

Abstract
Diagnosing lymphomas and leukemias requires challenging integration of microscopically-visible cellular morphology and antibody-identified cell type-specific molecule expression. Here, we merge these orthogonal techniques onto a single, high-throughput, highly-multiplexed, single cell assay. Using single cell mass cytometry, we simultaneously quantify 12 molecular components underlying cell morphological features with a multitude of phenotypes across 71 diverse hematologic diagnoses. This method, “single cell morphometric profiling,” reveals robust patterns of “morphometric” markers for each microscopically-identifiable cell type, distinct from other cell lineages. Individual features have particular diagnostic utilities, such as: VAMP-7 for flow cytometric side scatter; lamin B1 and rRNA highlighting progenitor populations comprising acute leukemias; and lamin A/C distinguishing mature T-cell lymphomas from normal T-cells. Computationally combined with machine learning, they form consistent two-dimensional maps for visualizing sample composition and tracing differentiation of normal and leukemic myeloid cells. Benchmarking against hematopathologists, we used morphometric machine learning for blast enumeration in myeloid leukemias – a classically difficult problem with existing technologies. Integrating morphometry with deep surface immunophenotyping yields a versatile platform amendable to both traditional cytometry plots and high-dimensional augmentation with new diagnostic capabilities, lending itself to automation for routine, systematic hematopathology diagnosis.
Keywords: CyTOF, mass cytometry, morphometry, clinical diagnostics, hematopathology, lymphoma, leukemia
INTRODUCTION
Lymphomas and leukemias are the most common type of cancer in children, and account for 10% of all cancer diagnoses in the United States.1 Advancements in prognostication and therapeutics have drastically increased the complexity of the diagnostic classification for these and other hematopoietic tumors. However, hematopathologists often make these diagnoses based upon light microscopy and immunophenotyping (flow cytometry and/or immunohistochemistry). These are heavily-manual, skill-dependent, non-scalable methods, whose disparate, jigsaw data must be mentally assembled into a unified diagnosis.
With microscopy, pathologists use visible differences in subcellular structures to identify cell types, abnormal cells, and their proportions. Cells with similar or overlapping morphologies cannot be distinguished this way, e.g. small B vs. T lymphocytes and acute myeloid vs. acute lymphoid leukemias. And with some cell morphologies and diagnoses, significant inter-observer variability and biases exist even among expert hematopathologists.2–4
In flow cytometry, cells are stained with customized mixtures of fluorescently-labeled antibodies against cell type-specific molecules (markers) – mostly cluster of differentiation (CD) glycoproteins on the cell surface. Each cell individually flows through a series of lasers and detectors to quantify the fluorescence, determine the presence or absence of each marker, and thereby identify its cell type by the pattern of markers (immunophenotype). This can distinguish B vs. T cells and myeloid vs. lymphoid cells, and works well for normal samples with an expected composition of known cell types and normal marker expression on all cells. However, it is limited by the antibodies selected for testing, and cannot distinguish cells with similar or overlapping immunophenotypes – particularly neoplasms which closely mimic their cells of origin. For instance, the cytometric profiles of normal monocytes, chronic monocytic leukemias, and acute monocytic leukemias all overlap.5–7 The same can be said for normal myelopoiesis, myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPN), and MDS/MPN neoplasms. These situations all currently require manual microscopy-based interrogation to resolve.5
To further complicate matters, “cell type-specific” molecules which behave well on healthy patient samples are not as specific in tumors, which can abnormally express or lack expression of virtually any surface molecule.5,8 Our dataset alone includes a CD20+ NK/T cell lymphoma, a CD3− T cell lymphoma, three CD19+ myeloid leukemias, and a CD56+ B cell leukemia. Immunotherapies such as the anti-CD20 agent rituximab and chimeric antigen receptor T cells (CAR-T) against CD19 further complicate immunophenotype-only approaches where the targeted antigen can be down-regulated as a mechanism of therapeutic escape.9 Interpretation is also complicated by large antibody panels, where 15-30 antibody reagents is fairly typical for characterizing blood cancers. Because diagnostic instruments can detect a practical simultaneous maximum of about 10, they must be split across several independent tests. If two markers of interest are in different tubes, determining whether both are co-expressed can be difficult and is sometimes impossible. To do this, hematopathologists often make assumptions based on prior knowledge and experience, which is not easily automated.
Therefore, neither microscopy or clinical cytometry alone is individually sufficient to make relatively common diagnoses, and even together require mental integration by a pathologist with years of continuous training and practice. However, combining both morphology and immunophenotype into a single platform could improve diagnostic capabilities beyond either modality alone. The subcellular features that pathologists examine microscopically are largely intracellular structures common to many cell types – e.g., chromatin, cytoplasm, and vesicles – but vary in amount and composition. Measuring the molecules underlying these structures provides us a way to “quantify” morphology, i.e. “morphometry,” and enables a cytometry workflow to indirectly “see” the features that pathologists see with microscopes.
But incorporating these morphometric markers into already-complex multi-tube fluorescent flow cytometry panels is unwieldy, both in technical performance and in interpretation. And thinking long-term, multiple immunotherapy clinical trials demand quantitative estimates of target antigen frequency on tumor cells. As development of these therapies accelerates, correlating across multiple flow cytometry tubes to both characterize tumor cells and quantify their therapeutic targets becomes more and more cumbersome. These increasing complexities, as well as the need to customize assays in a disease- and/or sample type-specific manner, beg for a more scalable, standardized, automatable approach.
Due to the high degree of multiplexing and quantitative dynamic range, approaches like mass cytometry/Cytometry by Time of Flight (CyTOF) hold promise to address these demands. Mass cytometry uses heavy metal isotope conjugates in the place of fluorophores to quantify over 40 antigens simultaneously on a single cell, across millions of cells in a single day.10–12 With this, an entire diagnostic panel can be combined into a single test to eliminate the need for customization and assay cross-correlation while permitting systematic and automatable analysis. Sample multiplexing – processing and running multiple samples simultaneously – allows batching, which reduces costs and stabilizes workflow despite fluctuations in workload and staffing.13,14 And key to a viable clinical test, it can be performed within the expected 24 hour turn-around-time and interpreted by a single hematopathologist, rather than taking weeks to months with a team of data scientists.
Here, we identify a set of cellular antigens that can be targeted by antibody probes to serve as quantitative surrogates for granularity, granular color, chromatin quality, nuclear shape, nucleolar size, cytoplasmic color, and cell size. Using this set of morphometric reagents, termed “scatterbodies,” as well as associated technical protocols and improvements, we characterize nucleated cells from healthy human bone marrow and contrast them to cells from 71 diverse disease bone marrow and blood samples to define normal and neoplastic populations by their single cell morphometric features. Samples include 26 acute myeloid leukemias (AML) enriched for monocytic differentiation and lack of CD34, five B lymphoblastic leukemias (B-ALL), three T lymphoblastic leukemias (TALL), three acute leukemias of ambiguous lineage (MPAL), nine mature B cell lymphomas, three myelomas, five mature T cell lymphomas, and ten non-acute myeloid neoplasms including myelodysplasias and systemic mastocytoses, among others. Individual scatterbodies show diagnostically useful characteristics, such as lamin B1 and rRNA enriched in normal and leukemic blasts regardless of lineage, and lamin A/C marking mature T cell lymphomas. Utilizing scatterbodies in combination, we demonstrate a reproducible dimensionality reduction technique based on linear discriminate analysis (LDA) to construct morphometric maps. These visualizations allow us to trace healthy and malignant hematopoietic differentiation as continuous rather than discretized processes, forming a basis for targeted cell enumeration which leverages decades of experience in cytometric cancer diagnosis and integrates seamlessly into current clinical laboratory workflows.
RESULTS
Scatterbodies quantify distinguishing structural features of hematopoietic cells
In order to quantitatively capture the cell morphology routinely assessed by light microscopy, we targeted multiple classes of structural components common to blood and immune cells (Fig. 1a). We ultimately selected eleven morphometric targets (Fig. 1a, b) as reflections of key morphologic features (Fig. 1c) which quantifiably differ between the major, diagnostically-relevant hematopoietic cell populations (Fig. 1d) – granulocytes, lymphocytes, monocytes, nucleated erythroid precursors, and hematopoietic blast cells. While keeping track of eleven cytometry dimensions can be challenging, the multiplexed single cell analysis of these features lends itself to being computationally combined into simple, reproducible two-dimensional representations – such as a stable “morphometric map” (Fig. 1e).
Figure 1: Scatterbodies capture morphologic differences of major hematopoietic cell populations in healthy human bone marrow.
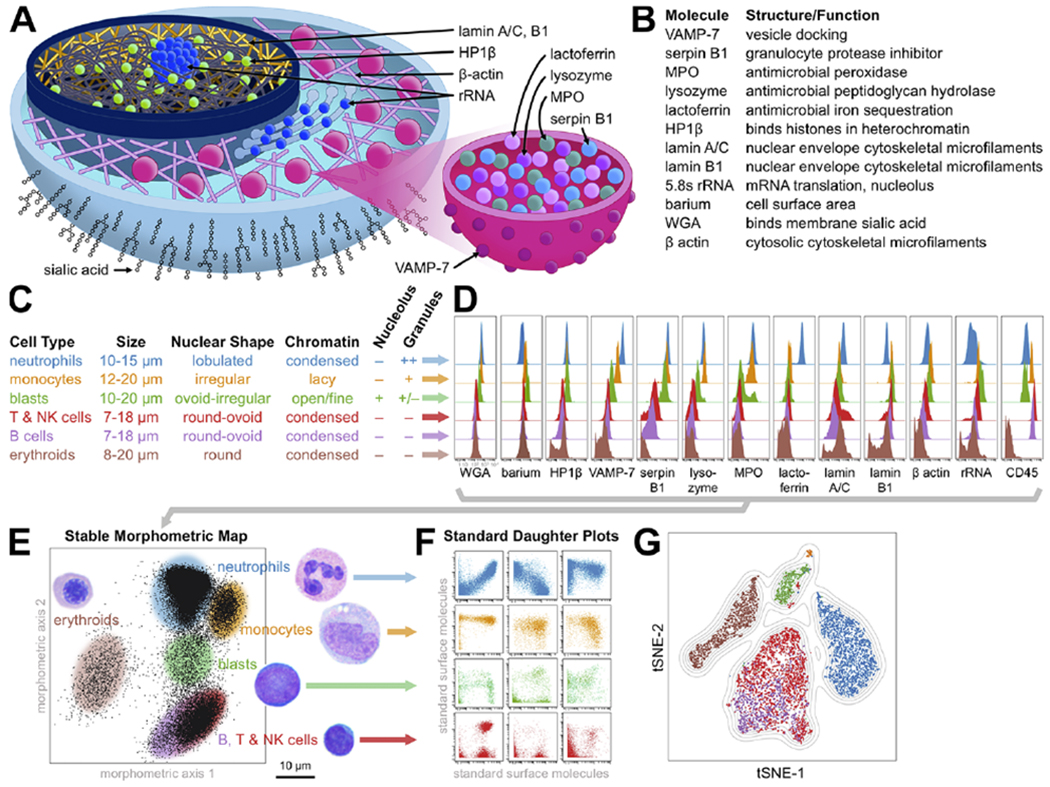
A) Diagram of scatterbody targets within a cutaway of a cell (left) and a granule within the cell (right). Note the presence of rRNA in both the nucleolus and ER. B) Summary table of scatterbodies. C-F) Summary of project design. C) Diagnostically important morphologic characteristics of major cell populations were morphometrically captured by scatterbodies. Cell sizes are from Carr et. al.39 D) Histograms of cell frequency distribution vs. scatterbody expression are compared for the major hematopoietic cell populations in a healthy human bone marrow. E) Reproducible axes built from combinations of scatterbodies form a stable morphometric “map,” whereby each population falls into a specific region on the map. Selecting a region from this combinatorial map (shaded ovals) is therefore similar to using multiple light microscopic features to identify cells (labels colored as in C, with adjacent images). F) Plotting cells from selected regions onto biaxial plots of standard surface molecules reveals immunophenotypic cell type composition and any abnormal cell types. G) t-SNE plot of the cell populations, generated using only scatterbodies and CD45, colored by cell identity as in C.
Selecting a morphometric region from this map (gating) would thus be like a pathologist using multiple morphologic features to visually identify a particular cell type – only quantitatively and reproducibly. Taking this “gated” region and plotting its contents along the standard surface CD molecules would then reveal its immunophenotypic cell type composition and any abnormal cell types (Fig. 1f). In this way, we could combine morphometry with standard immunophenotypic characterization.
We began by demonstrating differences in the quantities (expression) of morphometric targets between the major cell types in normal, healthy, human bone marrow (Fig. 1d). To accomplish this, we stained normal marrow samples with a mixture of both our morphometric reagents and classic cell type-specific CD antibodies (Supplementary Table 1). By assuming normal, conventional expression of these latter CD markers, we used them to isolate (gate) the major cell types within the samples in silico (Supplementary Fig. 1), and then independently evaluated their morphometric antigen expression.
The major hematopoietic populations in healthy bone marrow showed distinctive morphometric profiles, reflecting differences in cell morphology. Monocytes and neutrophils produce lysosomal/peroxisomal vesicles (granules) (Fig. 1a), microscopically visible as grainy cytoplasm and distinguishing them from non-granulocytes (lymphocytes and erythroids). Therefore, the antimicrobial enzymes and associated proteins within these granules – including serpin B1, lysozyme, MPO, lactoferrin – morphometrically separated granulated from non-granulated cells (Fig. 1d). VAMP-7 is a protein in the vesicular envelope which helps mediate docking to the cell membrane and release of granule contents (Fig. 1a).15 Neutrophils are more granular than monocytes and therefore expressed more VAMP-7 (Fig. 1d). As they mature, neutrophils also acquire lactoferrin in secondary granules, which have a paler, pink hue by microscopy than the primary granules of early granulocytes.
The remaining targets are present in all cells – not just granulocytes – but in varying amounts. Thus, it is natural that their differences are comparatively subtle, especially in arcsinh (log-like scale), although still several-fold. A meshwork of lamin A/C and B filaments forms the nuclear skeleton, and their quantities determine the mechanical properties and thereby the shape of the nucleus (Fig. 1a).16 An almost universal morphologic feature of blasts is so-called “fine chromatin,” which correlates with greater lamin B1 (Fig. 1d). Lamin A/C is greater in cell types with very round nuclei, such as nucleated erythroid precursors and plasma cells, consistent with experiments showing that overexpression induces nuclear hypolobation.17 The lamins are further characterized below.
5.8s rRNA is a ribosomal component necessary for the translation of mRNA into polypeptides and is a direct readout for ribosome copy number, predicted to be higher in cells with more endoplasmic reticulum (ER) volume and/or larger nucleoli – where ribosomes are assembled (Fig. 1a). Empirically, we found it to be greater in immature cells with prominent nucleoli such as blasts and associated with cells showing more abundant and/or basophilic cytoplasm (blue color by Wright-Giemsa stain) (Fig. 1d). The β-actin cytoskeletal meshwork interacts with granules to help regulate exocytosis – higher in granulocytes and monocytes (Fig. 1d). WGA lectin binds the sialic acids of surface membrane glycosylations, approximating cell surface area and thereby related to cell size (Fig. 1a). Larger cells like granulocytes and blasts have more surface membrane than lymphocytes and erythroids and thus bind more WGA (Fig. 1d).18 HP1β associates with transcriptionally silent regions of DNA in the nucleus and is associated with neutrophil differentiation.19 As lymphocytes and mature nucleated erythroid precursors are quite similar morphologically, we decided to separate them with CD45 (Fig. 1d, e). This well-established strategy takes advantage of one of the many uses for CD45, and avoids expending a valuable antibody slot for a more morphometric but single-purpose marker such as hemoglobin.
To confirm that our morphometric targets and CD45 could sufficiently capture the morphologic diversity of hematopoietic cells, we visualized the healthy samples using unsupervised t-Stochastic Neighbor Embedding (t-SNE) (Fig. 1g).20 While unstable (the plots are not identical from run to run), t-SNE agnostically separates cells using only the parameters fed to it, and displays them in a two dimensional plot. If fed sufficient morphometric parameters, then t-SNE would display all the morphologically distinct cell populations as separate dot clusters on the plot. If insufficient, then two or more of them would be inseparable. Using only our morphometric targets and CD45, t-SNE successfully separated the major, morphologically-distinct populations. Importantly, it did not separate B and T/NK cells, which are not morphologically distinct. Therefore, morphometric profiling quantitatively captured identifying morphologic features sufficient to classify the major cell types.
Single cell morphometric profiles are consistent across clinical patient samples with a broad spectrum of neoplasias
After demonstrating our method on normal, healthy human bone marrow samples, we sought to evaluate its utility in real-world patient samples containing neoplastic tumor cells. Such samples are typically refrigerated for days in anticoagulant with little oxygen – conditions which are known to affect light microscopic morphology.5
For this purpose, we initially selected 54 clinical samples spanning a broad range of the WHO hematopoietic tumor classification (AML, ALL, MDS, MPN, B and T cell lymphoma, myeloma). Almost every sample contained a mixture of malignant and background non-neoplastic cells. Some also contained dysplastic cells – differentiated cells which show abnormal morphologic features by light microscopy. We specifically enriched for tumors abnormally expressing cell type-“specific” CD markers from multiple cell types, abnormally lacking CD markers expected for their cell types, posing difficulties making the diagnosis by surface immunophenotype alone, and/or sharing immunophenotypic overlap with other diagnostic entities (Supplementary Table 2).5 In this way, we tested whether single cell morphometry was robust to neoplastic cell types with non-standard behaviors.
As with the healthy bone marrow samples, we had to define these patient sample populations using CD antibodies in order to evaluate their morphometric profiles independently (Supplementary Fig. 1). But due to all the CD marker abnormalities, we could not assume normal CD antigen expression – instead finding work-arounds using multiple isolation strategies and prior diagnostic knowledge. This approach could not be used consistently in a clinical diagnostic laboratory.
With cell lineages defined, we compared their morphometric profiles, providing primary data in the study repository. Normal cells from all 54 initial clinical samples showed consistent morphometric profiles (Supplementary Fig. 2) despite expected variation from differential storage times at 4° C. Neoplastic and dysplastic cells showed similar morphometry as their normal counterpart cell types, with interesting variations. To illustrate these points while representing the diversity of tumors and demonstrating the underlying morphometric principles, we chose eleven samples for detailed analysis.
Summarized as heat maps, patterns for each of the major cell populations came into focus (Fig. 2). Granule markers (VAMP-7, serpin B1, lysozyme, MPO, lactoferrin) distinguished monocytes and neutrophils (Fig. 2e, f, orange and blue dotted boxes) from nucleated erythroid precursors and lymphocytes (Fig. 2a, b, c, yellow dotted box). CD45 on lymphocytes (Fig. 2b, c) acted as a surrogate for lack of hemoglobin compared to erythroids (Fig. 2a), with subtler differences in lamin A/C, lamin B1, and β-actin. Lactoferrin differentiated neutrophils (Fig. 2f, bottom row of blue dotted box) from monocytes (Fig. 2e, bottom row of orange dotted box), with subtler differences in β-actin, VAMP-7, and CD45.
Figure 2: Morphometric profiles are consistent across clinical patient samples.
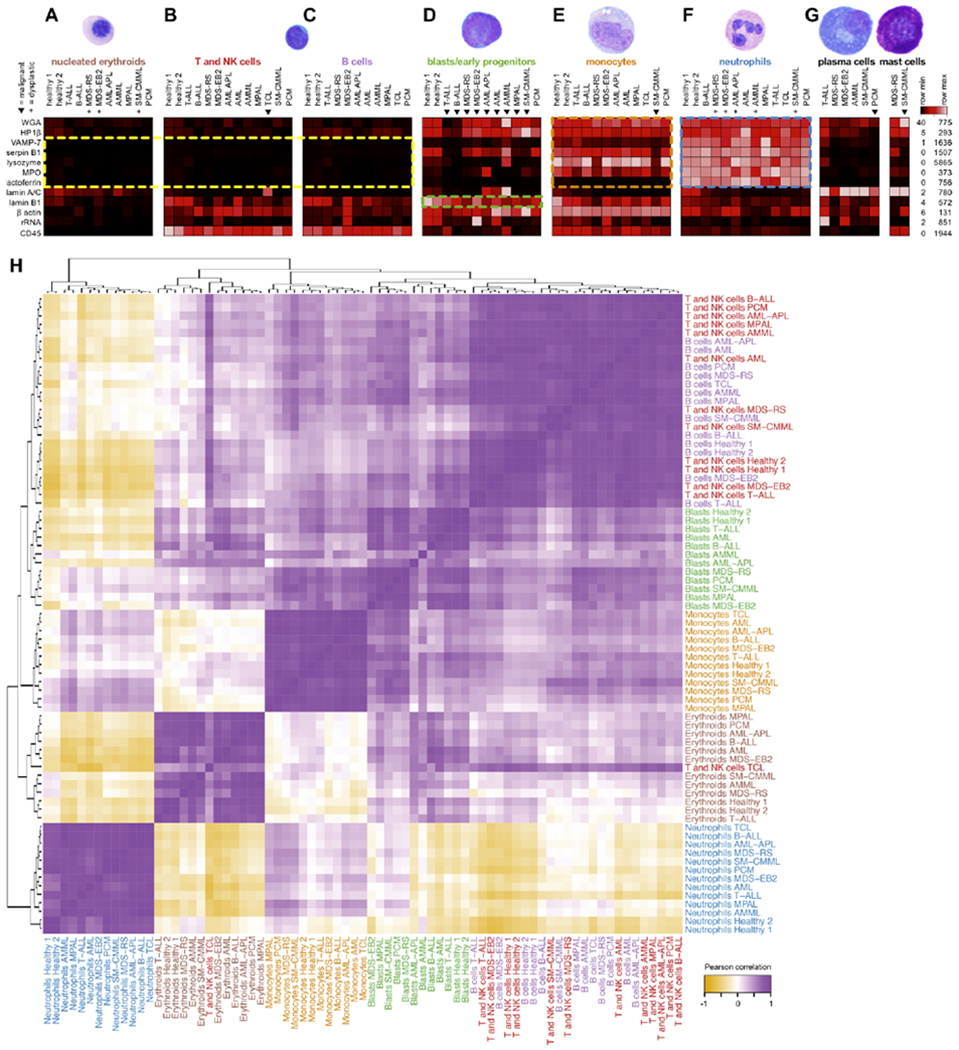
Scatterbody profiles of major populations (A-G, labels colored as in Fig. 1c) from the 11 main clinical samples and two healthy marrow donors. Clinical samples contain mixtures of normal and neoplastic populations, where triangles (▼) denote malignant populations as diagnosed by WHO criteria,5 and plus symbols (+) denote morphologically dysplastic (malformed) populations as determined by light microscopy. Not all samples contained significant numbers of all populations, and populations with fewer than 20 events (cells) are not shown. Each column represents the median values of scatterbodies of a single population from a single sample, scaled by row. Lamin A/C and lysozyme were scaled to a maximum of 500 counts due to plasma cell lamin A/C obscuring other populations and a lysozyme outlier >6-fold greater than all other populations. H) Pearson correlation of scatterbody profiles for hand-gated cell populations across all eleven representative samples and two healthy controls. Each scatterbody profile is defined as the vector of median expression values for all scatterbodies. The heatmap was hierarchically clustered and labels were colored by hematopoietic cell population.
Blasts are progenitors for all the other hematopoietic populations, with normal blasts showing a profile intermediate between lymphocytes and myeloid cells (neutrophils and monocytes). However, morphometric profiles of malignant blasts further reflect their lineage propensity, i.e. blasts from lymphoid leukemias show a more lymphoid pattern, while those from myeloid leukemias show a more myeloid profile (Fig. 2d, starred columns).
In order to verify these patterns quantitatively, we performed Pearson correlation of morphometric profiles between cell populations across patients, and hierarchically clustered the resulting matrix (Fig. 2h). Morphometric profiles for the same cell type (but from different patients) were highly correlated with each other and grouped together. This was even true of the malignant blasts, despite including lymphoid, myeloid, and ambiguous lineage leukemias widely varying in their expression of surface CD markers (Fig. 3d). As expected, B and T/NK cells largely grouped together owing to both similar morphology and morphometry. A lone exception – T cells from the TCL sample – demonstrated a diagnostically useful property, discussed below. In the larger data set, mature B and T cell lymphomas were morphometrically similar to non-neoplastic B and T cells (Supplementary Fig. 2). These overall findings reinforce that morphometric profiling consistently identifies cell types, even in disease samples, and in the absence of cell type-specific markers.
Figure 3: Individual morphometric features have unique diagnostic capabilities.
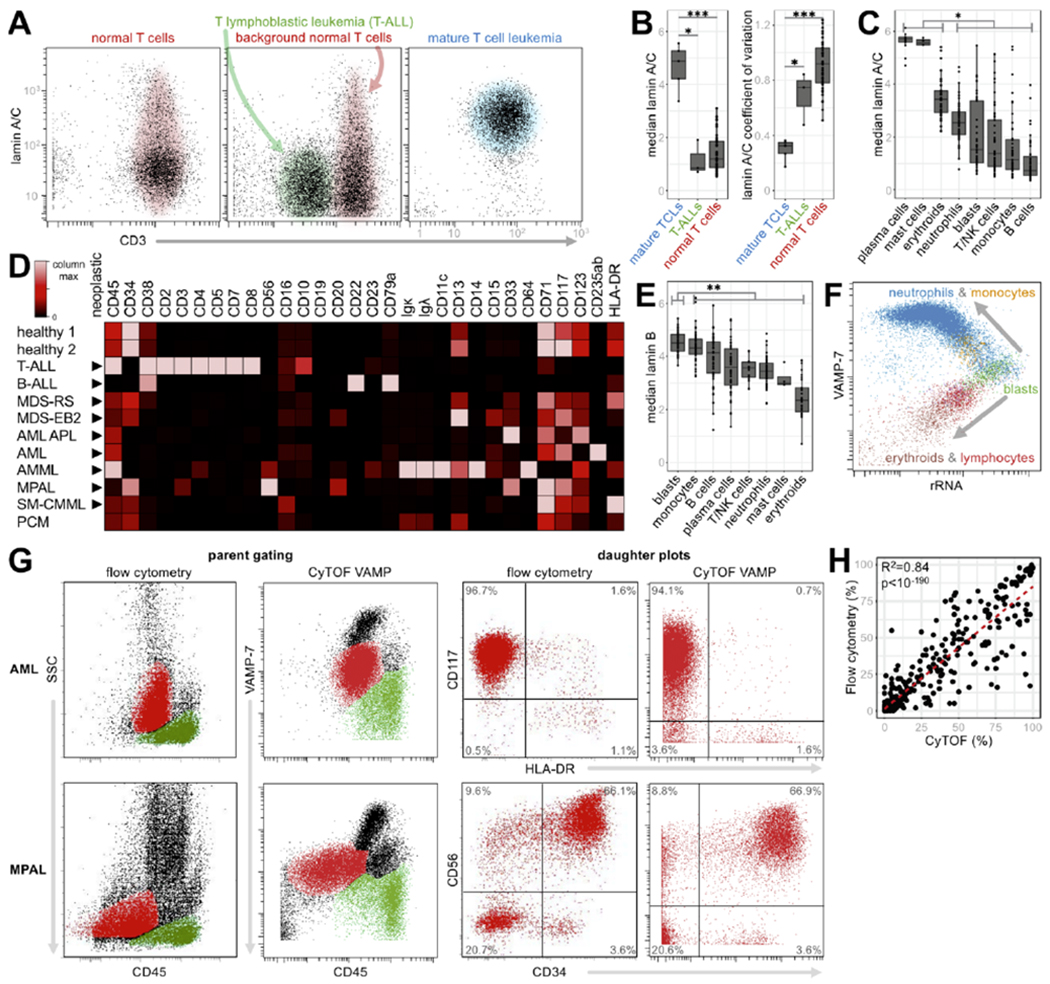
A) Lamin A/C expression on CD2+ cells in healthy marrow (left), a sample with T lymphoblastic leukemia (T-ALL, middle), and a mature T cell leukemia (TCL, right), with normal CD3+ T cells shaded pink, T-ALL green, and TCL blue. B) Median lamin A/C expression (asinh scale) and lamin A/C coefficients of variation (CV) in mature T cell lymphomas/leukemias (TCL), T-ALLs, and normal/reactive T cells across the initial set of 56 samples (two normal bone marrow samples + 54 clinical samples). Differences in distribution were evaluated between mature TCLs and each of T-ALLs and normal/reactive T cells. C) Median lamin A/C expression (asinh scale) in hematopoietic populations across the initial set of 56 samples. Differences in distribution were evaluated between plasma cells, mast cells, and erythroids, and each of the other five populations. D) Surface marker expression in blast populations is highly inconsistent. Triangles (▶) denote malignant populations. Each row represents the median values of surface markers, scaled from 0 to the maximum in the column, except CD16, CD10, CD19, CD20, CD23, and CD15 were scaled from 0 to 10 because maxima were below 10 counts (within the noise floor). Notably, the B-ALL sample is CD19− by both CyTOF and diagnostic flow cytometry after anti-CD19 CAR-T therapy. E) Median lamin B1 expression (asinh scale) in hematopoietic populations across the initial set of 56 samples. Differences in distribution were evaluated between blasts and each of the other hematopoietic populations across the initial set of 56 samples. Statistical significance was evaluated by the Wilcoxon signed rank test or Wilcoxon rank sum test (for distributions with <10 observations). Multiple hypothesis correction was performed using the Bonferroni method, * denotes p<0.05, ** denotes p<0.01, *** denotes p<0.001. F) rRNA decreases as cells differentiate from blasts (green) to VAMP-7+ granulated cells – monocytes (orange) and neutrophils (blue) – or VAMP-7 non-granulated cells – erythroids (brown) and lymphocytes (T cells in red and B cells in purple). G) VAMP-7 is functionally equivalent to SSC, enabling direct translation of mass cytometry data into general-purpose diagnostic hematopathology workflow. Parent plots of two samples (AML, MPAL) show ungated events by clinical flow cytometry SSC vs. CD45 (left column) and mass cytometry VAMP-7 vs. CD45 (second column). The blast gates (red events) and lymphocyte gates (green events) were defined on these plots with the aid of backgating. Events from the parent blast gates visualized on daughter plots for flow (third column) and mass cytometry (right column). Quadrants were set to quantify the number of events positive and negative for each marker. Fluors used in clinical flow cytometry plots were CD45 PerCP-Cy5.5, CD34 APC, CD56 PE, CD117 PE, and HLA-DR APC. H) Percent of events positive for each marker in every daughter plot across eleven samples (483 total data points) generated by parent gating using mass (x-axis) or flow (y-axis) cytometry. Correlation was evaluated by the Pearson method.
Individual morphometric features have unique diagnostic capabilities
After assessing the overall morphometric patterns of the major cell types, we examined the special, diagnostically-useful characteristics of individual morphometric targets.
We found lamin A/C to be useful for identifying mature T cell lymphomas. Hematopathologists currently lack a simple method to visually or cytometrically identify these clonal T cell populations, contrasting with the relative ease of detecting B cell lymphomas by κ/λ cytometry.21,22 Intrigued that normal/reactive peripheral blood, bone marrow, and lymph node T cells express a spectrum of lamin A/C (Supplementary Fig. 3), we surmised that neoplastic populations might show more homogeneous lamin A/C. Thus, their morphometry would reflect their clonal origin.
Strikingly, all five of our mature T cell lymphomas expressed uniform and brighter lamin A/C (Fig. 3a, b, Supplementary Fig. 3). These samples not only had significantly higher median expression of lamin A/C in their neoplastic T cells as compared to T-ALL blasts and normal T cells, but also significantly lower coefficients of variation (CV), reflecting tighter distributions that one might expect in clonal populations – suggesting lamin A/C can act as a proteomic marker of T cell clonal expansion.
This feature is useful for diagnosing mature T cell lymphomas, which can be overlooked or difficult to distinguish from normal T cells in clinical flow cytometry. It also appears useful for distinguishing T prolymphocytic leukemia (T-PLL) from T lymphoblastic leukemia (T-ALL) in a way that standard clinical flow cytometry cannot (Supplementary Fig. 3, clinical vignette).
Lamin A/C is also significantly more abundant in nucleated erythroid precursors, normal and neoplastic plasma cells, and normal and neoplastic mast cells than all other hematopoietic populations (Fig. 3c, Fig. 2a, g, Supplementary Fig. 2). This facilitates alternative gating strategies for these populations. In particular, nucleated erythroid cells are critical to evaluating myelodysplasia microscopically, but are not routinely evaluated in clinical cytometry. CD45 vs. lamin A/C separates these cells into two populations, away from other marrow cells – one less mature, dim CD45+ CD71+ variably CD235+ brightly lamin A/C+, and one more mature, CD45− CD71+ CD235+ moderately lamin A/C+ (Supplementary Fig. 2). As a morphometric anchor for tracing erythroid maturation, lamin A/C may show utility for identifying or confirming dyserythropoiesis in myelodysplastic syndromes (Supplementary Fig. 4).
In addition to practical cytometric T cell clonality, another exigency in diagnostic cytometry is a consistent identifier for neoplastic blasts – the malignant cells in acute leukemias. Clinically, the main blast cell indicators are CD34 and CD117.5 However, neither is universally expressed, and both can be expressed on mast cells, as well as early erythroid and platelet precursors.
In fact, for the eleven clinical samples in Fig. 3, identifying blasts by surface markers required six different strategies because no cell type-specific markers were consistent (Fig. 3d). Thus, we sought to find a marker correlating with the morphologically “fine” nuclear chromatin most blasts possess.5 Among the 39 samples with blast populations >20 cells, normal and neoplastic blasts consistently expressed high levels of lamin B1 (Fig. 2d, green dotted box, Fig. 3e, Supplementary Fig. 2). The median expression of lamin B1 in these blasts was significantly higher than that of every other hematopoietic population. Remarkably, this was consistent not only with typical CD34+ myeloid blasts (normal, MDS-EB2 in Fig. 2d), but also in a more differentiated CD34− T lymphoblastic leukemia (T-ALL in Fig. 2d) and classically difficult acute myeloid leukemias such as CD34− CD117− acute myelomonocytic leukemia (AMML in Fig. 2d). Given the immunophenotypic and lineage diversity of the leukemias shown here, this single cell morphometric strategy with lamin B1 quantification represents the most consistent blast identifier to date.
We also found blasts to be strong expressers of rRNA, although less than plasma cells and not as consistently as for lamin B1 (Supplementary Fig. 2). Interestingly, loss of rRNA correlated with maturation of neutrophils, erythroid cells, and lymphocytes. Moreover, this relationship appeared relatively linear, operating as a single antigen “pseudo-time” axis able to track cell development along multiple lineages. For example, in combination with VAMP-7 (described below), we were able to visualize the evolution of blasts into myeloid and lymphoid cells in a healthy bone marrow sample (Fig. 2f).
Clinically, lamin B1 and rRNA appear useful for identifying blast populations independently of surface antigens (Supplementary Fig. 8). This is particularly relevant in post-CAR-T specimens lacking canonical markers (Supplementary Fig. 5, clinical vignette).
Overall, the above findings thus exemplify how our morphometric targets – general structural proteins not specific to any cell type – can address specific diagnostic difficulties and provide insights across multiple cell lineages.
VAMP-7 is a surrogate for side scatter in classifying unknown hematopoietic malignancies
Of the individual morphometric targets, perhaps the most diagnostically impactful was VAMP-7, owing to its strong resemblance to light-based side scatter (SSC). Current fluorescence-based diagnostic flow cytometry is built upon decades of experience defining the major cell populations by their characteristic locations on a scatterplot of CD45 vs. SSC (Supplementary Fig. 6).23–25 By selecting these regions (gating) and assessing cell surface lineage marker expression on daughter plots, diagnosticians determine whether each gate contains abnormal cells. However, high content single cell proteomic methods like mass cytometry and CITE-seq cannot measure light-based SSC, hindering their use as general-purpose diagnostic tools.26,27
SSC is largely derived from light orthogonally reflecting off of intracellular granules, of which VAMP-7 is a functional component. Across our initial set of 54 clinical samples, we found VAMP-7 vs. CD45 plots to be strikingly similar to SSC vs. CD45 plots (Supplementary Fig. 7). Therefore, we queried whether it could be a valid alternative to a non-fluorescence cytometry diagnostic workflow (Fig. 3g).
To accomplish this, we directly compared clinical flow cytometry data from Stanford’s hematopathology service – gated in the standard way using SSC vs. CD45 – to mass cytometry data – generated from the same clinical specimen and gated by VAMP-7 vs. CD45 (Supplementary Fig. 8). For our eleven selected samples, we processed the data similarly – treating them as “unknown” diagnoses and prospectively gating the major hematopoietic populations without prior knowledge. From each gate, we produced hundreds of dot plots for independent side-by-side comparisons by four board-certified hematopathologists (study authors AT, JO, MJ, RO). Aside from slight differences in antibody clones and different storage times between flow and mass cytometry analysis, we found the plots to be diagnostically equivalent – that is, both analyses captured the same (sub)populations, showed the same immunophenotypic characteristics, and resulted in the same disease diagnoses.
To validate this more quantitatively, we further evaluated the percentage of cells in each gate positive for all comparable markers, resulting in a total of 483 points of comparison between the parent gating strategies (Fig. 3h). The results from flow and morphometric mass cytometry were highly correlated (R2=0.84, p<10−190).
Thus, VAMP-7 can be adopted in mass cytometry panels as a surrogate for SSC. Moreover, it illuminates a clear path for clinical validation, laboratory data processing workflow, pathologist interpretation, and adoption of the CyTOF as a clinical laboratory instrument.
Multiparameter morphometric maps for optimal population identification
While VAMP-7 was sufficient to recapitulate SSC for laboratory diagnostics, we hypothesized that new two-dimensional visualizations could be generated to integrate multiple dimensions of cell morphometry – similarly to how a pathologist examines multiple morphologic features on every cell. Among other possibilities, this could result in better separation of normal and dysplastic hematopoietic populations. To function as a morphometric map for clinical diagnostics, this visualization needed to be reproducible, interpretable, and capable of integrating new data.
To these ends, we used supervised dimensionality reduction by linear discriminant analysis (LDA) to generate morphometric maps (MM) (Fig. 4a, see methods for details). Originally intended as a classifier, LDA is a machine learning approach that uses defined training data to find optimal linear combinations of parameters (i.e. morphometric targets) to separate classes (i.e. hematopoietic cell populations). If subsequent data is scaled in a consistent manner, these linear combinations can then be used to identify the class of new data. However, our purpose was visualization rather than simple classification, so we used these linear combinations for reducing the data to two dimensions that optimally separate the major hematopoietic populations.
Figure 4: Morphometric maps recapitulate and improve upon SSC vs. CD45 gating.
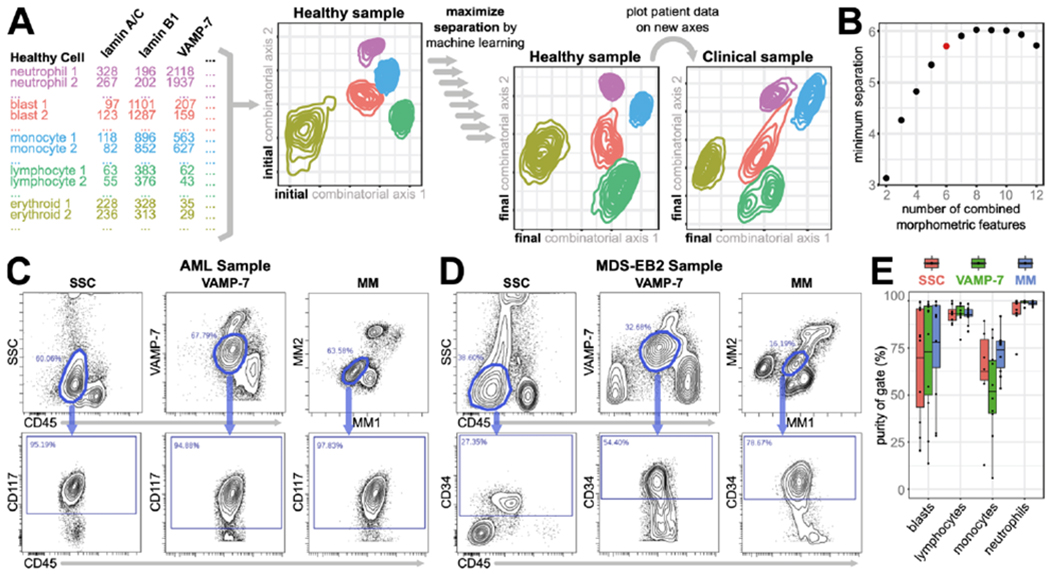
A) Workflow for generation of morphometric axes. Scatterbody values for single cells from each population were plotted onto combinatorial axes, which were improved by training on separating populations. Sample data were then plotted on the final combinatorial axes. B) The number of markers used to generate different morphometric axes vs. the minimum Euclidean distance between all population means for the highest-scoring marker combination. Red dot signifies the elbow point. C and D) Parent plots of the AML (C, top row) and MDS-EB2 (D, top row) patient samples with tight gates drawn on putative blast populations using CD45 vs. SSC (left columns), CD45 vs. VAMP-7 (middle columns), and MM axes (right columns). Daughter plots (bottom rows) depict and quantify the purity of the parent gates. E) Quantification of gate purity for the major hematopoietic populations from the 11 exemplative samples, using CD45 vs. SSC (salmon), CD45 vs. VAMP-7 (green), or MM axes (blue).
We began by identifying the major cell populations from a healthy bone marrow sample – erythroids, lymphocytes, monocytes, blasts, and neutrophils – based on cell type-specific CD antigens. Using the morphometric data for the individual cells in those populations, the cells were plotted on two-dimensional graphs whose axes were built from linear combinations of morphometric targets. We trained the algorithm to maximally separate the major cell types on these “morphometric maps.” We implemented a hybrid of forward and reverse stepwise subset selection to find the optimal combination of single cell features for capturing each cell’s morphometric profile with the minimum number of required markers (Fig. 4b).28 The algorithm selected six markers – lactoferrin, lamin A/C, lamin B1, lysozyme, VAMP-7, and CD45 – for generating map axes. While the axes were trained on a healthy control, subsequent visualizations of new data (e.g. unknown/disease samples) could thus be achieved with simple matrix multiplication, making the method reproducible, computationally efficient, and without the need for user-defined parameters to plot new data.
As a baseline, we compared the populations from our morphometric map (MM) to those by SSC vs. CD45 and VAMP-7 vs. CD45, using the same strategy of evaluating percentage of gated cells positive for surface markers as in Fig. 3f. We found LD gating was highly correlated with both SSC- and VAMP-7-based gating (R2 = 0.86; 0.96, p<10−200, Supplementary Fig. 9) – demonstrating that MM gating was a viable alternative.
We then asked if there were any advantages to MM axes over SSC or VAMP-7 vs. CD45. An important use case for this technique is separating abnormal blast cells away from contaminating monocytic and granulocytic cells – in order to obtain an accurate surface CD marker profile and sensitively detect abnormalities thereof. To test the ability of this morphometric map (MM) to accomplish this, we compared the purity of blast cell identification using MM axes with those by CD45 vs. SSC and CD45 vs. VAMP-7. Using our eleven representative disease samples, we naively gated (without the knowledge of cell type-specific markers) neoplastic blasts using CD45 vs. SSC, CD45 vs. VAMP-7, or MM axes. In an uncomplicated AML sample with a distinct leukemic population, all three methods performed well, with purities >94% as revealed by the expression of the stem cell factor receptor CD117 in selected cells (Fig. 4c). However, in the MDS-EB2 sample with 11% blasts, SSC gating performed poorly (27.3% purity), VAMP-7 gating performed moderately (54.4% purity), and MM gating achieved the highest purity (78.7%) based on the expression of the expression hematopoietic stem cell antigen CD34 (Fig. 4d).
Applying this metric to blasts, lymphocytes, monocytes, and neutrophils across our eleven representative samples, we found that MM gating performed the same or better compared to SSC and VAMP-7 in all four populations – showing higher and more consistent purity, particularly in the blast and monocyte gates (Fig. 4e, Supplementary Fig. 8). These results confirm that we can not only recapitulate, but also improve upon traditional light scatter-based morphological assessments by combining information from multiple single cell structural features into new morphometric axes with a supervised dimensionality reduction approach.
A myeloid morphometric map for contrasting normal granulopoiesis and myelodysplasia
Another important use case for the morphometric LDA approach is agnostically visualizing continuous and branching processes, particularly bone marrow myelopoiesis in myelodysplastic syndromes (MDS). Differentiation of blasts into neutrophils and monocytes is a gradual spectrum of differentiation. However, because traditional “gating” defines distinct cell populations with discrete cutoffs, it is ill-equipped for such continuous processes. In MDS, such abnormal differentiation can result in neutrophils and monocytes blending with one another in terms of both visual morphology and immunophenotype. Therefore, dividing them into separate gates creates artificial classifications whereas visualizing them together may achieve a more global view of the process and sensitive detection of dysplasia. Current visualizations of myeloid (dys)maturation are meandering and difficult to discern in many clinical samples (Supplementary Fig. 10).8,29,30
Using the same approach as we employed to generate a global morphometric map for major hematopoietic populations (Fig. 4a, b), we created a new set of myeloid differentiation (MD) axes to visualize maturation and differentiation of myeloid cells (Fig. 5a). As input to the algorithm, we fed single cell structural expression values of monocytes, blasts, and mature neutrophils from a healthy control sample. By optimally separating these populations, we hypothesized that we could spread the progression of differentiating cells across a new plot, facilitating easy visualization of the various stages of differentiation. The algorithm selected seven markers to generate the new axes: lactoferrin, lamin A/C, lamin B1, lysozyme, serpin B1, VAMP-7, and CD45. Strikingly, six of the seven markers were also used to generate the morphometric map in Fig. 4, though the differential contributions of each marker resulted in a starkly different separation and visualizations. Myeloid cells, identified using only VAMP-7 vs. CD45, were then visualized on the new axes and colored by their population of origin (Fig. 5a).
Figure 5: Myeloid morphometric maps for visualizing myelopoiesis and quantifying blasts in myeloid leukemias.
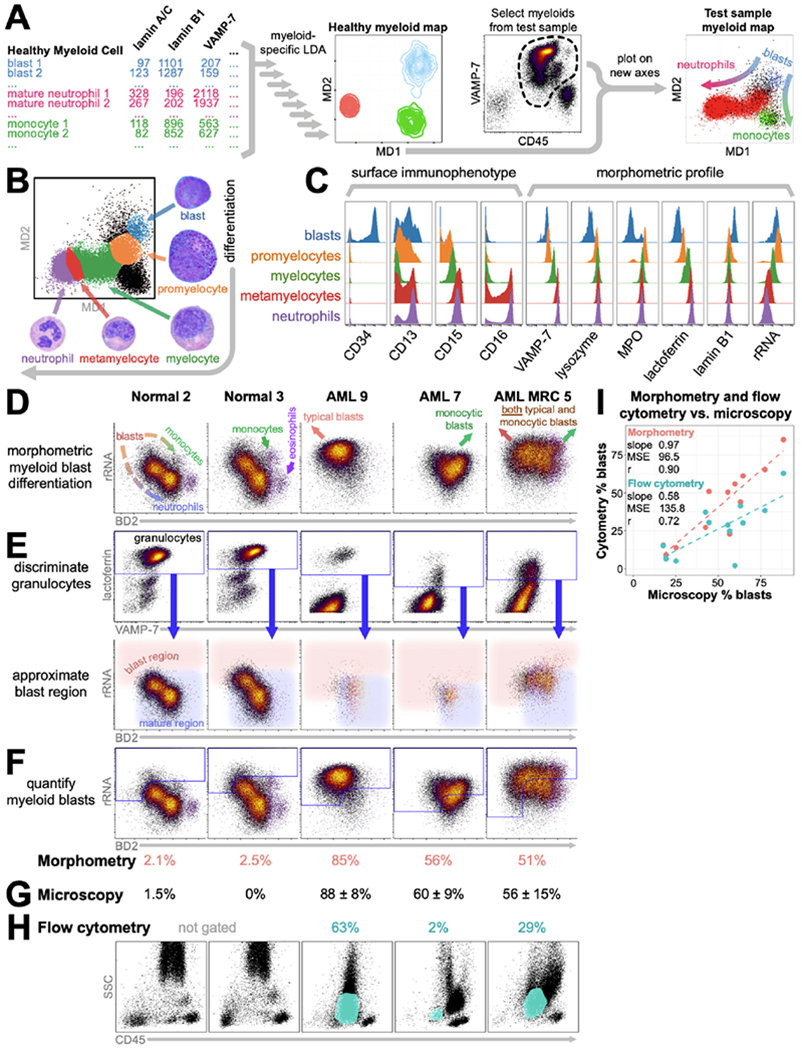
A) Workflow for generation of myeloid differentiation (MD) axes. B) Myeloid cells visualized on the myeloid differentiation (MD) axes. Gates (colors) are drawn for the five continuous phenotypes described for neutrophil differentiation, backgated by surface markers. Images depict the corresponding cell morphologies. C) Histograms of surface marker and scatterbody expression for the five gates drawn in B. D) Myeloid cells from two normal marrows and three representative myeloid leukemias, colored by density on a plot of rRNA vs. blast differentiation (BD2). E) Back-gating lactoferrin+ myeloid cells onto rRNA vs. BD2 enables approximation of the blast region – with lower BD2 and/or higher rRNA than lactoferrin+ granulocytes. F) The percentage of myeloid cells within this region (blue outline) is sb, scaled as described in the methods to yield the morphometric blast count (pink numbers). G) Morphologic blast counts by light microscopy ± one standard deviation. H) Flow cytometry blast counts (teal numbers) scaled from the percentage of CD45+ events within the blast gate (teal regions) on the SSC vs. CD45 plots. I) Blast counts by morphometry and flow cytometry plotted against those by light microscopy. MSE: mean squared error. r: Pearson correlation.
We then asked if these new axes would be useful for visualizing cells undergoing differentiation. We assessed the localization of the five states of neutrophil differentiation (blast → promyelocyte → myelocyte → metamyelocyte → neutrophil) on the MD axes based on the expression of canonical markers (CD34, CD13, CD15, and CD16), and found that each state localized to a unique position on the MD axes (Fig. 5b), facilitating manual identification directly in a single MD morphopmetric plot. This visualization of morphometric progression provides a simple way to visualize granulopoiesis without the use of canonical surface markers, recapitulating the morphologic progression that can be seen visually via microscopy by trained hematopathologists.
Using this MD-based gating, we evaluated the expression of surface markers and scatterbodies on each of these differentiation states in a healthy control sample (Fig. 5c). As has been previously described, each population had distinctive surface profiles, with coordinated increases in expression of CD15 and CD16 and loss and subsequent gain of CD13 as cells progress through the maturation process.23 Likewise, coordinated patterns in scatterbody expression can be seen as cells mature, including increased expression of granule-associated proteins (lysozyme, lactoferrin, and VAMP-7) and decreased expression of lamin B1 and rRNA content.
We applied our new axes to representative myeloid neoplasms to assess the dysregulation of their maturation processes (Supplementary Fig. 11). Plotting them on MD axes and coloring by canonical surface markers revealed that the axes placed populations with similar surface immunophenotypes in similar relative locations on the map (Supplementary Fig. 11a). Thus, they reflected the same underlying states of differentiation as surface markers, but independently and more intuitively – in two dimensions, without requiring extensive knowledge of cell populations and their surface marker combinations. Furthermore, morphometric maps appeared to better-reflect the morphologic distribution of cells than SSC vs. CD45 (Supplementary Fig. 11b–f).
Data-driven combinations of single cell structural features can thus be used to create morphometric maps which serve as quantitative and reproducible frameworks for assessing hematopoietic cell distributions. Robust to dysplastic cell types, these frameworks place surface immunophenotypes within a morphologic context and capture morphologic features and patterns typically requiring visual identification by expert observers.
A morphometric map to quantify blasts in myeloid leukemias with maturation
We next used our morphometric machine learning approach to perform blast enumeration in maturing myeloid leukemias – a recurring and classically difficult problem in both diagnostic cytometry and microscopy. Blast “counts” – the percentage of nucleated bone marrow cells which are blasts or blast equivalents – are required to diagnose and guide therapy for most myeloid neoplasms.5 The “gold” standard is light microscopy, which can suffer from inter-observer variability and biases.3,4 This is especially problematic when there is a gradual spectrum of maturation from blasts to mature cells such as neutrophils and monocytes – forcing pathologists to apply a subjective cutoff. Flow cytometry can approximate the count when blasts express sensitive and specific surface markers such as CD34 and CD117. However, the difficult cases with spectra of maturation often have complete or partial lack of CD34 and CD117 – forcing ad hoc strategies for enumeration (Fig. 5h, Supplementary Figs. 8, 11).
We therefore sought a morphometric method to reproducibly and systematically enumerate blasts in myeloid leukemias. We prospectively stained a new set of three normal marrows, 13 AMLs, and one MDS, chosen to enrich for CD34− and/or CD117− cases and to span a wide range of blast counts. To improve consistency between samples, we implemented a barcoded internal control to normalize for scatterbody staining variability (see methods and supplementary text). Using the same approach described above (Fig. 5a), we trained new blast differentiation (BD) axes to optimally separate monocytes, neutrophils, and blasts in the three healthy bone marrow samples, using only scatterbodies as input. As myeloid neoplasms had highly variable expression of MPO, lysozyme, and serpin B1, these markers were excluded from the analysis (Supplementary Fig. 2).
Plotting myeloid cells on rRNA vs. BD2 resulted in a morphometric map (Figure 5d) placing blasts at the upper left, maturing to neutrophils towards the lower right, and to monocytes towards the right. Importantly, normal monocytes (Figure 5d, Normal 2 and 3, green arrows) showed similar or lower rRNA levels than background maturing granulocytes. However, examination of our monocytic leukemias and unpublished immunohistochemical data revealed that leukemic blasts – both monocytic and non-monocytic – generally showed brighter rRNA than non-leukemic monocytes (Figure 5d, AML samples, MJ, unpublished data, Figure 1d, 3f). Furthermore, typical leukemic blasts were shifted towards the upper left of normal blasts (Figure 5d, AML 9) while monocytic blasts were shifted towards the upper right (Figure 5d, AML 7). Many cases show expansions in both types (Figure 5d, AML MRC 5, Supplementary Fig. 12).
To reduce blast enumeration to practice in a format requiring little a priori knowledge, we devised a strategy of using lactoferrin+ granulocytes (myelocytes, metamyelocytes, neutrophils) to set the maturity cut-off for blast identification within the rRNA vs. BD2 plot (Figure 5e). This allowed approximation of the blast region independent of CD34, CD117, or monocytic differentiation/maturation (Further discussion, Supplementary Figs. 12, 13).
Quantifying the myeloid events within this gate yielded blast counts across patient samples substantially closer to light microscopy than flow cytometry, slope 0.97 versus 0.58 and Pearson correlation 0.90 versus 0.72 (Figure 5f–h). Difficulty quantifying blasts by flow cytometry often occurs when leukemic blasts mature towards, immunophenotypically mimic, and intermix with other cell populations – thus making them difficult to identify. In the AML 9 sample, the CD34− CD117− blasts were phenotypically difficult to separate from background granulocytes (Figure 5h). In the AML 7 sample, the leukemic blasts had phenotypes similar to monocytes. And the AML MRC 5 sample featured both issues simultaneously. Our morphometric map for blast quantification was able to mitigate both sources of misidentification.
This example further demonstrates how supervised machine learning can be used to tailor a new combination of our multi-purpose morphometric markers to a specific diagnostic problem, improving our diagnostic capabilities beyond standard immunophenotyping and light microscopy alone.
DISCUSSION
Multiplexed single cell morphometric profiling is a quantitative, reproducible, high-throughput method demonstrated here by mass cytometry, which enables the measurement of molecules and structures correlating with key cellular features used by pathologists for classification and diagnosis in hematopathology. As fundamental components of cellular function, the structural targets identified here behave consistently across the major healthy and dysfunctional hematopoietic populations, unlike cell surface antigens that are notoriously inconsistent in many neoplasms.5
Individually, our morphometric targets have unique and valuable cell identification utilities in diagnostically relevant settings. VAMP-7 diagnostically substitutes for light-based side scatter (Fig 3e, f), providing a path to validating clinical mass cytometry with diagnostic flow cytometry. Lamin B1 marks both benign and malignant blasts regardless of lineage (Fig. 3d). Lamin A/C helps identify clonal mature T cell populations (Fig. 3a, b), while providing new ways to gate erythroid precursors, mast cells, and plasma cells (Fig 3c).
In combination, they can be used to build morphometric maps for multiparameter cell identification, similarly to a pathologist examining multiple morphologic features to identify normal and abnormal cells (Fig. 4, 5). Morphometric maps also provide a cell surface antigen-independent framework for tracing benign and malignant myeloid differentiation (Fig. 5). To build these maps, we developed a dimensionality reduction technique employing linear discriminant analysis (LDA), a machine learning approach for supervised classification. Lacking any stochastic step, this method produces visualizations which can be applied reproducibly and consistently across samples, in contrast to non-linear dimensionality reduction methods like t-SNE and UMAP, which create new maps with each run (Supplementary Fig. 14) and would challenge standardized clinical workflows.20,31 Furthermore, our approach incorporates the flexibility to build purpose-specific maps driven by the biology of the disease tissue of interest, generating tailored axes for both general immune monitoring (Fig. 4) and characterization of myelodysplasia (Fig. 5) here from a single analysis panel and run. Overlaying these maps with standard surface immunophenotyping markers, we can combine both morphology and cytometry into a single platform with capabilities beyond either platform separately. They can provide a more standardized approach to visualizing continuous and branching processes such as myelopoiesis. And patient-specific maps might improve our ability to distinctly separate and thus detect minimal residual disease.
Diagnostically, our single cell morphometric approach overcomes many of the challenges translating next generation single cell technologies, like mass cytometry, to clinical diagnostics – particularly the lack of light-based side scatter.26 Unlike other analytical pipelines, morphometric gating integrates seamlessly into current general-purpose diagnostic workflows. As it functions independently of lineage-specific surface markers, it is robust to edge cases of neoplasms with bizarre or ambiguous surface immunophenotypes. This was put into practice here by the independent analysis performed by Stanford hematopathologists comparing mass cytometry and flow cytometry plots (Supplementary Fig. 8) with little prior experience with mass cytometry data. Furthermore, with the development of immunotherapies which target canonical markers (e.g., CD19 for CAR-T cells), surface markers are increasingly unreliable for diagnosis. Thus, there is an ever-increasing need for robust and independent identifiers of neoplastic cell types.
The high degree of multiplexing in mass cytometry has inherent advantages in the clinical laboratory: 1. Antibodies do not need to be repeated across multiple tubes (e.g. CD45 in every clinical flow cytometry tube). 2. A single broad immunophenotyping tube can diagnose most samples, without requiring a pathologist to select or design a panel. 3. T raditional low-plex clinical flow cytometry requires cross-correlation of cell populations across multiple tubes, often using assumptions and experience. At times, this may be impossible and require running additional tubes with clinically-unvalidated and regulatorily-noncompliant combinations of fluorescent antibodies. As every probe in our mass cytometry panel is measured on every event, any combination and/or strategy of (sub)gating, back-gating, and plotting can be done directly in silico. 4. While flow cytometry can acquire raw data (cells per second) faster than mass cytometry, splitting a panel across multiple tubes and thoroughly washing the fluidics between these tubes (to prevent tube-to-tube cross-contamination) can be slower than running the panel as a single sample.
The main operational disadvantage of a mass cytometry-based platform is the time-consuming intracellular staining protocol. Other practical costs include those associated with instrument itself, nebulizers, argon gas, establishing robust operating procedures, technologist training, and clinical validation. Independent validation is important to ensure reproducibility between laboratories. And rather than focus on any specific disease, this study aimed to demonstrate the versatility and consistency of our method across a wide range of hematopoietic diseases and difficult cases. Thus, morphometric profiles need to be explored in greater depth for each disease entity to reveal further insights.
In the short term, these costs can be offset by using recently-developed single cell barcoding methods to process samples in batch.13,14 After barcoding each sample with a unique combination of heavy metals, samples are mixed together before staining and acquisition. Each cell is then traced back to its originating sample by its barcode. The staining can even be automated by robotics.32 A key to this is that all samples are stained with a single-tube universal panel. It reduces labor costs compared to clinical flow cytometry, where each panel is customized to the patient’s disease, sample cellularity, and clinical situation, and processing is performed one sample at a time, one tube at a time. Using a commercial inter-laboratory control to normalize scatterbodies also enables comparisons across days and standardization of combinatorial morphometric maps between laboratories.
In the long term, scatterbodies have exciting diagnostic and therapeutic potential. The haphazard surface immunophenotypes of myeloid neoplasms make it difficult to trace back to their cellular origins. Placing them onto a coherent underlying framework – using our novel combinatorial myeloid axes – we can potentially detect small dyspoietic populations and identify the earliest progenitors which are resistant to standard therapies. This opens new avenues for targeting these stem populations, as well as for closing off routes of therapeutic escape or clonal evolution. Additionally, these reagents may be useful for distinguishing and comparing cell types in other tissues and species.
Unlike laser-based measurements, our single cell morphometric profiling should be compatible with any antibody-based single cell technology, including CITE-seq or solid phase, slide-based immunohistochemistry, e.g. multiplexed ion beam imaging (MIBI) or imaging mass cytometry.27,33–35 In particular, these solid phase approaches could eventually be validated against clinical flow, as we did with mass cytometry here. Ultimately, this could facilitate both automated processing as well as morphometric profiling despite the lack of an adequate liquid sample – either due to a “dry tap” as with marrow fibrosis, or due to the lack of suitable preservation and transportation infrastructure as with low-resource settings.
Beyond the assay advantages of next-generation cytometry platforms, the potential for automating analysis with morphometrically-driven classifiers and standardized analysis creates an opportunity to democratize advanced hematopathology across medical centers and offers further economies in clinical operations. For instance, building a classifier to automatically identify major cell subsets would reduce time spent by highly-paid clinical laboratory scientists (CLS)/technologists in rote tasks such as manual gating. As mentioned in the introduction, the mental tube-to-tube cross-correlation required in low-plex clinical flow cytometry is overcome by increased multiplexing. Without the requirement of this mental component, a computer can generate much of the pathology report once the disease cell populations are identified. And as part of routine diagnosis, these populations could be automatically stored into a database without disruption to workflow. Furthermore, by combining each case with final diagnosis, genetic aberration(s), clinical outcome, and relapse (therapy-resistant) cell population data, machine learning-based classifiers could automatically predict diagnoses and prognoses for future samples, as well as identify unique clusters suggesting new diagnostic entities.
For the 70 years following Dr. Paul Ehrlich’s development of blood cell staining, the complete blood count (CBC) was performed manually by clinical laboratory technologists, painstakingly counting cells one-by-one using light microscopes. In the 70 years since Wallace Coulter invented the Coulter counter, it has reached the point where 80% of CBCs at our institution are performed and resulted by automated hematology analyzers without human intervention, and only 1% require a pathologist. As the complexities of classifying and treating human blood cancers increase, the heavily-manual process of diagnosing human blood cancers from light microscopy and flow cytometry cries for the development of more automated testing platforms. Single cell morphometry is a viable and cost-effective path to achieving this goal, leveraging some of the latest technologies and techniques in highly-multiplexed instrumentation and data analysis.
METHODS
Antibodies.
A summary of all mass cytometry antibodies, reporter isotopes, and concentrations can be found in Supplementary Table 1. Some primary conjugated antibodies are directly available from Fluidigm. β-actin, β2 microglobulin, and CD298 were conjugated with monoisotopic cisplatin as previously described.36 Otherwise, antibodies were labeled using MaxPAR antibody conjugation kits (Fluidigm Sciences) and titrated on human whole blood or bone marrow or Ficoll-purified PBMCs per the below staining protocol.
Polymer-free palladium wheat germ agglutinin (PFP WGA).
WGA has previously been characterized as a cell membrane binder and marker for cell size.18 We efficiently and inexpensively conjugate it with natural abundance palladium.14 100 mM ethylenediamine palladium chloride (DCED-palladium, Sigma-Aldrich 574902) in DMSO was “activated” by incubating for 3 days at 37° C prior to storage at −20° C. 500 μg WGA (Sigma-Aldrich L9640) was washed with Fluidigm R buffer in a 10 kDa microfilter and reduced with 300 μL 4 mM TCEP for 30 minutes at 37° C. After washing with Fluidigm C buffer, it was reacted for 60 minutes at 37° C with 200 μL of 1.5 mM activated DCED palladium freshly diluted in Fluidigm C buffer, followed by washing with Fluidigm W buffer per standard protocol and diluted to 0.5-1 mg/mL with antibody stabilization buffer and sodium azide.
Glutaraldehyde staining protocol.
Standard paraformaldehyde (PFA) fixation is insufficient for many antibodies to withstand methanol permeabilization, which renders them diagnostically unacceptable (Supplementary Fig. 15). To overcome this problem, we fixed with glutaraldehyde (GA) and validated properly against diagnostic flow cytometry. Unless otherwise indicated, all centrifugations were performed at 4° C and other steps below at room temperature. Whole blood and bone marrow were treated with RBC lysis buffer (BioLegend 420301), spun for 5 minutes at 300 x g, and 1-2 x 106 leukocytes washed with wash solution (low barium PBS, 0.5% BSA, 0.02% sodium azide, 20 U/mL heparin, 25 U/mL Sigma-Aldrich E8263 benzonase). Cells were resuspended in wash solution, incubated with 5 μL Fc blocker (BioLegend 422302) for 10 minutes, brought to 100 μL with surface antibodies, and incubated for 30 minutes, followed by viability staining with 1 nM monoisotopic Pt 194 cisplatin (Fluidigm 201194) in low barium PBS (lbPBS) for precisely 5 minutes. After washing with 3 mL wash solution, cells were fixed in 1 mL 0.8% GA (pH ~7.24) in lbPBS on ice. After spinning for 5 minutes at 500 x g, the supernatant was removed, and cells washed with 3 mL CSM (lbPBS, 0.5% BSA, 0.02% sodium azide) and permeabilized with 1 mL cold methanol (Sigma Aldrich 34860) for 10 minutes on ice. 2 mL CSM was added to wash, and cells washed again with 3 mL CSM. After resuspending in CSM, 1 μL 20 U/μL heparin37 and 5 μL Fc blocker were added, incubating for 20 minutes before bringing to 100 μL with intracellular antibodies and incubating for 30 minutes. Cells were then washed with 3 mL CSM, resuspended in 1 mL 0.8% GA in lbPBS with 0.03 μL iridium intercalator (Fluidigm 201192B) and 5 μg PFP WGA, and incubated at 4° C overnight.
Barium-based doublet and debris/antibody aggregate removal.
We collected barium 138 signal during acquisition and selected singlets from barium vs. DNA rather than event length vs. DNA, after removing diluted events at the leading edges of sample pushes (supplementary text). Here we use a novel and cost-free method for gating singlets and removing nonspecific debris/antibody aggregate reactivity. Events at the leading edges of sample pushes were removed by gating on barium 138 vs. time, and singlets gated by barium vs. DNA, followed by gating on viable cells (Supplementary Fig. 16). Given that doublets are perhaps best reduced by barcoding and limiting event rate to <300/s, postacquisition they are visualized and removed by barium vs. DNA better than by the current standard, event length vs. DNA. Barium vs. DNA also resolves a large degree of nonspecific debris/aggregate reactivity.
Samples.
Healthy bone marrow samples were ordered from AllCells and delivered and processed the same day. Seventy-one patient samples were collected for diagnosis in EDTA or heparin tubes (marrow, peripheral blood) or RPMI (solid tissue) and stored at 4° C. Research aliquots were obtained <3 days after collection as post-diagnostic excess material under IRB-30899 and IRB-40765, stained and run only once, deidentified, and relabeled with codes as described in the supplement.
Microscopic images.
Bone marrow aspirates were freshly smeared on glass slides, air dried, and stained on an automated stainer for 3 minutes in methanol, 3:00 Wright’s-Giemsa stain (Beckman Coulter Tru-Color Wright’s-Giemsa stain 7547178), 2:30 stain-buffer combination (50 mL Wright’s-Giemsa stain diluted with 90 mL phosphate buffer, pH 6.4), 0:30 deionized water, 3:00 drying, 1:00 methanol, 1:30 Wright-Giemsa stain, 1:00 stain-buffer combination, 0:30 deionized water, 3:00 drying. Digital images were taken with a 100x oil objective and Olympus DP22 camera, white balanced and cropped in Adobe Photoshop, and scaled identically.
Data acquisition and processing.
Clinical flow cytometry was performed in a CLIA-certified CAP-accredited diagnostic laboratory according to laboratory standard operating procedures on BD FACS Canto II instruments. Acquisition was performed on BD FACSDiva 8.01 and data processed in FCS Express 4.08.0016. Mass cytometry was performed on a DVS Sciences CyTOF 2 running DVS Sciences Instrument Control Software 6.0.626. Mass cytometry FCS files were bead normalized as described38, concatenated if necessary, and uploaded to CytoBank (cytobank.org) for gating (Supplementary Fig. 16). Counts were asinh-transformed with a cofactor of 5. Samples from figures 1–4 – two healthy controls and eleven patient samples (B-ALL 4Cr9, MDS-RS 1Ar0, MDS-EB2 1Ar1, AML APL 1An7, AML 4Cn5, AMML 1An6, MPAL 3Cn3, T-ALL 3An1, TCL 1An7, SM-CMML 1Ar0, PCM 2Ar) – underwent normalization of scatterbody channels to control for technical variation (Supplementary Fig. 17). For each scatterbody, a specific cell population was identified that was present in all samples, had unimodal expression of the scatterbody, and had stronger expression of the scatterbody than other hematopoietic cell populations (Fig. 1d). The peak of the distribution for the population was identified in all samples using a Gaussian kernel density estimate. For each sample, a coefficient, βn, was identified to solve the equation: where is the peak of the distribution of the reference sample (Healthy 1) and is the peak of the distribution of the sample undergoing normalization. The scatterbody expression value of every event in sample n was multiplied by βn, resulting in an alignment of peaks. All markers in these samples were subsequently scaled to the 99.5th percentile for comparability between markers.
For comparison to diagnostic flow cytometry, mass cytometry data was gated and plotted similarly. For our eleven main samples and two healthy donor marrows, four backgates were used to set the blast, lymphocyte, and monocyte gates on our mass cytometry VAMP-7 vs. CD45 plot and LD axes. Granulocytes were also gated. From those gates, thousands of daughter plots were generated to mirror our main clinical lymphoma and leukemia panels. As clinical flow cytometry is not typically run or gated so comprehensively, only ~900 had direct counterparts in the diagnostic flow plots. These were used for statistical analysis and evaluated side-by-side with the diagnostic flow plots by five board-certified hematopathologists.
Generation of LD axes.
Combinatorial morphometric map axes were generated using supervised linear dimensionality reduction with linear discriminant analysis (LDA).28 LDA creates linear combinations of predictors (i.e. scatterbodies) to maximize separation between classes (i.e. gated hematopoietic cell populations). LD axes in figure 4 were built to separate blasts, monocytes, erythroids, neutrophils, and monocytes (as defined by surface marker gating in the Healthy 2 sample) to facilitate morphometric gating of these populations. LD axes in figure 5 were built to separate blasts, monocytes, and mature neutrophils (CD15+ CD16++ neutrophils) to facilitate visualization of myeloid differentiation. These linear combinations can be visualized as new axes, in a similar fashion to principal component analysis (PCA). The ideal subset of scatterbodies used to generate these new axes was selected using a hybrid of forward and reverse stepwise selection, as an exhaustive search of every possible subset would be computationally intensive. This implementation scores each subset by calculating the Euclidean distance between every pair of population means in the two new axes and assigning the minimum distance as that subset’s score. This scoring method therefore rewards axes that maximize separation between the two nearest population means, ensuring the new axes can be used to cleanly visualize and gate all five populations. This approach was chosen over a cross-validation/classification approach as the ultimate goal of the algorithm was visualization of neoplastic samples, not classification. After exploring the subset space, the ideal combination was selected by identifying the elbow point of the “number of markers” vs. “highest score” plot. The coefficients (Supplementary Table 3) used to generate the new axes in the training data were then applied to all cells in all samples with simple matrix multiplication, facilitating plotting of all samples on the same two axes.
Comparison of blast enumeration by microscopy, flow cytometry, and morphometry.
Three normal marrows were used to train BD axes, with blasts neither quantified nor assessed by flow cytometry due to absence of suspicion for acute leukemia or myeloid neoplasia. Thirteen AML samples and one MDS sample were prospectively chosen to enrich for CD34− and/or CD117− cases and to span a wide range of blast counts. Clinical samples shown in figure 5 are REACTIVE-2A, REACTIVE-3A, AML-9Ar, AML-7An, and AML-MRC-5Ar. Samples were live cell barcoded with Pt 195 per Hartmann et al. and washed twice with wash solution.14 A standard control (Streck CD-CHEX PLUS) was barcoded with Pt 196 and washed twice with CSM. Sample and control were mixed together and stained as described above, except surface staining was done for 1 hour and all incubations done on ice until permeabilization. Scatterbodies in both the controls and samples were asinh transformed. Control samples were normalized to their 98th percentile of expression of each scatterbody, and then the same coefficient was applied to normalize each control’s complementary sample. Samples were gated for non-erythroid cells on lamin A/C vs. CD45 and myeloid cells on VAMP-7 vs. CD45 (Supplementary Fig. 12) prior to blast gating (Figure 5d–f). Four board-certified attending hematopathologists (SFP, MJ, JO, AT) performed separate 200-cell light microscopic differentials on cases with >5% blasts, enumerating: 1. Blasts/blast equivalents (mb); 2. non-blast myeloid cells (mm), e.g., neutrophils, eosinophils; and 3. non-myeloid cells (mn), primarily erythroid precursors and lymphocytes – where mb + mm + mn = 100%. To eliminate potential effects from differential lysis/sampling of non-myeloid cells, flow and mass cytometry blast counts were scaled. The flow cytometry blast gate was set using the laboratory’s standard operating procedure (first attempting to backgate on CD34 vs. CD38 and CD117 vs. CD15, and then by the technologist’s best judgment if unsuccessful, Figure 5h, Supplementary Fig. 8), reporting events in the blast gate (fb) and lymphocyte gate (fl) as a percentage of CD45+ events, where CD45+ events do not include erythroid precursors. Scaling fb to be comparable to mb thus follows the formula fb · (100 – mn)/(100 – fl). Morphometric blasts (sb) were quantified as a percentage of myeloid cells (which do not include erythroid precursors or lymphocytes) and thus scaled as sb · (1 – mn/100).
Statistical Analyses.
In Figs. 3b, c, and e, individual data points are represented by black dots, central black lines depict the median, upper and lower hinges depict the interquartile range (IQR), and whiskers depict range of data within hinges +/− 1.5 IQR. Sample sizes are as follows: mature TCL (N = 5), T-ALLs (N = 3), and normal/reactive T cells (N = 45); neutrophils (N = 53), monocytes (N = 44), T/NK cells (N = 54), B cells (N = 51), erythroids (N = 31), plasma cells (N = 13), mast cells (N = 4), blasts (N = 39). In Fig. 4e, individual data points are represented by black dots, central black lines depict the mean, upper and lower hinges depict the interquartile range (IQR), and whiskers depict range of data within hinges +/− 1.5*IQR. For comparisons of median expression values and coefficients of variation (CVs) of specific populations in Fig. 3, the Wilcoxon signed rank test or Wilcoxon rank sum test (for distributions with <10 observations) was performed with P-values adjusted by the Bonferroni method for multiple hypothesis correction as necessary. The tests compared the distribution of patient sample medians (or CVs) between specific cell populations. In Fig. 3b, the medians and CVs of Lamin A/C expression in neoplastic T cells of five patient samples with mature T cell neoplasms were compared with blasts of three patient samples with T-ALLs and T cells of all patient samples with normal T cells. In Fig. 3c, comparisons of median Lamin A/C expression were made between all pairwise combinations of plasma cells, mast cells, or erythroids against neutrophils, blasts, T and NK cells, monocytes, or B cells in all patient samples (15 tests total). In Fig. 3e, median Lamin B1 expression values in blasts from all patient samples that contained blasts were compared to median Lamin B1 expression values of every other hematopoietic population from the same samples.
Data availability.
Processed, deidentified clinical flow cytometry and Cytobank mass cytometry plots are provided as supplementary files. Deidentified clinical flow cytometry and mass cytometry data will be deposited on flowrepository.org with paper publication for all samples analyzed in the paper.
Code availability.
All code used for analyses was written in the R programming language (r-project.org). Code to reproduce all main figures is available at github.com/davidrglass
Supplementary Material
Acknowledgements
A.G.T. is supported by a Damon Runyon Cancer Research Foundation – DRCRF (DRG-118-16) and Stanford Department of Pathology Seed Grant; D.R.G. was supported by a Stanford Graduate Fellowship, and Bio-X Stanford Interdisciplinary Graduate Fellowship. F.J.H was supported by the EMBO organization (EMBO Long-Term Fellowship), the Novartis Foundation for medical-biological Research and the Swiss National Science Foundation (SNF Early Postdoc Mobility). S.C.B. was supported by the DRCRF Fellowship (DRG-2017-09), the NIH 1DP2OD022550-01, 1R01AG056287-01, 1R01AG057915-01, 1-R00-GM104148-01, 1U24CA224309-01, 5U19AI116484-02, U19 AI104209, The Bill and Melinda Gates Foundation, and a Translational Research Award from the Stanford Cancer Institute. We also thank Dr. Leeat Keren for her helpful comments and suggestions about the manuscript during its preparation.
REFERENCES
- 1.American Cancer Society. Cancer Facts & Figures 2018. Atlanta: American Cancer Society; 2018 (2018). [Google Scholar]
- 2.Font P et al. Interobserver variance in myelodysplastic syndromes with less than 5 % bone marrow blasts: unilineage vs. multilineage dysplasia and reproducibility of the threshold of 2 % blasts. Ann. Hematol 94, 565–573 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Hodes A et al. The challenging task of enumerating blasts in the bone marrow. Semin. Hematol 56, 58–64 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Goasguen JE et al. Morphological evaluation of monocytes and their precursors. Haematologica 94, 994–997 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th Edition). (IARC, 2017). [Google Scholar]
- 6.Gorczyca W Flow Cytometry Immunophenotypic Characteristics of Monocytic Population in Acute Monocytic Leukemia (AML-M5), Acute Myelomonocytic Leukemia (AML-M4), and Chronic Myelomonocytic Leukemia (CMML). Methods Cell Biol. 75, 665–677 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Kern W, Bacher U, Haferlach C, Schnittger S & Haferlach T Acute monoblastic/monocytic leukemia and chronic myelomonocytic leukemia share common immunophenotypic features but differ in the extent of aberrantly expressed antigens and amount of granulocytic cells. Leuk. Lymphoma 52, 92–100 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Xu Y, McKenna RW, Karandikar NJ, Pildain AJ & Kroft SH Flow cytometric analysis of monocytes as a tool for distinguishing chronic myelomonocytic leukemia from reactive monocytosis. Am. J. Clin. Pathol 124, 799–806 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Alcantara M, Tesio M, June CH & Houot R CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia 32, 2307–2315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bendall SC et al. Single-Cell Mass Cytometry of Differential a Human Hematopoietic Continuum. Science (80-.). 687, 687–697 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baranov VI et al. Mass Cytometry: Technique for Real Time Single Cell Multitarget Immunoassay Based on Inductively Coupled Plasma Time-of-Flight Mass Spectrometry. Anal. Chem 81, 6813–6822 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Bendall SC, Nolan GP, Roederer M & Chattopadhyay PK A deep profiler’s guide to cytometry. Trends Immunol. 33, 323–332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zunder ER et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc 10, 316–333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann FJ, Simonds EF & Bendall SC A Universal Live Cell Barcoding-Platform for Multiplexed Human Single Cell Analysis. Sci. Rep 8, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lacy P Mechanisms of degranulation in neutrophils. Allergy Asthma . Clin. Immunol 2, 98–108 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manley HR, Keightley MC & Lieschke GJ The Neutrophil Nucleus: An Important Influence on Neutrophil Migration and Function. Front. Immunol 9, 2867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowat AC et al. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J. Biol. Chem 288, 8610–8618 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stern AD, Rahman AH & Birtwistle MR Cell size assays for mass cytometry. Cytom. Part A 91, 14–24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popova EY, Claxton DF, Lukasova E, Bird PI & Grigoryev SA Epigenetic heterochromatin markers distinguish terminally differentiated leukocytes from incompletely differentiated leukemia cells in human blood. Exp. Hematol 34, 453–462 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Jamieson AR et al. Exploring nonlinear feature space dimension reduction and data representation in breast CADx with Laplacian eigenmaps and t -SNE. Med. Phys 37, 339–351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Dongen JJM et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98–3936. Leukemia 17, 2257–317 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Tembhare P et al. Flow cytometric immunophenotypic assessment of T-cell clonality by νβ repertoire analysis: detection of T-cell clonality at diagnosis and monitoring of minimal residual disease following therapy. Am. J. Clin. Pathol 135, 890–900 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen DT & Diamond Lawrence W., Braylan RC Flow Cytometry in Hematopathology: A Visual Approach to Data Analysis and Interpretation, Second Edition (Humana Press, 2007). doi: 10.1007/978-1-59745-162-8_1. [DOI] [Google Scholar]
- 24.Kalina T et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 26, 1986–2010 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Dongen JJM et al. EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia 26, 1908–l975 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Behbehani GK Applications of Mass Cytometry in Clinical Medicine: The Promise and Perils of Clinical CyTOF. Clin. Lab. Med 37, 945–964 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hastie T , Tibshirani R & Friedman J The Elements of Statistical Learning: Data Mining, Inference, and Prediction. (Springer, 2001). [Google Scholar]
- 29.Kussick SJ et al. Four-color flow cytometry shows strong concordance with bone marrow morphology and cytogenetics in the evaluation for myelodysplasia. Am. J. Clin. Pathol 124, 170–181 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Chung JW et al. A combination of CD15/CD10, CD64/CD33, CD16/CD13 or CD11b flow cytometric granulocyte panels is sensitive and specific for diagnosis of myelodysplastic syndrome. Ann. Clin. Lab. Sci 42, 271–280 (2012). [PubMed] [Google Scholar]
- 31.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–47 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Bjornson-Hooper ZB et al. A comprehensive atlas of immunological differences between humans, mice and non-human primates. bioRxiv 574160 (2019) doi: 10.1101/574160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keren L et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 174, 1373–1387.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Mei HE, Leipold MD & Maecker HT Platinum-conjugated antibodies for application in mass cytometry. Cytom. Part A 89, 292–300 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Rahman AH, Tordesillas L & Berin MC Heparin reduces nonspecific eosinophil staining artifacts in mass cytometry experiments. Cytom. Part A 89, 601–607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kleinsteuber K et al. Standardization and quality control for high-dimensional mass cytometry studies of human samples. Cytom. Part A 89, 903–913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carr JH & Rodak BF Clinical Hematology Atlas, Second Edition (Elselvier, 2004). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed, deidentified clinical flow cytometry and Cytobank mass cytometry plots are provided as supplementary files. Deidentified clinical flow cytometry and mass cytometry data will be deposited on flowrepository.org with paper publication for all samples analyzed in the paper.