

Abstract
Sex differences in brain development and postnatal behavior are largely determined by genetic sex and in utero gonadal hormone secretions. In humans however, determining the weight that each of these factors contribute remains a challenge, as social influences should also be considered. Cases of disorders of sex development (DSD) provide unique insight to infer how mutations in genes responsible for gonadal formation can perturb the subsequent developmental hormonal milieu and elicit changes in normal human brain maturation. Specific forms of DSDs such as complete androgen insensitivity syndrome (CAIS), congenital adrenal hyperplasia (CAH), and 5-alpha-reductase deficiency syndrome have variable effects between males and females, as the developmental outcomes of such conditions are largely dependent on sex chromosome composition. Medical and psychological works focused on CAH, CAIS and 5-α-reductase deficiency have helped form the foundation for understanding the roles of genetic and hormonal factors necessary for guiding human brain development. Here we highlight how the three aforementioned DSDs contribute to brain and behavioral phenotypes that can uniquely affect 46, XY and 46, XX in dramatically different fashions.
Introduction:
Research investigating the role that hormones and genetics contribute towards guiding sexual differentiation and behavior in rodents has been extensively reviewed elsewhere (Arnold 2009; Ngun et al. 2011). Strong evidence from such studies suggests that testosterone, either directly or indirectly, is responsible for organizing male-typical features, including gender role behavior, sexual orientation and perhaps gender identity. Evidence provided from the rodent model has laid the groundwork for our understanding of how in utero hormonal milieu and chromosomal composition influence brain development and contribute to generating observed sex differences. Unfortunately, similar research in humans is lacking in adequate depth from a biological standpoint. Investigating sex differences in human brain structure and cognitive development is challenging, as humans are a socially influenced species, making it difficult to tease apart the effects of environmental influences from biological contributions. Studying unique cases of DSDs where chromosomal composition and hormonal alterations are present has greatly improved our understanding of biological factors that contribute to sex differences within the human brain, from cognitive to structural. Here, we review and highlight how specific disorders of sex development have improved our understanding of the origins of certain sex differences within the brain, including gender-role and identity, sexual orientation, cognitive abilities as well as actual structural variations, that correspond with gender differences in humans.
The etiology of CAH, CAIS and 5-α-reductase deficiency:
Congenital adrenal hyperplasia:
CAH is the most common condition that contributes to incidences of ambiguous genitalia in 46, XX newborns. It is a recessive autosomal disorder that affects approximately 1 in 15,000 live births worldwide (Merke and Bornstein 2005; Therrell 2001). 95% of known CAH cases result from mutations in the CYP21A2 gene on chromosome 6, encoding the 21-hydroxylase (21-OH) enzyme (Merke and Bornstein 2005). Mutations in 21-OH generates two types of classical congenital adrenal hyperplasia; simple virilizing and the more severe form known as salt-wasting. By virtue of the mutation, many steroid hormone precursors within the adrenals become elevated due to improper metabolism into the necessary glucocorticoids, namely cortisol (White and Speiser 2000). In addition to causing a cortisol and aldosterone deficiency, some of the hormone precursors are shuttled into the androgen synthesis pathway, leading to increased levels of testosterone in both XX and XY fetuses (Ghayee and Auchus 2007; Speiser and White 2003). As a result of CAH, female patients are often born with various degrees of genital virilization, typically consisting of a partially fused rugated labia majora, a common urogenital sinus and clitoromegaly—providing an early diagnostic sign of CAH in XX patients (Speiser and White 2003; Witchel and Azziz 2011). 46, XY patients have minor to unnoticeable physical signs of the condition at birth, with the exception of some cases of hyperpigmentation of the genitals, and above average penile growth. The subtle nature of the clinical presentations in males results in a lower frequency of initial CAH diagnosis, and subsequently higher death rates for salt wasting cases (Therrell 2001; Witchel and Azziz 2011). In regards to treatment, both XX and XY CAH patients are supplemented with life-long mineralocorticoid and glucocorticoid replacement therapies in the forms of fludrocortisone and hydrocortisone, to supplement their cortisol deficiency and reduce the over production of hormone precursors (Merke and Bornstein 2005). Prevention of female genital virilization in the unborn fetus can be achieved by administering the steroid dexamethasone to the pregnant mother. This practice is quite controversial as the therapy must begin before fetus sex is determined, thereby exposing 7 out of 8 unaffected fetuses to DEX treatment, with potential unknown side effects on the developing brain (Heland et al. 2015) (Fig.1).
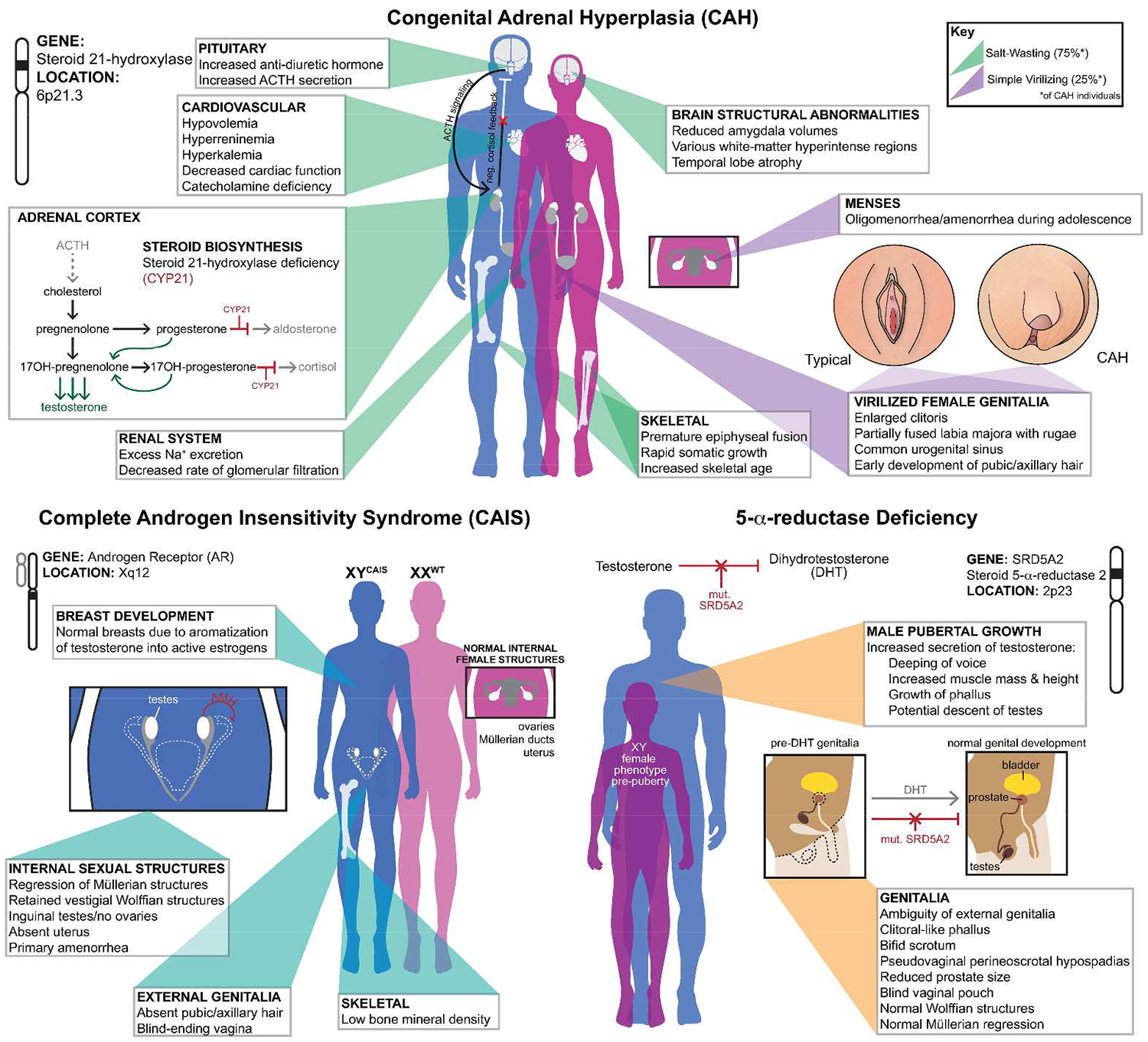
Medical overview of congenital adrenal hyperplasia (CAH), complete androgen insensitivity syndrome (CAIS) and 5-α-reductase deficiency.
Complete androgen insensitivity syndrome:
CAIS is the most common disorder of sex development causing sex reversal exclusively on a 46, XY chromosomal background. The prevalence of this disorder among live births worldwide is estimated to range between 1:20,000 and 1:99,000 (Boehmer et al. 2001; Mongan et al. 2015). 95% of all CAIS cases can be explained by inactivating mutations of the androgen receptor (AR) gene (Audi et al. 2010; Mongan et al. 2015). Over 500 mutations in AR have been associated with CAIS, the majority occurring either within the DNA-binding domain (DBD) or the ligand-binding domain (LBD), rendering the protein inactive (Gottlieb et al. 2012; Matias et al. 2000; Shao et al. 2015). 46, XY patients born with CAIS are universally assigned as females at birth due to the outward female phenotypic appearance of the genitalia. The diagnosis often occurs during pubertal years where patients present with primary amenorrhea, due to lack of internal female structures. Individuals with CAIS have a blind-ending vagina, absent ovaries, inguinal testes and no uterus, due to the regression of Müllerian structures via AMH secretion by the testes (Mongan et al. 2015). If inguinal testes are not removed before puberty, there is often normal breast development due to the high levels of circulating testosterone which is locally aromatized into active estrogens, stimulating breast growth. If gonads have been resected prior to puberty, estradiol supplementation is often administered to bring the 46, XY CAIS patients into the normal female range of the given hormone (Mongan et al. 2015) (Fig. 1).
5-α-reductase deficiency:
First identified in 1974, this DSD is an autosomal recessive condition caused by mutations within the SRD5A2 gene located on chromosome 2, and manifests as phenotypically hypo-masculinized genitals in 46, XY individuals (Imperato-McGinley et al.1974; Jenkins et al. 1992). This is a very rare disorder, with most documented cases occurring in areas of the world with high rates of consanguine mating, including the Dominican Republic, the islands of New Guinea and areas of the Middle East such as Turkey and Iraq (al-Attia 1997; Can et al. 1998; Di Marco et al. 2013; Imperato-McGinley et al. 1991; Sobel and Imperato-McGinley 2004). SRD5A2 encodes a steroid 5-α-reductase-2 enzyme, which is responsible for converting testosterone (T) into the more potent and developmentally necessary form, dihydrotestosterone (DHT). DHT is critical for the masculinization of genital tissue, and aids in proper penile growth and prostate development (Imperato-McGinley and Zhu 2002; Jenkins et al. 1992). Due to the improper conversion of T into DHT, 46, XY individuals born with this condition present with undescended inguinal testis, normal internal male structures and a reduction in prostate size (Imperato-McGinley and Zhu 2002). Externally however, this mutation leads to various degrees of genital ambiguity, appearing as complete female to nearly that of typical male infants. Those with genital ambiguity often present with a bifid scrotum, enlarged clitoris, blind vagina pouch, and various severities of hypospadias (Imperato-McGinley and Zhu 2002). During teenage years the undescended testis begin to secrete high levels of T and trigger male puberty, characterized by the deepening of the voice, increased muscle mass and height. In addition, there can be a dramatic increase in the growth of the phallus and occasional descent of the testes into the scrotal region (Imperato-McGinley and Zhu 2002) (Fig. 1).
Sexual Behaviors and Gender-Identity:
Two of the largest measurable sex differences observed between humans are gender identity and sexual orientation (Hines 2005). Deciphering what brings about these differences is a subject of much debate; however, many in the scientific community attribute them as being mediated in part by sex chromosomes and more directly by gonadal hormone secretions in utero. The theory of brain organization arose from mid-20th century works, where it was observed that administering testosterone to female rodents during a critical period for fetal brain organization brought about lifelong sexual and behavioral shifts (Phoenix et al. 1959). Work in humans addressing this subject is much more limited compared to studies in other mammals. Cases of DSDs provide us with unique insights into determining if and how male sex hormones are capable of establishing these sex differences with regards to sexual orientation and gender identity.
A study conducted by the Williams Institute of UCLA using large data sets from western societies, found that roughly 3.5% of the general population within the United States identified as having non-heterosexual orientation, and roughly 8.2% were determined as having experienced same-sex encounters (Gates 2011). Numerous studies have assessed the prevalence of non-heterosexual behaviors amongst 46, XX women with CAH from various regions of the world. This condition is the closest mimic of rodent studies that address the role of early androgen exposure and variations seen in adult behavior. Generally speaking, it has been found in the majority of studies that non-heterosexual behaviors are increased in women with CAH, typically compared to their unaffected female relatives or appropriate controls. In a study conducted by Zucker et al. focusing on the most severe salt-wasting form of CAH, as well as the milder simple virilizing form, it was found that around 27% of women with CAH had bisexual/homosexual sexual fantasies. Only 3.3% of individuals in the study had actual non-heterosexual partners which was not significantly different than controls (Zucker et al. 1996). A more recent study out of France found similar findings as Zucker, where they identified within their CAH cohort (individuals with varying degrees of genital virilization as measured on the Prader scale) that 20% exhibited bisexual and homosexual fantasies and only 5.8% had opposite sexual partners (Gastaud et al. 2007). Non-heterosexual behaviors and fantasies amongst women with CAH have been identified at varying percentage levels, as highlighted in an exhaustive account of such studies compiled by Meyer-Bahlburg and colleagues (Meyer-Bahlburg et al. 2008). In contrast however, other studies conducted on the topic have also found no differences in sexual orientation (Kuhnle et al. 1995; Lev-Ran 1974); however, some critics of these works have determined that the sample size in certain studies were too underpowered to detect a difference (Meyer-Bahlburg et al. 2008). Using a large CAH cohort, it was again revealed that women with CAH show more homosexual and bisexual behaviors, as measured by their sexual fantasies and actual sex partners. The study also successfully demonstrated that the level of non-heterosexual tendencies increased with the severity of the condition, a correlation associated with the level of in utero androgen exposure. Within the study cohort it was found that 47% of patients with the salt-wasting form of the condition experienced bisexual or homosexual fantasies, 33% for those with the simple virilizing form, and surprisingly 24% for those with the non-classical form of CAH (Meyer-Bahlburg et al. 2008). Biologically, it appears that in utero androgen exposure on an XX background most significantly affects sexual fantasies in women with CAH more so than directly altering their actual sexual orientation as measured by sexual partners, which may be influenced by additional constraints. These findings provide a strong argument for a biological influences on eliciting such outcomes; however, these studies do not eliminate the possibility of social and other psychological contributions for these observations, as reviewed by Rebecca Jordan-Young (Jordan-Young 2012).
In addition to sexual orientation, gender identity (one’s internal sense of being male or female, independent of gonadal sex) has also been a topic of investigation in cases of DSD. Among the compiled studies that have focused on 46, XX CAH and gender identity, it was found that the vast majority, (95%) of patients who were assigned to the female gender were satisfied and non-dysphoric in adulthood (Dessens et al. 2005). However 5% of CAH females exhibited gender dysphoria and/or identified as the male gender as reviewed by Dessens and others (Dessens et al. 2005). This 5% of dysphoria in CAH females is far above the estimated average of 0.3% of adults who identify as transgender within western societies, specifically the U.S. (Gates 2011). In instances where 46, XX CAH were assigned to the male gender at birth, a small sampled study showed that 12% of cases experienced gender dysphoria and associated themselves with the female gender (Dessens et al. 2005). However, later studies assessing outcomes in severely virilized 46, XX cases who were assigned as males at birth drew different conclusions about satisfaction of male-gender assignment and identity (Lee et al. 2010). In a small study population of twelve 46, XX CAH cases who were initially assigned as males, it was found that ten persisted in the assigned gender and lived male-typical and satisfying lives. Two of the male-assigned individuals within the study that were adolescently re-assigned to the female gender, subsequently re-assigned themselves back to the male gender in adulthood (Lee and Houk 2010; Lee et al. 2010). Unlike results found in studies examining sexual orientation, dysphoria in gender identity was neither correlated with the severity of the CAH condition, nor the degree of genital virilization, demonstrating that genital appearance at birth is not the best predictor for gender outcomes in cases of 46, XX DSD (Berenbaum and Bailey 2003). Collectively, these data demonstrate that we have yet to fully understand gender identity determinants in CAH individuals, and the general population for that matter, from a strictly biological perspective. It is however apparent that androgen exposure on XX genetic backgrounds can increase instances of dissatisfactions of ones gender assignment at rates far above estimated percentages found within general populations.
Some of the most convincing evidence linking testosterone to establishing male gender identity and sexual preference comes from work focused on individuals with complete androgen insensitivity syndrome and those with 5-α-reductase deficiencies. When assessing sexual orientation in cases of CAIS, it has been shown that 46, XY women are almost always exclusively heterosexual, preferring male sex partners, in addition to experiencing heterosexual fantasies and interests (Hines et al. 2003a; Wisniewski et al. 2000). During adolescence years CAIS women reported feeling 100 percent heterosexual and a minority (n=1) in a particular study stated same-sex attraction, but only in adulthood. This occurrence as the authors note may be influenced by other causes as opposed to their inability to respond to androgens during development (Wisniewski et al. 2000). In contrast, recent literature has begun to challenge this strongly established view of nearly exclusive heterosexual behaviors in women with CAIS. In a recent report by Brunner and colleagues, it was found that 46, XY women when compared to other females with infertility conditions showed increased tendencies of non-heterosexual preference and behavior—evidence that is out of line with previously established views of fully heterosexual outcomes in women with CAIS. (Brunner et al. 2016). These findings, while limited to a small sample size (n=11 CAIS), raise an interesting topic for further investigation that could identify additional influences that guide sexual preference and perhaps identity in 46, XY women, aside from their inability to respond to male-sex hormone.
Using a more scientifically unbiased approach to assess sexual response, and perhaps orientation in women with CAIS, a recent study focused on neural activation in response to viewing sexual images. Supporting the notion of predominantly heterosexual feelings in CAIS, it was found that women with this condition more closely resembled 46, XX heterosexual females than 46, XY males (Hamann et al. 2014). Functional MRI (fMRI) scans revealed that both 46, XY women with CAIS and 46, XX women had decreased amygdala activation when viewing nude male images or couples engaging in coitus. The results of this study were the first to establish that the brains of CAIS women more functionally resembled that of a typical female, further indicating that testosterone responsiveness (and not the Y chromosome), is the most likely driver for establishing gynephilic preference, and androphilic-typical brain activation towards sexual stimuli in humans (Hamann et al. 2014).
In regards to gender identity, 46, XY women with CAIS almost always report feeling comfortable in their female assignment, as shown by their typical female responses to assessments measuring psychosocial traits (Hines et al. 2003a). A thorough review of CAIS studies identified that out of the 156 individuals assessed in the research, there were no instances of male-gender identity reported (Mazur 2005). These findings add strength to the biological notion that hormonal response influences male-gender identity more so than chromosomal composition. Once again however, recent reports have begun to suggest that this theory is not as concrete as previously thought. A growing body of literature and case reports exists which have identified gender-dysphoria and cross-gender feelings in CAIS individuals at rates higher than have been established (Brunner et al. 2016; Kulshreshtha et al. 2009; T’Sjoen et al. 2011). The role of social and initial medical decisions associated with CAIS cases may be a contributing factor in these results because as stated by Tom Mazur, “The best predictor of adult gender identity in CAIS, PAIS (partial androgen insensitivity syndrome) and micropenis is initial gender assignment” (Mazur 2005).
In addition to CAIS, research focused on the understudied and rare 5-α-reductase deficiency syndrome has also helped expand the role of testosterone in establishing sex differences in regards to sexual orientation and gender identity in humans. Individuals affected with this condition are typically reared as girls; however, during the pubertal years when testosterone secretions from the testes are elevated and male puberty ensues, the vast majority change initial gender assignment (Sobel and Imperato-McGinley 2004). This outcome in persons with 5-α-reductase deficiency appears to be worldwide, and not necessarily a product of specific social influences, as gender-role and identity changes have been reported in cases from the Mediterranean, Middle East, Caribbean, Africa and New Guinea (al-Attia 1996; Al-Attia 1987; Sobel and Imperato-McGinley 2004). After change in gender role and gender identity these 46, XY individuals typically lead heterosexual lives, preferring female partners, despite having been initially reared as female themselves (Sobel and Imperato-McGinley 2004). This gender switch in individuals during male puberty raises the notion that despite insufficient DHT production necessary for typical male genitalia development, the organizational effects on the brain were established and able to influence male-gender role and eventually their identity, which seem to become solidified upon pubertal testosterone surges. 5-α-reductase deficiency syndrome is an unusual circumstance where teasing apart the effects of environmental influence and biology becomes extremely difficult. The activational effects of testosterone at puberty coupled with likely in utero organization do appear to be factors in establishing the change in gender role and identity. However, the physical change that also ensues during the transition process cannot be ruled out as another contributing factor for the observed change.
Highlight of Cognitive Sex Differences and Interpretation from Cases of DSD:
Sex differences in regards to cognitive traits have been extensively studied by numerous groups and have identified several measures where males and females show large deviations in mental task abilities. The origin of these sex differences have been attributed to chromosomal effects (Ngun et al. 2011), hormonal effects (McCarthy 2010) and social influences (Jordan-Young 2012). Regardless of the reason, cognitive differences between men and women are detected in the vast majority of studies. Cases of DSD again provide a unique platform to assess the possible contributing factors responsible for generating sex differences, as studies in individuals with CAH and CAIS have established unique hormonal and chromosomal influences. Spatial awareness and mental rotation ability reveal the strongest cognitive difference between men and women—with males out performing females (Geiser C 2007; Voyer et al. 1995).
Spatial task assessments in cases of DSD are predominantly focused on individuals with congenital adrenal hyperplasia and to a more limited extent, CAIS. 46, XX females with CAH, on average out-perform their unaffected matched controls on mental rotation tasks and spatial relations tests (Berenbaum et al. 2012; Hampson E 1998; Hampson and Rovet 2015). Studies of this nature have also found that girls with CAH versus their matched controls exhibit no enhancement in spatial ability, adding to the uncertainty of androgen influence on increasing spatial awareness on an XX background (Hines et al. 2003b). Some of these inconsistencies in the data may be attributed to variations seen between sub-types of CAH as opposed to an inaccurate determination of enhancements in spatial ability. It was found that spatial task enhancements show the strongest effect in girls with the severe salt wasting form of CAH, whereas those individuals with the simple virilizing form of the condition did not show a statistically significant difference (Hampson and Rovet 2015). In comparison, 46, XY males with CAH, appear to be de-masculinized on these features and perform worse than their matched controls on both measures (Berenbaum et al. 2012; Hines et al. 2003b). These findings are quite interesting as they allude to the possibility that high in utero androgen exposure generates different cognitive outcomes depending on sex chromosome complement. Males with CAH a priori would be expected to perform at least equally as opposed to underperform control males if the effect was exclusively due to excess testosterone. There may be both an optimal time and level of testosterone exposure during development that enhances such abilities, and if these levels too high in utero it may generate these observed outcomes in males with CAH.
Recently, individuals with CAIS were assessed using both traditional rotation tests and fMRI studies to determine how their brains responded to rotation tasks as compared to non-CAIS males and females. It was found that 46, XY CAIS women were slower to figure out the correct response in rotation test assessments as compared to their 46, XY male counterparts (van Hemmen et al. 2016). When focusing on neuroimaging, CAIS individuals were more similar to control women, showing less inferior parietal lobe activations during rotation tasks than they were to 46,XY males (van Hemmen et al. 2016). This study is quite remarkable as it demonstrates a strong role of androgenic influences on establishing mental rotation abilities on an XY background, despite the fact that males with CAH (elevated T levels) perform worse than controls. Again, these findings suggest that timing and dose of testosterone during development may be critical to establish or enhance specific cognitive traits in humans. CAH individuals have more confounding long-term health factors, whereas in CAIS these additional variables are minimized. Thus, this leaves the testosterone responses in the brain as the most reasonable explanation for the observed spatial performance differences. To the best of our knowledge, no spatial performance examinations have been conducted on individuals with 5-α-reductase deficiency.
Another cognitive task where mild sex differences arise is the ability to target objects using hand-eye coordination, another male-typical feature (Watson and Kimura 1991). In studies focusing on this trait, it was found that 46, XX CAH females were able to target both darts and ball throwing more accurately than their control counterparts (Collaer et al. 2009; Hines et al. 2003b). In males with CAH this trait seems to be unaffected, as both controls and affected individuals were statistically insignificant from one another in targeting abilities (Collaer et al. 2009; Hines et al. 2003b). It appears again that an in utero androgenic influence on an XX genetic background enables enhanced targeting coordination during adolescences and adulthood. This feature, unlike spatial ability, doesn’t appear to be sensitive to the dose or timing of testosterone during development, as CAH males did not underperform their controls on these measures. This is another trait that has yet to be examined in individuals with CAIS and 5-α-reductase deficiency. Such work would greatly improve our understanding of androgenic and sex chromosome influence on shaping specific cognitive sex differences within the human brain.
Additional cognitive traits which have been assessed and show sex differences include male-dominate tasks involving spatial perception, maze navigation, mathematics, and various mechanical skills, and female-dominate tasks involving working memory and verbal fluency (Collaer and Hines 1995; Halpern 2012; Woolley et al. 2010). Many of these traits have been assessed in XX and XY CAH cases; however, the cognitive differences that show the strongest sex and androgen influence are the highlighted spatial and targeting abilities. The works conducted in cases of DSD have helped expand our understanding of the influence of testosterone on both XX and XY genetic backgrounds with regards to altering spatial abilities and hand-eye coordination. These works have also demonstrated that the effect is most likely hormonally influenced, as CAIS individuals despite being XY cannot respond to androgen and therefore have spatial and cognitive traits in resemblance of XX females.
Brain structural variations in DSD cases:
Unlike research focused on sexual behavior and cognitive differences, there exists surprisingly little work focused on the structural variations of the brain as a result of DSD. A small but growing body of literature has addressed this topic mostly in individuals with CAH. Early work established that both 46, XY males and 46, XX females with 21-hydroxylase deficiency exhibited significantly smaller amygdala volumes when compared to aged matched controls, a known sexually dimorphic brain structure (Goldstein et al. 2001; Merke et al. 2003). It was found that regardless of age, girls with CAH exhibited a bilateral reduction in amygdala volume, whereas CAH affected males in the study had a 20% unilateral reduction in only the left amygdala, which is also the same reduction percentage for the left amygdala in CAH females. Increases of amygdala volumes over human development have been observed in males which is attributed to arise from androgenic stimulation (Giedd et al. 1997). Interestingly, it was found in 46, XY boys with CAH, this increase with developmental age was only present in the right, seemingly unaffected amygdala; whereas XY controls in the study group followed previously established bilateral size increases (Merke et al. 2003). The exact causes of these brain anomalies are still being addressed; however, there seems to be a clear sex difference in response, as males and females with similar imbalances as a result of CAH show different patterns of amygdala reductions. A study focusing primarily on 46, XX non-CAH individuals taking long-term corticosteroid replacement therapies also identified a 20% reduction in the left amygdala volume and an 11% reduction in right hemisphere amygdala (Brown et al. 2008). Collectively these findings indicate that the reduced amygdala volume is most likely due to an imbalance of glucocorticoids rather than excess androgens during development, as amygdala size is actually reduced in CAH patients despite high levels of in utero testosterone, whereas healthy male controls show increases in volume of the amygdala via androgen stimulation. These findings however do not offer a direct biological mechanistic explanation for observations of reductions in brain regions, but supports that imbalances in glucocorticoids and other steroids are the likely causative agents.
White-matter abnormalities:
Another brain structural metric that has been assessed in XX and XY individuals with CAH are abnormalities in white matter distributions. MRI studies of adult patients with salt wasting, simple virilizing and even non-classical congenital adrenal hyperplasia have identified white matter hyperintensities in various brain regions (Kaga et al. 2013; Mnif et al. 2013; Nass et al. 1997; Sinforiani et al. 1994; Winfeld et al. 2013). Areas of the brain that seem to be most frequently affected in CAH patients are the periventricular regions, areas of the cerebellum and the corpus callosum. Incidences of temporal lobe atrophy have also been documented from MRI studies of CAH patients (Mnif et al. 2013; Nass et al. 1997). The vast majority of cases assessed showing abnormalities in white matter across brain regions scored within the normal ranges of neurological function, indicating that despite these differences, the effect does not seem to impair proper brain functioning. These studies have all identified sub-clinical white matter variations, however the etiology and results of such findings remains unclear. It has been hypothesized that the logical causes of these outcomes results from hormonal imbalances during development, long-term glucocorticoid replacement therapies after birth, and even due to the genetic mutation itself. It has also been proposed that the imbalance of hormones during development contributes to improper oligodendrocyte differentiation and in improper myelination (Bergamaschi et al. 2006). In addition to adult studies, there have also been several documented cases of newborns with CAH exhibiting white matter anomalies (Kaga et al. 2013; Winfeld et al. 2013). While it does appears that replacement therapies over a long-time frame may affect white matter, findings in newborns with CAH raise the notion that long-term glucocorticoid replacement therapies may not be the sole explanation for the disturbances in white matter. Investigating these unusual findings in more detail will enable a better understanding of the role that glucocorticoids or lack thereof have on shaping the structure of the developing male and female brain. Currently however, the exact cause and mechanism behind these results remain speculative.
Concluding Remarks:
Research focused on cases of DSD have helped the scientific community better understand the interplay between gonadal hormones and sex chromosome complement in regards to generating some of the sex differences observed in humans. These works have shed light on the likelihood that testosterone exposure as opposed to sex chromosomes is a larger contributing factor for guiding ones sexual orientation, and to a lesser extent gender identity. We see that 46, XX CAH individuals that have been exposed to in utero testosterone experience a greater degree of dissatisfaction in gender assignment in addition to above average levels of homosexual and bisexual fantasies, a proxy for sexual preference. As previously mentioned, other variables are present in CAH cases such as life-long medical interventions and psychosocial confounds. These variables may constitute an environmental factor that when coupled with biological pre-dispositions, generate variations in sexual orientation and gender identity. To say that sexual orientation is determined solely by in utero hormonal milieu is unlikely. We see that the vast majority of CAH women despite having been exposed to above average levels of testosterone still identify as heterosexual as measured by both partners and sexual fantasies. The science of sexual orientation is still weakly understood at the mechanistic level, however, considerable amounts of research have formulated many possibilities as to the causes of same-sex attraction (Bailey et al. 2016; LeVay 2012).
The strongest evidence which adds support for the influence of testosterone in structuring gender identity comes from the work focused on 46, XY CAIS, where nearly all individuals researched indicate feelings typical of female gender. In addition to self-reports and clinical evaluations, recent fMRI studies have also demonstrated that CAIS women not only feel female, but also neurologically respond more similar to 46, XX women than 46, XY men when observing sexual images. However, new studies are continually emerging that suggest gender-identity and sexual orientation in individuals with CAIS is not as clear as once thought, and the rates of non-heterosexual and gender dysphoria may be much higher than currently stated. In addition to CAH and CAIS, 5-α-reductase deficiencies have also demonstrated the strong role of testosterone’s ability to hormonally organize the human brain and influence adult gender identity and behavior. If early in utero exposure had no influence in guiding brain gender, it would be expected that there would be considerable difficulty with the female to male transition observed in pubertal years in those with 5-α-reductase deficiency. What we observe however, is an overwhelming majority of individuals with this condition comfortably transitioned into the new gender role at puberty, a world-wide observation occurring throughout many different types of social environments. Despite the convincing findings for the role of testosterone in generating these observations, the influence of social and other environmental variables are also factors that require consideration.
Cognitive conclusions:
Studying cases of DSDs has also provided insight into some of the biological parameters that generate sex differences in cognitive abilities such as visuospatial awareness and targeting ability. From studies using 46, XX CAH individuals it has been well established that in utero androgen exposures seems to enhance the ability to mentally rotate objects as well as improving hand-eye coordination during targeting tasks. This trait appears to be dependent on sex chromosome complement in addition to hormone exposure as 46, XY males with CAH actually perform worse than their matched controls, which is unexpected giving the fact that CAH males would have equal or elevated levels of circulating testosterone. This raises the notion as earlier mentioned that proper timing and dosage is also likely important for enhancing such abilities and simply having above average levels of testosterone during development would not generate a “super-male”. CAIS provides another insight into this matter demonstrating that the ability to respond to testosterone on an XY background is critical to establish baseline spatial performance abilities. fMRI studies demonstrate that 46, XY CAIS had less inferior parietal lobe neuro-activation when performing spatial rotation tasks, a feature that resembles 46, XX females more than control genetic males. These fMRI studies on CAIS individuals once again minimizes social influences, and allows for a more unbiased assessment of the requirement of testosterone over genetic composition for shaping these cognitive performance sex differences.
Structural Conclusions:
From the MRI studies that have been conducted in patients with CAH it is clear that DSDs affect more than gonadal development. As highlighted, the central nervous system is highly sensitive to various hormones and imbalances of such can greatly affect downstream behavior as well as overall brain structure. Variations in amygdala volume seem to be present in some individuals with CAH; however the effect is different depending on sex chromosome composition. Specifically, 46, XY males with CAH show unilateral reductions in the left amygdala, whereas 46, XX females with CAH show bilateral reductions in overall volume. Alterations on amygdala volume seem to be consistent with long-term glucocorticoid replacement therapies, as findings in non-CAH patients on such hormone regiments also show amygdala abnormalities. The documentations of white matter irregularities seem to be unaffected by chromosomal sex, and cause similar variations in both males and females with CAH. The explanations of these results are not in consensus and more research will be needed before causations can be associated with the unusual white matter findings. While limited, these discoveries have opened a new area of potential investigation focusing on the role of glucocorticoid influences on the developing brain in addition to the more frequently studied gonadal hormonal contributions. Unfortunately, no extensive structural studies have been conducted in patients with CAIS or 5-α-reductase deficiencies. These findings would be invaluable in determining the direct effect of testosterone on the structures that have been found to be altered in some cases of CAH by MRI studies. Future work focusing on outcomes in individuals with DSD will continue to aid in deciphering the contributions of chromosomal sex and hormones on shaping the sexually dimorphic human brain.
Significance Statement:
Sex differences in the brain and behavior of humans have been identified on numerus measures. The cause of these sex differences has been associated with chromosomal constitution and circulating hormones, such as estrogen and testosterone. Research on individuals with DSD has improved the scientific community’s understanding of the potential contributions of the X and Y chromosome as well as the role of gonadal hormone secretions in generating these differences. This review highlights research that has expanded our understanding of the origins of specific sex differences observed in humans such as sexual orientation, gender identity and cognitive performance.
Footnotes
Conflicts of Interest: The authors of this study declare no conflict of interest that would have any bearing on the topics discussed in this review. MSB was the primary contributor to this article, conducting the literature review and manuscript preparation. AL prepared the graphic designs and both AL and NV assisted in the literature review and manuscript preparation. EV guided the topics of discussion and oversaw the finalization of this review.
References
- al-Attia HM. 1996. Gender identity and role in a pedigree of Arabs with intersex due to 5 alpha reductase-2 deficiency. Psychoneuroendocrinology 21(8):651–657. [DOI] [PubMed] [Google Scholar]
- al-Attia HM. 1997. Male pseudohermaphroditism due to 5 alpha-reductase-2 deficiency in an Arab kindred. Postgrad Med J 73(866):802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Attia HM, Kazin S, George S,. 1987. 5a-reductase deficiency in two Omani siblings. Emirates Medical Journal 5:236–242. [Google Scholar]
- Arnold AP. 2009. The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Hormones and behavior 55(5):570–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audi L, Fernandez-Cancio M, Carrascosa A, Andaluz P, Toran N, Piro C, Vilaro E, Vicens-Calvet E, Gussinye M, Albisu MA, Yeste D, Clemente M, Hernandez de la Calle I, Del Campo M, Vendrell T, Blanco A, Martinez-Mora J, Granada ML, Salinas I, Forn J, Calaf J, Angerri O, Martinez-Sopena MJ, Del Valle J, Garcia E, Gracia-Bouthelier R, Lapunzina P, Mayayo E, Labarta JI, Lledo G, Sanchez Del Pozo J, Arroyo J, Perez-Aytes A, Beneyto M, Segura A, Borras V, Gabau E, Caimari M, Rodriguez A, Martinez-Aedo MJ, Carrera M, Castano L, Andrade M, Bermudez de la Vega JA. 2010. Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development. The Journal of clinical endocrinology and metabolism 95(4):1876–1888. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Vasey PL, Diamond LM, Breedlove SM, Vilain E, Epprecht M. 2016. Sexual Orientation, Controversy, and Science. Psychological science in the public interest : a journal of the American Psychological Society 17(2):45–101. [DOI] [PubMed] [Google Scholar]
- Berenbaum SA, Bailey JM. 2003. Effects on gender identity of prenatal androgens and genital appearance: evidence from girls with congenital adrenal hyperplasia. The Journal of clinical endocrinology and metabolism 88(3):1102–1106. [DOI] [PubMed] [Google Scholar]
- Berenbaum SA, Bryk KL, Beltz AM. 2012. Early androgen effects on spatial and mechanical abilities: evidence from congenital adrenal hyperplasia. Behavioral neuroscience 126(1):86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi R, Livieri C, Uggetti C, Candeloro E, Egitto MG, Pichiecchio A, Cosi V, Bastianello S. 2006. Brain white matter impairment in congenital adrenal hyperplasia. Arch Neurol 63(3):413–416. [DOI] [PubMed] [Google Scholar]
- Boehmer AL, Brinkmann O, Bruggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MC, Niermeijer MF, Brunner HG, Rouwe CW, Waelkens JJ, Oostdijk W, Kleijer WJ, van der Kwast TH, de Vroede MA, Drop SL. 2001. Genotype versus phenotype in families with androgen insensitivity syndrome. The Journal of clinical endocrinology and metabolism 86(9):4151–4160. [DOI] [PubMed] [Google Scholar]
- Brown ES, Woolston DJ, Frol AB. 2008. Amygdala volume in patients receiving chronic corticosteroid therapy. Biol Psychiatry 63(7):705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner F, Fliegner M, Krupp K, Rall K, Brucker S, Richter-Appelt H. 2016. Gender Role, Gender Identity and Sexual Orientation in CAIS (“XY-Women”) Compared With Subfertile and Infertile 46,XX Women. J Sex Res 53(1):109–124. [DOI] [PubMed] [Google Scholar]
- Can S, Zhu YS, Cai LQ, Ling Q, Katz MD, Akgun S, Shackleton CH, Imperato-McGinley J. 1998. The identification of 5 alpha-reductase-2 and 17 beta-hydroxysteroid dehydrogenase-3 gene defects in male pseudohermaphrodites from a Turkish kindred. The Journal of clinical endocrinology and metabolism 83(2):560–569. [DOI] [PubMed] [Google Scholar]
- Collaer ML, Brook CG, Conway GS, Hindmarsh PC, Hines M. 2009. Motor development in individuals with congenital adrenal hyperplasia: strength, targeting, and fine motor skill. Psychoneuroendocrinology 34(2):249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaer ML, Hines M. 1995. Human behavioral sex differences: a role for gonadal hormones during early development? Psychol Bull 118(1):55–107. [DOI] [PubMed] [Google Scholar]
- Dessens AB, Slijper FM, Drop SL. 2005. Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Arch Sex Behav 34(4):389–397. [DOI] [PubMed] [Google Scholar]
- Di Marco C, Bulotta AL, Varetti C, Dosa L, Michelucci A, Baldinotti F, Meucci D, Castagnini C, Lo Rizzo C, Di Maggio G, Simi P, Mari F, Bertelloni S, Renieri A, Messina M. 2013. Ambiguous external genitalia due to defect of 5-alpha-reductase in seven Iraqi patients: prevalence of a novel mutation. Gene 526(2):490–493. [DOI] [PubMed] [Google Scholar]
- Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, Bougneres P. 2007. Impaired sexual and reproductive outcomes in women with classical forms of congenital adrenal hyperplasia. The Journal of clinical endocrinology and metabolism 92(4):1391–1396. [DOI] [PubMed] [Google Scholar]
- Gates GJ. 2011. How many people are lesbian, gay, bisexual, and transgender?. The Williams Institute. [Google Scholar]
- Geiser CLW, Eid Michael. 2007. A note on sex differences in mental rotation in different age groups. Intelligence 36(6):556–563. [Google Scholar]
- Ghayee HK, Auchus RJ. 2007. Basic concepts and recent developments in human steroid hormone biosynthesis. Rev Endocr Metab Disord 8(4):289–300. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Castellanos FX, Rajapakse JC, Vaituzis AC, Rapoport JL. 1997. Sexual dimorphism of the developing human brain. Prog Neuropsychopharmacol Biol Psychiatry 21(8):1185–1201. [DOI] [PubMed] [Google Scholar]
- Goldstein JM, Seidman LJ, Horton NJ, Makris N, Kennedy DN, Caviness VS Jr., Faraone SV, Tsuang MT. 2001. Normal sexual dimorphism of the adult human brain assessed by in vivo magnetic resonance imaging. Cereb Cortex 11(6):490–497. [DOI] [PubMed] [Google Scholar]
- Gottlieb B, Beitel LK, Nadarajah A, Paliouras M, Trifiro M. 2012. The androgen receptor gene mutations database: 2012 update. Hum Mutat 33(5):887–894. [DOI] [PubMed] [Google Scholar]
- Halpern D 2012. Sex Differences in Cognitive Abilities. New York: Psychology Press. [Google Scholar]
- Hamann S, Stevens J, Vick JH, Bryk K, Quigley CA, Berenbaum SA, Wallen K. 2014. Brain responses to sexual images in 46,XY women with complete androgen insensitivity syndrome are female-typical. Hormones and behavior 66(5):724–730. [DOI] [PubMed] [Google Scholar]
- Hampson ERJ, Altmann Deborah. 1998. Spatial Resoning in Children With Congenital Adrenal Hyperplasis Due to 21-Hydroxylase Deficiency. Developmental Neuropsychology 14(2):299–320. [Google Scholar]
- Hampson E, Rovet JF. 2015. Spatial function in adolescents and young adults with congenital adrenal hyperplasia: clinical phenotype and implications for the androgen hypothesis. Psychoneuroendocrinology 54:60–70. [DOI] [PubMed] [Google Scholar]
- Heland S, Hewitt JK, McGillivray G, Walker SP. 2015. Preventing female virilisation in congenital adrenal hyperplasia: The controversial role of antenatal dexamethasone. Aust N Z J Obstet Gynaecol. [DOI] [PubMed] [Google Scholar]
- Hines M 2005. Brain Gender Oxford University Press. [Google Scholar]
- Hines M, Ahmed SF, Hughes IA. 2003a. Psychological outcomes and gender-related development in complete androgen insensitivity syndrome. Arch Sex Behav 32(2):93–101. [DOI] [PubMed] [Google Scholar]
- Hines M, Fane BA, Pasterski VL, Mathews GA, Conway GS, Brook C. 2003b. Spatial abilities following prenatal androgen abnormality: targeting and mental rotations performance in individuals with congenital adrenal hyperplasia. Psychoneuroendocrinology 28(8):1010–1026. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. 1974. Steroid 5alpha-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science (New York, NY) 186(4170):1213–1215. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Miller M, Wilson JD, Peterson RE, Shackleton C, Gajdusek DC. 1991. A cluster of male pseudohermaphrodites with 5 alpha-reductase deficiency in Papua New Guinea. Clin Endocrinol (Oxf) 34(4):293–298. [DOI] [PubMed] [Google Scholar]
- Imperato-McGinley J, Zhu YS. 2002. Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency. Mol Cell Endocrinol 198(1–2):51–59. [DOI] [PubMed] [Google Scholar]
- Jenkins EP, Andersson S, Imperato-McGinley J, Wilson JD, Russell DW. 1992. Genetic and pharmacological evidence for more than one human steroid 5 alpha-reductase. J Clin Invest 89(1):293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan-Young RM. 2012. Hormones, context, and “brain gender”: a review of evidence from congenital adrenal hyperplasia. Soc Sci Med 74(11):1738–1744. [DOI] [PubMed] [Google Scholar]
- Kaga A, Saito-Hakoda A, Uematsu M, Kamimura M, Kanno J, Kure S, Fujiwara I. 2013. Brain white matter abnormality in a newborn infant with congenital adrenal hyperplasia. Clin Pediatr Endocrinol 22(4):77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnle U, Bullinger M, Schwarz HP. 1995. The quality of life in adult female patients with congenital adrenal hyperplasia: a comprehensive study of the impact of genital malformations and chronic disease on female patients life. Eur J Pediatr 154(9):708–716. [DOI] [PubMed] [Google Scholar]
- Kulshreshtha B, Philibert P, Eunice M, Khandelwal SK, Mehta M, Audran F, Paris F, Sultan C, Ammini AC. 2009. Apparent male gender identity in a patient with complete androgen insensitivity syndrome. Arch Sex Behav 38(6):873–875. [DOI] [PubMed] [Google Scholar]
- Lee PA, Houk CP. 2010. Review of Outcome Information in 46,XX Patients with Congenital Adrenal Hyperplasia Assigned/Reared Male: What Does It Say about Gender Assignment? Int J Pediatr Endocrinol 2010:982025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PA, Houk CP, Husmann DA. 2010. Should male gender assignment be considered in the markedly virilized patient With 46,XX and congenital adrenal hyperplasia? J Urol 184(4 Suppl):1786–1792. [DOI] [PubMed] [Google Scholar]
- Lev-Ran A 1974. Sexuality and educational levels of women with the late-treated adrenogenital syndrome. Arch Sex Behav 3(1):27–32. [DOI] [PubMed] [Google Scholar]
- LeVay S 2012. Gay, Straight, and the Reason Why. New York: Oxford University Press. [Google Scholar]
- Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Basler S, Schafer M, Egner U, Carrondo MA. 2000. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem 275(34):26164–26171. [DOI] [PubMed] [Google Scholar]
- Mazur T 2005. Gender dysphoria and gender change in androgen insensitivity or micropenis. Arch Sex Behav 34(4):411–421. [DOI] [PubMed] [Google Scholar]
- McCarthy MM. 2010. How it’s made: organisational effects of hormones on the developing brain. J Neuroendocrinol 22(7):736–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merke DP, Bornstein SR. 2005. Congenital adrenal hyperplasia. Lancet 365(9477):2125–2136. [DOI] [PubMed] [Google Scholar]
- Merke DP, Fields JD, Keil MF, Vaituzis AC, Chrousos GP, Giedd JN. 2003. Children with classic congenital adrenal hyperplasia have decreased amygdala volume: potential prenatal and postnatal hormonal effects. The Journal of clinical endocrinology and metabolism 88(4):1760–1765. [DOI] [PubMed] [Google Scholar]
- Meyer-Bahlburg HF, Dolezal C, Baker SW, New MI. 2008. Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Arch Sex Behav 37(1):85–99. [DOI] [PubMed] [Google Scholar]
- Mnif MF, Kamoun M, Mnif F, Charfi N, Kallel N, Rekik N, Naceur BB, Fourati H, Daoud E, Mnif Z, Sfar MH, Younes-Mhenni S, Sfar MT, Hachicha M, Abid M. 2013. Brain magnetic resonance imaging findings in adult patients with congenital adrenal hyperplasia: Increased frequency of white matter impairment and temporal lobe structures dysgenesis. Indian J Endocrinol Metab 17(1):121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongan NP, Tadokoro-Cuccaro R, Bunch T, Hughes IA. 2015. Androgen insensitivity syndrome. Best Pract Res Clin Endocrinol Metab 29(4):569–580. [DOI] [PubMed] [Google Scholar]
- Nass R, Heier L, Moshang T, Oberfield S, George A, New MI, Speiser PW. 1997. Magnetic resonance imaging in the congenital adrenal hyperplasia population: increased frequency of white-matter abnormalities and temporal lobe atrophy. J Child Neurol 12(3):181–186. [DOI] [PubMed] [Google Scholar]
- Ngun TC, Ghahramani N, Sanchez FJ, Bocklandt S, Vilain E. 2011. The genetics of sex differences in brain and behavior. Frontiers in neuroendocrinology 32(2):227–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phoenix CH, Goy RW, Gerall AA, Young WC. 1959. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 65:369–382. [DOI] [PubMed] [Google Scholar]
- Shao J, Hou J, Li B, Li D, Zhang N, Wang X. 2015. Different types of androgen receptor mutations in patients with complete androgen insensitivity syndrome. Intractable Rare Dis Res 4(1):54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinforiani E, Livieri C, Mauri M, Bisio P, Sibilla L, Chiesa L, Martelli A. 1994. Cognitive and neuroradiological findings in congenital adrenal hyperplasia. Psychoneuroendocrinology 19(1):55–64. [DOI] [PubMed] [Google Scholar]
- Sobel V, Imperato-McGinley J. 2004. Gender identity in XY intersexuality. Child Adolesc Psychiatr Clin N Am 13(3):609–622, viii. [DOI] [PubMed] [Google Scholar]
- Speiser PW, White PC. 2003. Congenital adrenal hyperplasia. N Engl J Med 349(8):776–788. [DOI] [PubMed] [Google Scholar]
- T’Sjoen G, De Cuypere G, Monstrey S, Hoebeke P, Freedman FK, Appari M, Holterhus PM, Van Borsel J, Cools M. 2011. Male gender identity in complete androgen insensitivity syndrome. Arch Sex Behav 40(3):635–638. [DOI] [PubMed] [Google Scholar]
- Therrell BL. 2001. Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 30(1):15–30. [DOI] [PubMed] [Google Scholar]
- van Hemmen J, Veltman DJ, Hoekzema E, Cohen-Kettenis PT, Dessens AB, Bakker J. 2016. Neural Activation During Mental Rotation in Complete Androgen Insensitivity Syndrome: The Influence of Sex Hormones and Sex Chromosomes. Cereb Cortex 26(3):1036–1045. [DOI] [PubMed] [Google Scholar]
- Voyer D, Voyer S, Bryden MP. 1995. Magnitude of sex differences in spatial abilities: a meta-analysis and consideration of critical variables. Psychol Bull 117(2):250–270. [DOI] [PubMed] [Google Scholar]
- Watson NV, Kimura D. 1991. Nontrivial sex differences in throwing and intercepting: Relation to psychometrically-defined spatial functions. Personality and Individual Differences 12(5):375–385. [Google Scholar]
- White PC, Speiser PW. 2000. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 21(3):245–291. [DOI] [PubMed] [Google Scholar]
- Winfeld M, Patel P, Shah B, Nass R, Milla S. 2013. Early occurrence of cerebral white matter abnormality detected in a neonate with salt-wasting congenital adrenal hyperplasia. J Pediatr Endocrinol Metab 26(1–2):13–17. [DOI] [PubMed] [Google Scholar]
- Wisniewski AB, Migeon CJ, Meyer-Bahlburg HF, Gearhart JP, Berkovitz GD, Brown TR, Money J. 2000. Complete androgen insensitivity syndrome: long-term medical, surgical, and psychosexual outcome. The Journal of clinical endocrinology and metabolism 85(8):2664–2669. [DOI] [PubMed] [Google Scholar]
- Witchel SF, Azziz R. 2011. Congenital adrenal hyperplasia. J Pediatr Adolesc Gynecol 24(3):116–126. [DOI] [PubMed] [Google Scholar]
- Woolley DG, Vermaercke B, Op de Beeck H, Wagemans J, Gantois I, D’Hooge R, Swinnen SP, Wenderoth N. 2010. Sex differences in human virtual water maze performance: novel measures reveal the relative contribution of directional responding and spatial knowledge. Behav Brain Res 208(2):408–414. [DOI] [PubMed] [Google Scholar]
- Zucker KJ, Bradley SJ, Oliver G, Blake J, Fleming S, Hood J. 1996. Psychosexual development of women with congenital adrenal hyperplasia. Hormones and behavior 30(4):300–318. [DOI] [PubMed] [Google Scholar]