

SUMMARY
Cargo trafficking along microtubules is exploited by eukaryotic viruses, but no such examples have been reported in bacteria. Several large Pseudomonas phages assemble a dynamic, tubulin-based (PhuZ) spindle that centers replicating phage DNA sequestered within a nucleus-like structure. Here, we show that capsids assemble on the membrane and then move rapidly along PhuZ filaments toward the phage nucleus for DNA packaging. The spindle rotates the phage nucleus, distributing capsids around its surface. PhuZ filaments treadmill toward the nucleus at a constant rate similar to the rate of capsid movement and the linear velocity of nucleus rotation. Capsids become trapped along mutant static PhuZ filaments that are defective in GTP hydrolysis. Our results suggest a transport and distribution mechanism in which capsids attached to the sides of filaments are trafficked to the nucleus by PhuZ polymerization at the poles, demonstrating that the phage cytoskeleton evolved cargo-trafficking capabilities in bacteria.
In Brief
Bacteriophage-encoded tubulin-like proteins form treadmilling filaments in their bacterial host to transport viral capsids to the subcellular compartment where phage DNA is replicated and transcribed.
Graphical Abstract
INTRODUCTION
Microtubules play a key role in intracellular cargo trafficking in eukaryotes. Many viruses exploit microtubule trafficking to translocate from the surface of the plasma cell membrane to the cell interior, such as to the nucleus to initiate viral replication (Cohen et al., 2011; Greber and Way, 2006; Portilho et al., 2016; Sodeik et al., 1997). Newly assembled viral particles also traffic along microtubules to reach the surface of the cell for egress (Greber and Way, 2006; Ward, 2011). In addition to serving as a conduit for movement of mature particles in and out of cells, microtubules have been shown to be important for other steps of viral replication, including providing transportation for capsids (Dohner et al., 2002; Sodeik et al., 1997) or viral RNA-protein complexes (Nturibi et al., 2017) or by contributing to efficient capsid formation (Iwamoto et al., 2017). In contrast to eukaryotes, cargo trafficking along tubulin filaments has not been reported in bacteria.
We recently described a family of Pseudomonas phages that assemble a bipolar spindle composed of a tubulin-like protein (PhuZ) (Erb et al., 2014; Kraemer et al., 2012; Zehr et al., 2014). PhuZ is expressed by phage 201Φ2–1 early during infection of P. chlororaphis, where it forms three-stranded filaments (Erb et al., 2014; Kraemer et al., 2012; Zehr et al., 2014, 2018) that spatially organize viral replication. Like eukaryotic microtubules, PhuZ filaments are polarized, with kinetically distinct plus and minus ends. PhuZ polymers display dynamic instability in vitro and in vivo; that is, filaments can cease growing at their plus ends, catastrophically depolymerize, and then be rescued to start growing again (Erb et al., 2014; Kraemer et al., 2012). Notably, a different behavior, treadmilling, in which polymerization at the plus end of the filament is matched by depolymerization at the minus end, has been observed with in vitro-assembled PhuZ polymers (Erb et al., 2014), but it has remained unclear whether filaments of the spindle can also treadmill in vivo.
The PhuZ spindle plays a key role in phage reproduction by positioning replicating phage DNA in the center of the cell (Erb et al., 2014; Kraemer et al., 2012). Phage 201Φ2–1 DNA is encased by a proteinaceous shell composed of gp105, forming a nucleus-like structure, referred to as the phage nucleus, that compartmentalizes proteins according to function (Chaikeeratisak et al., 2017b). Early during infection, the dynamically unstable filaments of the PhuZ spindle push the nucleus from the cell pole to the midcell where it then oscillates in position (Erb et al., 2014; Chaikeeratisak et al., 2017b). Proteins involved in DNA replication and transcription localize inside the phage nucleus, while ribosomes and proteins involved in metabolic processes localize in the cytoplasm on the outside (Chaikeeratisak et al., 2017b). The nucleus likely provides protection for phage DNA by excluding proteins of the host defense systems (restriction systems, nucleases, and CRISPR-Cas) that target invading phage DNA (Chaikeeratisak et al., 2017a, 2017b; Mendoza et al., 2018). The phage spindle and nucleus are conserved among the related large phages ΦPA3 and ΦKZ that replicate in P. aeruginosa (Aylett et al., 2013; Chaikeeratisak et al., 2017a).
Curiously, phage capsids assemble on the plasma membrane but must dock on the surface of the phage nucleus in order to package phage DNA (Chaikeeratisak et al., 2017a, 2017b). How capsids relocate from the membrane to the surface of the phage nucleus was unclear and potentially explained by three models (Figure 1A): (1) via “random diffusion” through the cell until capsids make contact with the nuclear shell; (2) via “DNA spooling,” in which strands of phage DNA emanating from the phage nucleus are captured by the packaging ATPase at the base of the capsid and the process of spooling DNA into the capsid transports them to the shell surface; or (3) via “capsid trafficking,” in which the PhuZ spindle plays a role in transporting capsids to the phage nucleus.
Figure 1. Phage Capsids Traffic along PhuZ Filaments to the Phage Nucleus for DNA Encapsidation by 60 mpi.

(A) Three possible models of capsid trajectory toward the phage nucleus.
(B) 3D-SIM images showing appearance of the phage nucleus at various developmental stages of infected P. chlororaphis cells. The phage nucleus shows uniform staining in the first 50 min of infection. At 60 min post infection (mpi), bright puncta appear surrounding the phage nucleus. Scale bars, 0.5 micron.
(C) Rapid time-lapse imaging of GFP-tagged capsid (gp200; green) and mCherry-tagged shell (gp105; red) in P. chlororaphis infected with phage 201Φ2–1 over a 34 s interval. Capsids (green) assemble near the cell membrane, and immediately after detachment, they independently migrate along the same straight-line trajectory toward the phage shell (red). Arrows indicate individual capsids. See also Data S1 (see Movie 1).
(D) Rapid time-lapse imaging of GFP-tagged capsids (gp200; green) and mCherry-tagged wild-type PhuZ (gp059; red) during an interval of 20 s in phage 201Φ21-infected P. chlororaphis cells. Blue arrow indicates a capsid traveling along the PhuZ spindle from the cell pole to phage nucleus. See also Data S1 (see Movie 3).
(E) Still images of phage 201Φ2–1-infected P. chlororaphis cells expressing GFP-tagged capsid (gp200; green) and mCherry-tagged wild-type PhuZ (gp059; red) at 45 and 50 mpi.
(F) Still images of phage 201Φ2–1-infected P. chlororaphis cells expressing GFP-tagged internal core protein (gp246; green) and mCherry-tagged capsid (gp200; red) at 50 mpi.
Dashed lines indicate cell borders. Scale bars in (C)–(F), 1 micron. See also Figures S1 and S2.
Here, we explore the mechanisms underlying capsid movement during phage infection. Using time-lapse microscopy and cryo-electron tomography (cryo-ET), we show that capsids move rapidly and directionally along PhuZ filaments but become trapped along mutant PhuZ filaments that are defective in guanosine triphosphate (GTP) hydrolysis. Photobleaching studies demonstrate that filaments of the spindle treadmill toward the nucleus, providing a simple mechanism by which capsids might be transported.
RESULTS
Capsids Move Directionally along PhuZ Filaments
To investigate models for capsid movement, we used rapid time-lapse imaging of GFP-tagged capsids and followed their movement from the cell membrane to the phage nuclear shell. We first identified the stage of infection during which capsids relocate by using DAPI staining and super-resolution fluorescence microscopy to determine approximately when DNA packaging occurs. DAPI staining of P. chlororaphis cells infected with phage 201Φ2–1 showed that nuclear staining intensity increased as DNA replicated until 50 min post infection (mpi) and that viral particles containing packaged DNA accumulated around the phage nucleus at 60 mpi (Figures 1B and S1A). We therefore used time-lapse imaging to study capsid trafficking between 40 and 60 mpi by simultaneously imaging fluorescently labeled capsids (gp200) and shells (gp105). P. chlororaphis cells expressing gp200-GFP and mCherry-gp105 were infected with phage 201Φ2–1, and at approximately 45 mpi when capsid assembly occurs, cells were imaged every 2 s for 2 min. In Figure 1C, multiple capsids (green foci) were observed to assemble on the cell membrane, where they remained motionless for several seconds (Data S1, see Movie 1) (Chaikeeratisak et al., 2017b). Capsids then independently followed a linear trajectory, seemingly traveling on the same intracellular track directly to the phage nucleus where they docked (Figure 1C; Data S1, see Movie 1). Movement was directional and rapid, with capsids traveling over 1 micron in length in fewer than 30 s. These results suggest that capsids move via either model 2 (DNA spooling) or model 3 (capsid trafficking) (Figure 1A). We obtained similar results with phage ΦPA3 infecting P. aeruginosa. Rapid time-lapse imaging of GFP-tagged PA3-capsids (gp136) and mCherry-tagged PA3-shell (gp053) showed that the trajectory of the capsids was a straight line moving rapidly toward midcell (Figure S1B; Data S1, see Movie 2).
To determine whether the PhuZ spindle is involved in capsid movement, we imaged GFP-tagged capsids with either mCherry-tagged wild-type PhuZ or a PhuZD190A mutant that blocks GTP hydrolysis. We infected P. chlororaphis cells expressing gp200 (capsid)-GFP and either wild-type mCherry-PhuZ or mutant mCherry-PhuZD190A and visualized capsid assembly and movement in time-lapse microscopy beginning at 45 mpi. Our capsid-PhuZ co-localization experiments demonstrated that, as capsids traveled through the cell toward the phage nucleus, they co-localized with PhuZ polymers and appeared to travel on the same path along the PhuZ filaments in both time-lapse (Figure 1D; Data S1, see Movie 3) and still images (Figure 1E, left). By 50 mpi, most capsids were docked on the phage nucleus (Figure 1E, right). Capsids co-localized with another protein involved in capsid assembly (internal core protein gp246), suggesting that the capsids that dock on the surface of the phage nucleus are fully assembled (Figure 1F).
PhuZ Mutants Defective in GTP Hydrolysis Block Capsid Migration and Rotation of the Phage Nucleus
In P. chlororaphis cells expressing the mutant mCherry-PhuZD190A, phage 201Φ2–1 capsids lined the filaments at both 50 mpi and 70 mpi (Figure 2A, arrows). In time-lapse microscopy, the capsids appeared to be immobilized on the sides of the PhuZ filaments (Figures 2C and S2C; Data S1, see Movie 4). Similar results were found for closely related phage ΦPA3, which replicates in P. aeruginosa by a similar mechanism (Chaikeeratisak et al., 2017b). ΦPA3 capsids individually travelled along similar trajectories (Figure S1B; Data S1, see Movie 2) and co-localized with and migrated along wild-type PhuZ filaments (Figure S1C; Data S1, see Movie 5). In the presence of catalytically defective PhuZD190A, ΦPA3 capsids became statically attached to filaments (Figures 2B, 2D, S2A, S2B, and S2D; Data S1, see Movie 6).
Figure 2. Phage Capsids Are Trapped along Mutant PhuZ Spindles in Both Phage 201𝚽2–1 and Phage 𝚽PA3, Resulting in Reduced Encapsidation.

(A) Fluorescence images of P. chlororaphis expressing GFP-tagged 201-capsid (gp200; green) and catalytically defective mCherry-tagged 201-PhuZD190A (gp059; red) infected with phage 201Φ2–1 at 50 and 70 mpi.
(B) Fluorescence images of P. aeruginosa expressing mCherry-tagged PA3-capsid (gp136; false color, green) and catalytically defective GFP-tagged PA3-PhuZD190A (gp028; false color, red) infected with phage ΦPA3 at 75 mpi.
(C) Time-lapse imaging of P. chlororaphis expressing GFP-tagged 201-capsid (gp200; green) with mCherry-tagged 201-PhuZD190A (gp059; red) over an interval of 88 s. See also Data S1 (see Movie 4).
(D) Time-lapse imaging of P. aeruginosa expressing mCherry-tagged PA3-capsid (gp136; false color, green) with GFP-tagged PA3-PhuZD190A (gp028; false color, red) over an interval of 299 s. See also Data S1 (see Movie 6).
(E) Distribution of GFP-tagged 201-capsids (gp200) in P. chlororaphis cells expressing either wild-type (left) or mutant (right) PhuZD190A infected with phage 201Φ2–1 at 50 mpi. Average Z-projection images (n = 32) of GFP intensity (top); distribution plots of GFP intensity of individuals (n = 32) (bottom).
(F) Graph showing percentage of detected GFP-tagged capsids in phage 201Φ2–1-infected P. chlororaphis cells expressing either wild-type (green) or mutant PhuZD190A (hatched bar) versus the fraction of cell length from the midcell to the cell pole.
(G) 3D-SIM images of encapsidated phage particles in phage 201Φ2–1-infected P. chlororaphis cells expressing either wild-type (top) or mutant (bottom) PhuZD190A at various time points. Arrows indicate positions of the phage nuclei.
(H) Box plot showing the number of encapsidated phage particles counted in phage 201Φ2–1-infected P. chlororaphis cells expressing either wild-type (top) or mutant (bottom) PhuZ at 80 mpi.
Asterisks indicate the average number of the encapsidated particles counted per strain. Dashed lines indicate the border of the cells. Scale bars, 1 micron. See also Figure S2.
DAPI staining was used as a way to assess, independently of GFP fusions, whether the expression of the PhuZD190A mutant interfered with capsid trafficking, which is required for DNA packaging at the phage nucleus. As previously reported (Chaikeeratisak et al., 2017a, 2017b), capsids containing DNA accumulated on the phage nucleus by 60 mpi (Figure 1B). However, in the presence of PhuZD190A, the number of encapsidated phage particles decreased approximately 5-fold (from an average of 10.5 to 2.1 particles), suggesting that capsid DNA packaging is delayed (Figures 2G and 2H). Taken together, our results suggest that capsids rely upon the PhuZ spindle to migrate rapidly to the phage nucleus and the GTP hydrolysis mutations in PhuZ trap capsids along static filaments and therefore delay DNA packaging.
To further confirm the difference in capsid positioning between cells expressing the wild-type or PhuZD190A mutant at 50 mpi, we quantitated gp200 (capsid)-GFP position from 32 infected cells. We plotted capsid position versus normalized cell length and found that the distribution of capsids is biased toward the midcell near the nucleus in cells expressing wild-type PhuZ, with approximately 63% of capsids occurring within 30% of the cell midpoint (Figures 2E and 2F). However, in the presence of the PhuZD190A mutant, the capsids are more evenly distributed throughout the cell (Figures 2E and 2F). The effect of the PhuZD190A catalytic mutation on capsid distribution is consistent with the PhuZ spindle playing a role in capsid migration.
We then used cryo-focused ion beam milling (cryo-FIB) coupled with cryo-ET to visualize capsids associated with filaments at high resolution in a near-native state (Chaikeeratisak et al., 2017b). Since capsids only transiently interacted with filaments as they translocated to the phage nucleus in wild-type cells, we performed cryo-FIB-ET on cells expressing PhuZD190A mutants in which capsids appeared to become trapped. In samples of P. aeruginosa cells expressing mutant PhuZD190A and infected with phage ΦPA3 for 70 min, 18 empty capsids (green) were clustered around PhuZ filaments (blue) in a ~200 nm slice of the sample (Figure 3A, 3B, and S2E). At this time point, capsids are normally filled with DNA in wild-type cells (Figure S2F), yet these capsids are empty, suggesting that capsids associated with the mutant PhuZ filaments are unable to traffic to the nucleus for DNA packaging, in agreement with fluorescence microscopy data (Figure 2G). Since capsids appear in close proximity (3–4 nm) to the filament (Figures 3C–3H), we attempted to identify amino acids that are conserved on the outer surface of PhuZ filaments that might possibly serve as a point of attachment. Three aspartic acid residues (D235, D259, D263) that are conserved among PhuZ proteins encoded by ΦPA3, ΦKZ, and 201Φ2–1 were mutated to alanine (Figure S3D). One of them, D235A, completely blocked filament assembly (data not shown). The mutants D259A and D263A retained their ability to assemble filaments, while the double mutant, D259A-D263A, appeared slightly impaired in filament formation. We examined the ability of capsids to traffic along all three of these mutant filaments using time-lapse microscopy (Figure S3C). We also examined DNA packaging into capsids (a hallmark of successful trafficking) and nucleus rotation in the three mutants (Figure S3B). In each case, capsids were still able to move along the mutant filaments, mature capsids filled with DNA were observed, and the phage nucleus rotated at midcell. Attempts to detect a direct interaction between PhuZ and capsid proteins using yeast two-hybrid systems also yielded a negative result (Figure S3A).
Figure 3. Cryo-electron Tomography Revealing Capsids Trapped along Mutant PhuZ Filaments during Phage 𝚽PA3 Infection in P. aeruginosa at 70 mpi.
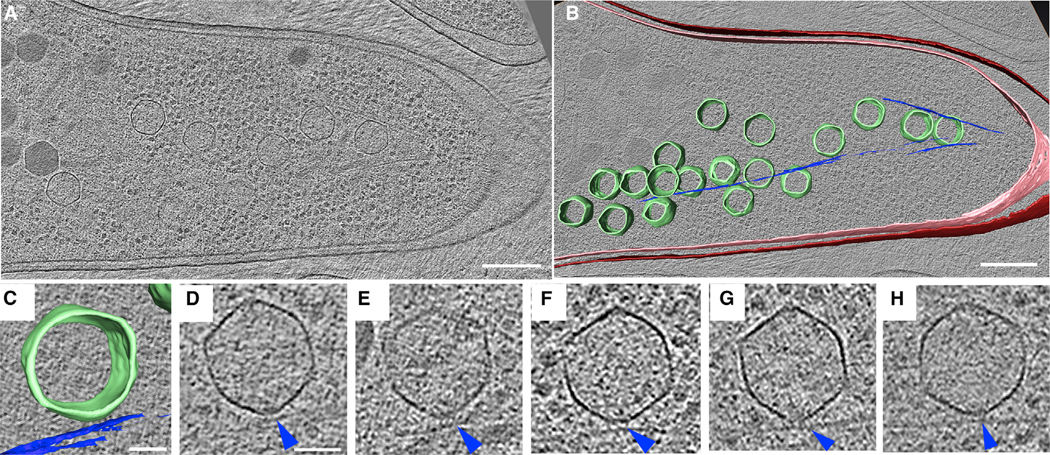
(A) A slice through a tomogram of a cryo-focused ion beam–thinned phage-infected cell at 70 mpi. Scale bar, 200 nm.
(B) Annotation of the tomogram in (A) showing extracted structures, including capsids (green), cytoplasmic membrane (pink), outer membrane (red), and mutant PhuZD190A spindles (blue). Scale bar, 200 nm.
(C and D) Zoomed-in view (C) of one of the capsids stuck on the mutant filament from the tomogram shown in (A) and its corresponding tomogram slice (D). Scale bar, 50 nm.
(E–H) Slices of tomograms of capsids trapped along the mutant filaments from tomograms of other phage-infected cells taken for this study, with blue arrow pointing towards the mutant spindle.
See also Figure S2.
During phage 201Φ2–1 infection, the phage nucleus is pushed from the cell pole to the cell midpoint where, at approximately 40–45 mpi, it suddenly begins to rotate in position (Chaikeeratisak et al., 2017b) (Figures 4A and S4A; Data S1, see Movie 7). Figure 4A and Data S1 (see Movie 7) show an example of a phage nucleus (green) being pushed transversely by the PhuZ spindle (red) and undergoing approximately two revolutions within 60 s, with an average linear velocity of 43.6 ± 7.6 nm/s (n = 10). In contrast, phage nuclei formed in the presence of mutant PhuZD190A are both mispositioned and do not rotate (Figures 4B and S4B; Data S1, see Movie 8). At 45 mpi, 46% (n = 611) of the wild-type nuclei underwent rotations in the presence of wild-type filaments, while in the presence of the PhuZD190A mutant, less than 6% (n = 286) rotated (Figure 4D). The sudden onset of phage nucleus rotation, which occurs at approximately the same time as capsid movement begins, suggests a switch in the dynamics of the PhuZ spindle from primarily centering the phage nucleus to rotating it.
Figure 4. Rotation of the Phage Nucleus Exerted by PhuZ Spindle Distribute Phage Capsids around the Nucleus.

(A and B) Rapid time-lapse imaging of phage 201Φ2–1-infected P. chlororaphis expressing GFP-tagged shell (gp105) with either mCherry-tagged wild-type PhuZ (gp059) (A) or mCherry-tagged mutant PhuZD190A (gp059) (B) during 60 s intervals. In the presence of wild-type filaments, the shell (green) rotates counter-clockwise when the PhuZ filaments push the shell transversely; the shell successfully rotates twice within 42 s. The mutant PhuZD190A is unable to catalyze GTP hydrolysis and appears static, resulting in a mispositioned and motionless shell within the infected cell. See also Data S1 (see Movies 7 and 8).
(C) Rapid time-lapse microscopy of phage 201Φ2–1-infected P. chlororaphis expressing GFP-tagged capsid (gp200) and mCherry-tagged wild-type PhuZ (gp059) in a 26 s interval. A capsid (arrow) travels along the filament from cell pole toward the phage nucleus, which rotates counterclockwise. The capsid docks on the surface of the nucleus at 26 s. Dashed lines indicate cell borders. Scale bars, 1 micron. See also Data S1 (see Movie 9).
(D) Graph showing the percentage of rotating nuclei in P. chlororaphis infected cells in the presence of either wild-type PhuZ (wt) or mutant PhuZD190A (mt). The graph shows that the number of rotating nuclei in the presence of wild-type PhuZ (46.2%) is significantly higher (p < 0.01) than that in the presence of mutant PhuZD190A (5.9%). Data were collected from infected cells at 50 mpi from at least three different fields and are represented as mean ± SE (n; wt = 611 and mt = 286).
See also Figure S4.
Rotation is dependent upon the dynamic GTPase activity of the PhuZ spindle, raising the question of what role rotation might play in phage reproduction. In our time-lapse co-localization experiments, capsids trafficked along the spindle and docked on the rotating shell (Figure 4C; Data S1, see Movie 9). The process of rotation appears to distribute capsids widely around the nuclear shell. We obtained identical results with phage ΦPA3 in P. aeruginosa: capsids traveled along PhuZ filaments and docked on the rotating phage nucleus, where they then rotated together (Figures S4C and S4D; Data S1, see Movies 10 and 11), showing that this mechanism is conserved among this family of large Pseudomonas phages.
GTP-Dependent Treadmilling of PhuZ Filaments Is Responsible for Capsid Movement and Rotation of the Phage Nucleus
The mechanism underlying capsid movement along filaments was unclear. In eukaryotic cells, cargo trafficking along microtubules is typically dependent upon motor proteins such as kinesin and dynein that use ATP hydrolysis to drive unidirectional transport. However, phage and bacteria are generally thought to lack such motor proteins. Therefore, we explored the possibility that capsid movement is driven by filament treadmilling. In this model, insertion of new subunits occurs at one end of the filaments located near the cell poles, thereby driving capsids attached to the sides of the filaments toward midcell. To determine whether spindle filaments treadmill, we used photobleaching to mark one end of the spindle after approximately 45–50 mpi, when capsids are migrating to the nucleus. As shown in Figures 5A and 5B, bleached zones (arrows) near the ends of the spindles moved rapidly toward the nucleus (Data S1, see Movies 12 and 13). The median rate of bleached-zone movement through the cell was 51.5 nm/s (n = 40; Figures 5C and 5E), suggesting that filaments treadmill at a constant rate with no evidence of significant pausing even when taking images more frequently (Figure S5B) or expanding the length of the cells with antibiotics to increase the distance of travel (Figure 5C). In all cases, regardless of filament length, bleached zones moved at a constant rate toward midcell. When both sides of a bipolar spindle were bleached, the two bleached zones migrated in unison toward the cell center with similar rates, arriving at the nucleus at the same time (Figure 5B; Data S1, see Movie 13). Quantitation of the rate of capsid migration toward the nucleus revealed that they move with a median rate of 50.3 nm/s (n = 10; Figures 5D and 5E), very similar to the rate of treadmilling (51.5 nm/s, n = 40). In the presence of the PhuZD190A mutant, the average rates of filament treadmilling and capsid migration both fall to near zero (Figures 5E and S4E; Data S1, see Movie 14). These results suggest that filaments of the spindle require GTP hydrolysis to treadmill and transport capsids toward the nucleus (Figure 5F), providing a potential mechanism for capsid migration driven by net filament growth near the poles and net loss near the nucleus. Nucleus rotation also occurred at a similar rate and was dependent upon PhuZ GTP hydrolysis, suggesting that treadmilling is responsible for both capsid movement and rotation, thereby temporally coupling these two processes.
Figure 5. PhuZ Filaments Treadmill Unidirectionally toward the Nucleus at a Constant Rate.

(A) A single photobleaching event at the cell pole(arrow) moves toward the phage nucleus. See also Data S1 (see Movie 12).
(B) A double photobleaching event shows that bleach spots made at both cell poles (arrows) flux down the filaments toward the phage nucleus. Scale bars, 1 micron. See also Data S1 (see Movie 13).
(C) Graph showing rates of bleach-spot movement (distance in microns versus time in seconds) in wild-type ΦPA3-PhuZ filaments, mutant ΦPA3PhuZD190A filaments, and wild-type ΦPA3-PhuZ filaments treated with the antibiotic ticarcillin to produce elongated cells.
(D) Graph showing rates of capsid movement (distance in microns versus time in seconds) when co-expressed with either wild-type ΦPA3-PhuZ or the mutant ΦPA3-PhuZD190A.
(E) Box-and-whisker plots showing average speeds of movement of bleach spots on wild-type PA3 PhuZ filaments (WT filament), on wild-type PA3 PhuZ filaments in ticaricillin-treated cells (ticaricillin-treated filament), or on mutant PA3 PhuZD190A filaments (MT filament). Average speed of capsid movement in cells with wild PA3 PhuZ filaments (WT capsid) or mutant PA3 PhuZD190A filaments (MT capsid).
(F) Model of PhuZ filament treadmilling, indicating that addition of new subunits causes the bleached subunits (gray) to flux toward the nucleus. See also Figures S4 and S5 and Data S1 (see Movie 14).
DISCUSSION
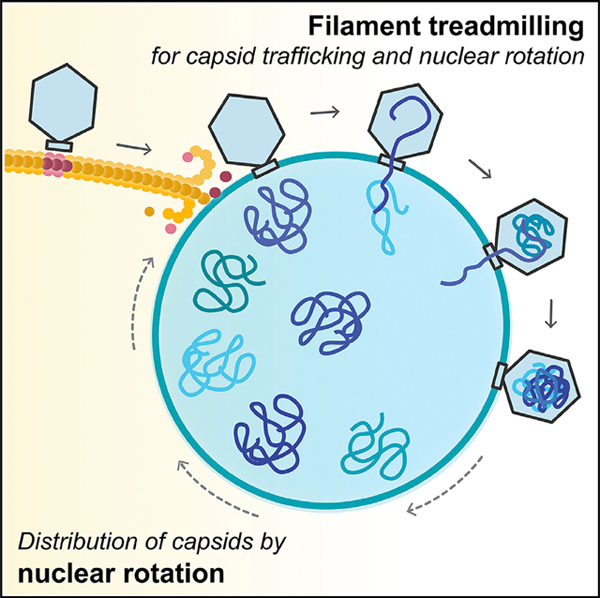
Our results suggest a model in which capsids assemble on the host cell membrane and migrate along filaments of the PhuZ spindle to reach the phage nucleus (Figure 6). Delivering capsids directly to the surface of the shell increases the rate at which these large phage particles dock and begin the process of DNA encapsidation. Rotation of the nucleus by the spindle likely serves to distribute the capsids more evenly around the shell surface, maximizing efficient DNA packaging (Figure 6E). These phage genomes are 316 kilobases (kb) (phage 201Φ2–1) and 309 kb (phage ΦPA3) in size, and therefore occupy substantial space inside the phage nucleus. If two particles docked immediately adjacent to one another, only one might be able to package DNA. Capsid trafficking is driven by GTP-dependent PhuZ polymerization and is conserved between phages 201Φ−1 and ΦPA3 and likely other phages containing PhuZ homologs (Figure S5). PhuZ is conserved in a number of large phage genomes, including those that infect Vibrio and Erwinia (Figure S5C). These results show that phages evolved a mechanism for trafficking cargo using a cytoskeletal element in bacteria.
Figure 6. Model of Capsid Trafficking and the Role of Nucleus Rotation in Distributing Capsids around the Phage Nucleus.

The spindle’s functions are complex and change as phage development proceeds.
(A) At the onset of infection, dynamically unstable filaments assemble with minus ends anchored at the cell poles, and plus ends oriented toward midcell push the growing phage nucleus to the cell midpoint where it oscillates in position.
(B) Model of dynamically unstable filaments showing cycles of polymerization, depolymerization, and recovery at the plus end of the polymer.
(C) Later in infection, the function of the spindle switches from centering the nucleus to rotating it in position and transporting capsids to the phage nucleus for DNA packaging. Treadmilling provides the driving force and temporally couples both processes.
(D) Model of treadmilling filaments showing addition of new subunits at the minus end near the cell pole drives photobleached subunits (purple) toward midcell.
(E) Rotation of the phage nucleus serves to distribute capsids evenly around its surface.
(F) Capsids are delivered to the surface of the phage nucleus for DNA packaging.
Our results suggest a simple transport mechanism in which treadmilling filaments are responsible for rapid and directional capsid movement. Newly formed capsids release from the membrane, diffuse a short distance, and attach to the sides of filaments (Figures 6C–6F). As new subunits are added to filament ends located at the cell poles, capsids are transported by the polymer to the phage nucleus. Capsids are released from filaments when they reach the depolymerizing ends adjacent to the nucleus, allowing them to dock on the phage nuclear surface (Figures 6E and 6F). This model is supported by our finding that PhuZ subunits flux toward the nucleus at a rate almost identical to the rate of capsid movement.
Upon DNA injection and formation of the phage nucleus, spindle assembly begins when PhuZ monomers nucleate to form a hexameric structure to which additional monomers are added (Zehr et al., 2014). The filaments of the PhuZ spindle use dynamic instability to position and maintain the phage nucleus at midcell (Figures 6A and 6B) (Erb et. al., 2014; Kraemer et. al., 2012). We previously demonstrated that PhuZ filaments are intrinsically polar, with the plus end of the filament exhibiting rapid growth and dynamic instability in vitro. In vivo, the minus end of the filament is localized to the cell pole, and the dynamically unstable plus end is oriented toward midcell (Figures 6A and 6B). Later during phage replication, the behavior of the spindle changes from positioning the nucleus at midcell (Figures 6A and 6B) to rotating it in position concomitant with the transport of capsids (Figures 6C and 6D), suggesting developmental regulation of spindle dynamics. PhuZ filaments flux toward the phage nucleus, suggesting that, given the previous demonstration of polarity, the minus ends located at the cell poles are capable of constant growth. While filament minus ends can also grow, albeit at a higher critical concentration than the plus ends, what is unexpected here is that minus-end growth and plus-end dynamic instability would exist simultaneously. While the cellular concentration of PhuZ monomers are unknown, proteomics experiments show that the total amount of PhuZ protein increases as infection progresses (Figure S5A), which could lead to altered filament dynamics over time. In isolated microtubules, the higher tubulin concentrations needed for minus-end growth would lead to rapid plus-end growth and less frequent catastrophic collapse, although both can occur simultaneously (Walker et al., 1988). However, the in vivo situation here is far more complex, leading to several plausible explanations: (1) the effective concentration of PhuZ could be higher at the poles than the midcell; (2) resistance imposed by centering the nucleus could accelerate depolymerization at the midcell; (3) bundling of the filaments toward the poles could stabilize the minus ends and enhance the effective on-rate. Previous work suggested that there is a phage-encoded mechanism for anchoring PhuZ filaments at the poles (Erb et. al., 2014) and that this could facilitate adding new subunits, much as the microtubule-associated protein XMAP215 does (Ayaz et al., 2014; Brouhard et al., 2008). In vivo, PhuZ filaments are clearly bundled along much of their length but splay near the midcell, becoming much harder to visualize (Erb et. al., 2014). In vitro studies have shown that non-dynamic PhuZ filaments are much more likely to bundle than their dynamic counterparts (Erb et al., 2014; Kraemer et al., 2012; Zehr et al., 2014). Thus, either the statistics of having many available minus ends or a structure altered by bundling could also accelerate minus-end addition. Given that the functions of the spindle change over time, it’s not surprising that spindle behavior and dynamics are complex.
Microtubules also display different behaviors depending upon the specific cell type, buffer conditions, or accessory factors. While minus ends grow at a slower rate and higher threshold concentration than the plus ends, the specific conditions can affect the two ends in different ways. This variability in conditions leads to the observation of dynamic instability (Brouhard and Rice, 2018; Mitchison and Kirschner, 1984; Zhang et al., 2015) at both ends of a microtubule and treadmilling-like behavior (Grego et al., 2001; Margolis and Wilson, 2019; Rodionov et al., 1999; Rodionov and Borisy, 1997; Shaw et al., 2003; Waterman-Storer and Salmon, 1997a, 1997b). In fact, microtubules have been observed to treadmill in both the minus-to-plus and plus-to-minus directions in vitro (Grego et al., 2001). Similarly, our results show that PhuZ filaments are capable of displaying two distinct behaviors coupled to the spindle’s biological functions: dynamic instability, which is essential for centering the phage nucleus, and treadmilling, which is necessary for transporting phage capsids and rotating the nucleus. These two types of behaviors are consistent with our prior studies, further illuminate the central role of the PhuZ cytoskeleton in phage reproduction, and highlight the plasticity of tubulins in assembling unique polymers tailored to performing specific cellular activities.
The nature of the proposed interaction between capsids and PhuZ polymers is unclear. In principle, capsids might associate with the sides of PhuZ filaments directly or indirectly via another protein. Precedence for capsids directly interacting with microtubules has been reported for several eukaryotic viruses. For example, the Hepatitis C virus interacts with tubulins, affects microtubule dynamics, and has been proposed to move through the cytoplasm by associating with dynamic microtubules (Roohvand et al., 2009). Our results suggest a common solution to viral transport that occurs in evolutionarily divergent domains of life.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joe Pogliano (jpogliano@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strain, growth condition, and bacteriophage preparation
Bacterial strains used in this study are listed in Key Resource Table and Table S1. To amplify the strain, Pseudomonas chlororaphis strain 200-B was grown on Hard Agar (HA) containing 10 g Bacto-Tryptone, 5 g NaCl, and 10 g agar in 1L ddH2O and incubated at 30°C overnight. Pseudomonas aeruginosa strains PA01 and PA01-K2733 (pump-knockout strain) were grown on Luria-Bertani (LB) media containing 10g Bacto-Tryptone, 5g NaCl, 5g Bacto-yeast extract in 1L ddH2O and incubated at 30°C overnight. Lysates for phages 201Φ2–1 and ΦPA3 were made by infecting respective saturated host cultures with 10μl of high titer lysate, incubating for 15 minutes at room temperature, mixing with HA top agar (0.35%; phage 201Φ2–1) or LB top agar (0.35%; phage ΦPA3) and pouring over corresponding HA or LB plates. Plates were incubated at 30°C overnight. Plates that formed web-lysis were then flooded with 5mL of phage buffer and incubated at room temperature for 5 hours. The phage lysates were then aspirated, clarified by centrifugation at 15,000 rpm for 10 minutes, and stored at 4°C with 0.01% chloroform by volume.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| NEB 5-alpha Competent E. coli | New England Biolabs | Cat#C2987H |
| Pseudomonas aeruginosa (PAO1) | Pogliano strain collection | N/A |
| Pseudomonas aeruginosa K2733 | Keith Poole of Queen’s University Kingston, ON, Canada |
N/A |
| Pseudomonas chlororaphis 200-B | Drs. Julie Thomas and Steven Hardies of UT Health Sciences San Antonio | N/A |
| Pseudomonas aeruginosa phage PhiPA3 | Dr. Marcy Erb | N/A |
| Pseudomonas chlororaphis phage 201phi2–1 | Drs. Julie Thomas and Steven Hardies of UT Health Sciences San Antonio | N/A |
| Saccharomyces cerevisiae Y2H Gold | Clontech | Cat#630489 |
| Saccharomyces cerevisiae Y187 | Clontech | Cat#630489 |
| See Table S1 for a complete list of strains used in this paper | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FM 4–64 | Thermo Fisher Scientific | Cat#T13320 |
| DAPI (4’,6-Diamidino-2-Phenylindole, Dihydrochloride) |
Thermo Fisher Scientific | Cat#D1306 |
| Ticarcillin disodium salt | Sigma-Aldrich | Cat#T5639 |
| Gentamycin sulfate | Sigma-Aldrich | Cat#G1914 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530L |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat#N0447L |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat#E5520S |
| T4 DNA ligase | New England Biolabs | Cat#M0202L |
| Nucleospin plasmid extraction kit | Macherey-Nagel | Cat#740588 |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | Cat#740609 |
| Matchmaker Gold Yeast Two-Hybrid System | Clontech | Cat#630489 |
| Oligonucleotides | ||
| See Table S2 for a complete list of oligonucleotides used in this paper | N/A | |
| Recombinant DNA | ||
| pHERD30T plasmid | Dr. Hongwei D Yu of Marshall University | N/A |
| See Tables S3 and S4 | N/A | |
| Software and Algorithms | ||
| DeltaVision SoftWoRx 6.5.2 | GE HealthCare | http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/7.0.0/SoftWoRx.htm |
| FIJI/ImageJ | NIH | https://imagej.nih.gov/ij/ |
| CellProfiler 2.0 | Lamprecht et al., 2007 | cellprofiler.org |
| Amira | Commercial software by Thermo Scientific (formerly FEI) |
https://www.fei.com/software/amira-3d-for-lifesciences/; RRID:SCR_014305 |
| TomoSegMemTV | Martinez-Sanchez et al., 2014 | https://sites.google.com/site/3demimageprocessing/tomosegmemtv |
| IMOD | Kremer et al., 1996 | http://bio3d.colorado.edu/imod/; RRID:SCR_003297 |
| Microsoft Excel | Microsoft Office | https://products.office.com/en-us/excel |
| Adobe Photoshop | Adobe | https://www.adobe.com/products/photoshop.html |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Adobe Acrobat | Adobe | https://www.adobe.com/products/acrobat.html |
Plasmid constructions and bacterial transformation
Fluorescent-tagged phage proteins were constructed into the pHERD 30T vector backbone using oligonucleotides as listed in Table S2. Phage genes were directly amplified from high-titer lysates of phages 201Φ2–1 or ΦPA3 using PCR amplification. Amplicons and vector backbones were then ligated together to generate phage gene-containing plasmids via isothermal assembly, immediately followed by transformation into E. coli DH5α plated on LB supplemented with gentamycin sulfate (15μg/mL). See also Table S4 for more detail on plasmid construction. Constructs were confirmed with sequencing and subsequently electroporated into either P. chlororaphis strain 200-B or P. aeruginosa strains PA01 and/or PA01-K2733. P. chlororaphis strain was grown on HA supplemented with gentamycin sulfate (25μg/mL) and P. aeruginosa strains PA01 and PA01-K2733 were grown on LB supplemented with gentamycin sulfate either at 300 μg/mL or 15μg/mL, respectively. See Tables S1 and S3 for a list of plasmids and strains.
METHOD DETAILS
Single cell-infection assay
The bacterial cells were grown on 1.2% agarose pads, supplemented with desired arabinose to induce fluorescent-tagged protein expression to label wild-type proteins from phage. The cells were then incubated at 30°C for 3 hours without a coverslip in a humid chamber. 5 μl of high-titer lysate (1010 pfu/ml) was added to the corresponding host cells on agarose pads to begin the phage infection, and then the cells were further incubated to allow phage infections occur. For ticarcillin treated experiments, 10μl of 50mg/ml TIC stock was added to cells 1.5 hours post inoculation on agarose pads and infection was performed thereafter as mentioned above. At desired time points after phage infection, a coverslip was put on the slide and fluorescent microscopy was then initiated. Data of static images and time-lapse imaging were collected and processed as described below.
Fixed and live cell static-image fluorescence microscopy
The fixed cell imaging protocol was adopted from “Fixed Cell Imaging” methodology in Chaikeeratisak et al., 2017. The DeltaVision Spectris Deconvolution microscope (Applied Precision, Issaquah, WA, USA) was used to visualize the fixed and live cells. For static images, the cells were imaged at 8 stacks in the Z-axis from the focal plane with 0.15 μm increments. Images were further processed by the deconvolution algorithm in DeltaVision SoftWoRx Image Analysis Program. Further image analysis and processing was performed in Fiji.
Time-lapse fluorescence microscopy
For time-lapse imaging, the cells were prepared and infected as indicated above. Cells were imaged from a single stack at the focal plane for desired length of time at selected intervals using Ultimate Focusing mode. Timelapse images were later deconvolved and analyzed as stated above.
3D-Structured Illumination Microscopy
Pseudomonas cells were grown, infected, and fixed as indicated above. Cells were stained with 1 μg/mL DAPI and then imaged using an Applied Precision/GE OMX V2.2 Microscope. Microscopic raw data were sequentially taken by SI-super-resolution light path to collect 3 mm thickness of samples with 125 nm increments in the z-axis with compatible immersion oils (Applied Precision). 3D-structured illumination microscopy (SIM) images were then rendered by standard OMX SI reconstruction parameters in DeltaVision SoftWoRx Image Analysis Program.
Fluorescence Recovery After Photobleaching
For time-lapse imaging, the cells were prepared and infected as indicated above. Filaments were photobleached using a laser (QLM module, API) for 0.05 sec at 31.3% power and then followed with time lapse imaging – images taken every 2 seconds for 1 minute with the Applied Precision/GE OMX V2.2 Microscope. Images were deconvolved with DeltaVision SoftWoRx.
Yeast Two-hybrid assay
Interaction between the proteins of interest was investigated using the Yeastmaker Yeast Transformation System 2. Partial gene and full-length gene were cloned into the bait (pGBKT7) and prey (pGADT7) vector (Clontech), as described in Table S1. The resulting recombinant constructs were transformed into the yeast Saccharomyces cerevisiae strain Gold and Y187 (Clontech) using the Yeast-maker™ Yeast Transformation System 2 (Clontech). Both yeast colonies containing the protein of interest were paired in a single 1.5mL centrifuge tube containing 500μL of 2X YPDA and vortexed to mix. Colonies were incubated shaking at 200 rpm at 30°C overnight. The mated cultures were plated on selective agar plates DDO and QDO/X/A to investigate the positive interaction. All constructs were tested for toxicity and autoactivation prior to the experiments. Y2HGold containing pGBKT7–53 with Y187 containing pGADT7-T was used as the positive control and Y2HGold containing pGBKT7-Lam with Y187 containing pGADT7-T was used as the negative control.
Tomography Sample Preparation and Data Acquisition
Infection of P. aeruginosa cells with phage ΦPA3 was done as indicated above. At 70 mpi, cells were scraped off from the surface of the pad using ¼ LB media. 7 μl of cells were deposited on holey carbon coated QUANTIFOIL® R 2/1 copper grids that were glow discharged using Pelco easiGlow™ glow discharge cleaning system. Manual blotting from the side of the grid opposite to the cells using Whatman No. 1 filter paper removed excess liquid such that cells form a monolayer on the surface of the grid. Cells were then plunge-frozen in a mixture of ethane/propane using a custom-built vitrification device (Max Planck Institute for Biochemistry, Munich).
Grids were then mounted into modified FEI Autogrids™ to avoid any mechanical damage to the delicate grids during subsequent transfer steps. Then, these clipped grids were transferred into Scios (Thermo Fisher Scientific, formerly FEI), a dual-beam (cryo-FIB/SEM) microscope equipped with a cryogenic stage. Thin sections of 100–250 nm, or lamellae, were prepared as previously described in Chaikeeratisak et al., 2017 containing 10–12 cells each.
Tilt-series were collected from typically −65° to +65° with 1.5° or 2° tilt increments using SerialEM4 in a 300-keV Tecnai G2 Polara microscope (FEI) equipped with post-column Quantum Energy Filter (Gatan) and a K2 Summit 4k x 4k direct detector camera (Gatan). Images were recorded at a nominal magnification of 34,000 with a pixel size of 0.61 nm. The dose rate was set to 10–12 e/physical pixel at the camera level. Frame exposure was set to 0.1 seconds, with a total exposure in a frame set to be determined by an algorithm targeting an average count number. The total dose in a tomogram was typically ~100–120 e/A2 with a defocus of −5 μm. The dataset for this study consists of 16 tomograms from 7 FIB-milled lamellas. Reconstruction of tilt-series was done in IMOD (Kremer et al., 1996) using patch tracking method. Semi-automatic segmentation of the membranes was done using TomoSegMemTV (Martinez-Sanchez et al., 2014), an open-source software based on tensor voting, followed by manual segmentation with Amira software (FEI Visualization Sciences Group). Filaments were traced manually using Amira as well.
QUANTIFICATION AND STATISTICAL ANALYSIS
To visualize the consensus location of gp200 (capsids), 32 single cell images from each experiment, wild type and mutant, were aligned and stacked using FIJI image analysis program (Schindelin et al., 2012). Then, the average GFP signals of all images in the stack were projected onto a single image. To visualize gp200 distribution of each cell, the GFP signal from each single cell image was combined in to a 1-pixel line and stacked. Performing on 8-bit GFP channel images, the location of gp200 from mid-cell was automatically measured by CellProfiler 2.0 image analysis program (Lamprecht et al., 2007) and normalized by cell length. Locations of bleach spots and capsids were measured from microscopy images in FIJI and plotted as distance over time in Microsoft Excel.
Analyses of the number of cells or phage particles were done from at least 3 independent biological experiments. Data represent as mean values or mean ±SEM. Pairwise comparison between the data were performed using Student’s t test for unpaired data with unequal variance. (A p value less than 0.05 indicates a significant difference) All statistical analysis, data processing, and data presentation were conducted using KaleidaGraph and Microsoft Excel.
Supplementary Material
Highlights.
Capsids traffic along a viral encoded tubulin filament
Treadmilling of the filament provides the mechanism of capsid movement through the cell
Rotation of phage nucleus by the filament distributes capsids for efficient DNA packaging
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health grants GM031627, R35GM118099 (D.A.A.), GM104556 (J.P. and D.A.A.), GM129245 (J.P. and E.V.), R01-GM57045 (K.P.), and 1DP2GM123494–01 (E.V.) and by the Howard Hughes Medical Institute (D.A.A.). V.C. was supported by the Thailand research fund and the office of the higher education commission (MRG6180027). P.N. was supported by the Thailand research fund and the office of the higher education commission (MRG6080081). We used the UCSD Cryo-EM Facility (supported by NIH grants to Dr. Timothy S. Baker and the Agouron Institute) and the San Diego Nanotechnology Infrastructure of UCSD (supported by the NSF grant ECCS-1542148). We would like to thank Ethan Garner and Arshad Desai for helpful discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.05.032.
REFERENCES
- Ayaz P, Munyoki S, Geyer EA, Piedra FA, Vu ES, Bromberg R, Otwinowski Z, Grishin NV, Brautigam CA, and Rice LM (2014). A tethered delivery mechanism explains the catalytic action of a microtubule polymerase. eLife 3, e03069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylett CH, Izoré T, Amos LA, and Löwe J. (2013). Structure of the tubulin/FtsZ-like protein TubZ from Pseudomonas bacteriophage FKZ. J. Mol. Biol 425, 2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ, and Rice LM (2018). Microtubule dynamics: an interplay of biochemistry and mechanics. Nat. Rev. Mol. Cell Biol 19, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouhard GJ, Stear JH, Noetzel TL, Al-Bassam J, Kinoshita K, Harrison SC, Howard J, and Hyman AA (2008). XMAP215 is a processive microtubule polymerase. Cell 132, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikeeratisak V, Nguyen K, Egan ME, Erb ML, Vavilina A, and Pogliano J. (2017a). The Phage Nucleus and Tubulin Spindle Are Conserved among Large Pseudomonas Phages. Cell Rep. 20, 1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikeeratisak V, Nguyen K, Khanna K, Brilot AF, Erb ML, Coker JK, Vavilina A, Newton GL, Buschauer R, Pogliano K, et al. (2017b). Assembly of a nucleus-like structure during viral replication in bacteria. Science 355, 194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Au S, and Pante N. (2011). How viruses access the nucleus. Biochim. Biophys. Acta 1813, 1634–1645. [DOI] [PubMed] [Google Scholar]
- Dohner K, Wolfstein A, Prank U, Echeverri C, Dujardin D, Vallee R, and Sodeik B. (2002). Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 13, 2795–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb ML, Kraemer JA, Coker JK, Chaikeeratisak V, Nonejuie P, Agard DA, and Pogliano J. (2014). A bacteriophage tubulin harnesses dynamic instability to center DNA in infected cells. eLife 3, e03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber UF, and Way M. (2006). A superhighway to virus infection. Cell 124, 741–754. [DOI] [PubMed] [Google Scholar]
- Grego S, Cantillana V, and Salmon ED (2001). Microtubule treadmilling in vitro investigated by fluorescence speckle and confocal microscopy. Biophys. J 81, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Cai D, Sugiyama M, Suzuki R, Aizaki H, Ryo A, Ohtani N, Tanaka Y, Mizokami M, Wakita T, et al. (2017). Functional association of cellular microtubules with viral capsid assembly supports efficient hepatitis B virus replication. Sci. Rep 7, 10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer JA, Erb ML, Waddling CA, Montabana EA, Zehr EA, Wang H, Nguyen K, Pham DS, Agard DA, and Pogliano J. (2012). A phage tubulin assembles dynamic filaments by an atypical mechanism to center viral DNA within the host cell. Cell 149, 1488–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, and McIntosh JR (1996). Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol 116, 71–76. [DOI] [PubMed] [Google Scholar]
- Lamprecht MR, Sabatini DM, and Carpenter AE (2007). CellProfiler: free, versatile software for automated biological image analysis. Biotechniques 42, 71–75. [DOI] [PubMed] [Google Scholar]
- Margolis RL, and Wilson L. (2019). Microtubule treadmills—possible molecular machinery. Nature 293, 705. [DOI] [PubMed] [Google Scholar]
- Martinez-Sanchez A, Garcia I, Asano S, Lucic V, and Fernandez JJ (2014). Robust membrane detection based on tensor voting for electron tomography. J. Struct. Biol 186, 49–61. [DOI] [PubMed] [Google Scholar]
- Mendoza SD, Berry JD, Nieweglowska ES, Leon LM, Agard D, and Bondy-Denomy J. (2018). A nucleus-like compartment shields bacteriophage DNA from CRISPR-Cas and restriction nucleases. bioRxiv. 10.1101/370791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, and Kirschner M. (1984). Dynamic instability of microtubule growth. Nature 312, 237–242. [DOI] [PubMed] [Google Scholar]
- Nturibi E, Bhagwat AR, Coburn S, Myerburg MM, and Lakdawala SS (2017). Intracellular Colocalization of Influenza Viral RNA and Rab11A Is Dependent upon Microtubule Filaments. J. Virol 19, e01179–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portilho DM, Persson R, and Arhel N. (2016). Role of non-motile microtubule-associated proteins in virus trafficking. Biomol. Concepts 7, 283–292. [DOI] [PubMed] [Google Scholar]
- Rodionov VI, and Borisy GG (1997). Microtubule Treadmilling in Vivo. Science 275, 215–218. [DOI] [PubMed] [Google Scholar]
- Rodionov V, Nadezhdina E, and Borisy G. (1999). Centrosomal control of microtubule dynamics. Proc Natl Acad Sci USA 96, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roohvand F, Maillard P, Lavergne JP, Boulant S, Walic M, Andreo U, Goueslain L, Helle F, Mallet A, McLauchlan J, et al. (2009). Initiation of hepatitis C virus infection requires the dynamic microtubule network: role of the viral nucleocapsid protein. J. Biol. Chem 284, 13778–13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Sallfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SL, Kamyar R, and Ehrhardt DW (2003). Sustained microtubule treadmilling in Arabidopsis cortical arrays. Science 300, 1715–1718. [DOI] [PubMed] [Google Scholar]
- Sodeik B, Ebersold MW, and Helenius A. (1997). Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol 136, 1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RA, O’Brien ET, Pryer NK, Soboeiro MF, Voter WA, Erickson HP, and Salmon ED (1988). Dynamic instability of individual microtubules analyzed by video light microscopy: rate constants and transition frequencies. J. Cell Biol 107, 1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BM (2011). The taking of the cytoskeleton one two three: how viruses utilize the cytoskeleton during egress. Virology 411, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, and Salmon ED (1997a). Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J. Cell Biol 139, 417–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, and Salmon ED (1997b). Microtubule dynamics: Treadmilling comes around again. Curr. Biol 7, R369–R372. [DOI] [PubMed] [Google Scholar]
- Zehr EA, Kraemer JA, Erb ML, Coker JK, Montabana EA, Pogliano J, and Agard DA (2014). The structure and assembly mechanism of a novel three-stranded tubulin filament that centers phage DNA. Structure 22, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehr EA, Rohu A, Liu Y, Verba KA, Pogliano J, Grigorieff N, and Agard DA (2018). Mechanistic Origins of Dynamic Instability in Filaments from the Phage Tubulin, PhuZ. bioRxiv 10.1101/311498. [DOI] [Google Scholar]
- Zhang R, Alushin GM, Brown A, and Nogales E. (2015). Mechanistic Origin of Microtubule Dynamic Instability and Its Modulation by EB Proteins. Cell 162, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.