 or ρ < −0.8) and significant (P < 0.01) correlation. The size of each node is proportional to the number of connections (i.e., degree). Numbers around each rectangle and each two-way arrow represent the inner associations of each subcommunity, and the remaining numbers represent the external associations between subcommunities. (b) Comparison of node-level topological features among four different subcommunities. The top and bottom boundaries of each box indicate the 75th and 25th quartile values, and lines within each box represent the median values. Different letters indicate significant differences (P < 0.05), determined by the nonparametric Mann-Whitney U test.
Modular structure of the co-occurrence network.
The average path length, average clustering coefficient, and observed modularity were all greater than their respective corresponding Erdös-Rényi random networks (Table 2), indicating nonrandom network structure. The entire network can be divided into six major modules, in which modules I and II accounted for 23.51% and 23.05% of the entire network, respectively (Fig. 6a). Most of the modules were specific (relatively more abundant) to a particular sampling transect (Fig. 6b). For instance, the OTUs in modules II and V were more specific to the S-N transect (without vegetation) and were represented by Helicobacteraceae and Desulfobulbaceae. Module III was specific to the S-M transect (mainly covered with Scirpus mariqueter) and was represented by the JTB 255 marine benthic group (JTB255-MBG). Likewise, the OTUs in modules I and IV mostly occurred in samples from the S-F transect (mainly covered with Phragmites australis). Across all modules, only modules IV and V consisted of rare taxa (see Fig. S5 in the supplemental material). The taxonomic distribution was more diverse in modules I to III, while the Anaerolineaceae accounted for a relatively high proportion of the six major modules (see Fig. S6 in the supplemental material). In addition, the OTUs affiliated with the Anaerolineaceae were identified as potential keystone species in modules I, II, and VI, whereas potential keystone species in modules III, IV, and V were affiliated with JTB255-MBG, Haliangiaceae, and Saprospiraceae, respectively.
FIG 6.

Co-occurrence patterns of OTUs. The nodes were colored according to different types of modularity class (a). Ternary plots displaying the relative abundance of OTUs from modules I to VI in the three transects (b). A connection stands for a strong (Spearman’s ρ > 0.8 or ρ < −0.8) and significant (P < 0.01) correlation. The size of each node is proportional to the number of connections (i.e., degree). Major modules have more than 80 nodes. Other modules include small modules (n = 223) with ≤10 nodes per module. Each circle represents one individual OTU. For each OTU, abundance was averaged over all samples at each transect.
Ecological processes structuring the assembly of the abundant and rare biospheres.
We found the soil sand content and electrical conductivity (EC) to largely explain the variation in the abundant subcommunity, as demonstrated by the distance-based multivariate linear model (DistLM) (see Table S2 in the supplemental material). These variables accounted for 60.52% of the variation explained (Table S2). For the rare biosphere, these variables only accounted for 8.47% of the variation (Table S2). The null modeling analysis provided a good description of the frequency of occurrence of microbial taxa in each individual community (R2 = 0.721) (Fig. 7a). Almost all of the abundant taxa were distributed below the prediction zone, whereas the rare taxa were mainly found in the neutral zone (Fig. 7b; see also Table S3 in the supplemental material). The nearest taxon index (NTI) and net relatedness index (NRI) values were calculated to determine whether the OTUs were more closely related to co-occurring related taxa than the random expectation (see Fig. S7 in the supplemental material). These results showed that taxa in both biospheres were phylogenetically clustered (NTI > 2), and significant differences (analysis of variance [ANOVA]; P < 0.001) exist in the stochastic/deterministic balance structuring these subcommunities (Fig. S7a and b).
FIG 7.

Relative influences of assembly processes structuring the entire bacterial community (a) and the abundant and rare biospheres (b). The solid black line represents the 95% confidence interval above and below the prediction.
DISCUSSION
Distributional patterns of the soil bacterial abundant and rare biospheres.
In the present study, distinct fractions of bacterial communities in soil (abundant and rare biospheres) significantly separated across the sampling transects (Fig. 1). The rare biosphere held higher α-diversity than the abundant one (see Fig. S1 in the supplemental material), thus corroborating the idea that rare taxa contribute greatly to the overall community diversity and metabolic functioning (5, 18). In addition, we found that the rare taxa significantly differed across the sampling transect, whereas a large proportion of the abundant taxa were ubiquitous across all samples (Fig. 1a). This aligns with similar findings of previous studies in lakes, ocean, and polluted soil ecosystems (6, 18, 19). Similarly to this salt marsh, all of these previously studied areas are known to experience dynamic environmental fluctuations. Hence, it is possible to envision that the abundant taxa constitute a more stable community than the rare biosphere. Several reasons could account for that; for example, (i) abundant taxa occupy wider niches than rare ones (see Fig. S4 in the supplemental material), thus utilizing an array of resources that support their persistence, (ii) rare taxa encompass more metabolically diverse organisms that respond quickly to environmental changes (22), and (iii) rare taxa are more likely to go extinct or drop below a level of detection. Moreover, the rare taxa also had higher β-diversity than the abundant biosphere (Fig. 3), which might indicate external microbial inputs through dispersal (8, 17). However, the structure of the abundant subcommunity was more similar to that of the whole community, accounting for a significant proportion of community dissimilarity. This is consistent with Shade et al. (23) and confirms that the higher proportion of abundant taxa in the entire community greatly contributed to β-diversity.
Diverse species have distinct life strategies and occupy various ecological niches (3). The taxonomic and functional distributions of the abundant and rare taxa were found to be significantly different (see Fig. S2 and S3 in the supplemental material). For example, Anaerolineae species, which are associated with the degradation of specific carbohydrates and other cellular materials in salt marshes (21, 24), were prevalent and comprised an abundant OTU (Fig. S2). However, Fig. S3 shows no significant difference related to biogeochemical cycling between these two biospheres. This might be attributed to potential limitations of our functional prediction. Nevertheless, the bacterial secretion system and the ABC transporters, which play roles in a wide range of physiological processes (25–27), were more prevalent in the rare biosphere (Fig. S3). In addition, greater differences in α-diversity and β-diversity across the three sampling transects were detected in the rare biosphere than in the abundant counterpart (Table 1, Fig. 3 and 4). These findings corroborate the idea that the rare biosphere is more sensitive to changes in the environment, with a potentially key role in ecosystem functioning. Unlike the abundant taxa, the rare biosphere displayed the lowest diversity in sediments farthest from the water-land junction (S-F) (Fig. 1, Fig. S1). This difference possibly resulted from the relatively weaker disturbance frequency and intensity of hydrodynamic exchanges at these sites. In addition, the S-F sediments were relatively richer in nutrients (21). Both contributed to the growth of abundant taxa and to detected differences between the two biospheres.
Abundant taxa occupy central nodes in the co-occurrence network.
Microorganisms with specific ecological niches may interact with each other and form complex interactive networks (12, 28), thus influencing microbial distributions and ecological functions (29). In this study, a correlation-based network analysis was conducted to inquire into the interrelationship of abundant and rare taxa. We found both of these groups to roughly cluster separately, i.e., members of both biospheres were less connected with each other (Fig. 5a). We infer that the rare taxa were more transect-specific than the abundant taxa and therefore statistically co-occurred less frequently with abundant taxa. The topological features of abundant taxa were significantly higher than those of the rare ones, except closeness centrality (Fig. 5b). This suggests that abundant taxa occupied more central locations within the network (17), which nicely aligns with findings reported in other previous studies (6, 20).
In co-occurrence analysis, the division of the network into modules can provide insights into the different groups of nodes that perform different functions (30). The composition of the modules in this study varied along the transects (see Fig. S6 in the supplemental material), which provides evidence for the existence of distinct ecological niches over the spatial scale. Furthermore, these niches were preferentially occupied by different bacterial taxa. For example, modules II and V (higher abundance in S-N) were composed of OTUs affiliated with Helicobacteraceae and Desulfobulbaceae (Fig. S6), which are taxa known to be associated with sulfur cycling (31, 32). The JTB 255 marine benthic group (JTB255-MBG), which is important for carbon fixation, dominated module III (higher abundance in S-M) (Fig. S6). Interestingly, we did not find any potential keystone taxa belonging to the rare biosphere, which stands in contrast to a previous study (6). This indicates that a different dynamic occurs in taxa belonging to the rare and abundant biospheres, thus resulting in distinct patterns of species co-occurrence, with potential direct effects on their respective roles in ecosystem functioning.
Stochasticity and environmental selection differently structure the rare and abundant biospheres.
The mean NTIs for both biospheres were significantly greater than zero (Fig. S7a), suggesting that deterministic processes are important in structuring the bacterial community assembly in this system (33)—a finding also corroborated by the null modeling analysis. This was confirmed by the significant correlation between the subcommunities and physicochemical factors (see Table S2 in the supplemental material). In contrast to the previous findings for reservoirs and lakes (19, 34), Table S2 shows that both the abundant and rare biospheres were significantly related to similar local physicochemical factors, i.e., sand content and EC. This might be attributed to the specific features of the salt marsh. The sediment formation process of salt marshes reflects gradual changes in soil texture over time (35). The sand content is also closely related to nutrient dynamics and microbial functioning (17). Salinity, based on EC measurements, is an intrinsic factor of salt marshes and has been demonstrated to be a major driver of the distribution of bacterial communities in intertidal zones (21, 35). Stochastic processes also exerted significant effects on bacterial community assembly (Fig. 7a). However, most of the rare taxa were found in the prediction zone, whereas almost all the abundant taxa were below the prediction zone (Fig. 7b; see also Table S3 in the supplemental material). The rare taxa were also phylogenetically more clustered (Fig. S7a), which suggests that members of the rare biosphere are ecologically more similar within the subcommunity than were members of the abundant biosphere.
Furthermore, the variation explained by the physicochemical parameters was higher for the abundant subcommunity than for the rare subcommunity (Table S2). This might be attributed to niche breadth differences between both biospheres. The niche breadth of rare taxa was relatively narrower than that of abundant taxa (Fig. S4). Also, most of the rare taxa might consist of oligotrophic bacteria, which are metabolically more diverse and occupy distinct niches in the soil (36). On the other hand, it is expected that abundant taxa are composed of heterotrophs that more rapidly utilize readily available resources to stimulate growth (37). Moreover, the interplay of ecological processes mediating the assembly of abundant and rare taxa also differed. In this study, the assembly of rare taxa showed a higher relative influence of stochastic processes (Fig. 7b and Table S3). It is possible to infer that rare taxa may be more strongly influenced by demographic stochasticity and dispersal due to their relatively small population sizes (7). In line with that, the diversity of the rare biosphere across the transects was greater than that of the abundant taxa (Fig. 4). These results are consistent with previous studies on surface seawater (19) but stand in contrast to those of the study by Liu et al. (34) that showed rare bacterial subcommunities of inland lakes and reservoirs to be primarily governed by local environmental variables. Such differences might likely be attributed to the degree of local variation in environmental conditions across distinct systems. For example, in the salt marsh system studied here, the close proximity to the sea imposes a frequent gradient of disturbance due to the tidal regime, whereas in Liu et al. (34), lower variation is likely to occur in inland freshwater ecosystems. Generally, the higher relative influence of stochastic processes would be correlated with a higher spatial turnover rate. However, our study showed opposite results (Fig. 2). According to Woodcock et al. (38), we proposed that the lower slope of the rare subcommunity might be related to the sampling method and sequencing depth. Although the majority of the bacterial taxa were identified in the current study, the distorting effect of undersampling and sequencing depth might still exist. This might also explain the relatively weak role of rare taxa in the co-occurrence network in the study. More samples and deeper sequencing depth are required in future studies.
Our study provides insights into the distribution of bacterial communities by partitioning both the abundant and rare biospheres in soils from a typical salt marsh ecosystem. Despite encompassing higher diversity, the rare biosphere was found to hold a limited distribution and larger variations across the transects. Both deterministic selection and stochastic processes simultaneously affected the community assembly of the rare and abundant biospheres, albeit at different levels. The co-occurrence network analysis revealed a few abundant taxa to occupy central nodes, thus suggesting their potential role as keystone species. Taken together, this study advances our knowledge on how distinct ecological processes and biotic interactions structure the distribution of abundant and rare taxa in the system and promotes a better understanding of community assembly and its potential implications for ecosystem functioning.
MATERIALS AND METHODS
Sampling locations and data collection.
In brief, 15 sediment samples were collected from the salt marsh located in south Hangzhou Bay (30°21′56″N, 121°12′39″E) across three transects that were 300, 1,000, and 2,500 m from the water-land junction, corresponding to S-N, S-M and S-F, respectively (21). These three transects had distinct vegetation types, i.e., bare sites, sites dominated by Scirpus mariqueter, and sites dominated by Phragmites australis, respectively. Soil physicochemical analyses were performed to determine pH, organic carbon (OC), electrical conductivity (EC), total nitrogen (TN), ammonium, soil particle composition, available phosphorus (AP), and available potassium (AK) in each individual sample. Detailed information and data are available in Yao et al. (21). Total DNA extraction of 60 sediment samples (15 samples × 4 replicates) was carried out using the Power Soil DNA isolation kit (Mo Bio, Carlsbad, CA). Bacterial communities were profiled by targeting the V4 hypervariable region (515F-806R) of the 16S rRNA gene using the Illumina MiSeq platform (MajorBio Co., Ltd., Shanghai, China) following a 2-bp × 300-bp read configuration.
Sequence analysis.
The acquired sequences were processed using QIIME v.1.8 (http://qiime.org/index.html). The raw data were quality processed according to Caporaso et al. (39), and we used USEARCH and the UCHIME algorithm (40) to remove chimeric sequences. The sequences with high quality, at a 97% similarity level, were clustered into different operational taxonomic units (OTUs) using UPARSE (40) and then classified into groups on the basis of their taxonomy. Sequences identified as archaea and chloroplasts were removed from the analysis. Singletons were also removed from the OTU table prior to diversity analysis. After quality filtering, 16,660 sequences of each sample were selected to normalize the sequencing effort across samples at random for downstream analysis at the same sequencing depth. Finally, we applied Tax4Fun, which implements nearest neighbor recognition with minimal 16S rRNA sequence similarity, to link the 16S rRNA gene sequence to the functional annotation of the sequenced prokaryotic genome (41). In brief, the SILVA128-based OTU table was used as an input in Tax4Fun to generate a functional prediction based on the relative abundance of Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology (KO) annotations (42).
Statistical analysis.
All OTUs with relative abundance values above 0.5% in at least one sample were defined as “abundant,” while those below 0.01% across all the samples were defined as “rare.” Alpha diversity values were determined in QIIME v.1.8 using multiple indices (except Pielou’s evenness, which used the diversity function in the “vegan” R package). The spatial impacts on α-diversity were tested by one-way analysis of variance using SPSS v.13.0 (SPSS, Inc., IL, USA). We measured β-diversity based on Bray-Curtis dissimilarity. The β-diversity values were split into two components, namely the balanced variation (richness) and the unidirectional abundance gradients (turnover) within the distinct subcommunities, using the “bray.part” function in the “betapart” R package (43). A spatial decline analysis, which quantified the Bray-Curtis dissimilarities between each pair of samples, was conducted to identify the spatial patterns in bacterial communities. Then, we plotted the geographic distance against dissimilarity (44). Bacterial community composition was visualized by principal-coordinate analysis (PCoA) based on Bray-Curtis dissimilarity. Analysis of similarity (ANOSIM) was conducted to investigate differences in the bacterial communities among groups using PAST v.3.2.3 (45). Levin’s niche breadth indices for abundant and rare bacterial communities were calculated using the “niche.width” function in the “spaa” R packages (46, 47).
Co-occurrence network analysis.
Only the OTUs present in more than 6 samples were used to run network analyses to improve statistical confidence. In total, 11,272 OTUs were used. We used the “picante” R package to calculate all possible pairs of Spearman’s rank correlations (ρ) between the retained OTUs. Only those correlations with values that were robust (|ρ| > 0.8) and of statistical significance (P < 0.01) were incorporated into the networks (14). Network visualization was carried out using Gephi v.0.9.2 and modular analysis (48). Subsequently, topological properties were analyzed by means of Gephi based on node level and were examined by nonparametric Mann-Whitney U test to measure their statistical differences across different taxa. Nodes with a degree value of >70 and a betweenness centrality value of >5,000 were identified as representing keystone species in the co-occurrence network (49). The “igraph” R package randomly generated a total of 1,000 Erdös-Rényi networks with an equal number of nodes and edges as the real one (50).
Community assembly.
We applied the Sloan neutral community model to identify the relative importance of the neutral processes in structuring the abundant and rare biospheres of bacterial communities (51) using the R codes reported by Burns et al. (52). The Akaike information criterion (AIC) of the neutral and binomial model was compared to determine whether local communities are random subsets of the source metacommunity (13, 35). Computation of the AIC of each model was conducted in R based on 1,000 bootstrap replicates. The AIC of the neutral model (−91,078) was lower than that of the binomial model (−73,624), suggesting that the random sampling of the source community was not the main factor structuring bacterial assembly. Additionally, the nearest taxon index (NTI) and net relatedness index (NRI) (53) for each subcommunity were conducted on the null model “taxa.labels” (999 randomization runs). Higher values of NTI and NRI suggested stronger phylogenetic clustering patterns in the communities, while lower counterparts indicated communities with weaker phylogenetic clustering or overdispersion (33).
Data availability.
The raw sequences were deposited in the NCBI Sequence Read Archive under accession number SRP156339.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Z. Zhu for comments on an earlier version of this paper.
The work was supported by the National Natural Science Foundation of China (grants 41977348, 41601517, and 41706132), the Natural Science Foundation of Ningbo (grant 2019A610445), the Natural Science Foundation of Ningbo University (grant XYL17023), and the K.C. Wong Magna Fund of Ningbo University.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Madsen EL. 2011. Microorganisms and their roles in fundamental biogeochemical cycles. Curr Opin Biotechnol 22:456–464. doi: 10.1016/j.copbio.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Salles JF, Silva M, Dini-Andreote F, Dias ACF, Guillaumaud N, Poly F, van Elsas JD. 2017. Successional patterns of key genes and processes involved in the microbial nitrogen cycle in a salt marsh chronosequence. Biogeochemistry 132:185–201. doi: 10.1007/s10533-017-0296-y. [DOI] [Google Scholar]
- 3.Pedrós-Alió C. 2012. The rare bacterial biosphere. Annu Rev Mar Sci 4:449–466. doi: 10.1146/annurev-marine-120710-100948. [DOI] [PubMed] [Google Scholar]
- 4.Elshahed MS, Youssef NH, Spain AM, Sheik C, Najar FZ, Sukharnikov LO, Roe BA, Davis JP, Schloss PD, Bailey VL, Krumholz LR. 2008. Novelty and uniqueness patterns of rare members of the soil biosphere. Appl Environ Microbiol 74:5422–5428. doi: 10.1128/AEM.00410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch MD, Neufeld JD. 2015. Ecology and exploration of the rare biosphere. Nat Rev Microbiol 13:217–229. doi: 10.1038/nrmicro3400. [DOI] [PubMed] [Google Scholar]
- 6.Xue Y, Chen H, Yang JR, Liu M, Huang B, Yang J. 2018. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J 12:2263–2277. doi: 10.1038/s41396-018-0159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jousset A, Bienhold C, Chatzinotas A, Gallien L, Gobet A, Kurm V, Küsel K, Rillig MC, Rivett DW, Salles JF, van der Heijden MG, Youssef NH, Zhang X, Wei Z, Hol WH. 2017. Where less may be more: how the rare biosphere pulls ecosystems strings. ISME J 11:853–862. doi: 10.1038/ismej.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dini-Andreote F, de Cássia Pereira e Silva M, Triadó-Margarit X, Casamayor EO, van Elsas JD, Salles JF. 2014. Dynamics of bacterial community succession in a salt marsh chronosequence: evidences for temporal niche partitioning. ISME J 8:1989–2001. doi: 10.1038/ismej.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nemergut DR, Schmidt SK, Fukami T, O’Neill SP, Bilinski TM, Stanish LF, Knelman JE, Darcy JL, Lynch RC, Wickey P, Ferrenberg S. 2013. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77:342–356. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao JQ, Cao XF, Zhao L, Wang J, Gao Z, Wang MC, Huang Y. 2016. The importance of neutral and niche processes for bacterial community assembly differs between habitat generalists and specialists. FEMS Microbiol Ecol 92:fiw174. doi: 10.1093/femsec/fiw174. [DOI] [PubMed] [Google Scholar]
- 11.Wu W, Logares R, Huang B, Hsieh CH. 2017. Abundant and rare picoeukaryotic sub-communities present contrasting patterns in the epipelagic waters of marginal seas in the northwestern Pacific Ocean. Environ Microbiol 19:287–300. doi: 10.1111/1462-2920.13606. [DOI] [PubMed] [Google Scholar]
- 12.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 13.Jiao S, Yang YF, Xu YQ, Zhang J, Lu YH. 2020. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J 14:202–216. doi: 10.1038/s41396-019-0522-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu A, Ju F, Hou L, Li J, Yang X, Wang H, Mulla SI, Sun Q, Burgmann H, Yu CP. 2017. Strong impact of anthropogenic contamination on the co-occurrence patterns of a riverine microbial community. Environ Microbiol 19:4993–5009. doi: 10.1111/1462-2920.13942. [DOI] [PubMed] [Google Scholar]
- 15.Haruta S, Kato S, Yamamoto K, Igarashi Y. 2009. Intertwined interspecies relationships: approaches to untangle the microbial network. Environ Microbiol 11:2963–2969. doi: 10.1111/j.1462-2920.2009.01956.x. [DOI] [PubMed] [Google Scholar]
- 16.Layeghifard M, Hwang DM, Guttman DS. 2017. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol 25:217–228. doi: 10.1016/j.tim.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiao S, Chen W, Wei G. 2017. Biogeography and ecological diversity patterns of rare and abundant bacteria in oil-contaminated soils. Mol Ecol 26:5305–5317. doi: 10.1111/mec.14218. [DOI] [PubMed] [Google Scholar]
- 18.Nolte V, Pandey RV, Jost S, Medinger R, Ottenwälder B, Boenigk J, Schlötterer C. 2010. Contrasting seasonal niche separation between rare and abundant taxa conceals the extent of protist diversity. Mol Ecol 19:2908–2915. doi: 10.1111/j.1365-294X.2010.04669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mo Y, Zhang W, Yang J, Lin Y, Yu Z, Lin S. 2018. Biogeographic patterns of abundant and rare bacterioplankton in three subtropical bays resulting from selective and neutral processes. ISME J 12:2198–2210. doi: 10.1038/s41396-018-0153-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Hou F, Xie W, Wang K, Zhou X, Zhang D, Zhu X. 9 October 2019. Interaction and assembly processes of abundant and rare microbial communities during a diatom bloom process. Environ Microbiol doi: 10.1111/1462-2920.14820. [DOI] [PubMed] [Google Scholar]
- 21.Yao Z, Du S, Liang C, Zhao Y, Dini-Andreote F, Wang K, Zhang D. 2019. Bacterial community assembly in a typical estuarine marsh with multiple environmental gradients. Appl Environ Microbiol 85:e02602-18. doi: 10.1128/AEM.02602-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Logares R, Audic S, Bass D, Bittner L, Boutte C, Christen R, Claverie JM, Decelle J, Dolan JR, Dunthorn M, Edvardsen B, Gobet A, Kooistra WH, Mahe F, Not F, Ogata H, Pawlowski J, Pernice MC, Romac S, Shalchian-Tabrizi K, Simon N, Stoeck T, Santini S, Siano R, Wincker P, Zingone A, Richards TA, de Vargas C, Massana R. 2014. Patterns of rare and abundant marine microbial eukaryotes. Curr Biol 24:813–821. doi: 10.1016/j.cub.2014.02.050. [DOI] [PubMed] [Google Scholar]
- 23.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, Fierer N, Gilbert JA. 2014. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 5:e01371-14. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang B, Wang LY, Zhou Z, Mbadinga SM, Zhou L, Liu JF, Yang SZ, Gu JD, Mu BZ. 2016. High frequency of Thermodesulfovibrio spp. and Anaerolineaceae in association with Methanoculleus spp. in a long-term incubation of n-alkanes-degrading methanogenic enrichment culture. Front Microbiol 7:1431. doi: 10.3389/fmicb.2016.01431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. 2006. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the dictyostelium host model system. Proc Natl Acad Sci U S A 103:1528–1533. doi: 10.1073/pnas.0510322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beeckman DS, Vanrompay DC. 2010. Bacterial secretion systems with an emphasis on the chlamydial type III secretion system. Curr Issues Mol Biol 12:17–41. [PubMed] [Google Scholar]
- 27.Fath MJ, Kolter R. 1993. ABC transporters: bacterial exporters. Microbiol Rev 57:995–1017. doi: 10.1128/MMBR.57.4.995-1017.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Röttjers L, Karoline F. 2018. From hairballs to hypotheses—biological insights from microbial networks. FEMS Microbiol Rev 42:761–780. doi: 10.1093/femsre/fuy030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barberan A, Bates ST, Casamayor EO, Fierer N. 2012. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J 6:343–351. doi: 10.1038/ismej.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong W, Jousset A, Guo S, Karlsson I, Zhao Q, Wu H, Kowalchuk GA, Shen Q, Li R, Geisen S. 2018. Soil protist communities form a dynamic hub in the soil microbiome. ISME J 12:634–638. doi: 10.1038/ismej.2017.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obi CC, Adebusoye SA, Ugoji EO, Ilori MO, Amund OO, Hickey WJ. 2016. Microbial communities in sediments of Lagos Lagoon, Nigeria: elucidation of community structure and potential impacts of contamination by municipal and industrial wastes. Front Microbiol 7:1213. doi: 10.3389/fmicb.2016.01213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuever J. 2014. The family Desulfobulbaceae, p 75–86. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes: Deltaproteobacteria and Epsilonproteobacteria. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 33.Stegen JC, Lin X, Konopka AE, Fredrickson JK. 2012. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J 6:1653–1664. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu L, Yang J, Yu Z, Wilkinson DM. 2015. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J 9:2068–2077. doi: 10.1038/ismej.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dini-Andreote F, Brossi MJL, van Elsas JD, Salles JF. 2016. Reconstructing the genetic potential of the microbially-mediated nitrogen cycle in a salt marsh ecosystem. Front Microbiol 7:902. doi: 10.3389/fmicb.2016.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia X, Dini-Andreote F, Salles JF. 2018. Community assembly processes of the microbial rare biosphere. Trends Microbiol 26:738–747. doi: 10.1016/j.tim.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 37.Pedros-Alio C. 2006. Marine microbial diversity: can it be determined? Trends Microbiol 14:257–263. doi: 10.1016/j.tim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 38.Woodcock S, Curtis TP, Head IM, Lunn M, Sloan WT. 2006. Taxa–area relationships for microbes: the unsampled and the unseen. Ecol Lett 9:805–812. doi: 10.1111/j.1461-0248.2006.00929.x. [DOI] [PubMed] [Google Scholar]
- 39.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aßhauer KP, Wemheuer B, Daniel R, Meinicke P. 2015. Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31:2882–2884. doi: 10.1093/bioinformatics/btv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. 2014. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42:199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baselga A. 2010. Partitioning the turnover and nestedness components of beta diversity. Global Ecol Biogeogr 19:134–143. doi: 10.1111/j.1466-8238.2009.00490.x. [DOI] [Google Scholar]
- 44.Collins SL, Micheli F, Hartt L. 2000. A method to determine rates and patterns of variability in ecological communities. Oikos 91:285–293. doi: 10.1034/j.1600-0706.2000.910209.x. [DOI] [Google Scholar]
- 45.Cáceres MD, Legendre P. 2009. Associations between species and groups of sites: indices and statistical inference. Ecology 90:3566–3574. doi: 10.1890/08-1823.1. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J. 2004. Quantitative ecology. Science Press, Beijing, China: (In Chinese.) [Google Scholar]
- 47.Levins R. 1968. Evolution in changing environments. Princeton University Press, Princeton, NJ. [Google Scholar]
- 48.Jacomy M, Bastian M, Heymann S. 2009. Gephi: an open source software for exploring and manipulating networks, p 361–362. Proceedings of the 3rd International Conference on Weblogs and Social Media. AAAI, Menlo Park, CA. [Google Scholar]
- 49.Guimera R, Nunes Amaral LA. 2005. Functional cartography of complex metabolic networks. Nature 433:895–900. doi: 10.1038/nature03288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Csardi G, Nepusz T. 2006. The igraph software package for complex network research. InterJ Complex Syst 1695:1–9. [Google Scholar]
- 51.Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. 2006. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol 8:732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- 52.Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, Bohannan BJ. 2016. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J 10:655–664. doi: 10.1038/ismej.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Webb CO. 2000. Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. Am Nat 156:145–155. doi: 10.1086/303378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequences were deposited in the NCBI Sequence Read Archive under accession number SRP156339.