

Abstract
Cells can store memories of prior experiences to modulate their responses to subsequent stresses, as seen for the protein kinase A (PKA)-mediated general stress response in yeast, which is required for resistance against future stressful conditions. Using microfluidics and time-lapse microscopy, we quantitatively analyzed how the cellular memory of stress adaptation is encoded in single yeast cells. We found that cellular memory was biphasic. Short-lived memory was mediated by trehalose synthase and trehalose metabolism. Long-lived memory was mediated by PKA-regulated stress-responsive transcription factors and cytoplasmic messenger ribonucleoprotein (mRNP) granules. Short- and long-lived memory could be selectively induced by different priming input dynamics. Computational modeling revealed how the PKA-mediated regulatory network could encode previous stimuli into memories with distinct dynamics. This biphasic memory-encoding scheme might represent a general strategy to prepare for future challenges in rapidly changing environments.
One-sentence summary:
Yeast use different PKA-dependent mechanisms to encode short- and long-term memories of stresses.
Editor’s summary:
Remembering stress with PKA
A prior stress can prime organisms to successfully survive subsequent stresses. In yeast, the kinase PKA is inhibited by stress, resulting in changes in gene expression that mediate the general stress response. Using PKA inhibition as a mimic for stress, Jiang et al. examined how yeast encoded the memories of previous stresses. Short-term memory required metabolism of the sugar trehalose and was more sensitive to the amplitude of the stress than its duration. In contrast, long-term memory was sensitive to stress duration and required stress-induced transcription factors and the storage of messenger ribonucleoproteins (mRNPs) in granules. These results show how yeast use PKA to “remember” previous exposures to stress in the short term or over a longer period of time.
Introduction
Cells survive rapidly changing environments through adaptation mediated by sophisticated signaling and gene regulatory networks. How these networks operate dynamically to process complex extracellular signals and elicit appropriate responses remains a challenging question (1–3). Advances in microfluidics and single-cell imaging technologies allow us to track the responses of individual living cells in a precisely controlled changing environment, providing a unique opportunity to elucidate the underlying principles for dynamic signal processing in cells (4). In this study, we exploited these technologies to systematically probe the regulatory network that enables cells to encode memory of prior environmental cues in order to modulate their adaptive responses to future challenges.
History-dependent cellular behaviors have been found in many organisms (5–11). For instance, plant cells obtain enhanced resistance to various future diseases and abiotic stresses, once primed by mild stresses or chemical compounds (12, 13). In another example, pre-treatment of human macrophages with interferon-γ substantially boosts subsequent responses triggered by lipopolysaccharide (LPS) against various pathogens and in tumor cell killing (14, 15). In this study, we referred these history-dependent responses in single cells as “cellular memory”, which is of course fundamentally different from the neuronal memory in animals.
In the yeast Saccharomyces cerevisiae, a given stress can activate its specific response pathway as well as a common signaling pathway shared by several different stresses, called the general stress response (GSR) pathway (16). This pathway is primarily mediated by protein kinase A (PKA). In response to stresses, PKA is rapidly inhibited, leading to activation of downstream transcription factors, such as Msn2 and Msn4, and the induction of hundreds of stress responsive genes (17–20). The GSR pathway is not required for survival against immediate stress threats, but instead, is required for resistance against future stressful conditions (21–23). However, the mechanisms that mediate memory encoding of environmental changes remain unclear.
In this study, we used GSR as a model to quantitatively analyze how PKA-dependent regulatory processes operate dynamically to encode the memory of environmental changes. We combined microfluidics with time-lapse microscopy to precisely control the dynamics of priming inputs and to quantify the memory effect on stress adaptation in single cells. We found that cellular memory shows two phases, a fast-decaying phase mediated by trehalose metabolism and a long-lasting phase mediated by stress-activated transcription factors and messenger ribonucleoprotein (mRNP) granules. Moreover, the memory dynamics could be modulated by priming inputs. Whereas a high amplitude transient input specifically induced the fast-decaying phase of memory, a prolonged input was needed to elicit the long-lasting memory effect. We further developed a computational model based on the molecular processes identified experimentally. Our model quantitatively revealed the regulatory scheme that encodes the information of previous environmental inputs into distinct memory dynamics, implying a general strategy to optimize resource allocation and prepare for future challenges under rapidly changing environments.
Results
PKA encodes biphasic cellular memory
Probing the effect of cellular memory has long been challenging, in part due to the difficulty in generating well-controlled sequential environmental changes in cell cultures. We took advantage of advances in microfluidic technologies that allow the precise control of extracellular conditions and tracking of the responses of single cells over extended periods (24, 25). In this study, we modified the channel design of an existing microfluidic device (18) to include separate control of three media inlets, one each for the priming input, the normal growth medium, and the stress treatment (Fig. 1A). To increase the experimental throughput, we also aligned four individual channels into a single device to enable simultaneous running of multiple experiments, each with its own cell population and stimulus condition. Using this device, we first exposed the cells to a pulse of priming input followed by a “break time” with the normal growth medium. We then treated these primed cells with a sustained environmental stress and evaluated their adaptation responses. The device was mounted on a time-lapse microscope to track the responses of a large number of single cells throughout the entire experiment.
Fig. 1. PKA encodes biphasic cellular memory.
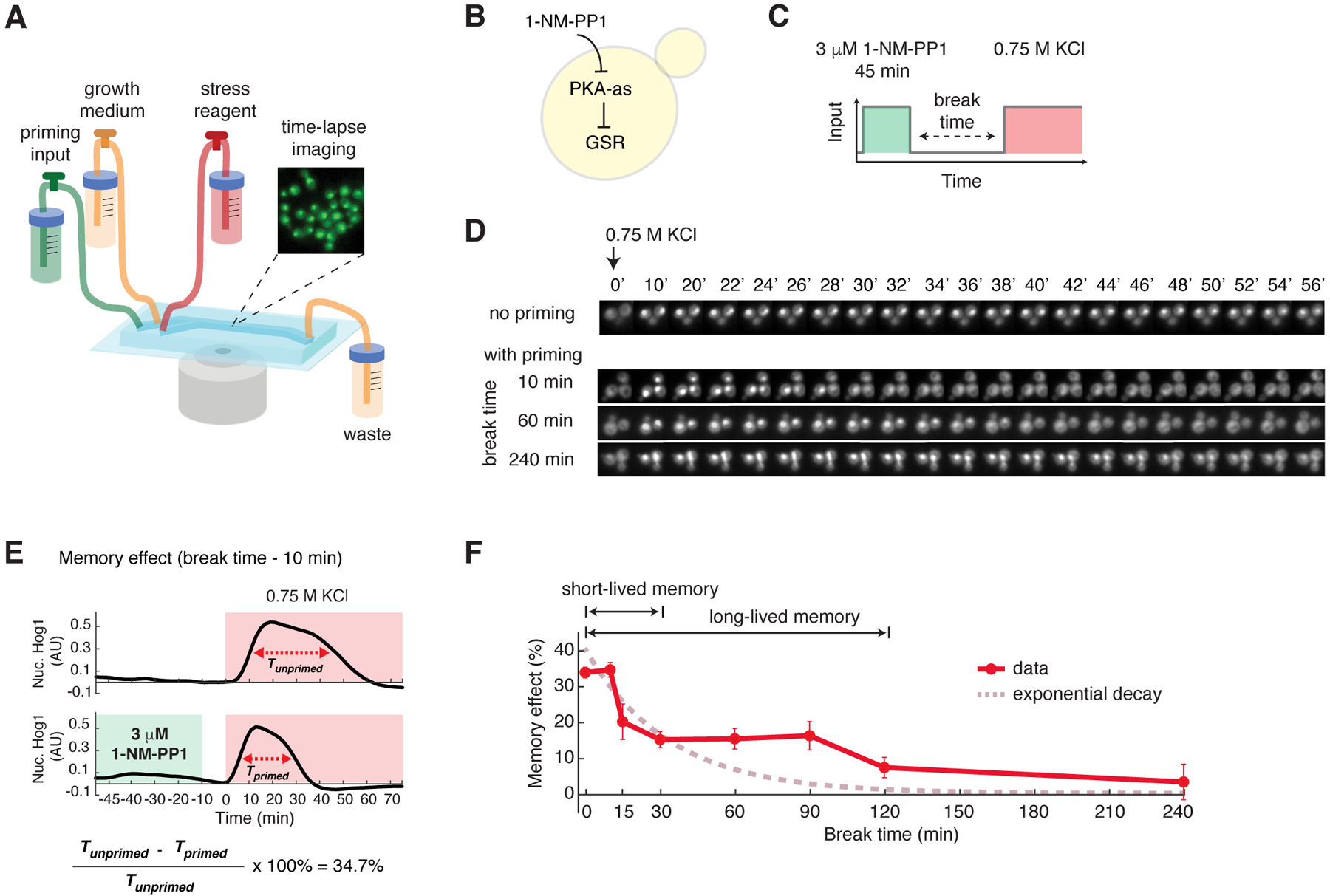
(A) A diagram of the microfluidic system used in priming experiments. The microfluidic device contained three inlets for priming input, growth medium, and stress reagent, respectively. Yeast cells were immobilized in the culturing chamber for time-lapse microscopic imaging. The medium flow from the inlets to the waste was driven by gravity. The three inlets could be opened or closed to allow medium switching during the experiment. (B) A diagram describing the analog sensitive PKA strain used in this study. (C) A schematic of the experimental design. Within the microfluidic device, cells were first exposed to a pulse of 1-NM-PP1 priming input, which was followed by a break time of normal growth medium. These primed cells were then exposed to sustained 0.75 M KCl treatment. (D) Representative time-lapse images of Hog1 nuclear translocation in the priming experiments with different break times. (E) Time traces of Hog1 translocation in response to 0.75 M KCl (red shaded) without (top) or with (bottom) 45 min priming with 3 μM 1-NM-PP1 (green shaded) followed by 10 min break time. The duration of nuclear localization was quantified using the full width at half maximum (FWHM) in single cells (Tprimed and Tunprimed). The memory effect was calculated using Tprimed and Tunprimed. (F) Biphasic memory dynamics in response to the high-amplitude prolonged priming input as indicated in (C). The plot shows the relationship of memory effect versus break time. Data points are averages of at least three independent experiments. Error bars are standard error of the mean (SEM). A theoretical exponential decay curve (dashed curve in dark pink) was superimposed with the data. The Hog1 time trace for each break time is shown in Figure S1.
For the priming input, we used a chemical genetics strategy in which we introduced analog-sensitive mutations into the PKA isoforms so that they remain fully functional but can be specifically inhibited by the small molecular inhibitor 1-NM-PP1 (26). We have previously employed this strategy to control PKA activity, mimicking an upstream signaling event that specifically activates GSR but not other stress specific pathways (18, 27–29) (Fig. 1B). Moreover, combined with time-lapse microscopy and microfluidics, it enabled us to generate precisely controlled temporal patterns of PKA inhibition as priming inputs and to evaluate their effects on cells’ adaptation to the subsequent environmental stress.
For the subsequent stress treatment, we chose hyperosmotic stress (0.75M KCl) because the stress adaptation process in individual cells can be reliably quantified using a specific reporter, the stress-activated protein kinase Hog1, which we tagged with YFP. In response to osmotic stress, Hog1 rapidly translocates to the nucleus to induce an increase in intracellular osmolyte; once the osmolyte balance is restored and the cell recovers from the stress, Hog1 exits the nucleus (30). The timing of Hog1 nuclear export strongly correlates with the restoration of cell volume (reflecting the turgor pressure recovery and cellular adaptation) (31). Thus, the duration of Hog1 nuclear localization serves as a proxy for the time needed for the cell to recover from a stress treatment. A shorter duration represents a faster adaptation whereas a longer duration represents a slower adaptation. We note that other pathways besides Hog1 that respond and adapt to hyperosmotic stress can also be used as indicators for stress recovery (32, 33). We used Hog1 nuclear localization in this study because it has been well characterized and is easy to quantify with time-lapse microscopy (31, 34, 35). The priming input of PKA inhibition did not trigger Hog1 nuclear localization (Fig. S1).
Using Hog1 nuclear localization as a reporter, we observed that a 45-min priming input with 3 μM PKA inhibitor (Fig. 1C), which causes full PKA inhibition (based on the level of Msn2 translocation (18, 27, 29)), shortened the time needed to recover from hyperosmotic stress. Furthermore, the effect of this priming input decayed with increasing break times (Fig. 1D). To quantify the effect of priming input and the dynamics of its decay, we measured and compared the durations of Hog1 nuclear translocation with and without the priming input (Tprimed, Tunprimed) for each break time. For instance, the priming input with a 10-min break time decreased the adaptation time by 34.7% from 40 min to 26 min (Fig. 1E). We defined this percentage decrease in recovery time as a quantitative measure of the “memory effect” and used it throughout our report (data processing workflow shown in Fig. S2).
When we plotted the memory effect as a function of break time, we observed that the decay of memory effect was biphasic. About half of the memory effect was lost rapidly within 30 minutes, whereas the remaining memory effect plateaued until 90 minutes and then declined slowly (Fig. 1F; Hog1 time traces shown in Fig. S1). These biphasic memory dynamics were in contrast with the scenario where the memory is primarily mediated by the expression of stress-resistance genes (21, 22), which would follow an exponential decay trajectory as gene products are degraded during the break time (Fig. 1F, exponential decay curve). We termed the fast-decaying component “short-lived” memory and the longer-lasting component “long-lived” memory. We also evaluated the effect of priming inputs on Hog1 adaptation in single cells but did not observe overt changes in cell-to-cell variability (Fig. S3, A and B). As a control, we performed the same experiments in WT cells without PKA analog-sensitive mutations and did not observe a memory effect from the 1-NM-PP1 input, confirming that analog treatment affected stress recovery specifically by inhibiting PKA activity (Fig. S4).
To test whether the memory effect showed two phases with a natural stress as priming input, we used 0.5M KCl as the priming input and observed similar biphasic memory dynamics (Fig. S5, A and B; Fig. S6), indicating that the memory pattern we found was not specific to chemical inhibition of PKA. In addition, to determine whether similar memory dynamics were observed with a different secondary stress, we evaluated the effect of PKA inhibition as the priming input on the cells’ adaptation to glucose limitation. The stress-activated transcription factor Msn2 displays an adaptive nuclear translocation response to glucose limitation and hence can be used as a reporter for stress recovery (18, 36). Similar to hyperosmotic stress response, we observed that the priming input shortened the adaptation time substantially, and the memory effects decayed with a biphasic pattern (Fig. S7, A to D; Fig. S8). These results suggest that the biphasic pattern of memory dynamics may be a general property for yeast stress responses.
Short- and long-lived memory can be selectively induced by different priming input dynamics
We next considered how changing the dynamics of priming inputs affected cellular memory. We used the three types of priming inputs used here are referred to as: “high-amplitude prolonged” (3 μM 1-NM-PP1, 45 min); “high-amplitude transient” (3 μM, 15 min); and “low-amplitude prolonged” (0.75 μM, 45 min). Cells exposed to a 15-min priming input with 3 μM 1-NM-PP1 (Fig. 2A) exhibited only the fast-decaying component of memory effect and the long-lasting phase was abolished, in contrast to the response in cells exposed to a 45-min priming input with the same amplitude (Fig. 2B, compare the blue and dark pink curves). When cycloheximide (CHX) was added to inhibit protein synthesis during the priming experiment, this short-lived memory effect was not affected (Fig. 2C), suggesting that short-lived memory did not depend on gene expression, but might instead be mediated by metabolites or post-translational protein modifications. Although the effect of this 15-min priming input on stress adaptation was small (~ 4 min decrease in adaptation time), it was statistically significant (Fig. S9, A and B).
Fig. 2. Short- and long-lived memory can be selectively induced by different priming input dynamics.
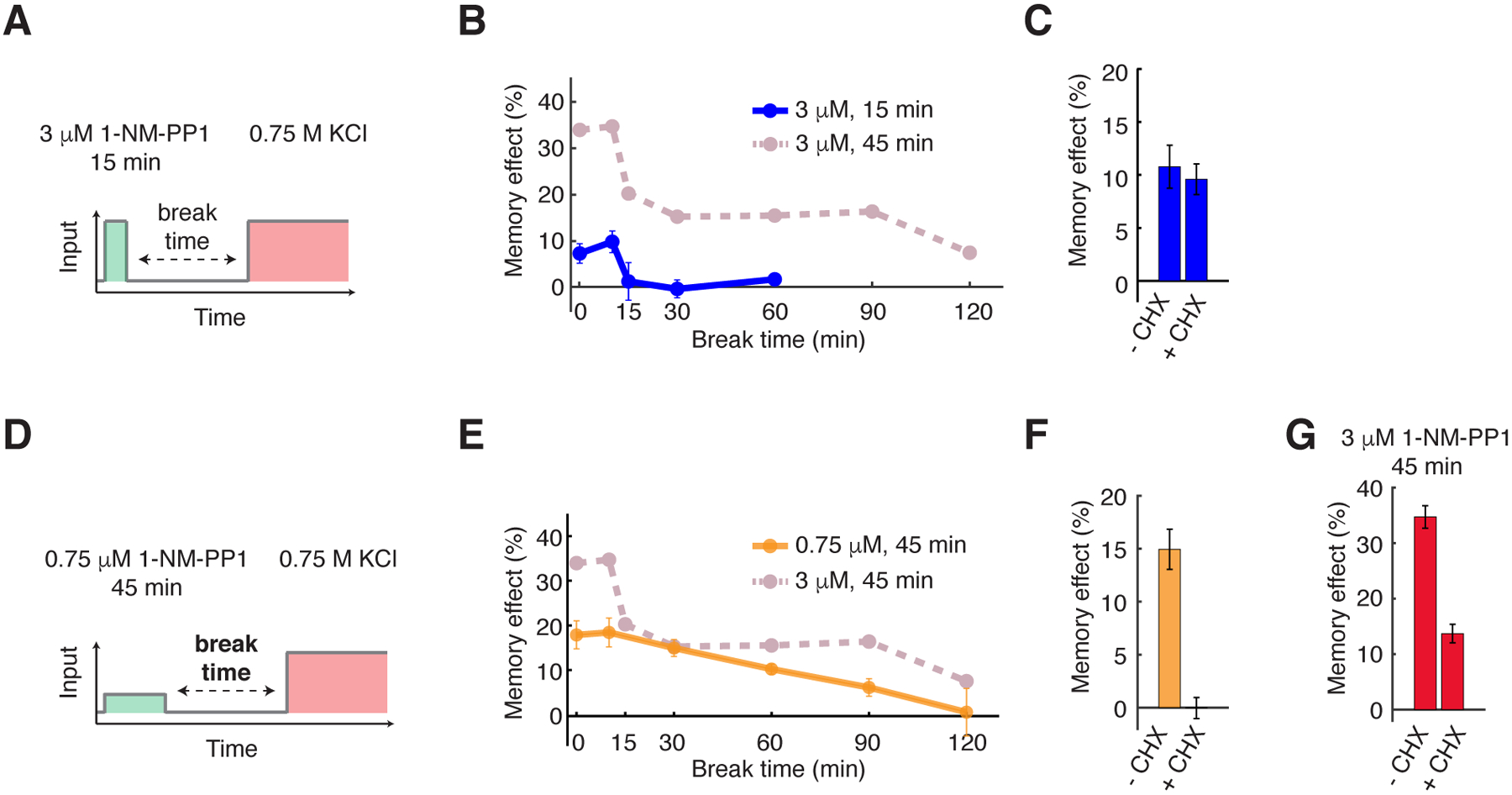
(A) A schematic of the priming experiment with high-amplitude transient priming input (15 min, 3 μM 1-NM-PP1). (B) The dynamics of memory effect in response to the high-amplitude transient priming input. Dashed line in dark pink reproduces the memory dynamics from Fig. 1F and was included in the plot for comparison. (C) The bar graph shows the memory effects in response to 15 min, 3 μM 1-NM-PP1 priming input with a 10 min break time, with or without 10 μg/mL cycloheximide treatment (CHX) to inhibit translation. CHX was added at the beginning of priming inputs and removed after the break time. 10-min break time was chosen to focus on short-lived memory. (D) A schematic of the priming experiment with low-amplitude prolonged priming input (45 min, 0.75 μM 1-NM-PP1). (E) The dynamics of memory effect in response to the low-amplitude prolonged priming input. Dashed line in dark pink reproduces the memory dynamics from Fig. 1F and was included in the plot for comparison. (F) The bar graph shows the memory effects in response to 45 min, 0.75 μM 1-NM-PP1 priming input with a 30 min break time, with or without the CHX treatment. 30-min break time was chosen to focus on long-lived memory. (G) The bar graph shows the memory effects in response to 45 min, 3 μM 1-NM-PP1 priming input with a 10 min break time, with or without the CHX treatment. 10-min break time was chosen because both short-lived and long-lived memories coexist at this break time. Data points are averages of at least three independent experiments. Error bars represent SEM.
To further evaluate the effect of input amplitude on cellular memory, we treated cells with a 45-min priming input of lower amplitude (0.75 μM 1-NM-PP1), which partially inhibited PKA based on the level of Msn2 translocation (18, 27, 29) (Fig. 2D). Under this condition, the fast-decaying memory component and the plateau phase (0 – 90 minutes) of long-lived memory were both abolished (Fig. 2E, compare the orange and dark pink curves). Instead, cells displayed a slow exponential decay trajectory, as would be expected if the memory effect was primarily mediated by gene expression. Accordingly, CHX abolished this memory effect (Fig. 2F), confirming that it required gene expression. Furthermore, the memory effect in response to 45-min priming input with high amplitude (3 μM 1-NM-PP1) was decreased by more than 2-fold with the CHX treatment (Fig. 2G), indicating that the memory under this condition, which consisted of both short-lived and long-lived memory components (Fig. 1F), depended partially on gene expression.
Together, these results demonstrated that the short- and long-lived components of cellular memory could be dissected by modulating the amplitude and duration of priming inputs and might be mediated by different molecular processes. Short-lived memory might be mediated through a fast translation-independent process, whereas an input duration-dependent slow gene expression process might mediate the long-lasting memory, which could be further stabilized by another input amplitude-dependent mechanism.
Short-lived memory is mediated by trehalose synthesis and metabolism
To determine the molecular process that mediated short-lived memory, we deleted 5 known or putative PKA phosphorylation targets (Gph1, Ctt1, Nth1, Gcy1, and Tps1) involved in metabolic or stress-response pathways (37–39). Among these PKA targets, deletion of the trehalose synthase Tps1 (40, 41) abolished short-lived memory (Fig. 3A). Trehalose is a simple carbohydrate produced in many organisms that acts as membrane protectant and protein stabilizer to enhance cell survival under stressful conditions (42). We used the tps1Δ hxk2Δ strain because tps1Δ is unable to grow on fermentable carbon sources, such as glucose, but this growth is restored in a tps1Δ hxk2Δ double mutant (43). Because the deletion of HXK2 alone did not affect the memory effect (Fig. 3A), the effect of the double mutant was primarily due to the deletion of TPS1. We further confirmed that intracellular trehalose level was rapidly increased in response to the PKA inhibition input and that this increase was lost with Tps1 deletion (Fig. 3B). Because trehalose plays an important role in cellular protection against osmotic stress (42), the increased level of trehalose by the priming input could temporarily enhance the acquired resistance to the subsequent osmotic stress, accounting for the observed short-lived memory.
Fig. 3. Short-lived memory is mediated by trehalose synthesis and metabolism.
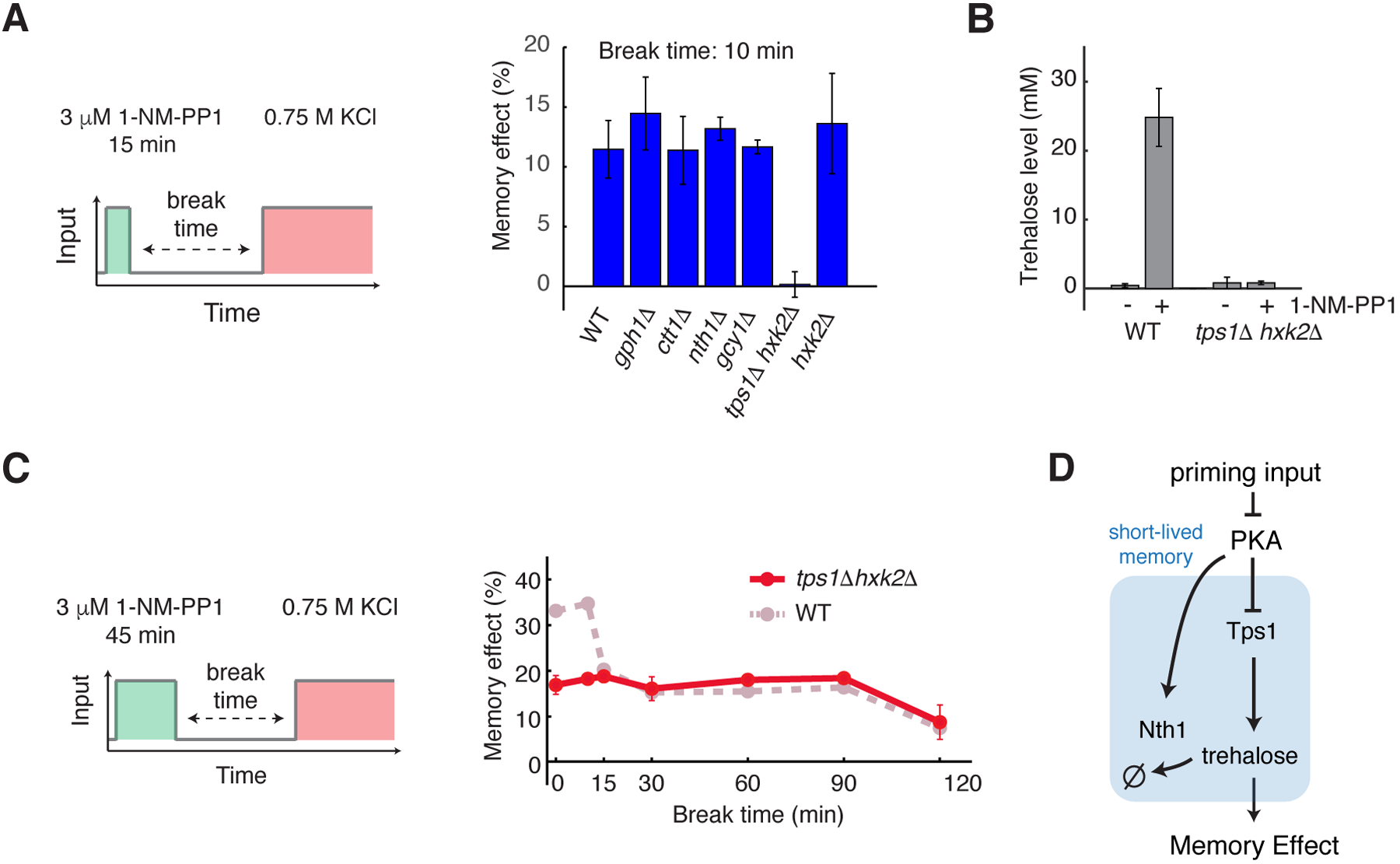
(A) The bar graph shows the memory effects in response to the high-amplitude transient priming input (15 min, 3 μM 1-NM-PP1) with a 10 min break time in WT and mutant strains. (B) The bar graph shows trehalose levels in WT and tps1Δ hxk2Δ, with and without PKA inhibition. Trehalose levels were measured after a 20-min treatment of 3 μM 1-NM-PP1. (C) Memory dynamics in tps1Δ hxk2Δ in response to the high-amplitude prolonged priming input (45 min, 3 μM 1-NM-PP1). Left, schematic illustrating the treatment procedure of the priming experiment. Right, memory dynamics in tps1Δ hxk2Δ cells. Dashed line in dark pink reproduces the memory dynamics in WT cells from Fig. 1F and was included in the plots for comparison. Data points are averages of at least three independent experiments. Error bars show SEM. (D) A diagram illustrating the network motif that gives rise to short-lived memory.
To validate the central role of trehalose in short-lived memory, we manipulated its degradation. The activity of the trehalose-degrading enzyme Nth1 is also regulated by PKA-dependent phosphorylation (38, 44). We observed that, whereas short-term memory was abolished in the absence of Tps1, it became prolonged when NTH1 was deleted (Fig. S10, A and B; compare the blue and grey blue curves), suggesting that the activation of Nth1 accounted for the fast decay rate of the trehalose level. In other words, the duration of short-lived memory was encoded by the activity of Nth1.
To test whether Tps1 and trehalose also contributed to long-lived memory, we examined memory dynamics in response to the high-amplitude prolonged input (3 μM 1-NM-PP1, 45 min). The absence of Tps1 abolished the fast-decaying component of memory without affecting long-lived memory (Fig. 3C, compare the red and dark pink curves), indicating that trehalose synthesis and metabolism contribute exclusively to short-lived memory. In summary, these results showed that short-lived memory was mediated by PKA-dependent regulation of Tps1 and Nth1, which control trehalose synthesis and metabolism (Fig. 3D).
Long-lived memory is mediated by stress-activated transcription factors and mRNP granules
We next investigated the processes underlying long-lived memory, which was gene expression-dependent (Fig. 2F). To determine the transcription factors that induced this response, we deleted 6 PKA-regulated stress-responsive transcription factors (Gis1, Sko1, Hot1, Yap1, and Msn2/4) (17, 45–49). Whereas the deletion of Msn2/4 abolished long-lived memory in response to the 45 minutes, 0.75 μM 1-NM-PP1 priming input (Fig. S11A), the mutant showed only a partial loss of memory in response to the 3 μM 1-NM-PP1 priming input (Fig. S11B), suggesting that another transcription factor might play a compensatory role under this condition. Because the deletion of Yap1 also diminished long-lived memory in response to the 0.75 μM 1-NM-PP1 priming input (Fig. S11A), we generated the msn2Δ msn4Δ yap1Δ triple mutant and observed a complete loss of long-lived memory in the triple mutant (Fig. S11B). These results identified Msn2/4 and Yap1 as the primary transcription factors that mediate the transcriptional response generating long-lasting memory. Moreover, we observed that the short- and long-lived memories are both abolished in the triple mutant (Fig. 4A and B). This loss of short-lived memory in the triple mutant was consistent with previous studies (29, 50), which showed that the expression of TPS1, required for short-lived memory, depends on the transcription factors deleted in the triple mutant.
Figure 4. Long-lived memory is mediated by stress-activated transcription factors and mRNP granules.
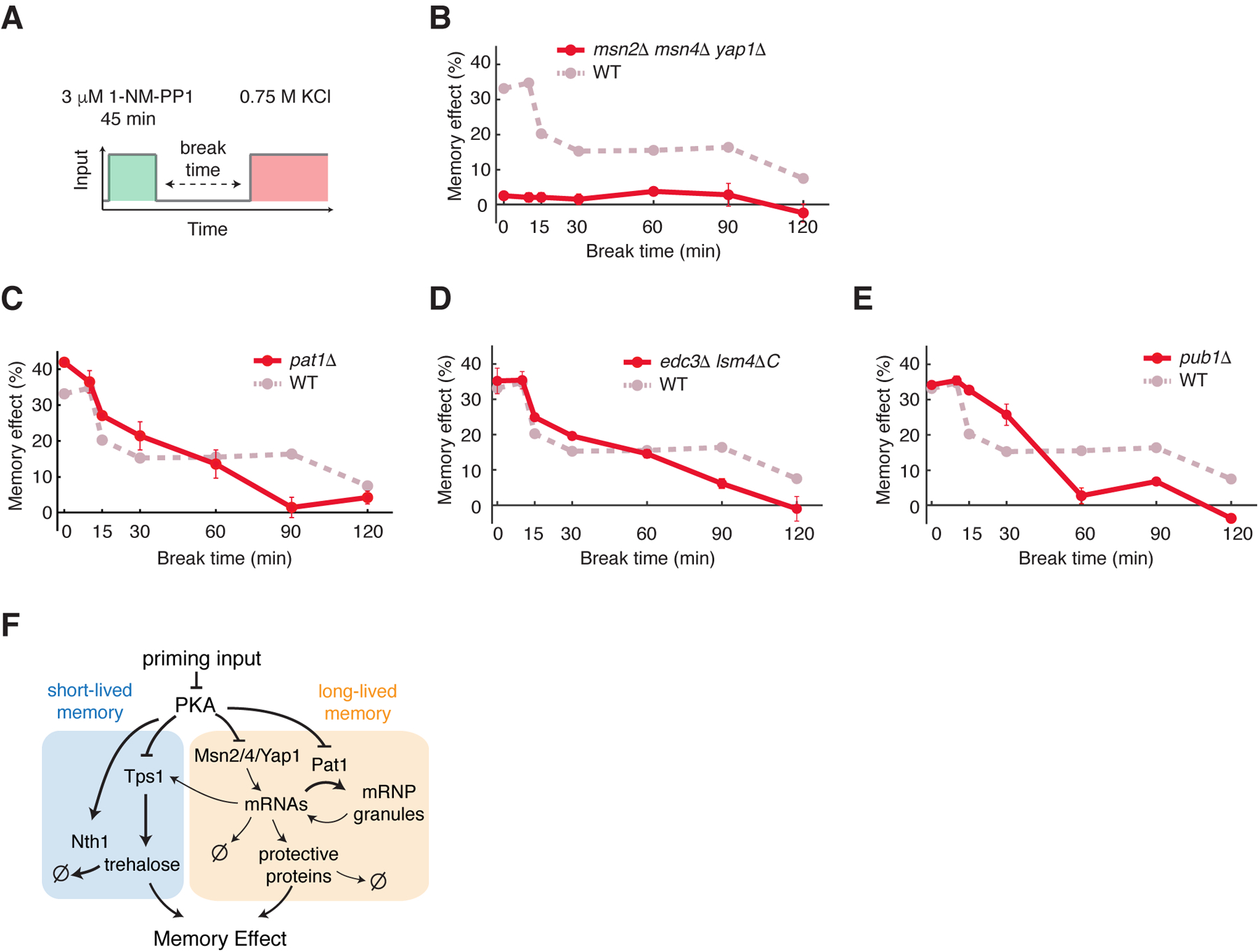
(A) A schematic of the priming experiment with the high-amplitude prolonged priming input (45 min, 3 μM 1-NM-PP1). (B to E) Memory dynamics in msn2Δ msn4Δ yap1Δ (B), pat1Δ (C), edc3Δ lsm4ΔC (D), and pub1Δ (E) cells in response to the high-amplitude prolonged priming input. Data points are averages of at least three independent experiments. Error bars represent SEM. Dashed lines in dark pink reproduce the memory dynamics in WT cells from Fig. 1F and was included in the plots for comparison. (F) A diagram illustrating the PKA-regulated network that mediates short-lived and long-lived memory.
We then investigated the mechanism that stabilizes long-lived memory underlying the plateau phase of memory that is maintained up to 90 minutes after the removal of priming inputs (Fig. 1F). In response to stress or PKA inhibition, cells accumulate various stress-responsive gene mRNAs in cytoplasmic mRNP granules, called processing-bodies (PBs) and stress granules (SGs), which regulate mRNA translation, decay, and storage (51–54). PKA regulates the formation of these mRNP granules by phosphorylating the PB scaffolding protein Pat1 (51), which is essential for the formation of PBs (51, 55). We observed that the plateau phase of long-lived memory was abolished in the pat1Δ strain and the memory effect exhibited a continuous decay (Fig. 4C, compare the red to the dark pink curves). We also observed a similar change in memory dynamics in edc3Δ lsm4ΔC cells, which are defective in PB formation (56)(Fig. 4D). These results indicate that PBs are required for maintaining the plateau phase. PBs and SGs are discrete but functionally interacting compartments. Some mRNAs in PBs can be stored in a translationally silenced state during stress and then return to translation through SGs upon stress removal (53, 55). To determine the role of SGs in memory, we examined memory dynamics in the absence of the SG component Pub1 (55). Similar to pat1Δ and edc3Δ lsm4ΔC cells, pub1Δ cells no longer exhibited the plateau phase of memory (Fig. 4E, compare the red and dark pink curves). We note that pub1Δ and WT cells grew at a similar rate throughout the experiments, indicating that the plateau phase observed in WT cells was not related to cell growth rate. These results suggested that the plateau phase of long-lived memory depended on the PB- and SG-mediated mRNA storage pathway. Notably, pat1Δ, edc3Δ lsm4ΔC, and pub1Δ showed higher memory levels than those of WT for shorter break times (Fig. 4C, D and E, 15 – 30 mins), in agreement with the role of PBs and SGs in mRNA translational silencing in addition to storage.
Furthermore, to confirm the localization of the mRNAs of stress responsive genes to mRNP granules, we used the MS2 coat protein (MS2-CP) fused to GFP (57, 58) to visualize the mRNAs of two well-characterized PKA-regulated stress responsive genes, DCS2 and SIP18 (28, 29) in living cells. In response to the 3 μM 1-NM-PP1 input, DCS2 and SIP18 mRNAs formed foci that co-localized with PBs as indicated by the PB marker Dcp2-mCherry (59) as well as some distinct granules (Fig. S12). We have previously observed a similar localization pattern for Msn2/4 targets, such as GLC3 and HXK1, during glucose starvation (54). This localization pattern coincided with poor protein production from these mRNAs during stress, suggesting that these stress-induced mRNAs were translationally silenced and stored in mRNP granules to confer long-lasting cellular memory. Together, these findings uncovered that two processes, gene transcription and mRNA storage by mRNP granules, operated together to generate long-lived memory (Fig. 4F).
A computational model can simulate and predict memory dynamics
To quantitatively understand the dynamic encoding of memory, we constructed a computational model. In the model, the network is composed of two memory-encoding motifs, one for short-lived memory and the other for long-lived memory (Fig. 4F). The short-lived memory motif comprises a fast process, in which PKA regulates the activities of Tps1 and Nth1 by phosphorylation (38, 39). In response to PKA inhibition, Tps1 is activated, boosting trehalose production; at the same time, Nth1 is inhibited, slowing down trehalose degradation. As a result, the trehalose level increases rapidly. Subsequently, when the PKA inhibition input is removed, Tps1 is inhibited while Nth1 is activated, leading to a rapid decline in trehalose levels during the break time. This feedforward loop enables quick tuning of trehalose levels, accounting for the fast-changing component of memory effect. The long-lived memory motif consists of two processes that function together to regulate gene expression dynamics, a transcriptional response mediated by transcription factors Yap1 and Msn2/4 (60) and a mRNA storage process mediated by Pat1 (53). For the model output, we assumed that the memory effect linearly depended on the sum of the amounts of trehalose and stress resistance gene products in most of the kinetic regimes, unless the substance concentrations reached extremely high levels. We validated this assumption using a computational model developed by Muzzey et al. (31), which explicitly simulates the adaptation dynamics of the osmotic stress signaling pathway and has been well constrained by experimental data (Fig. S13, A to C).
With computationally obtained best-fit parameters, our model reproduced all the experimental data (Fig. S14). In particular, in our model, the high-amplitude transient input (Fig. 5A, left panel) specifically induces the trehalose production process (Fig. 5A, middle panel), generating the fast-decaying memory (Fig. 5A, right panel, compare with data in Fig. 2B). The low-amplitude prolonged input (Fig. 5B, left panel) only induced the transcriptional response but not mRNP granule formation, because the input was too weak, (Fig. 5B, middle panel), resulting in a slow exponential decay of the memory effect after input removal (Fig. 5B, right panel, compare with data in Fig. 2E). The high-amplitude prolonged input (Fig. 5C, left panel), however, led to co-activation of the fast trehalose production and the slow transcriptional response with mRNA storage by mRNP granules (Fig. 5C, middle panel). Once the transcriptional response was initiated, a portion of the newly synthesized mRNAs for stress resistance genes was stored in mRNP granules due to PKA regulation of Pat1. Upon input removal, these mRNAs were slowly released and translated, resulting in a long-lasting (up to ~90 minutes) memory plateau (Fig. 5C, right panel, compare with data in Fig. 1F). Consistent with the model, we observed that the high-amplitude prolonged input induced the formation of mRNP granules, as indicated by DCS2 mRNA foci, whereas the high-amplitude transient input or low-amplitude prolonged input could not (Fig. 5, A to C, right panel insets). These live-cell mRNA results support our model in which the formation and function of mRNP granules depended on both the input amplitude and duration. In summary, our model suggested that the three PKA-regulated processes - trehalose metabolism, gene transcription and mRNP granule formation - operated and coordinated in a specific temporal order to enable the biphasic memory dynamics observed experimentally.
Fig. 5. Computational modeling reveals the network-mediated encoding of cellular memory.
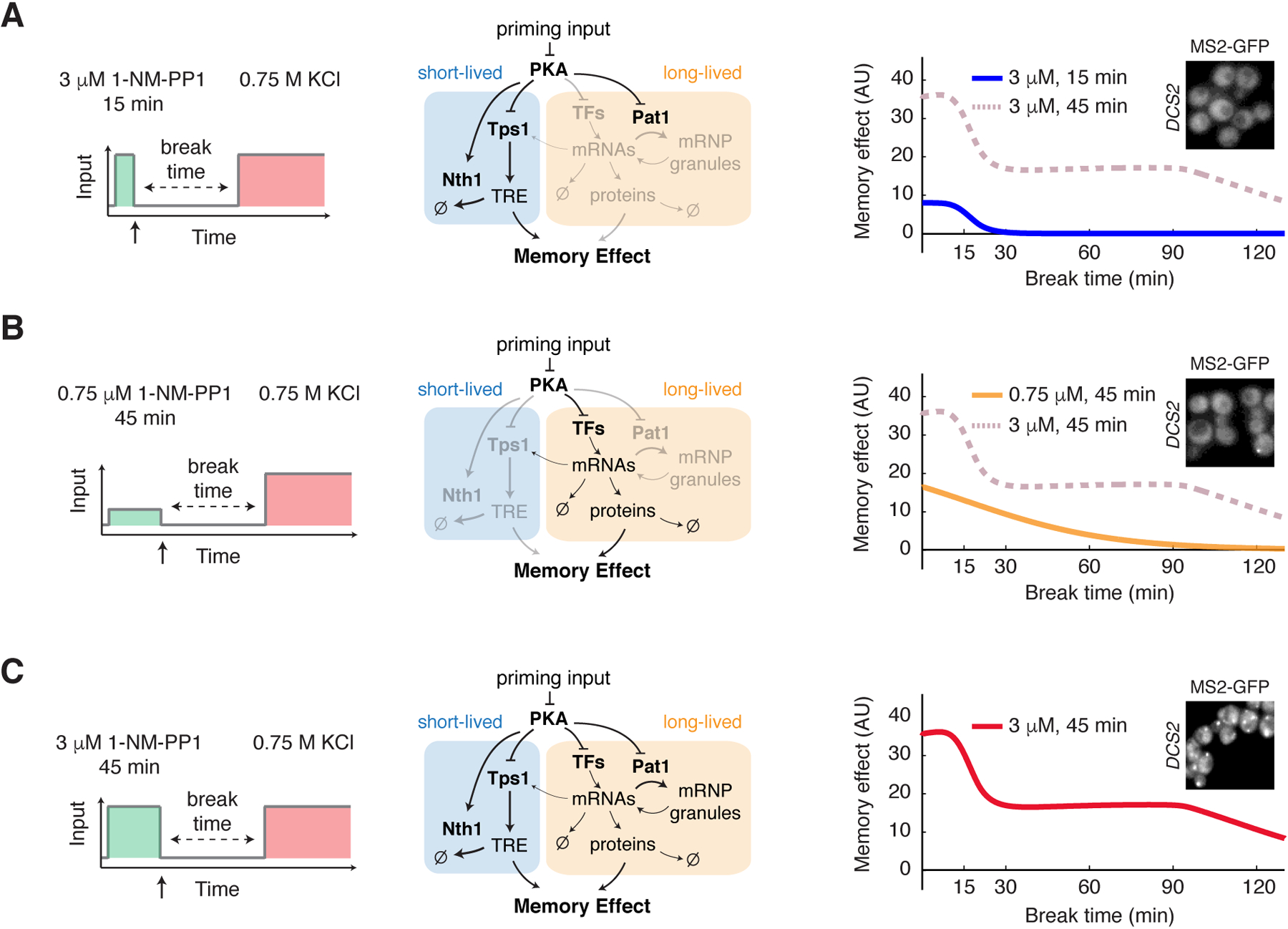
(A) Model simulation of memory dynamics in response to the high-amplitude transient priming input (15 min, 3 μM 1-NM-PP1). Left, schematic illustrating the treatment procedure. Middle, diagram highlighting the part of the network that is activated by the input (black – activated; light gray – not activated). Right, the plot shows the simulated dynamics of memory effect. Dashed line in dark pink reproduces the simulated dynamics in response to the high-amplitude prolonged priming input (45 min, 3 μM 1-NM-PP1) and was included in the plot for comparison. (B) Model simulation of memory dynamics in response to the low-amplitude prolonged priming input (45 min, 0.75 μM 1-NM-PP1). (C) Model simulation of memory dynamics in response to the high-amplitude prolonged priming input (45 min, 3 μM 1-NM-PP1). Insets, granule formation of DCS2 mRNA in response to indicated inputs. Representative images were acquired at the end of input treatments, as indicated by the arrows on left panels.
Moreover, our model largely reproduced the memory dynamics observed in the mutants of key regulatory factors. In the absence of Tps1, the priming input (Fig. S15A, left panel) could not induce the production of trehalose (Fig. S15A, middle panel), resulting in a loss of the fast decaying component of memory, whereas the long-lived memory remained intact (Fig. S15A, right panel, compare with data in Fig. 3C). Upon deletion of the transcription factors Msn2/4 and Yap1, the priming input (Fig. S15B, left panel) could no longer induce the expression of stress resistance genes, resulting in a loss of long-lived memory. Meanwhile, the absence of these transcription factors led to the loss of Tps1 expression required for the short-lived memory (Fig. S15B, middle panel). As a result, both short- and long-lived memories were abolished (Fig. S15B, right panel, compare with data in Fig. 4A). When Pat1 was deleted, the newly synthesized mRNAs, induced by the priming input (Fig. S15C, left panel), could no longer be stored in mRNP granules (Fig. S15C, middle panel), but instead underwent immediate translation, resulting in a higher initial level of stress resistance than that of WT cells (during break time 0 – 30 min). This stress resistance, however, decayed more quickly, because the gene products are being directly and continuously degraded, and cells can no longer maintain a plateau of long-lived memory (Fig. S15C, right panel, compare with data in Fig. 4C).
To test our model, we first used it to predict how the memory effect changes as a function of priming input duration while keeping the break time constant (Fig. 6A). We chose a 60-min break time to focus on the long-lived memory, which depended on input duration (Fig. 2B and Fig. 5A). Our model predicted that the memory effect would increase sharply between 15 and 30-min of input duration, the time window needed to sufficiently activate gene expression and mRNP granules, and would gradually reach saturation with increasing input duration beyond 30 minutes (Fig. 6A, “Prediction”). The data from our experiments agreed with the model prediction, with a curve shape similar to that of the simulated one (Fig. 6A, “Experiment”).
Figure 6. Model prediction and experimental validation.
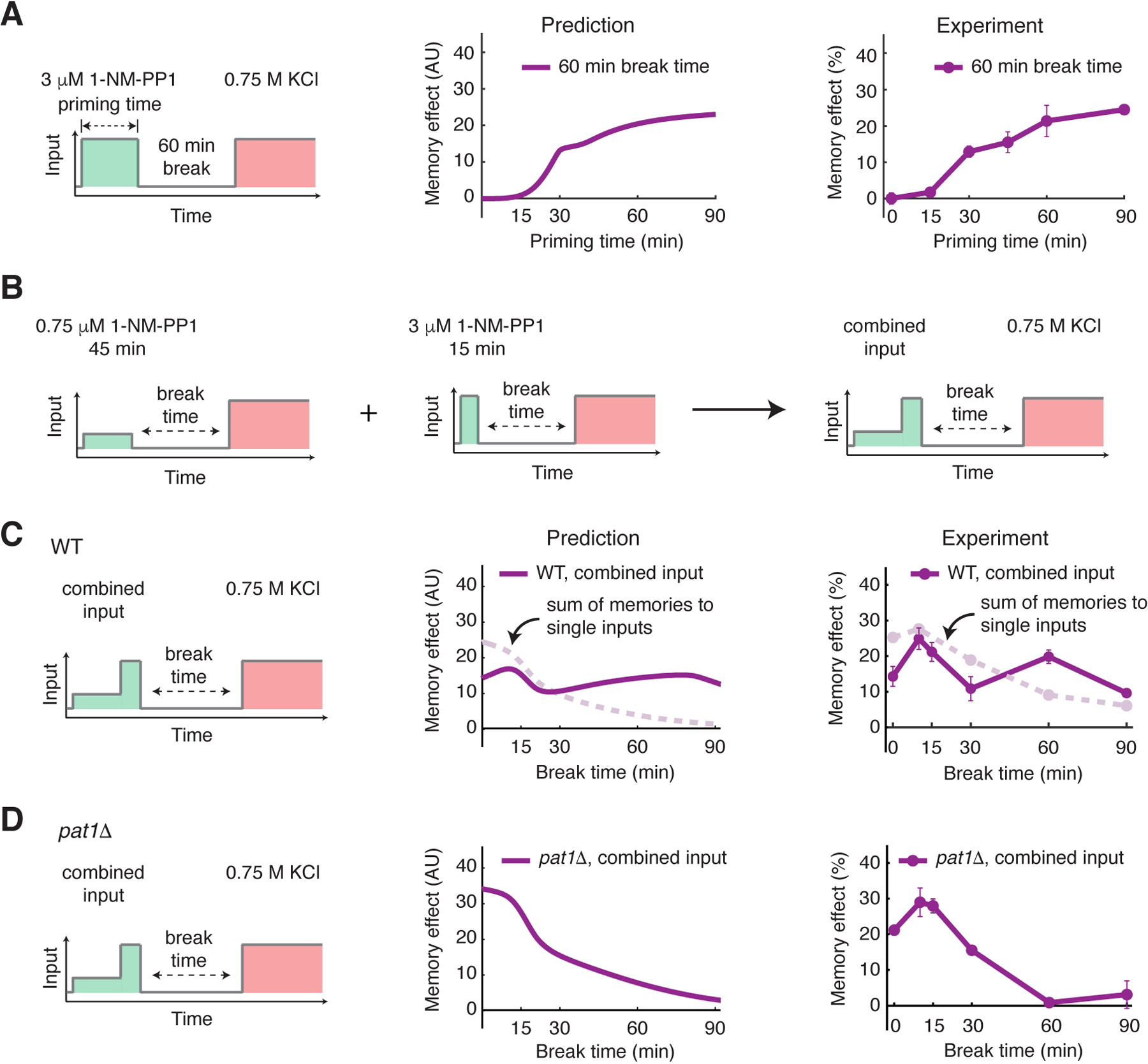
(A) Model prediction and experimental validation of memory effect as a function of priming input duration (60 min break time). Left, schematic illustrating the treatment procedure. Middle, plot showing the predicted memory effects with increasing priming input durations. Right, plot showing the experimentally measured memory effects with increasing priming input durations. (B) A diagram illustrating the temporally combined input. Two single inputs (45 min, 0.75 μM 1-NM-PP1 and 15 min, 3 μM 1-NM-PP1) were applied sequentially to produce the combined input (45 min, 0.75 μM 1-NM-PP1 followed by 15 min, 3 μM 1-NM-PP1). (C) Model prediction and experimental validation of memory dynamics in WT cells in response to the combined input. Left, schematic illustrating the treatment procedure. Middle, plot showing the predicted memory dynamics in response to the combined input. Dashed line in light pink reproduces the sum of memories to two single inputs from Figs. 5A and 5B and was included in the plot for comparison. Right, plot showing the experimentally measured memory dynamics in WT cells in response to the combined input. Dashed line in light pink reproduces the sum of memories to two single inputs from Figs. 2B and 2E and was included in the plot for comparison. (D) Model prediction and experimental validation of memory dynamics in pat1Δ cells in response to the combined input.
Furthermore, we used the model to predict the memory dynamics in response to a combined pattern of priming input. Neither low-amplitude prolonged input nor high-amplitude transient input could induce the plateau phase of memory (Fig. 2B and E), yet our model predicted that these two inputs, when applied sequentially (Fig. 6B), should be capable of generating a memory plateau phase that is Pat1 dependent (Fig. 6C, “Prediction”). In this scenario, the low-amplitude prolonged input would first produce a high amount of mRNAs, and the subsequent high-amplitude transient input would induce mRNP granules to store newly synthesized mRNAs, enabling the plateau phase. This prediction was intriguing because it illustrated that the memory effect to the combined input was not simply the sum of the effects to the two single inputs (Fig. 6C, “Prediction”, compare the purple and light pink curves) due to the mRNP-dependent storage mechanism. Therefore, because the memory-encoding processes were largely independent, the memory effects would become additive when the mRNA storage process was removed in the pat1Δ mutant (Fig. 6D, “Prediction”). In agreement with the model, our experiments showed a plateau phase of memory in response to the combined input (Fig. 6C, “Experiment”). Moreover, in the absence of Pat1, the plateau phase was abolished and the memory dynamics largely resembled the sum of memory effects to the two single inputs (Fig. 6D, “Experiment”), consistent with the model prediction.
These results validated our model and demonstrated its predictive power. Given that the memory-encoding processes were kinetically separated, the model-guided analyses suggest the possibility of rationally designing patterns of priming input and reprogramming the temporal order of regulatory processes to generate the desired forms of memory dynamics.
Discussion
Cellular memory allows cells to adjust their responses to environmental cues based on their prior experience. In this study, we used GSR in yeast Saccharomyces cerevisiae as a model system and demonstrated that the memory effect on stress adaptation was biphasic, comprised of a fast decaying component (short-lived memory), which was mediated by post-translational regulation of the trehalose metabolism pathway, and a long-lasting component (long-lived memory), which was mediated by transcription factors and mRNP granules. These memory-encoding processes were mediated by PKA, an evolutionarily conserved kinase with a central role in many molecular and cellular processes that is also associated with diverse diseases (61, 62). Due to the functional pleiotropy of PKA, it has been difficult to target pharmacologically to achieve therapeutic specificity. Here, we found that different PKA signaling dynamics, depending on the input amplitude and duration, selectively induced specific downstream pathways or processes, leading to distinct memory dynamics. These results raise the possibility of perturbing the dynamics of signaling hubs for specific therapeutic outcomes (63, 64).
Among the PKA-controlled pathways, our results highlighted the contributions of two major biological processes to acquired stress resistance. The first one was metabolism of trehalose, which is critical for mediating yeast stress responses. For instance, under non-optimal temperatures, Tsl1, a regulatory factor of the trehalose synthase Tps1, mediates the heterogeneous cellular phenotypes in growth, survival (65), and bimodal gene expression (66). Because Tsl1 is also encoded by a GSR gene (67), it would be interesting to expand our approach to evaluate the priming effects of PKA input on single-cell responses to temperature stress. This analysis could shed light on how priming inputs influence cell-to-cell variability and fate decision in stress response.
The second process that we revealed was mRNP granule-regulated gene expression. Cytoplasmic mRNP granules, such as PBs and SGs, play important roles in controlling the translation, degradation and storage of mRNAs upon environmental changes (53, 68). Although the biochemical and biophysical characteristics of mRNP granules have been elucidated (69), their functional roles in cell physiology remain largely unclear. mRNP granules have been related to cellular memory formation. For example, transient overexpression of proteins with intrinsically disordered domains (highly enriched in RNA-binding proteins) can induce inheritance of biological traits in yeast cells over many generations (70). Another report showed that the aggregation of Whi3, a yeast RNA-binding protein, can encode memory of previous unsuccessful mating attempt and modulate cells’ mating capacity (71). Moreover, a synthetic biology study revealed phase-separated protein droplets can memorize transient spatial stimuli in mammalian cells (72). In accord with these findings, our results provide additional independent evidence that the PKA-regulated mRNP granules contribute to a long-lasting memory of previous environmental challenges and facilitate the adaptation to future stresses. Further analyses will systematically determine the identities of these stored mRNAs that mediate the memory effect, the detailed mechanisms that direct these mRNAs to mRNP granules, and the prevalence of this mRNP-dependent memory effect in regulating other cellular responses, such as the hormetic effect on aging (73).
In addition, through our modeling analysis, we obtained a quantitative understanding about the dynamics of cellular memory and the regulatory network that controls these dynamics. Previous systems biology studies focused on cellular memory originated from bistability, in which different initial conditions lead to distinct steady states, resulting in history-dependent cellular behaviors or outcomes (74, 75). These systems often contain positive feedback loops that give rise to bistability underlying differentiation or fate decision processes (76–78). In contrast, in this study, we investigated the cellular memory in adaptive stress response, which arose from regulated storage and decay of gene products. The memory dynamics are governed by a network comprised of multiple parallel pathways with highly diversified inherent kinetics. This network architecture conferred signal processing capability and plasticity in shaping memory dynamics, enabling cells to determine their memory patterns in response to rapidly changing environments. In particular, this system coupled two low-pass filters with different thresholds to effectively separate the short-term responses to transient signals from the chronic ones to prolonged signals. Moreover, the system also featured a PKA-regulated mRNP granule formation process, which represented a new network motif for biological information storage. The initiation of the storage depended on input amplitude, whereas how long the storage could last depended on the amount of newly synthesized mRNAs being localized in the granules and hence depended on both the amplitude and the duration of priming inputs. In this way, mRNP granules enabled cells to integrate the information about input amplitude and duration and tune the dynamics of their memory. Guided by the dynamic regulatory schemes revealed by modeling, we further designed priming input patterns to reprogram the temporal order of fast- and slow-acting processes in the network and reshape the memory dynamics. For future studies, advanced fluorescent reporters and imaging technologies (68, 79) could be employed to track on a single-cell level the explicit spatiotemporal dynamics of key species in the model, such as mRNAs and mRNP granules. These data would enable us to constrain and improve our model and, ultimately, enhance the predictive power of the model. We anticipate that a quantitative and predictive understanding of memory control would produce opportunities for broader and more effective use of priming treatments as a low-cost non-genetic approach for stress management in agriculture, biotechnology, and clinical intervention.
Finally, we want to highlight the biological relevance of our findings. We revealed that, because the molecular processes governing the two memory components have distinct kinetic properties, short- and long-lived memories could be selectively induced by different dynamics of priming inputs. Whereas a high amplitude transient input induced fast decaying memory that enabled short-term stress resistance, a prolonged input elicited long-lasting memory conferring long-term stress resistance. This regulatory scheme is analogous to the fast-responding innate immune response and the long-lasting adaptive immune response in mammals. Moreover, mRNP granules were responsive to the amplitude and duration of inputs and could function to further tune the duration of the long-term memory component based on the input dynamics. Together, this integrated regulatory network enables cells to process the information of a previous stress encounter and determine the length of their memories. We speculate that this type of regulation may represent a strategy for cells to optimize resource allocation for future challenge preparation and may be widely applicable to organisms living in rapidly changing environments. Furthermore, given that this regulation is readily tunable, cells could evolve their memory dynamics through natural selection to match the environmental fluctuations in their habitats.
Materials and Methods
Strain Construction
Standard methods for the growth, maintenance, and transformation of yeast and bacteria and for manipulation of DNA were used throughout. All Saccharomyces cerevisiae strains used in this study are derived from the W303 background (MATa leu2–3,112 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 GAL+ psi+ ADE+). The strains used in this study are listed in Table S1.
Microfluidics
The previously reported Y-shape microfluidic device (18, 28) with two inlets has been modified to accommodate three inlets on a single device and used in this study. The device fabrication and the setup of microfluidic experiments were performed as described previously (18, 20, 24, 27, 28).
Time-lapse microscopy
All time-lapse microscopy experiments were performed using a Nikon Ti-E inverted fluorescence microscope with an Andor iXon X3 DU897 EMCCD camera and a Spectra X LED light source. A CFI Plan Apochromat Lambda DM 60X Oil Immersion Objective (NA 1.40 WD 0.13MM) was used for all experiments. Three positions were chosen for each microfluidics channel. For each position, phase contrast, YFP, mCherry, and iRFP images were taken sequentially every two minutes. When the acquisition of the image series started, cells loaded in the microfluidic device were maintained in synthetic complete medium (SC, 2% dextrose) for the first five minutes before 1-NM-PP1 was introduced. Media input was switched manually between SC medium, SC medium with 1-NM-PP1 and SC medium with KCl at the indicated time points. The exposure and intensity settings for each fluorescence channel were set the same as that used in our earlier work (28).
For priming experiments, cells were inoculated from a YPD plate into 2 ml SC liquid media two days before the experiment. On the second day, saturated cells were diluted by 1:20,000 into fresh SC media and grown overnight to reach OD = 0.6. These exponentially growing cells were diluted by 1:2 and grown for another 2 hours before being loaded into the microfluidic devices.
Image analysis
The images were processed using custom MATLAB code for single-cell tracking and fluorescence quantification. The whole cell was segmented using the phase contrast images and the nucleus was segmented using the iRFP images. The cytoplasm was the region of the cell that was not the nucleus. The nuclear to cytoplasmic ratios of Hog1-YFP were calculated using the mean fluorescence intensities of Hog1-YFP in the nucleus and in the cytoplasm. The ratios were subtracted by baseline which is the ratio right before KCl was introduced (close to 1) and then plotted against the time. The duration of Hog1 translocation for each condition was quantified using the full width at half maximum (FWHM) and used to calculate the memory effect for each break time (Fig. S2). We determined the sample size of our single-cell data based on similar studies published previously (18, 27–29).
Trehalose assay
Trehalose level was measured using the trehalose assay kit (Megazyme). Using this assay, trehalose was converted into gluconate-6-phosphate, generating NADPH in a two-step reaction; the NADPH concentration was determined by measuring the absorbance at 340 nm. To determine trehalose concentrations, 13 mL of yeast culture at OD≈0.5 was harvested and put on ice for 5 minutes before centrifuged for 5 min at 4 °C. Cells were washed with 0.1 M phosphate buffer (pH 5.9) to remove glucose in media and resuspended in 1 ml 0.25 M Na2CO3 solution, and OD was measured. Additional Na2CO3 solution was added to make the cell densities (OD) the same for 0 and 20 min samples. Samples (~1 mL) were boiled for 20 min to release intracellular trehalose. After cooling, the samples were centrifuged at 12 000 g for 3 min to remove cell debris. Two aliquots (300 μl) of supernatant were transferred to two new tubes, which were separately used to determine total glucose level and pre-existing glucose level. The following regents were then added to the cell lysates sequentially: 150 μl acetic acid (1 M), 650 μl distilled water, 100 μL imidazole buffer (2 M imidazole, 100 mM magnesium chloride and 0.02% w/v sodium azide; pH 7.0), 50 μL NADP 1/ATP (12.5 mg/mL NADP+ and 36.7 mg/mL ATP) and 10 μL suspension of HK/G-6-PDH (425 U/mL hexokinase and 212 U/mL glucose-6-phosphate dehydrogenase), 10 μL trehalase (490 U/mL). The mixtures were incubated at room temperature for 5 min for the reactions. Absorbance at 340 nm was recorded to determine the trehalose concentration in solution first. To estimate the intracellular concentration, we assumed that cell density at OD=1 is 1×107 cells/ml and yeast cell volume is 42 fl. Pre-existing glucose was determined in a control reaction without added trehalase.
Live-cell mRNA visualization
The MS2-CP strains for mRNA visualization were constructed as described previously (54). The promoter and the coding region of genes of interest were amplified by PCR and then inserted into a template vector, which contains 12x MS2 loop sequences in the integration vector pRS305. The plasmid was linearized by EcoRV and integrated into the W303 background yeast strain which has PKA analog-sensitive mutations (NH084) at the LEU2 locus. A plasmid that constitutively expresses MS2 coat proteins fused with GFP driven by MYO2 promoter (54) was also integrated into the same strain at the HIS3 locus. To visualize the colocalization of mRNAs and PBs, a pRS304 plasmid that expresses DCP2-mCherry under the native DCP2 promoter was integrated into the same strain at the TRP1 locus (NH0857).
To perform live-cell mRNA visualization, cells were cultured to OD 0.6 and then loaded into microfluidic devices for time-lapse microscopy. For each position, phase contrast, mCherry, GFP and iRFP images were taken sequentially every two minutes. After the start of image acquisition, cells were maintained in SD media for the first five minutes to obtain a baseline for each fluorescence channel prior to the introduction of 3 μM 1-NM-PP1 treatment. The exposure and intensity settings for each channel were set as follows: GFP 200 ms at 9% lamp intensity, mCherry 1s at 10% lamp intensity, and iRFP 300 ms at 15% lamp intensity.
Computational Modeling
Our model focused on the PKA-dependent memory-encoding network, comprised of the experimentally-identified processes that regulate the levels of metabolites or proteins needed for enhancing stress adaptation, including trehalose and stress resistance gene products. The input of the model is the PKA inhibition signal (priming input). For the model output, we assumed that the memory effect linearly depended on the sum of the amounts of trehalose and stress resistance gene products in most of the kinetic regimes, unless the substance concentrations reached extremely high levels. This assumption left out the explicit inclusion of the downstream Hog1 pathway in our model and simplified our analysis.
To validate this simplification, we tested it using a computational model developed by Muzzey et al. (31), which explicitly simulates the adaptation dynamics of the osmotic stress signaling pathway and has been well constrained by experimental data. In this model, there are four subsystems. H represents the reactions that link an osmotic with Hog1 nuclear translocation. D and I are the Hog1-dependent and Hog1-independent subsystems respectively. G represents the metabolic reactions involved in glycerol synthesis. The ODE model has three species and five parameters. where s1, s2, and s3 are the respective outputs of the H, D and G. The Hog1-independent subsystem, I, is a parameter in the ODEs, which is αi. u(t) is the input, which takes the Heaviside step form in our simulation. This model produces a perfect adaptation of the s1 species.
In our simplification, we assumed that our priming input changes the initial condition of this system by producing substances (trehalose and stress resistance gene products) before stress and hence reduces the adaptation time. To study the dependence of the adaptation time (FWHM of the peak) on the pre-produced substances, we either varied the initial condition of s2, which is the Hog1-dependent substances, or the value of αi, which is the Hog1-independent substances. All other parameters and initial values were kept the same as those described in (31). In both cases, we observed a near-linear relationship (Fig. S13, A to C). These analyses validated the simplification in our model.
In our model, the network consisted of three molecular processes, trehalose metabolism, stress resistance gene transcription and mRNP granule formation, all of which are PKA regulated. For trehalose metabolism, PKA regulates both trehalose production and degradation by phosphorylating Tps1 and Nth1, respectively. More specifically, PKA-mediated phosphorylation inhibits Tps1 activity and enhances Nth1 activity based on the previous reports. Because the regulation is primarily through phosphorylation, we assumed that it was a fast process. For stress resistance gene expression, PKA regulated mRNA transcription through transcription factors and regulated mRNA translation and degradation through mRNP granules. More specifically, the PKA inhibition input activated transcription factors and mRNP granules. Once the mRNP granules were activated, a portion of the newly synthesized mRNAs was stored in mRNP granules where their translation and degradation were paused. Upon input removal, these mRNAs were slowly released, resuming their translation and degradation. Because gene transcription was a multi-step process, we assumed that it is a relatively slow process; in contrast, because PKA regulated mRNP granules through phosphorylation, we assumed that it was a relatively fast process. Moreover, the level of Tps1 also depended on PKA-regulated gene expression, resulting in a connection between the trehalose metabolism pathway and the gene expression process. Based on the experimental observations, these three processes had different dependence on input amplitude and duration. Trehalose metabolism and mRNP granules could be activated only in response to high-amplitude inputs; by contrast, mRNA transcription could be induced by low-amplitude input but a prolonged duration was needed.
Computational modeling and all the simulations were done using MATLAB. The model contained 13 variables and 27 independent parameters. The effect of growth-dependent dilution was incorporated in the decay rates in our model. The function “lsqnonlin” was used for data fitting. The data of three dynamic inputs and three mutants (tps1Δ, pat1Δ, msn2/4Δ yap1Δ) were used for data fitting (Fig. S14). To highlight the role of mRNP granules, time points for long-lived memory plateau (Fig. 1F) were weighted by 20-fold for data fitting. Fitting starting with completely random guesses failed. Because our model is an abstract model in which each equation or parameter represents a combination of a series of biochemical reactions, the initial guesses for most of the parameters are not available from the literature. Instead, we estimated the initial guesses based on the experimental data. In particular, the memory dynamics data from different priming conditions and various mutants allowed us to estimate the initial guesses of parameters, step by step, for each small sub-network in the system. Here we used the parameters for the trehalose pathway as an example to illustrate how we obtained the initial guesses. Our data showed that high-amplitude transient input activated the short-lived memory that depended solely on Tps1 (Figs. 2, A and B, and 3A). We further showed that the trehalose degradation enzyme Nth1 was involved in regulating the decay rate of trehalose, and thereby the decay rate of memory effect (Fig. S10, A and B). The short-lived memory decayed at ~15 min (Fig. 2B), which allowed us to estimate the limits for trehalose decay rate (k18) and Nth1 activation rate (k1). In addition, the memory effect remained at a plateau for ~10 min after the removal of the priming input (Fig. 2B), suggesting a delay between the inactivation of Nth1 and the decay of trehalose. This delay could arise from a thresholding effect in the enzymatic reactions of trehalose degradation, governed by the trehalose decay equilibrium constant (KM8) and the Hill coefficient (n), and hence could be used for initial guesses of these parameters. Furthermore, the trehalose production rate (k19) could be estimated by dividing the memory level (~0.08 from Fig. 2B) by the duration of priming input (15 min) and the Tps1 level.
Among the Hill coefficients in the model, n3 governed the thresholding effect in Nth1-mediated trehalose degradation and was important for reproducing the ~10 min delay between Nth1 inactivation and trehalose decay (Fig. 2B), and n4 governed the thresholding effect in mRNP formation, both of which were important for reproducing the response to the low amplitude prolonged input (Fig. 2E). We chose the value of 4 for all these Hill coefficients because it generated enough non-linearity required for the thresholding behaviors while not exceeding the biologically relevant range. We did not include these Hill coefficients in model fitting to reduce the number of parameters for fitting.
We tested 10 random sets of initial guesses which are randomly chosen within 8-fold (1/8 to 8) of our first set of guesses and compared the squared norm of the residuals of the final fitting results and then selected the best-fit set of parameter values. We further expanded the range of parameter scan to 16-fold of the first initial guesses and tested additional 10 random sets of initial guesses. We did not find parameter sets that outperformed the chosen best-fit parameter set. Due to the large number of parameters in the model, the chosen best-fit parameter set might not be the global minimum mathematically. However, it did not prevent us from using this model to illustrate our hypothesized mechanism and to make testable predictions (Figs. 5, A to C and 6, A to D).
Under our experimental conditions, the stress adaptation responses were relatively uniform among cells, and priming inputs did not alter cell-to-cell variability (Fig. S3, A and B). Therefore, we did not consider stochasticity in our model.
The initial conditions are provided in Table S2. Reactions and rate constants are provided in Table S3.
Model Equations:
Supplementary Material
Figure S1. Time traces of Hog1 nuclear translocation with different break times.
Figure S2. Data processing workflow for quantification of memory effects as a function of the break time.
Figure S3. The effect of priming input on stress adaptation in single cells.
Figure S4. A priming input with 3 μM 1-NM-PP1 cannot induce the memory effect in cells with wild-type PKA.
Figure S5. Memory dynamics in response to 45 min 0.5 M KCl priming input.
Figure S6. Time traces of Hog1 nuclear translocation with different break times when 0.5 M KCl is used as the priming input.
Figure S7. Biphasic memory effects on the adaptation to glucose limitation.
Figure S8. Time traces of Msn2 nuclear translocation with different break times when glucose limitation is the second stress.
Figure S9. The effect of 15-min 3 μM 1-NM-PP1 on stress adaptation time is statistically significant.
Figure S10. Short-lived memory is regulated by the trehalose degrading enzyme Nth1.
Figure S11. Identification of the transcription factors Msn2/4 and Yap1 in mediating long-lived memory.
Figure S12. Colocalization of DCS2 or SIP18 mRNAs with PBs in response to 3 μM 1-NM-PP1.
Figure S13. Modeling the relationship between the level of priming products and Hog1 adaptation time.
Figure S14. Model fitting with all the experimental data.
Figure S15. Computational modeling reproduces the modulation of memory dynamics in the absence of key regulatory factors.
Table S1. Strains constructed in this study.
Table S2. Initial conditions of all species in the model.
Table S3. Best-fit parameter values used in the model.
Acknowledgments:
This work was supported by NIH R01 GM111458 (to N.H.) and NIH R35 GM128798 (to B.M.Z.).
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The code and strains associated with this work are available upon request.
References and Notes:
- 1.Purvis JE, Lahav G, Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Behar M, Hoffmann A, Understanding the temporal codes of intra-cellular signals. Curr Opin Genet Dev 20, 684–693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine JH, Lin Y, Elowitz MB, Functional roles of pulsing in genetic circuits. Science 342, 1193–1200 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett MR, Hasty J, Microfluidic devices for measuring gene network dynamics in single cells. Nat Rev Genet 10, 628–638 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schenk PM et al. , Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc Natl Acad Sci U S A 97, 11655–11660 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsumoto H, Hamada N, Takahashi A, Kobayashi Y, Ohnishi T, Vanguards of paradigm shift in radiation biology: radiation-induced adaptive and bystander responses. J Radiat Res 48, 97–106 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Lou Y, Yousef AE, Adaptation to sublethal environmental stresses protects Listeria monocytogenes against lethal preservation factors. Appl Environ Microbiol 63, 1252–1255 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durrant WE, Dong X, Systemic acquired resistance. Annu Rev Phytopathol 42, 185–209 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Scholz H, Franz M, Heberlein U, The hangover gene defines a stress pathway required for ethanol tolerance development. Nature 436, 845–847 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Netea MG, Latz E, Mills KH, O’Neill LA, Innate immune memory: a paradigm shift in understanding host defense. Nat Immunol 16, 675–679 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Hilker M et al. , Priming and memory of stress responses in organisms lacking a nervous system. Biol Rev Camb Philos Soc 91, 1118–1133 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Savvides A, Ali S, Tester M, Fotopoulos V, Chemical Priming of Plants Against Multiple Abiotic Stresses: Mission Possible? Trends Plant Sci 21, 329–340 (2016). [DOI] [PubMed] [Google Scholar]
- 13.A. P. G. Prime et al. , Priming: getting ready for battle. Mol Plant Microbe Interact 19, 1062–1071 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Gifford GE, Lohmann-Matthes ML, Gamma interferon priming of mouse and human macrophages for induction of tumor necrosis factor production by bacterial lipopolysaccharide. J Natl Cancer Inst 78, 121–124 (1987). [DOI] [PubMed] [Google Scholar]
- 15.Hayes MP, Freeman SL, Donnelly RP, IFN-gamma priming of monocytes enhances LPS-induced TNF production by augmenting both transcription and MRNA stability. Cytokine 7, 427–435 (1995). [DOI] [PubMed] [Google Scholar]
- 16.Gasch AP et al. , Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11, 4241–4257 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorner W et al. , Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev 12, 586–597 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hao N, O’Shea EK, Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat Struct Mol Biol 19, 31–39 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Pastor MT et al. , The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J 15, 2227–2235 (1996). [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, AkhavanAghdam Z, Tsimring LS, Hao N, Coupled feedback loops control the stimulus-dependent dynamics of the yeast transcription factor Msn2. J Biol Chem 292, 12366–12372 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berry DB, Gasch AP, Stress-activated genomic expression changes serve a preparative role for impending stress in yeast. Mol Biol Cell 19, 4580–4587 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan Q, Haroon S, Bravo DG, Will JL, Gasch AP, Cellular memory of acquired stress resistance in Saccharomyces cerevisiae. Genetics 192, 495–505 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry DB et al. , Multiple means to the same end: the genetic basis of acquired stress resistance in yeast. PLoS Genet 7, e1002353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen AS, Hao N, O’Shea EK, High-throughput microfluidics to control and measure signaling dynamics in single yeast cells. Nat Protoc 10, 1181–1197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y et al. , Multigenerational silencing dynamics control cell aging. Proc Natl Acad Sci U S A 114, 11253–11258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop AC et al. , A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Hao N, Budnik BA, Gunawardena J, O’Shea EK, Tunable signal processing through modular control of transcription factor translocation. Science 339, 460–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.AkhavanAghdam Z, Sinha J, Tabbaa OP, Hao N, Dynamic control of gene regulatory logic by seemingly redundant transcription factors. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansen AS, O’Shea EK, Promoter decoding of transcription factor dynamics involves a trade-off between noise and control of gene expression. Mol Syst Biol 9, 704 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brewster JL, Gustin MC, Hog1: 20 years of discovery and impact. Sci Signal 7, re7 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Muzzey D, Gomez-Uribe CA, Mettetal JT, van Oudenaarden A, A systems-level analysis of perfect adaptation in yeast osmoregulation. Cell 138, 160–171 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxena A, Sitaraman R, Osmoregulation in Saccharomyces cerevisiae via mechanisms other than the high-osmolarity glycerol pathway. Microbiology 162, 1511–1526 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Lee YJ, Jeschke GR, Roelants FM, Thorner J, Turk BE, Reciprocal phosphorylation of yeast glycerol-3-phosphate dehydrogenases in adaptation to distinct types of stress. Mol Cell Biol 32, 4705–4717 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mettetal JT, Muzzey D, Gomez-Uribe C, van Oudenaarden A, The frequency dependence of osmo-adaptation in Saccharomyces cerevisiae. Science 319, 482–484 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hersen P, McClean MN, Mahadevan L, Ramanathan S, Signal processing by the HOG MAP kinase pathway. Proc Natl Acad Sci U S A 105, 7165–7170 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Wever V, Reiter W, Ballarini A, Ammerer G, Brocard C, A dual role for PP1 in shaping the Msn2-dependent transcriptional response to glucose starvation. EMBO J 24, 4115–4123 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang PK, Tugendreich S, Fletterick RJ, Molecular analysis of GPH1, the gene encoding glycogen phosphorylase in Saccharomyces cerevisiae. Mol Cell Biol 9, 1659–1666 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schepers W, Van Zeebroeck G, Pinkse M, Verhaert P, Thevelein JM, In vivo phosphorylation of Ser21 and Ser83 during nutrient-induced activation of the yeast protein kinase A (PKA) target trehalase. J Biol Chem 287, 44130–44142 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trevisol ET, Panek AD, De Mesquita JF, Eleutherio EC, Regulation of the yeast trehalose-synthase complex by cyclic AMP-dependent phosphorylation. Biochim Biophys Acta 1840, 1646–1650 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Reinders A et al. , Structural analysis of the subunits of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae and their function during heat shock. Mol Microbiol 24, 687–695 (1997). [DOI] [PubMed] [Google Scholar]
- 41.De Virgilio C et al. , Disruption of TPS2, the gene encoding the 100-kDa subunit of the trehalose-6-phosphate synthase/phosphatase complex in Saccharomyces cerevisiae, causes accumulation of trehalose-6-phosphate and loss of trehalose-6-phosphate phosphatase activity. Eur J Biochem 212, 315–323 (1993). [DOI] [PubMed] [Google Scholar]
- 42.Hounsa CG, Brandt EV, Thevelein J, Hohmann S, Prior BA, Role of trehalose in survival of Saccharomyces cerevisiae under osmotic stress. Microbiology 144 (Pt 3), 671–680 (1998). [DOI] [PubMed] [Google Scholar]
- 43.Hohmann S et al. , The growth and signalling defects of the ggs1 (fdp1/byp1) deletion mutant on glucose are suppressed by a deletion of the gene encoding hexokinase PII. Curr Genet 23, 281–289 (1993). [DOI] [PubMed] [Google Scholar]
- 44.Souza AC, De Mesquita JF, Panek AD, Silva JT, Paschoalin VM, Evidence for a modulation of neutral trehalase activity by Ca2+ and cAMP signaling pathways in Saccharomyces cerevisiae. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas 35, 11–16 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Proft M et al. , Regulation of the Sko1 transcriptional repressor by the Hog1 MAP kinase in response to osmotic stress. EMBO J 20, 1123–1133 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pascual-Ahuir A, Posas F, Serrano R, Proft M, Multiple levels of control regulate the yeast cAMP-response element-binding protein repressor Sko1p in response to stress. J Biol Chem 276, 37373–37378 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Hinnebusch AG, Natarajan K, Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell 1, 22–32 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roosen J et al. , PKA and Sch9 control a molecular switch important for the proper adaptation to nutrient availability. Mol Microbiol 55, 862–880 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Charizanis C, Juhnke H, Krems B, Entian KD, The oxidative stress response mediated via Pos9/Skn7 is negatively regulated by the Ras/PKA pathway in Saccharomyces cerevisiae. Mol Gen Genet 261, 740–752 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Norbeck J, Blomberg A, The level of cAMP-dependent protein kinase A activity strongly affects osmotolerance and osmo-instigated gene expression changes in Saccharomyces cerevisiae. Yeast 16, 121–137 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Ramachandran V, Shah KH, Herman PK, The cAMP-dependent protein kinase signaling pathway is a key regulator of P body foci formation. Mol Cell 43, 973–981 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang C et al. , Context-dependent deposition and regulation of mRNAs in P-bodies. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Decker CJ, Parker R, P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4, a012286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zid BM, O’Shea EK, Promoter sequences direct cytoplasmic localization and translation of mRNAs during starvation in yeast. Nature 514, 117–121 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchan JR, Muhlrad D, Parker R, P bodies promote stress granule assembly in Saccharomyces cerevisiae. J Cell Biol 183, 441–455 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Decker CJ, Teixeira D, Parker R, Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol 179, 437–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haim-Vilmovsky L, Gerst JE, m-TAG: a PCR-based genomic integration method to visualize the localization of specific endogenous mRNAs in vivo in yeast. Nat Protoc 4, 1274–1284 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Bertrand E et al. , Localization of ASH1 mRNA particles in living yeast. Mol Cell 2, 437–445 (1998). [DOI] [PubMed] [Google Scholar]
- 59.Brengues M, Teixeira D, Parker R, Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310, 486–489 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith A, Ward MP, Garrett S, Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J 17, 3556–3564 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skalhegg BS, Tasken K, Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front Biosci 5, D678–693 (2000). [DOI] [PubMed] [Google Scholar]
- 62.Chiaradonna F, Balestrieri C, Gaglio D, Vanoni M, RAS and PKA pathways in cancer: new insight from transcriptional analysis. Front Biosci 13, 5257–5278 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Behar M, Barken D, Werner SL, Hoffmann A, The dynamics of signaling as a pharmacological target. Cell 155, 448–461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y, Roberts J, AkhavanAghdam Z, Hao N, Mitogen-activated protein kinase (MAPK) dynamics determine cell fate in the yeast mating response. J Biol Chem 292, 20354–20361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levy SF, Ziv N, Siegal ML, Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol 10, e1001325 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Charlebois DA, Hauser K, Marshall S, Balazsi G, Multiscale effects of heating and cooling on genes and gene networks. Proc Natl Acad Sci U S A 115, E10797–E10806 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yaakov G, Lerner D, Bentele K, Steinberger J, Barkai N, Coupling phenotypic persistence to DNA damage increases genetic diversity in severe stress. Nat Ecol Evol 1, 16 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Standart N, Weil D, P-Bodies: Cytosolic Droplets for Coordinated mRNA Storage. Trends Genet 34, 612–626 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Mitchell SF, Parker R, Principles and properties of eukaryotic mRNPs. Mol Cell 54, 547–558 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Chakrabortee S et al. , Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell 167, 369–381 e312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caudron F, Barral Y, A super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell 155, 1244–1257 (2013). [DOI] [PubMed] [Google Scholar]
- 72.Dine E, Gil AA, Uribe G, Brangwynne CP, Toettcher JE, Protein Phase Separation Provides Long-Term Memory of Transient Spatial Stimuli. Cell Syst 6, 655–663 e655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mattson MP, Hormesis defined. Ageing Res Rev 7, 1–7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Acar M, Becskei A, van Oudenaarden A, Enhancement of cellular memory by reducing stochastic transitions. Nature 435, 228–232 (2005). [DOI] [PubMed] [Google Scholar]
- 75.Nevozhay D, Adams RM, Van Itallie E, Bennett MR, Balazsi G, Mapping the environmental fitness landscape of a synthetic gene circuit. PLoS Comput Biol 8, e1002480 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiong W, Ferrell JE Jr., A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature 426, 460–465 (2003). [DOI] [PubMed] [Google Scholar]
- 77.Chang DE et al. , Building biological memory by linking positive feedback loops. Proc Natl Acad Sci U S A 107, 175–180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang L et al. , Bistable switches control memory and plasticity in cellular differentiation. Proc Natl Acad Sci U S A 106, 6638–6643 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aizer A et al. , Quantifying mRNA targeting to P-bodies in living human cells reveals their dual role in mRNA decay and storage. J Cell Sci 127, 4443–4456 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Time traces of Hog1 nuclear translocation with different break times.
Figure S2. Data processing workflow for quantification of memory effects as a function of the break time.
Figure S3. The effect of priming input on stress adaptation in single cells.
Figure S4. A priming input with 3 μM 1-NM-PP1 cannot induce the memory effect in cells with wild-type PKA.
Figure S5. Memory dynamics in response to 45 min 0.5 M KCl priming input.
Figure S6. Time traces of Hog1 nuclear translocation with different break times when 0.5 M KCl is used as the priming input.
Figure S7. Biphasic memory effects on the adaptation to glucose limitation.
Figure S8. Time traces of Msn2 nuclear translocation with different break times when glucose limitation is the second stress.
Figure S9. The effect of 15-min 3 μM 1-NM-PP1 on stress adaptation time is statistically significant.
Figure S10. Short-lived memory is regulated by the trehalose degrading enzyme Nth1.
Figure S11. Identification of the transcription factors Msn2/4 and Yap1 in mediating long-lived memory.
Figure S12. Colocalization of DCS2 or SIP18 mRNAs with PBs in response to 3 μM 1-NM-PP1.
Figure S13. Modeling the relationship between the level of priming products and Hog1 adaptation time.
Figure S14. Model fitting with all the experimental data.
Figure S15. Computational modeling reproduces the modulation of memory dynamics in the absence of key regulatory factors.
Table S1. Strains constructed in this study.
Table S2. Initial conditions of all species in the model.
Table S3. Best-fit parameter values used in the model.