

Abstract
Isobaric labeling empowers proteome-wide expression measurements simultaneously across multiple samples. Here an expanded set of 16 isobaric reagents based on an isobutyl-proline immonium ion reporter structure (TMTpro) is presented. These reagents have similar characteristics to existing tandem mass tag reagents but with increased fragmentation efficiency and signal. In a proteome-scale example dataset, we compared eight common cell lines with and without Torin1 treatment with three replicates, quantifying more than 8,800 proteins (mean of 7.5 peptides per protein) per replicate with an analysis time of only 1.1 h per proteome. Finally, we modified the thermal stability assay to examine proteome-wide melting shifts after treatment with DMSO, 1 or 20 μM staurosporine with five replicates. This assay identified and dose-stratified staurosporine binding to 228 cellular kinases in just one, 18-h experiment. TMTpro reagents allow complex experimental designs—all with essentially no missing values across the 16 samples and no loss in quantitative integrity.
Proteomics characterizes all proteins in a biological system and has become a vital tool for addressing important biological questions. Yet in many technical respects, proteomics still lags behind genomics; the primary challenges are speed of analysis, depth of proteome coverage and quality of quantitation. Although a variety of approaches have been introduced to address these issues1,2, they almost always involve a trade-off. An important need lingers for increased sample multiplexing capacity while simultaneously addressing all three of these fundamental challenges, setting the stage for proteomics to tackle a wide range of biological problems on an ever-increasing scale.
Isobaric labeling through tandem mass tags (TMT) enables proteome-wide relative quantification for many samples simultaneously in one experiment and is proving to be an effective strategy for balancing these issues3–6. TMT reagents are composed of an amine-reactive group, a mass normalization group and a reporter ion group (Fig. 1a). The amine-reactive group can label peptides (N termini and the ε-amino groups of lysines). The structure and nominal mass of each amine-reactive TMT reagent are identical. Yet each tag varies in the distribution of five heavy isotopes across the reporter ion and normalization groups. As such, the difference in reporter ion mass necessary for quantification among different tags is offset by the difference in the mass normalization group. Labeled peptides, regardless of isotope distribution, coelute as a single composite peak with the same m/z value in an MS1 scan. The fragmentation of the labeled peptides during the subsequent MS2 or MS3 higher-energy collisional dissociation (HCD) event generates reporter ion peaks of differing mass enabling quantification across samples4,7.
Fig. 1|. Overview of TMTpro reagents for sample multiplexing.
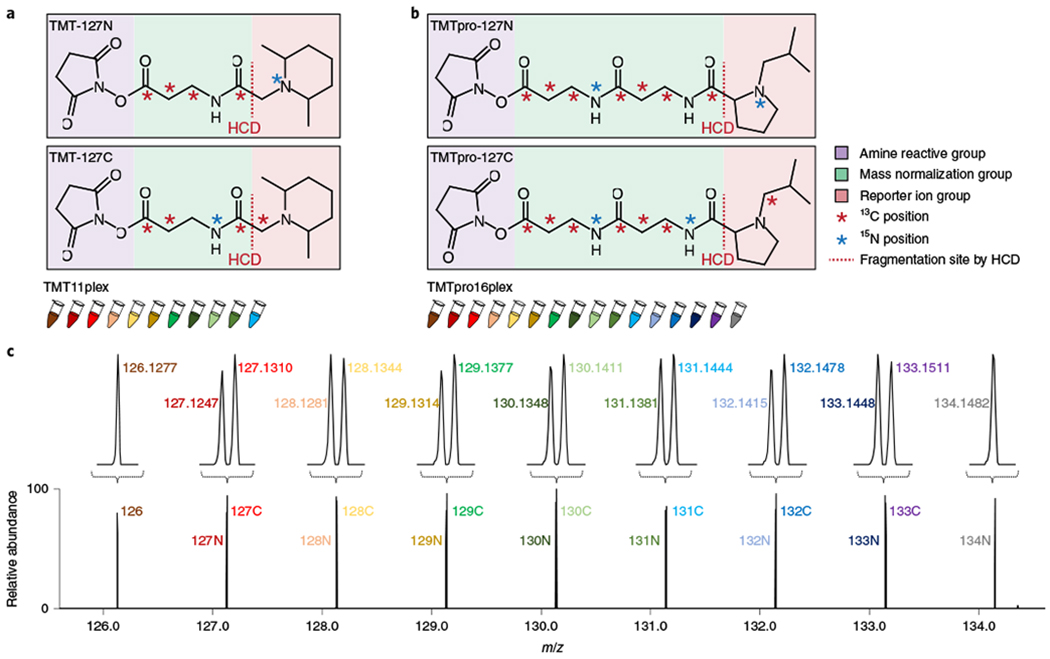
a,b, Chemical structures of TMT (a) and TMTpro reagents (b). TMT and TMTpro reagents are available as set of up to 11plex and 16plex, respectively. TMT-127N, TMT-127C, TMTpro-127N and TMTpro-127C are shown here as examples. See Supplementary Fig. 1 for structures for all TMTpro reagents. c, Illustration of TMTpro16plex reporter ions.
Sample multiplexing with TMT reagents was initially limited to just six samples4,7. Substituting a nitrogen-15 for a carbon-13 atom resulted in tiny, but resolvable, mass differences that allowed for a 10plex reagent set4,8. Filling out all combinations of positions in the reporter group with the existing five heavy atoms resulted in an 11plex reagent set9. However, any further increases would require more heavy isotopes and a new reporter ion/linker chemistry.
Here we present the next generation of TMT reagents termed TMTpro tags (Fig. 1b). These reagents have nine atoms enriched for stable heavy isotopes with the potential to provide up to 18 different reagents (Supplementary Figs. 1 and 2), of which a 16plex reagent set is presented here. Using the previous tag structure (five atoms for isotopic enrichment), a maximum of 11 reagents could be synthesized (Fig. 1a). Exploiting (1) the small, but measurable, mass difference between an extra neutron in a nitrogen-15 versus a carbon-13 atom (6 millimass units)10, (2) an optimized linker and (3) a proline-based reporter ion group, 16 next-generation isobaric labeling reagents were synthesized11 (Fig. 1b,c).
Results
Characterization of TMTpro isobaric labeling reagents.
We first tested the effect of the TMTpro reagents on protein identification and reporter ion signal using a light version of the TMT and TMTpro tags (that is, TMT0 and TMTpro0). We labeled trypsinized human cell lysates (Fig. 2a) and achieved complete labeling efficiencies of 95.4% (TMT0) and 97.6% (TMTpro0). Very small fractions of peptides remained partially labeled (TMT0: 1.3%; TMTpro0: 1.9%) or unlabeled (TMT0: 3.2%; TMTpro0: 0.4%). From 45 min gradients, the TMTpro labeling resulted in slightly lower numbers of peptides than TMT (10.3% fewer, Fig. 2b), but yielded increased average reporter ion signal at a lower optimized collision energy (45%, Fig. 2c). Optimal collision energy for TMTpro reagents for a high-resolution MS2 (hrMS2) method was also lower than TMT reagents (Supplementary Fig. 3). In addition to improved signal for the TMTpro reagents, shorter MS3 ion injection times were needed for TMTpro-labeled peptides (Supplementary Fig. 4a). A systematic increase in elution position (~1 min h−1 gradient) was observed (Supplementary Fig. 4b). All other figures of merit were nearly identical including the distributions for precursor m/z, MS2 ion injection time, SEQUEST scores (XCorr and deltaCn), mass error and peptide length (Supplementary Fig. 4c–h).
Fig. 2|. Comparison of TMT0- and TMTpro0-labeled samples.
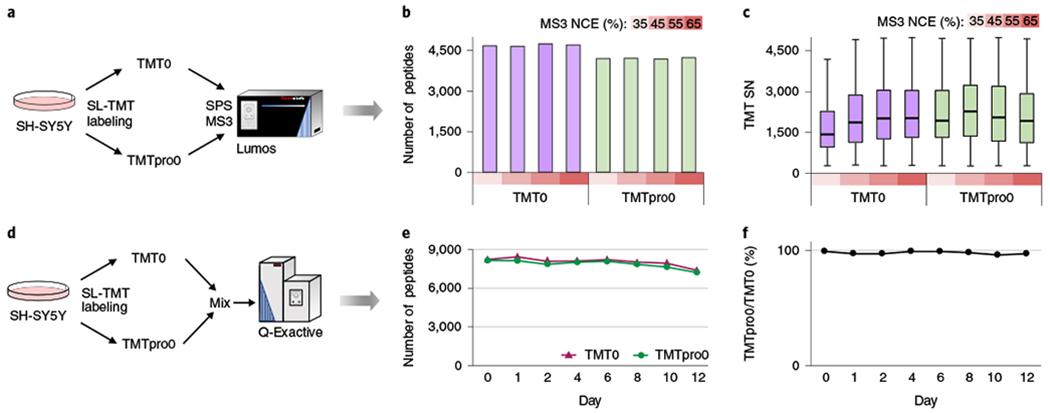
a, Trypsinized SH-SY5Y cell lysate was labeled with light (no heavy isotopes) TMT0 or TMTpro0, and analyzed by SPS–MS3. TMT0 has a monoisotopic modification mass of 224.1525. TMTpro0 has a monoisotopic modification mass of 295.1896. They both generate a reporter ion of a monoisotopic mass of 126.1277 by HCD. b, The number of TMT0- and TMTpro0-labeled peptides identified with increasing MS3 NCE. TMTpro labeling had an average of 10.3% fewer peptide identifications compared with TMT-labeling. c, Summed reporter ion signal-to-noise (SN) of TMT0- and TMTpro0-labeled peptides. Distributions are presented as box plots (center line, median; box limits correspond to the first and third quartiles; whiskers, 1.5× interquartile range). Numbers of peptides in each box are (from left to right): 4,656, 4,633, 4,723, 4,685, 4,187, 4,194, 4,168 and 4,220. TMTpro0-labeled peptides had a lower optimal MS3 NCE (45%) and generated higher reporter ion signal-to-noise than TMT0-labeled peptides at respective optimal MS3 NCE. d, Stability test for TMTpro0-labeled peptides. Trypsinized SH-SY5Y cell lysate was labeled with TMT0 and TMTpro0, respectively, mixed and analyzed at various time points (days 0–12) by HCD-hrMS2. The sample was stored at room temperature over the 12-d period. e, The number of TMT0- and TMTpro0-labeled peptides identified in the sample prepared in d. f, Proportion of TMTpro0-labeled peptides to TMT0-labeled peptides in the sample prepared in d.
We next tested each tag’s stability by labeling cell lysate with TMT0 and TMTpro0 and then mixing the two samples before analysis. Each liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis then was used to compare the number of peptides identified from each tagged proteome shot over 12 d at room temperature (Fig. 2d). We observed similar stability as measured by the numbers of peptides identified (Fig. 2e) and the proportion of TMTpro0- to TMT0-labeled peptides (Fig. 2f).
To investigate the quantitative precision of the TMTpro and TMT reagents, we compared protein expression differences between HCT116 and 293T cells using SPS–MS3 (ref. 12), finding highly similar quantitative performance (Supplementary Fig. 5). The average percentage coefficient of variation was <7% for both reagents (Supplementary Fig. 5b). Replicates clustered together regardless of tag (Supplementary Fig. 5c), and an excellent correlation was found between experiments (r = 0.89) (Supplementary Fig. 5d). Furthermore, we noted no significant ratio compression from the 11-to-16plex increase (Supplementary Fig. 5e). Similar results were observed when a hrMS2 method was used on a Q-Exactive HF-X instrument (Supplementary Fig. 6).
TMTpro reagents enabled the profiling of the human proteome in 1.1 h.
We next benchmarked the use of TMTpro for accommodating complex experimental designs in a single experiment. We reasoned that combining the new 16plex reagents with a real-time search (RTS) approach13,14, would allow us to quantify >8,000 proteins across an entire dataset while using only ~1 h of analysis time per proteome. The RTS application reduces analysis time by as much as 50% without a loss in quantification14. A multiplexing level of 16 samples and a total analysis time of 18 h (12 fractions × 90 min runs) would result in a final analysis time of just 1.1 h per proteome.
We investigated Torin1-altered protein abundance and phosphorylation states in eight human cell lines using a fully fractionated TMTpro16plex-labeled sample set (Fig. 3a). Torin1 is a highly selective ATP-competitive inhibitor of the molecular target of rapamycin (mTOR) kinase complexes. The mTORC1/C2 (complex 1/2) function as master regulators of cell growth and proliferation by coordinating these processes with the availability of nutrients and suppressing catabolic processes including autophagy15. Deregulation of the mTOR pathway has been implicated in many diseases (for example, cancer) and mTOR inhibitors have been intensively studies as potential immunosuppressants and anticancer agents16. Torin1 attenuates mTORC1 signals deemed insensitive to the widely studied mTOR inhibitor rapamycin, leading to translation suppression and autophagic flux activation17. We treated eight broadly used and well-characterized human cell lines with either Torin1 or DMSO (control) for 12 h in biological triplicate (Fig. 3a). Before downstream analysis, western blotting of cell extracts revealed a marked reduction in phosphorylated EIF4EBP1, indicating efficacy of treatment across all cell lines (Supplementary Fig. 7). We adapted the streamlined–TMT-based protocol, and analyzed both the fully fractionated whole cell proteome, as well as the phosphoproteome (‘mini-phos’ format) across the same samples18. Proteome and phosphoproteome can be analyzed using the same 100 μg starting material through the ‘mini-phos’ format18. Three 16plexes accommodated a total of 48 samples. For protein-level analysis, 24 fractions were collected from basic pH reversed-phase LC (bRPLC), but only 12 were analyzed by RTS–synchronous precursor selection (RTS–SPS)–MS3 (refs. 12–14) with 90 min gradients. The IMAC-enriched phosphopeptides were analyzed without pre-fractionation by standard SPS–MS3 (Fig. 3a)12.
Fig. 3|. Application of TMTpro reagents to eight human cell lines treated with Torin1.
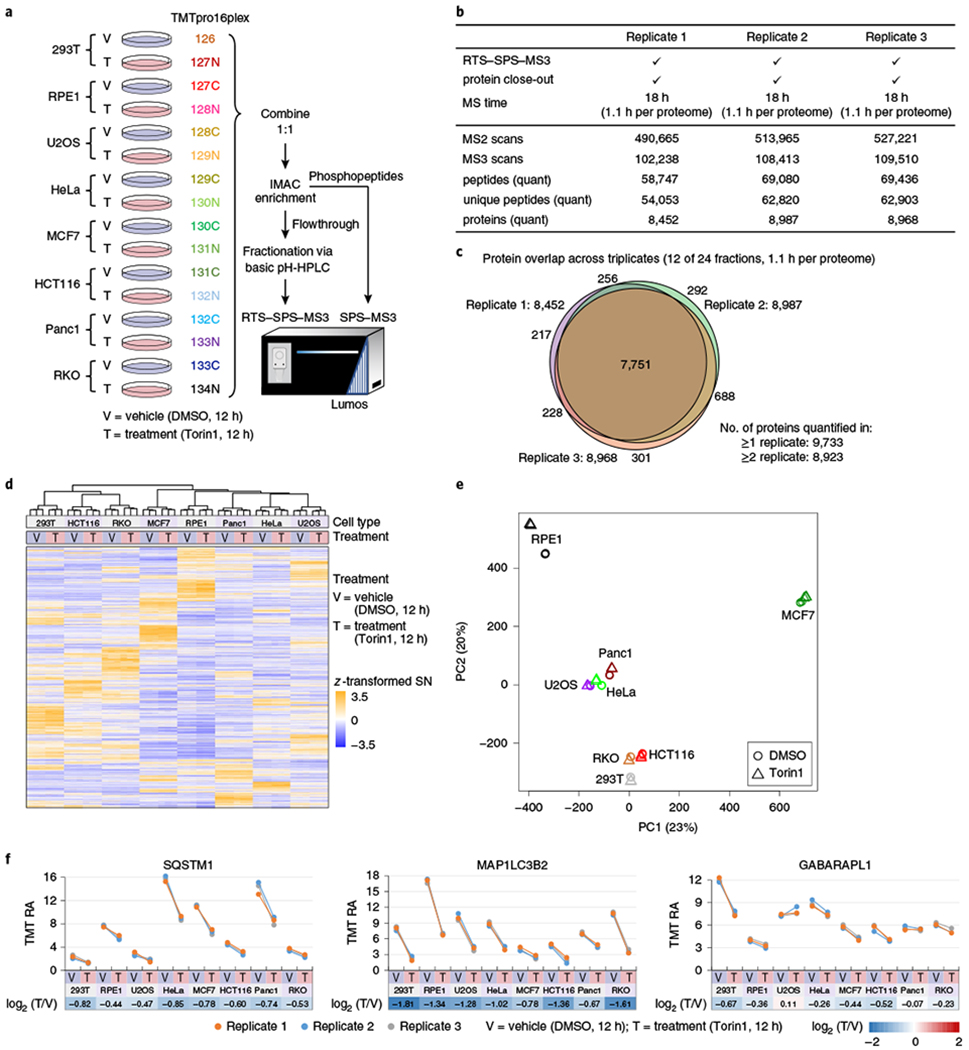
a, Workflow for sample preparation. Eight human cell lines were treated with DMSO or Torin1 (an mTOR inhibitor) in three cell culture replicates for 12h and labeled with TMTpro16plex reagents. Samples were combined 1:1 across all 16 channels. Phosphopeptides were enriched with IMAC, and the flowthrough was fractionated into 24 fractions via bRPLC. Fractions were analyzed by RTS-SPS-MS3 to reduce analysis time. b, RTS-SPS-MS3 analysis metrics for protein data (12 of 24 fractions, A set). RTS-SPS-MS3 and protein closeout (two peptides per protein for each fraction or up to 24 total peptides) were used. MS time on each 16plex replicate was 18 h (1.1 h per proteome). Numbers of MS2 scans, MS3 scans, peptides, unique peptides and proteins in each replicate are shown. c, Overlap of quantified proteins across three replicates (12 of 24 fractions). More than 8,800 proteins were quantified per replicate. In total, 9,733 proteins were quantified and 7,751 were quantified in all three replicates. d,e, Unsupervised HCA (d) and PCA (e) of quantified proteins showed that samples were profiled without measurable bias or batch effect (n = 3 cell culture replicates). f, Downregulation of SQSTM1, MAP1LC3B2 and GABARAPL1 with Torin1 treatment across eight cell lines. RA stands for relative abundance. TMT RA is the percentage of total S/N within a 16plex (see Methods).
Total data acquisition time for the whole proteome analysis was 18 h per replicate or 1.1 h per proteome (Fig. 3b). We quantified an average of 8,802 ± 304 proteins in each replicate, 7,751 proteins in all three replicates and 9,733 proteins in at least one replicate (Fig. 3c). We also analyzed the remaining 12 fractions (24 in total) and a total of over 10,000 proteins were quantified and of them more than 8,600 were quantified across all 48 samples using only ~2.2 h per proteome with no missing values (Supplementary Fig. 8a and Supplementary Table 1). A mean coefficient of variation of less than 6% was observed for all 16 conditions (Supplementary Fig. 8b). Unsupervised hierarchical clustering analysis (HCA) and principal component analysis (PCA) illustrated that samples were quantified without measurable bias or batch effect (Fig. 3d,e).
SQSTM1 is an autophagy receptor and recruits cargo for degradation19,20. MAP1LC3B2 and GABARAPL1 are conjugated to autophagosomal membranes and recognize/recruit SQSTM1 (refs. 19,20). These proteins are degraded along with autophagosomal contents on fusion with the lysosome; reduction of SQSTM1, as well as other autophagy receptors following 10 h Torin1 treatment has recently been demonstrated in 293T cell samples21. We successfully captured Torin1-dependent downregulation of SQSTM1 and autophagosome-conjugated ATG8 proteins in our 16plex analysis (Fig. 3f). In total, protein expression was reproducible in the replicates but varied widely across cell lines (Supplementary Fig. 8c). Thousands of different protein expression profiles were measured in this experiment exhibiting differences due to either cell line, Torin1 treatment or both (Supplementary Fig. 8c–e). Gene ontology analysis of the 1,916 protein changes specific to Torin1 treatment showed that downregulated proteins were enriched in ribosome, cell growth and translation-related pathways and structures (Supplementary Fig. 8e). Conversely, upregulated proteins were enriched in lysosome, catabolic process and extracellular vesicles (Supplementary Fig. 8e). These results agreed with known biology that inhibition of the mTOR pathway leads to cell growth and translation arrest, and induction of autophagy increases lysosome-related biological function and catabolism16,17.
Additionally, the 12 fractions from replicate 1 were reanalyzed using hrMS2 on a Q-Exactive HF-X instrument and SPS–MS3 on an Orbitrap Lumos instrument (Supplementary Fig. 9a and Supplementary Table 2). TMTpro reagents performed well with both methods, and presented linearly correlated quantitation between hrMS2, SPS–MS3 and RTS–SPS–MS3 methods (Supplementary Fig. 9). Similar to classic TMT reagents, TMTpro reagents showed ratio compression with hrMS2 (Supplementary Fig. 9). Compared with hrMS2 and RTS–SPS–MS3, SPS–MS3 quantified the least number of proteins (Supplementary Fig. 9a). Doubling the gradient time enabled SPS–MS3 to quantify a comparable number of proteins (Supplementary Fig. 9a). These data indicate that TMTpro performs well on platforms without RTS–SPS–MS3 capabilities.
The ‘mini-phos’ analysis enriched phosphopeptides using just 100 μg of protein input per channel. We localized and quantified more than 7,000 phosphorylation events in each replicate, 4,785 phosphorylation sites in all replicates and 9,566 sites overall (Supplementary Fig. 10a,b and Supplementary Table 3). Quantified phosphorylation sites were highly consistent among replicates, showing a median coefficient of variation of ~6% (Supplementary Fig. 10c). Greater than 99% of all sites also had corresponding protein expression measurements (Supplementary Fig. 10d), thereby improving the interpretability of phosphorylation site changes22. HCA and PCA showed that phosphorylation sites were quantified without obvious bias or batch effect (Supplementary Fig. 10e,f). Serine 1859 from CAD and serine 47 from DNAJC2 are known substrates of the mTOR pathway23,24. Phosphorylation of CAD S1859 was downregulated in all eight cell lines, and phosphorylation of DNAJC2 S47 was downregulated in five cell lines (Supplementary Fig. 10g). These protein and phosphorylation datasets serve as resources for the (phospho)proteome-wide consequences of mTOR inhibition by Torin1 across a wide panel of cell lines.
TMTpro reagents facilitated the complex two-dimensional (2D) proteome integral stability alteration (PISA) assay.
As a final example, we modified the recently presented PISA assay25 using TMTpro reagents. Thermal proteome profiling (TPP) is a method for de novo discovery of ligand-target protein interactions26. In a classic TPP assay, protein abundance across ten different temperatures is measured to generate protein melting curves and calculate melting points. Two TMT experiments are required to compare the melting curves/points between control- and ligand-treated lysates26. In a PISA assay, samples across an entire temperature gradient are combined25. The protein abundance in the pooled sample, which represents the area under the melting curve, is used to measure the effect of a ligand on protein thermal stability25. The sample pooling step in the PISA assay massively increases throughput (by about tenfold), as the ligand-treated and control samples can be compared in a single TMT experiment with up to five replicates. While a single TMT10plex can accommodate the five versus five arrangement, if two concentrations of a ligand are to be compared in a PISA assay, two TMT10plex experiments must be performed with the control as the bridge between experiments. However, using TMTpro, we can accommodate the control and both concentrations in a single 15plex experiment, thereby requiring 50% less handling and data acquisition time and resulting in no missing values across concentrations (Fig. 4 and Supplementary Fig. 11).
Fig. 4|. Application of TMTpro to a two-dimensional PISA assay.
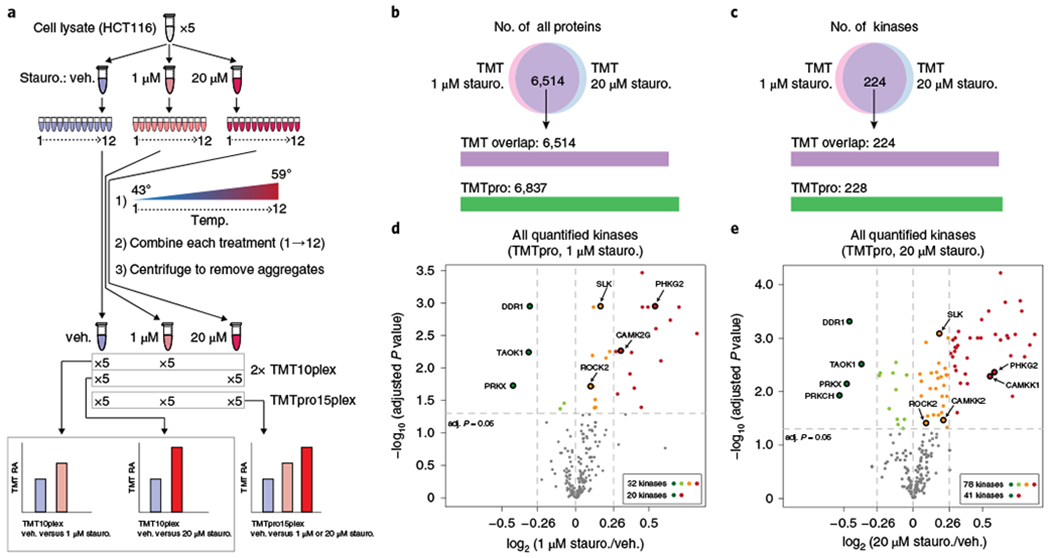
a, Workflow of sample preparation. HCT116 cell lysate was divided into three aliquots and treated with the broad-spectrum protein kinase inhibitor staurosporine (stauro.). The experiment was carried out in quintuplicate. Each sample was subsequently divided into 12 tubes and a heat gradient was applied. An equal volume from each tube was pooled, centrifuged, digested and labeled with the first ten channels of TMT or the first 15 channels of TMTpro. Soluble protein abundance in vehicle (veh.) and staurosporine-treated samples, which represents the area under the melting curve, were used to measure the effect of staurosporine on protein thermal stability. b,c, TMTpro-labeling quantified proteins (b) and kinases (c) with no missing values for the PISA assay. d,e, Volcano plots of all quantified kinases under staurosporine treatment (TMTpro). Thermal stabilities of 32 kinases were affected by 1 μM staurosporine and 20 showed ≥20% AUC changes (log2 ratio cutoff 0.26; d). Seventy-eight kinases were affected by 20 μM staurosporine and 41 showed ≥20% AUC changes (e). Destabilized kinases (dark green) and top five kinases showing high affinity to staurosporine among stabilized kinases (from database ‘The IUPHAR/BPS Guide to PHARMACOLOGY’) are labeled (n = 5 cell culture replicates, two-sided paired t-test, P values adjusted by the Benjamini-Hochberg method among all quantified kinases and filtered at 0.05).
We treated HCT116 cell lysate with DMSO, 1 or 20 μM staurosporine (a broad-spectrum kinase inhibitor) in quintuplicate and performed the PISA assay (Fig. 4a–c and Supplementary Table 4). Using TMTpro, we determined that the thermal stabilities of 32 and 78 kinases were significantly shifted using 1 and 20 μM staurosporine, respectively (Fig. 4d,e). Nearly all of the kinases affected by 1 μM staurosporine were also significantly altered by 20 μM staurosporine (Supplementary Fig. 11b). An inhibitor must first bind the target to modify its thermal stability. We found that kinases with altered abundance showed higher affinity to staurosporine among 442 screened human kinases27 (Supplementary Fig. 11c). Higher multiplexing facilitated a 2D-PISA (dose and temperature) assay similar to 2D-TPP experiments28,29, yet our analysis included five replicates of each treatment. Other arrangements are possible. For example, if two replicates are used, a combination of eight doses and/or compounds could be examined in just one experiment. In addition, we analyzed the TMTpro-labeled samples in this experiment using hrMS2 on a Q-Exactive HF-X instrument. Overall results were consistent between hrMS2 and RTS–SPS–MS3. hrMS2 quantified more kinases, but ratio compression resulted in a reduced number of significantly changing kinases (Supplementary Fig. 12 and Supplementary Table 5).
Discussion
The TMTpro reagents represent a powerful starting point for even further improvements to sample multiplexing in proteomics. For example, in combination with metabolic labeling using stable isotopes, TMTpro could examine turnover rates of newly synthesized proteins in greater depth than reported previously30,31, and TMTpro can also be extended easily to even higher order sample multiplexing, often termed hyperplexing32,33. Subcellular resolution of spatial proteomics can be greatly increased with TMTpro reagents as well34. For lower resolution instruments (Q-TOF, quadrupole or ion trap), nine unit-resolved reagents are available within the TMTpro set.
We emphasize that sample multiplexing in proteomics extends beyond simply reducing analysis time (increasing throughput) and thus an 11-to-16plex increase has profound consequences. Each multiplexed experiment represents essentially a closed system (that is, a biological assay) where each protein quantification event extends across all channels with no missing values. Inside the closed system, several noteworthy advantages emerge: (1) Proteome depth is remarkable due to fractionation often surpassing 8,000 proteins quantified from cells or tissues; (2) stable isotope labeling directly empowers multiplexing while at the same time providing highly accurate relative quantification; (3) statistical inference can be applied with greater power across the entire system; and (4) complex experimental designs can be accommodated (for example, an expanded number of treatments and replicates can be arranged in a single TMTpro experiment). As such, replicates, dose-response and time-course measurements, positive and negative controls can all be analyzed within the same experiment, at true proteome scale, with stronger statistical power and basically no missing values across all samples. In summary, this next generation of TMTs, TMTpro, will drive the field of quantitative proteomics to unprecedented multiplexing heights and cellular proteome depths.
Methods
Mammalian cell culture/cell extract preparation.
HeLa, 293T, HCT116, U2OS, RPE1 and MCF7 cells were purchased from ATCC. Panel cells were a gift from J. Mancias (Dana-Farber Cancer Institute), and RKO cells were a gift from K. Haigis (Beth Israel Deaconess Medical Center). Cells were maintained in DMEM with high glucose/pyruvate (Invitrogen) supplemented with 10% FBS (Hyclone). For MCF7 cultures, human insulin (Sigma) was added to a final concentration of 0.01 mg ml−1. RKO cultures were grown in RPMI1640 (Thermo) supplemented with 10% FBS. All cell lines were maintained in a 5% CO2 incubator at 37 °C. For Torin1 treatment, cells were grown to approximately 60% confluence in biological triplicate 10 cm plates, followed by 12 h treatment with DMSO (vehicle) or 150 nM Torin1 (Cell Signaling Technology). Cells were washed twice with cold PBS, then collected and lysed by scraping into 8 M urea lysis buffer (8 M urea, 200 mM EPPS, pH 8.5) with protease inhibitors (Complete Mini, Roche). Cell extracts were homogenized by vortexing, incubated on ice for 10 min, then diluted to 4 M urea with 200 mM EPPS. Extracts were treated with 20 unit ml−1 Benzonase (Millipore) for 15 min to disrupt DNA/chromatin and snap frozen. Protein concentration was determined with BCA assays for subsequent LC–MS sample preparation. For the PISA assay, HCT116 cells in a 15 cm dish were collected at approximately 80% confluency using trypsin, washed once with PBS and frozen in liquid nitrogen.
Western blotting.
Twenty micrograms of protein from the cell extract preparation described above (DMSO or Torin1-treated) were denatured in NuPAGE LDS sample buffer (Invitrogen) supplemented with 100 mM DTT, followed by SDS–PAGE using 4–12% Bis-Tris gels in MOPS running buffer (Invitrogen). Proteins were transferred to polyvinylidene fluoride membranes (0.45 mm Millipore), blocked with 5% milk in TBST (30 min) and incubated with primary antibodies overnight at 4 °C according to the manufacturer’s recommended dilutions. Anti-Phospho-4E-BP1 (Thr37/46) and anti-4E-BP1 were from Cell Signaling Technology (catalog nos. 2855, 9644). Anti-β-Actin was from Santa Cruz Biotechnology (catalog no. sc-69879). Following primary antibody incubation, membranes were washed three times with TBST, then incubated in secondary antirabbit or antimouse conjugated to horseradish peroxidase (Promega) for 30 min (1:5,000 dilution). Membranes were washed four times and signals were developed using enhanced chemiluminescence (Western Lightning ECL, Perkin Elmer).
PISA assay.
Cell pellets were thawed on ice briefly and resuspended in lysis buffer (1× PBS, 1 mM MgCl2, 0.5% NP-40, and protease inhibitor cocktail, pH 7.2). Cells were incubated for 15 min at 4 °C with gentle agitation. Lysates were cleared by centrifugation at 20,000g for 15 min. The soluble fraction was collected and protein concentration was determined by BCA assays (Pierce). Each lysate was diluted to 2 mg ml−1 in lysis buffer and divided into three aliquots. Each aliquot was mixed in a 1:1 ratio with lysis buffer with DMSO (vehicle), lysis buffer with 2 μM staurosporine (Sigma), or lysis buffer with 40 μM staurosporine to yield three reactions with a final protein concentration of 1 mg ml−1 and a final staurosporine concentration of 0 (vehicle), 1 or 20 μM, respectively. Reactions were incubated at room temperature for 10 min with gentle shaking. An equal volume from each reaction was distributed into 12 PCR tubes. Samples were heated in a thermocycler for 3 min across a temperature gradient from 43 to 59 °C (43.3, 43.6, 44.6, 46.2, 48.1, 50.2, 52.4, 54.5, 56.3, 57.8, 58.8 and 59 °C). Following heating, samples were equilibrated at room temperature for 5 min. An equal volume was removed from each PCR tube and pooled. These samples were centrifuged at maximum speed (~21,000g) for 2 h to remove aggregated proteins. An equal volume of the resulting soluble fraction (~75 μg protein) was collected and mixed with an equal volume of buffer comprising 400 mM EPPS, pH 8.5 and 2% SDS to yield a final concentration of 200 mM EPPS, 1% SDS, pH 8.5.
MS sample preparation.
Samples were reduced with 5 mM TCEP for 30 min, alkylated with 10 mM iodoacetamide for 30 min and then quenched with 10 mM DTT for 15 min. Streamlined–TMT18 and SP3 (ref. 35) protocols were used as a basis for cell line and PISA sample preparation, respectively. TMT and TMTpro11 (ThermoFisher Scientific) reagents were reconstituted with anhydrous acetonitrile and stored in 50 μl frozen aliquots at −80 °C. We typically used a kit within a month and finish each aliquot within three freeze–thaw cycles without observing a decrease in labeling efficiency.
TMT0- and TMTpro0-labeling of SH-SY5Y cell lysate.
One hundred micrograms of protein from syringe-lysed SH-SY5Y whole cell lysate were chloroform-methanol precipitated and reconstituted in 100 μl of 200 mM EPPS (pH 8.5). The sample was digested by Lys-C overnight at room temperature and then by trypsin for 6 h at 37 °C, both at a 1:100 protease-to-protein ratio. Fifty-eight micrograms of TMT0 or TMTpro0 (3 μl) and 7 μl of 100% anhydrous acetonitrile were added to 25 μg aliquots of peptides (25 μl). Samples were labeled for 60 min at room temperature and quenched with 3 μl of 5% hydroxylamine. Samples were acidified and desalted by StageTip before LC–MS/MS analysis.
Isobaric labeling of 11plex cell line samples.
One hundred micrograms of protein from each sample were chloroform-methanol precipitated and reconstituted in 100 μl of 200 mM HEPES (pH 8.5). Samples were digested by Lys-C overnight at room temperature and then by trypsin for 6 h at 37 °C, both at a 1:100 protease-to-protein ratio. Fifty-eight micrograms of TMTpro reagents (3 μl) and 7 μl of 100% anhydrous acetonitrile were added to each digest corresponding to 25 μg of protein (25 μl) (labels used: 126: 293T DMSO (Vehicle, V) Rep1; 127N: 293T V Rep2; 127C: 293T V Rep3; 128N: 293 Torin1 (T) Rep1; 128C: 293T Rep2; 129N: 293T T Rep3; 129C: HCT116 V Rep1; 130N: HCT 116V Rep2; 130C: HCT116 V Rep3; 131N: HCT116 T Rep1; 131C: HCT116 T, Rep2). Fifty-eight micrograms of TMT11 reagents (3 μl) and 7 μl of 100% anhydrous acetonitrile were added to another aliquot of digest, each corresponding to 25 μg of protein (25 μl; labels used: 126: 293T DMSO (Vehicle, V) Rep1; 127N: 293T V Rep2; 127C: 293T V Rep3; 128N: 293 Torin1 (T) Rep1; 128C: 293T Rep2; 129N: 293T T Rep3; 129C: HCT116 V Rep1; 130N: HCT116 V Rep2; 130C: HCT116 V Rep3; 131: HCT116 T Rep1, 131C: HCT116 T, Rep2). Samples were labeled for 60 min at room temperature. To check labeling efficiency, 2 μl (5.7% v/v) of each sample were pooled, desalted and analyzed by MS. After the labeling efficiency check, samples were quenched, pooled and desalted for subsequent LC–MS/MS analysis.
TMTpro16plex labeling of 48 cell line samples.
One hundred micrograms of protein from each sample were TCA-precipitated. After washes with acetone and methanol, the protein pellet was reconstituted in 100 μl of 200 mM HEPES (pH 8.5). Samples were digested by Lys-C overnight at room temperature and then trypsin for 6 h at 37 °C, both at a 1:100 protease-to-protein ratio. To each digest 234 μg of TMTpro16plex reagents (12 μl) and 30 μl of 100% anhydrous acetonitrile were added for labeling (labels used: 126: 293T Vehicle (V); 127N: 293T Torin1 (T); 127C: RPE1 V; 128N: RPE1 T; 128C: U2OS V; 129N: U2OS T; 129C: HeLa V; 130N: HeLa T; 130C: MCF7 V; 131N: MCF7 T; 131C: HCT116 V; 132N: HCT116 T; 132C: Panc1 V; 133N: Panc1 T; 133C: RKO V; 134N: RKO T). Samples were labeled for 60 min at room temperature. Two microliters (1.4% v/v) of each sample were pooled, desalted and analyzed by MS to check labeling efficiency. After labeling efficiency check, samples were quenched by adding 5 μl of 5% hydroxylamine and pooled. Pooled samples were then desalted with 200 mg Sep-Pak solid-phase extraction columns. Pierce High-Select Fe-NTA phosphopeptide enrichment kit was used to enrich phosphopeptides from the pooled mixture. The unbound fraction and column washes were combined, desalted and then fractionated with bRPLC. Fractions were collected in a 96-well plate and combined for a total of 24 fractions (A set and B set) before desalting and subsequent LC–MS/MS analysis.
Isobaric labeling of PISA samples.
The reduced and alkylated proteins were bound to SP3 beads, washed three times with 80% ethanol and subjected to on-bead digestion overnight at 37 °C in 200 mM EPPS, pH 8.5 while shaking with Lys-C protease at a 1:100 protease-to-protein ratio. Trypsin was added to a 1:100 protease-to-protein ratio and the samples were incubated for 6 h at 37 °C while shaking. The beads were removed from the samples and anhydrous acetonitrile was added to a final concentration of around 30%. Roughly 75 μg of peptides were labeled with ~150 μg of TMT or TMTpro in the presence of ~28% acetonitrile at room temperature for 60 min. The labeled peptides were then quenched with hydroxylamine, pooled and desalted by Sep-Pak (Waters). Samples were dried, resuspended in 5% acetonitrile and 10 mM ammonium bicarbonate, pH 8 and subjected to bRPLC. Fractions were collected in a 96-well plate and combined for a total of 24 fractions (A and B set) before desalting and subsequent LC–MS/MS analysis of nonadjacent 12 fractions (A or B set).
MS analysis.
Data for stability test for TMTpro0- and TMT0-labeled peptides were collected using a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (ThermoFisher Scientific) coupled with a Famos Autosampler (LC Packings) and an Accela600 LC pump (ThermoFisher Scientific). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ~20 cm of Accucore C18 resin (2.6 μm, 150 Å, ThermoFisher Scientific). For each analysis, we loaded ~1 μg onto the column. Peptides were separated using a 120 min gradient of 3 to 25% acetonitrile in 0.125% formic acid with a flow rate of ~300 nl min−1. The scan sequence began with an Orbitrap MS1 spectrum with the following parameters: resolution 70,000, scan range 300–1,500 Th, automatic gain control (AGC) target 1 × 106, and maximum injection time 250 ms. We selected the top 20 precursors for MS2 analysis that consisted of HCD with the following parameters: resolution 35,000, AGC 1 × 106, maximum injection time 100 ms, isolation window 1.6 Th, and normalized collision energy (NCE) 32%. The minimum AGC target was set at 2 × 103, which corresponds to a 2 × 104 intensity threshold. In addition, unassigned, singly and >8+ charged species were excluded from MS2 analysis and dynamic exclusion was set to automatic.
The isobaric labeled 11plex cell line samples, as well as the figures of merit evaluation samples, were analyzed on an Orbitrap Lumos mass spectrometer coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific) using the standard SPS–MS3 method12. Peptides were separated on a 35 cm column (inner diameter (i.d.) 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house. Samples were separated using a 150 min gradient (11plex-labeled cell line samples) or a 45 min gradient (samples for figures of merit evaluation) at 550 nl min−1. MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 1 × 106). Determined charge states between 2 and 5 were required for sequencing and a 120 s dynamic exclusion window was used. Data-dependent ‘top 10’ MS2 scans were performed in the ion trap with collision-induced dissociation (CID) fragmentation (Turbo; NCE 35% for TMT and 34% for TMTpro; maximum injection time 35 ms; AGC 1 × 104). MS3 quantification scans were performed using the multi-notch MS3-based TMT method12 (ten SPS ions; 50,000 resolution; NCE 65% for TMT and 45% for TMTpro for 11plex-labeled cell line samples; four NCEs as indicated in Fig. 2 for samples for figures of merit evaluation; maximum injection time 85 ms; AGC 2e5).
Data for TMTpro16plex-labeled 48 cell line samples were collected on an Orbitrap Lumos mass spectrometer (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific). Peptides were separated on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house using a 90 min gradient (0–2 min: 3–8% acetonitrile with 0.1% formic acid, 2–5 min: 8–10%, 5–70 min: 10–25%, 70–80 min: 25–30%, 80–85 min: 30–100%, 85–90 min: 100%) at 575 nl min−1. MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 4e5). Determined charge states between 2 and 4 were required for sequencing and a 120 s dynamic exclusion window was used. MS2 scans were performed in the ion trap with CID fragmentation (isolation window 0.7 Da; Turbo; NCE 35%; maximum injection time 35 ms; AGC 2 × 104). An online RTS algorithm was used to trigger MS3 scans for quantification14. MS3 scans were collected in the Orbitrap using a resolution of 50,000, NCE of 45%, maximum injection time of 86 ms and AGC of 1.5 × 105. The closeout was set at two peptides per protein per fraction, so that MS3s were no longer collected for proteins having two peptide-spectrum matches (PSMs) that passed quality filters14.
Phosphoproteomic samples for the TMTpro16plex-labeled 48 cell line experiment, were injected twice on an Orbitrap Lumos mass spectrometer (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific). Phosphopeptides were separated on a 35 cm column (i.d. 100 μm) packed in-house with reversed-phase materials (Accucore, 2.6 μm, 150 Å) using a 180 min gradient (0–2 min: 2–5% acetonitrile with 0.1% formic acid, 2–5 min: 5–6%, 5–140 min: 6–18%, 140–160 min: 18–22%, 160–170 min: 22–28%, 170–175 min: 28–100%, 175–180 min: 100%) at 500 nl min−1. MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 1 × 106). Determined charge states between 2 and 5 were required for sequencing and a 150 s dynamic exclusion window was used. Data-dependent ‘top 10’ MS2 scans were performed in the ion trap with HCD fragmentation (Rapid; NCE 30%; maximum injection time 60 ms; AGC 1 × 104) or CID fragmentation with MSA (Turbo; NCE 34%; maximum injection time 60 ms; AGC 1 × 104). MS3 quantification scans were performed using multi-notch MS3-based TMT method12 (ten SPS ions; 50,000 resolution; NCE 45%; maximum injection time 100 ms; AGC 1.5 × 105).
Data for PISA assay samples were collected on an Orbitrap Lumos mass spectrometer (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific). Peptides were separated on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house using a 90 min gradient (0–2 min: 7–8% acetonitrile with 0.1% formic acid, 2–5 min: 8–9%, 5–70 min: 9–18%, 70–80 min: 18–26%, 80–85 min: 26–100%, 85–90 min: 100%) at 500 nl min−1. MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 4 × 105). Determined charge states between 2 and 5 were required for sequencing and a 60 s dynamic exclusion window was used. MS2 scans were performed in the ion trap with CID fragmentation (isolation window 0.7 Da; Turbo; NCE 34%; maximum injection time 60 ms; AGC 1 × 104). An online RTS algorithm was used to trigger MS3 scans for quantification14. MS3 scans were collected in the Orbitrap using a resolution of 50,000, NCE of 45%, maximum injection time of 86 ms and AGC of 1.5 × 105. The closeout was set at two peptides per protein per fraction, so that MS3s were no longer collected for proteins having two PSMs that passed quality filters14.
High-resolution MS2 (hrMS2) analyses were performed on a Q-Exactive HF-X (ThermoFisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ~25 cm of Accucore C18 resin (2.6 μm, 150 Å, ThermoFisher Scientific). Peptides were separated using a 90 min gradient of 9 to 29% acetonitrile in 0.125% formic acid with a flow rate of ~600 nl min−1. The scan sequence began with an Orbitrap MS1 spectrum with the following parameters: resolution 120,000, scan range 350–1,400 Th, AGC target 3 × 106 and maximum injection time 50 ms. We selected the top 20 precursors for MS2 analysis that consisted of HCD with the following parameters: resolution 45,000, AGC 1 × 105, maximum injection time 96 ms, isolation window 0.5 Th and NCE of 30%. The minimum AGC target was set at 1 × 104. In addition, unassigned, singly and >5+ charged species were excluded from MS2 analysis and dynamic exclusion was set to 60 s.
SPS–MS3 analyses of the replicate 1 of the Torin1-treated samples were performed on an Orbitrap Lumos mass spectrometer coupled to a Proxeon EASY-nLC 1200 LC pump (ThermoFisher Scientific) using the standard SPS–MS3 method12. Peptides were separated on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house. Samples were analyzed twice, once using a 90 min and a second injection using a 180 min gradient at 520 nl min−1. MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 4e5). Determined charge states between 2 and 5 were required for sequencing and a 120 s dynamic exclusion window was used. Data-dependent ‘top 10’ MS2 scans were performed in the ion trap with CID fragmentation (Turbo; NCE 35%; maximum injection time 35 ms; AGC 1 × 104). MS3 quantification scans were performed using the multi-notch MS3-based TMT method12 (ten SPS ions; 50,000 resolution; NCE 45%; maximum injection time 86 ms; AGC 2 × 105).
MS data analysis.
Raw files were first converted to mzXML. Database searching included all human entries from UniProt (downloaded on 4 February 2014). The database was concatenated with one composed of all protein sequences in reverse order. Sequences of common contaminant proteins (for example, trypsin, keratins and so on) were appended as well. Searches were performed using a 50 ppm precursor ion tolerance and 0.9 Da (low-resolution MS2) or 0.03 Da (high-resolution MS2) product ion tolerance. TMT or TMTpro on lysine residues and peptide N termini (+229.1629 Da for TMT, 224.1525 Da for TMT0, 304.2071 Da for TMTpro and 295.1896 Da for TMTpro0) and carbamidomethylation of cysteine residues (+57.0215 Da) were set as static modifications (except when testing for labeling efficiency, in which the TMT or TMTpro modifications are set to variable), while oxidation of methionine residues (+15.9949 Da) was set as a variable modification. For phosphoprotein analysis, +79.9663 Da was set as a variable modification on serine, threonine and tyrosine residues.
PSMs were adjusted to a 1% false discovery rate (FDR)36. PSM filtering was performed using linear discriminant analysis as described previously37, while considering the following parameters: XCorr, ΔCn, missed cleavages, peptide length, charge state and precursor mass accuracy. Each run was filtered separately. Protein-level FDR was subsequently estimated. For each protein across all samples, the posterior probabilities reported by the linear discriminant analysis model for each peptide were multiplied to give a protein-level probability estimate. Using the Picked FDR method38, proteins were filtered to the target 1% FDR level. Phosphorylation site localization was determined using the AScore algorithm39. A threshold of 13 corresponded to 95% confidence that a given phosphorylation site was localized.
For reporter ion quantification, a 0.003 Da window around the theoretical m/z of each reporter ion was scanned, and the most intense m/z was used. Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMT or TMTpro reagents according to the manufacturer’s specifications (Supplementary Table 6). Briefly, the correction factors are applied to the raw peak intensity before calculating signal-to-noise. The correction is done by arranging a system of linear equations where the unknowns are the corrected channel intensities, their coefficients are the correction factors and the constant terms are the observed peak intensities. A solution is found by solving the system using matrix operations. Peptides were filtered to include only those peptides with a summed signal-to-noise ratio of ≥110 across 11 TMT11plex channels or ≥160 across 16 TMTpro16plex channels. An isolation purity of at least 0.7 (70%) in the MS1 isolation window was used for samples analyzed without online real-time searching or samples analyzed with hrMS2. For each protein, the filtered peptide TMT or TMTpro signal-to-noise values were summed to create protein quantification values. To control for different total protein loading within a TMT or TMTpro experiment, the summed protein quantities of each channel were adjusted to be equal in the experiment. Phosphosite quantifications were also normalized by correction factors generated in this process to account for protein loading variance. For each protein in a TMTpro experiment, the signal-to-noise was scaled to sum to 100 to enable comparisons across experiments, coefficient of variation calculation, HCA, PCA, paired t-test and two-way analysis of variance.
Bioinformatics analysis.
Two-way analysis of variance was performed in R 3.4.2 to assess the effects of cell type, treatment, interaction of cell type and treatment on protein abundance. P values were adjusted with Benjamini-Hochberg method and filtered at 1 × 10−5. Partial eta squared (partial η2), which measures the ratio of the variance explained by an effect and that effect plus its associated error variance, was reported as effect size and filtered at 0.7. Gene ontology analysis was performed with DAVID (v.6.8)40 with all quantified proteins used as the background. Other data analyses were also conducted in R v.3.4.2. Please refer to the Nature Research Reporting Summary for additional information.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank members of the Gygi Laboratory, particularly R. Rad, at Harvard Medical School. This work was funded in part by NIH grant nos. 1R01GM132129 (to J.A.P.) and 5R01GM067945 (to S.P.G), and the Mark Foundation for Cancer Research Fellow of the Damon Runyon Cancer Research Foundation DRG 2359-19 (to J.G.V.V.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592-020-0781-4.
Data availability
The MS data have been deposited in the ProteomeXchange Consortium with the dataset identifier PXD014369, PXD016491 and PXD016940. The list of human kinases was downloaded from UniProt on 9 June 2019 (https://www.uniprot.org/docs/pkinfam). Affinity data of 442 human kinases to staurosporine were downloaded from the database ‘The IUPHAR/BPS Guide to Pharmacology’ on 14 May 2019 (https://www.guidetopharmacology.org/).
Competing interests
The TMTpro reagents were commercialized by ThermoFisher Scientific in September 2019. C.E., P.N., R.V., A.M.R., R.D.B. and J.C.R. are employees of ThermoFisher Scientific. A.H.T. and I.P. were employees of Proteome Sciences. K.K. is an employee of Proteome Sciences. S.P.G. is a member of the scientific advisory board for ThermoFisher Scientific.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41592-020-0781-4.
Peer review information Allison Doerr was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
References
- 1.Li H et al. Current trends in quantitative proteomics—an update. J. Mass Spectrom 52, 319–341 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Pappireddi N, Martin L & Wuhr M A review on quantitative multiplexed proteomics. Chem. Bio. Chem 20, 1210–1224 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson A et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem 75, 1895–1904 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Rauniyar N & Yates JR 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res 13, 5293–5309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vasaikar S et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049 e1019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chick JM et al. Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayon L et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem 80, 2921–2931 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Paulo JA et al. Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics. Proteomics 15, 462–473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stepanova E, Gygi SP & Paulo JA Filter-based protein digestion (FPD): a detergent-free and scaffold-based strategy for TMT workflows. J. Proteome Res 17, 1227–1234 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McAlister GC et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem 84, 7469–7478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson A et al. TMTpro: design, synthesis, and initial evaluation of a proline-based isobaric 16-plex tandem mass tag reagent set. Anal. Chem 91, 15941–15950 (2019). [DOI] [PubMed] [Google Scholar]
- 12.McAlister GC et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erickson BK et al. Active instrument engagement combined with a real-time database search for improved performance of sample multiplexing workflows. J. Proteome Res 18, 1299–1306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schweppe DK et al. Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. Preprint at bioRxiv 10.1101/668533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J & Guan KL mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol 21, 63–71 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Saxton RA & Sabatini DM mTOR Signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Thoreen CC et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem 284, 8023–8032 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarrete-Perea J, Yu Q, Gygi SP & Paulo JA Streamlined tandem mass tag (SL-TMT) protocol: an efficient strategy for quantitative (phospho) proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res 17, 2226–2236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizushima N A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol 20, 521–527 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Dikic I & Elazar Z Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol 19, 349–364 (2018). [DOI] [PubMed] [Google Scholar]
- 21.An H et al. TEX264 Is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol. Cell 74, 891–908 e810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu R et al. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol. Cell Proteom 10, M111 009654 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Sahra I, Howell JJ, Asara JM & Manning BD Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barilari M et al. ZRF1 is a novel S6 kinase substrate that drives the senescence programme. EMBO J 36, 736–750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaetani M et al. Proteome integral solubility alteration: a high-throughput proteomics assay for target deconvolution. J. Proteome Res 18, 4027–4037 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Savitski MM et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Harding SD et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46, D1091–D1106 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becher I et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol 12, 908–910 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Dai L et al. Horizontal cell biology: monitoring global changes of protein interaction states with the proteome-wide cellular thermal shift assay (CETSA). Annu. Rev. Biochem 88, 383–408 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Savitski MM et al. Multiplexed proteome dynamics profiling reveals mechanisms controlling protein homeostasis. Cell 173, 260–274 e225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zecha J et al. Peptide level turnover measurements enable the study of proteoform dynamics. Mol. Cell Proteom 17, 974–992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dephoure N & Gygi SP Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signal 5, rs2 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hebert AS et al. Neutron-encoded mass signatures for multiplexed proteome quantification. Nat. Methods 10, 332–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulvey CM et al. Using hyperLOPIT to perform high-resolution mapping of the spatial proteome. Nat. Protoc 12, 1110–1135 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Hughes CS et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc 14, 68–85 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Elias JE & Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Huttlin EL et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savitski MM, Wilhelm M, Hahne H, Kuster B & Bantscheff M A scalable approach for protein false discovery rate estimation in large proteomic data sets. Mol. Cell Proteom 14, 2394–2404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beausoleil SA, Villen J, Gerber SA, Rush J & Gygi SP A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 24, 1285–1292 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.