

Distribution of the 25bp Intronic Deletion in MYBPC3 in South Asian populations
In 2001, a 25bp deletion in intron 32 (MYBPC3Δ25bp) was described in two unrelated South Asian (SA) families associated with hypertrophic cardiomyopathy (HCM) (Fig. A).1, 2 In these reports, MYBPC3Δ25bp was shown to segregate in an autosomal dominant manner with highly variable penetrance. Interestingly, MYBPC3Δ25bp was detected in 16 out of 229 unrelated healthy SA individuals, suggesting its prevalence in southern India and a possible role as a regional genetic polymorphism and potential “genetic modifier.” A new paper by Harper and colleagues describes the MYBPC3 splicing variant, c.1224–52G>A, in HCM.3
Figure. Genotype and potential contributing factors to MYBPC3Δ25bp pathogenicity.
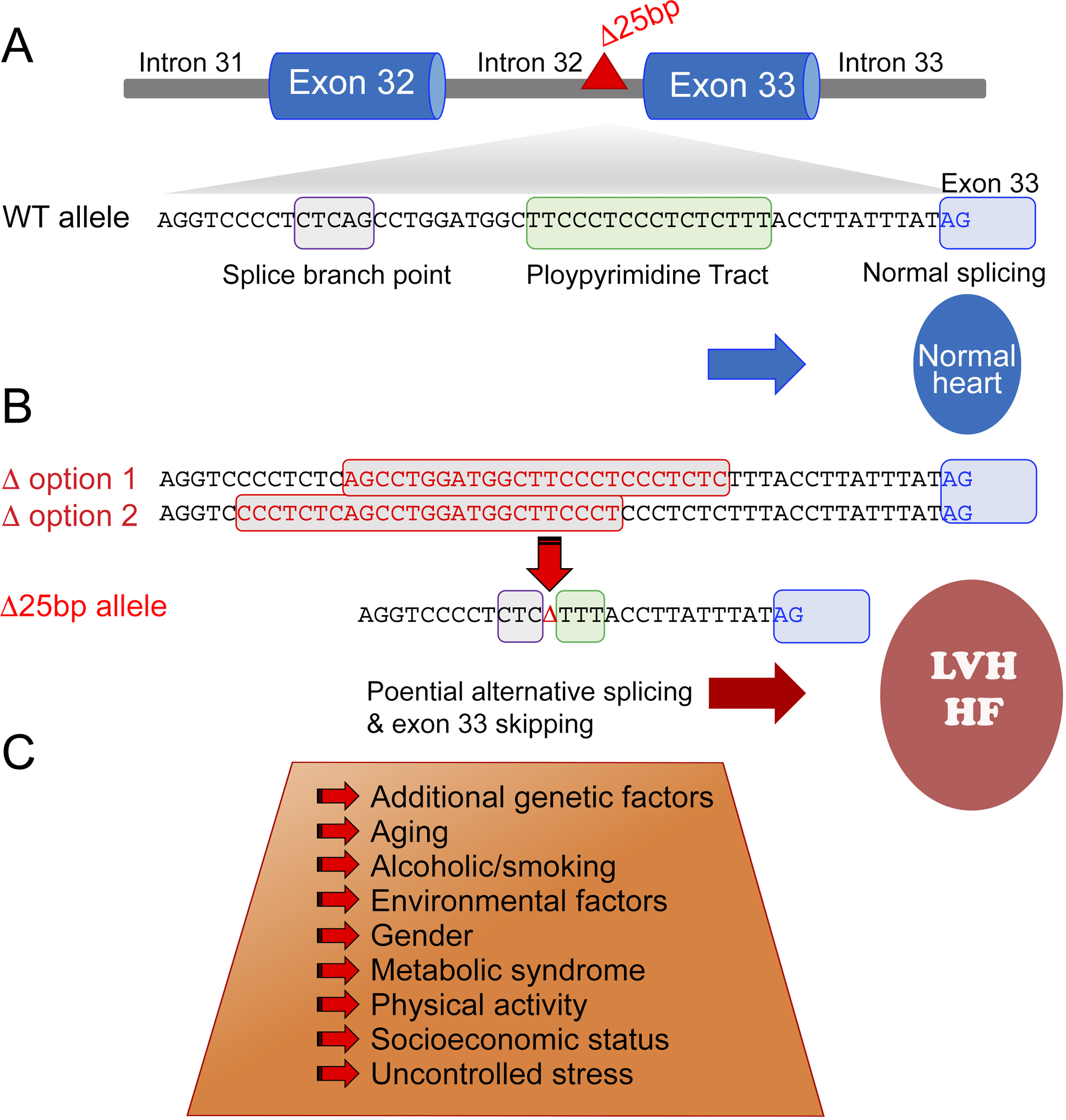
(A) MYBPC3Δ25bp maps to intron 32 between the splice branch point and polypyrimidine track. In the absence of MYBPC3Δ25bp, normal exon splicing occurs. (B) The presence of the MYBPC3Δ25bp variant causes one of two possible changes (red boxes) both resulting in the same partial deletion of the splice branch point and polypyrimidine track sequences. (C) The presence of additional risk factors may increase the potential for alternative splicing and exon 33 skipping leading to left ventricular hypertrophy (LVH) and heart failure (HF), a key focus of current and future work.
Prior population-based studies examined MYBPC3Δ25bp frequency and clinical correlates. A large case-control cohort reported findings from 6,273 individuals belonging to 107 ethnic populations across 35 Indian states and 2,085 indigenous individuals of 63 ethnic/racial groups from 26 countries, including all five continents.4 This study concluded that MYBPC3Δ25bp is a South Asian-specific common variant (3.8%) associated with a range of outcomes, including HF in the setting of multiple forms of cardiomyopathy, not simply HCM. Secondary cardiovascular risk factors, including metabolic syndrome, age, ventricular arrhythmia and other gene mutations were enriched in MYBPC3Δ25bp carriers.4 An ancestry-based study using 23andMe data confirmed MYBPC3Δ25bp as enriched in those with self-described SA ancestry (4.5%) and its infrequency in those of European ancestry (<0.05%).5 Consistent with these findings, Simonson et al. (2010)6 and Bashyam et al. (2012)7 reported similar prevalence of MYBPC3Δ25bp with those of SA ancestry of 8% and 6%, respectively. A more recent study of those of SA ancestry living in the U.S. determined a 6% frequency of MYBPC3Δ25bp.8 In aggregate, MYBPC3Δ25bp is estimated to be present in approximately 100 million SA descendants worldwide, including ~350,000 living in the U.S. As a risk allele, then, it is important to clarify the clinical correlates and coassociating factors.
Association of MYBPC3Δ25bp variant with cardiovascular disease
As a risk allele, MYBPC3Δ25bp is found in other cardiomyopathies, left ventricular hypertrophy (LVH) and chronic HF,4 as well as hypertension, ventricular tachyarrhythmias and aging.4 Three independent studies showed that carriers of MYBPC3Δ25bp had worse coronary heart disease outcomes compared to noncarriers.9–11 The 2018 study of South Asians living in the USA (US-SAs) took a genotype-first view to assess clinical findings associated with MYBPC3Δ25bp by examining carriers with electrocardiograms and echocardiograms.8 Mild shifts in LV function were noted in MYBPC3Δ25bp compared to controls. Approximately 10% of MYBPC3Δ25bp carriers had an additional variant, MYBPC3D389V. MYBPC3D389V was only observed in the presence of MYBPC3Δ25bp, and familial evaluation demonstrated the coinheritance of the two variants, consistent with their presence on the same allele. Those individuals with both D389V and Δ25bp had the most abnormal findings by echocardiography, compared to noncarriers and Δ25bp carriers.8
Molecular consequences of MYBPC3Δ25bp
Experimental evidence supports a mechanistic role for MYBPC3Δ25bp. Its expression in neonatal cardiomyocytes demonstrated skipping of exon 33.2 Strikingly, evaluation of expression in a MYBPC3Δ25bp–positive human heart confirmed exon 33 skipping in vivo.4 Exon 33 skipping results in the loss of 62 amino acids to produce modified C10 domain of cardiac myosin binding protein-C (cMyBP-CΔC10mut). Furthermore, this variant shifts the stop codon to the 3’ UTR and adds a novel 55 amino acids in the C10 domain at the carboxyl terminus. cMyBP-C interacts via its C10 domain with the myosin LMM region, which is essential for cMyBP-C adherence, localization and stabilization in the sarcomere. If the C10 domain is modified, or truncated, cMyBP-C will not localize in the sarcomere.12, 13
The presence of exon 33 skipping in human cardiac biopsy samples4, even if only partial, may alter cardiac properties. The underlying molecular mechanism could be attributed to haploinsufficiency or poison polypeptide.13, 14 In theory, cMyBP-CΔC10mut would not bind to myosin LMM, resulting in removal through the ubiquitin proteasome system. Alternatively, through nonsense-mediated mRNA decay, the mutant RNA could be removed, and no translation would occur to synthesize cMyBP-CΔC10mut, causing haploinsufficiency. A poison polypeptide effect can occur if cMyBP-CΔC10mut binds actin and the myosin S2 region through its N’-terminal interactions in the sarcomere. To test these two hypotheses, we recently overexpressed cMyBP-CΔC10mut in adult cardiomyocytes in vitro and confirmed that it mislocalized in the Z-line of the sarcomere, rendering it nonfunctional.13
To determine the in vivo consequence of cMyBP-CΔC10mut, a cardiac-specific transgenic mouse model was generated in which cMyBP-CΔC10mut was expressed under the control of cardiac-specific α-myosin heavy chain promoter.13 In the transgenic line, mRNA levels of both endogenous and mutant MYBPC3 were robustly detected (20-fold). Mutant protein comprised 31.6% of the total cMyBP-C protein, implying instability of the mutant compared to the wild type. It was revealed that (i) the expression of cMyBP-CΔC10mut was pathogenic in the heart, causing an HCM phenotype by 12 weeks of age, (ii) cMyBP-CΔC10mut was mislocalized in the sarcomere and caused a poison polypeptide effect, and (iii) the C10 domain was critical for cMyBP-C stability and localization. These experimental modeling studies concluded that cMyBP-CΔC10mut protein is toxic and leads to a cascade of events beginning with improper incorporation in the sarcomere, followed by contractile dysfunction, and, finally, the induction of pro-hypertrophic signaling and cardiac remodeling.13
A novel MYBPC3 mutant, MYBPC3 c.1224–52G>A, disrupts splicing
The Harper study evaluated the coinheritance of c.1224–52G>A (MYBPC3−52), another intronic variant in MYBPC3, in the etiology of HCM.3 Two large HCM cohorts were evaluated with a combined total of 5,394 HCM cases.15 Overall, very few SAs were included in these HCM cohorts and only 17 SA MYBPC3Δ25bp carriers. Panel gene analysis was used to identify pathogenic, likely pathogenic or variants of uncertain significance in the HCM cohorts. The authors focused on a previously identified, potentially pathogenic variant, MYBPC3−52, that arose from the introduction of a cryptic splice acceptor site in intron 13, which includes a 50 bp intronic sequence in the mRNA, causing a frameshift and premature termination at codon 438.15 MYBPC3−52 was identified in 11 of 38 MYBPC3Δ25bp carriers. Haplotype analysis identified strong linkage disequilibrium between MYBPC3−52 and MYBPC3Δ25bp, suggesting that MYBPC3−52 could be driving the HCM risk associated with MYBPC3Δ25bp. However, the MYBPC3−52 variant was also found in the absence of MYBPC3Δ25bp, indicating that these two variants are separable and that MYBPC3−52 may impart risk on its own. Expression analysis using RNA from lymphocytes from two MYBPC3−52 carriers documented aberrant splicing. Future studies using cardiac-derived RNA and protein expression will be helpful to discern pathogenic mechanisms. Since MYBPC3D389V was not included in the haplotype analysis, the role of D389V in the context of the MYBPC3−52 remains to be explored. Dissection of specific MYBPC haplotypes, including the D389V allele, may help define ancestral population structures and/or functional consequences of the MYBPC3Δ25bp allele and its risk.
This new work suggests a role for MYBPC3Δ25bp in HCM in the setting of primary mutations. It further highlights MYBPC3Δ25bp as a risk allele for HF and other adverse cardiovascular outcomes, as observed in the population studies.4 As a risk allele, the co-occurrence with pathogenic alleles, or even conventional risk factors like metabolic syndrome, is expected to worsen outcomes and disease trajectory. For example, the presence of MYBPC3Δ25bp would likely intensify other HCM mutations or further promote LVH in the setting of hypertension. Whether the presence of these additional disease factors actually promotes more abnormal splicing around MYBPC3Δ25bp requires experimental study.
Questions for future studies
South Asians have significantly greater risk for cardiovascular diseases,16–18 often developing heart disease up to a decade earlier than other ethnic groups and often with greater mortality.16–19 This increased risk is not solely explained by hypertension, diabetes, obesity, sedentary activity, smoking or diet. Instead, some unusually prevalent non-modifiable risk factors, such as elevated lipoprotein(a) and clotting factors, appear to be present.18 Added to this array of inherited predispositions is MYBPC3Δ25bp. Overall, MYBPC3Δ25bp is a discrete marker shown to define a population at substantially increased risk for pathological LVH and other cardiovascular diseases and, thus representing a unique opportunity to improve outcomes and define novel mechanisms of cardiovascular disease (Fig. B–C). However, future studies will be needed to tease out the complex genotype-phenotype correlations specific to South Asian descendants with the aim of dissecting MYBPC3Δ25bp risk factors using various tools, such as humanized mouse models, human induced pluripotent stem cells-derived cardiomyocytes and organoids.20
Sources of Funding:
Supported by National Institutes of Health grants R01 HL130356 (SS), R56 HL139680 (SS), R01 AR067279 (SS), R01 HL105826 (SS), and R01 HL143490 (SS), U01 HL131914 (EMM), and R01 HL128075 (EMM). This work was further supported by the American Heart Association 2019 Institutional Undergraduate Student 19UFEL34380251 (SS), Transformation 19TPA34830084 (SS) and Career Development 189CDA34110460 (MJP) awards, as well as the PLN Foundation (PLN crazy idea).
Nonstandard Abbreviations and Acronyms
- bp
base pair
- cMyBP-C
Cardiac myosin binding protein-C protein
- cMyBP-CΔC10mut
Modified C10 domain of cardiac myosin binding protein-C
- LVH
Left ventricular hypertrophy
- MYBPC3
Cardiac myosin binding protein-C gene
- MYBPC3Δ25bp
25bp deletion mutation in intron 32 of MYBPC3 gene
- MYBPC3−52
c.1224–52G>A in MYBPC3 gene
- HCM
Hypertrophic cardiomyopathy
- HF
Heart failure
Footnotes
Disclosures: Dr. Sadayappan provided consulting and collaborative research studies to the Leducq Foundation (CURE-PLAN), Red Saree Inc., Greater Cincinnati Tamil Sangam, AstraZeneca, MyoKardia, Merck and Amgen, but such work is unrelated to the content of this manuscript. Dr. McNally serves as consultant to AstraZeneca, Amgen, Pfizer, Tenaya Therapeutics and Invitae; these activities are unrelated to the content of this work.
References:
- 1.Sakthivel S, Waldmuller S, Saadi AV, Joseph PK, Rakesh PG, Padmakumar R, Tharakan JM, Richard P, Schwartz K, Rajamanickam C, Vosberg HP. Novel mutations in MYH7 and MYBPC3 of an Indian family causing hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:A105 (Abstract). [Google Scholar]
- 2.Waldmuller S, Sakthivel S, Saadi AV, Selignow C, Rakesh PG, Golubenko M, Joseph PK, Padmakumar R, Richard P, Schwartz K, et al. Novel deletions in MYH7 and MYBPC3 identified in Indian families with familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2003;35:623–36. [DOI] [PubMed] [Google Scholar]
- 3.Harper AR, Bowman M, Hayesmoore JBG, Sage H, Salatino S, Blair E, Campbell C, Currie B, Goel A, McGuire K, et al. A Re-evaluation of the South Asian MYBPC3(Delta25) Intronic Deletion in Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2020;13:e002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowdry AB, Mandegar MA, Benton GM, Naughton BT, Conklin BR. Population Sampling and in vitro Modeling of a 25bp Deletion in MYBPC3 Associated With Hypertrophic Cardiomyopathy. 23andMe, Inc 2012. https://blog.23andme.com/wp-content/uploads/2012/11/HCM-ASHG-TTAM.pdf
- 6.Simonson TS, Zhang Y, Huff CD, Xing J, Watkins WS, Witherspoon DJ, Woodward SR, Jorde LB. Limited distribution of a cardiomyopathy-associated variant in India. Ann Hum Genet. 2010;74:184–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bashyam MD, Purushotham G, Chaudhary AK, Rao KM, Acharya V, Mohammad TA, Nagarajaram HA, Hariram V, Narasimhan C. A low prevalence of MYH7/MYBPC3 mutations among familial hypertrophic cardiomyopathy patients in India. Mol Cell Biochem. 2012;360:373–82. [DOI] [PubMed] [Google Scholar]
- 8.Viswanathan SK, Puckelwartz MJ, Mehta A, Ramachandra CJA, Jagadeesan A, Fritsche-Danielson R, Bhat RV, Wong P, Kandoi S, et al. Association of Cardiomyopathy With MYBPC3 D389V and MYBPC3Delta25bpIntronic Deletion in South Asian Descendants. JAMA Cardiol. 2018;3:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S, Mishra A, Srivastava A, Bhatt M, Garg N, Agarwal SK, Pande S, Mittal B. Role of common sarcomeric gene polymorphisms in genetic susceptibility to left ventricular dysfunction. J Genet. 2016;95:263–72. [DOI] [PubMed] [Google Scholar]
- 10.Srivastava A, Garg N, Mittal T, Khanna R, Gupta S, Seth PK, Mittal B. Association of 25 bp deletion in MYBPC3 gene with left ventricle dysfunction in coronary artery disease patients. PLoS One. 2011;6:e24123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar S, Mishra A, Srivastava A, Garg N, Agarwal SK, Pande S, Mittal B. Role of sarcomeric gene polymorphisms on left ventricular dysfunction in coronary artery disease patients. Molecular Cytogenetics. 2014;7:P112. [Google Scholar]
- 12.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuster DWD, Lynch TL, Barefield DY, Sivaguru M, Kuffel G, Zilliox MJ, Lee KH, Craig R, Namakkal-Soorappan R, Sadayappan S. Altered C10 domain in cardiac myosin binding protein-C results in hypertrophic cardiomyopathy. Cardiovasc Res. 2019;115:1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hossain MB, Elbeck Z, Siga H, Knoll R. Myosin binding protein-C and hypertrophic cardiomyopathy: role of altered C10 domain. Cardiovasc Res. 2019;115:1943–1945. [DOI] [PubMed] [Google Scholar]
- 15.Bagnall RD, Ingles J, Dinger ME, Cowley MJ, Ross SB, Minoche AE, Lal S, Turner C, Colley A, Rajagopalan S, et al. Whole Genome Sequencing Improves Outcomes of Genetic Testing in Patients With Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2018;72:419–429. [DOI] [PubMed] [Google Scholar]
- 16.Volgman AS, Palaniappan LS, Aggarwal NT, Gupta M, Khandelwal A, Krishnan AV, Lichtman JH, Mehta LS, Patel HN, Shah KS, et al. Atherosclerotic Cardiovascular Disease in South Asians in the United States: Epidemiology, Risk Factors, and Treatments: A Scientific Statement From the American Heart Association. Circulation. 2018;138:e1–e34. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan S South Asians and Cardiovascular Disease: The Hidden Threat. Cardiology. 2019;48:17–19. [Google Scholar]
- 18.Sharma M, Kartha CC, Mukhopadhyay B, Goyal RK, Gupta SK, Ganguly NK, Dhalla NS. India’s March to Halt the Emerging Cardiovascular Epidemic. Circ Res. 2017;121:913–916. [DOI] [PubMed] [Google Scholar]
- 19.Tu JV, Chu A, Rezai MR, Guo H, Maclagan LC, Austin PC, Booth GL, Manuel DG, Chiu M, Ko DT, et al. The Incidence of Major Cardiovascular Events in Immigrants to Ontario, Canada: The CANHEART Immigrant Study. Circulation. 2015;132:1549–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker RC, Sadayappan S. Designing Human In Vitro Models for Drug Development. J Am Coll Cardiol. 2020;75:587–589. [DOI] [PubMed] [Google Scholar]