

Abstract
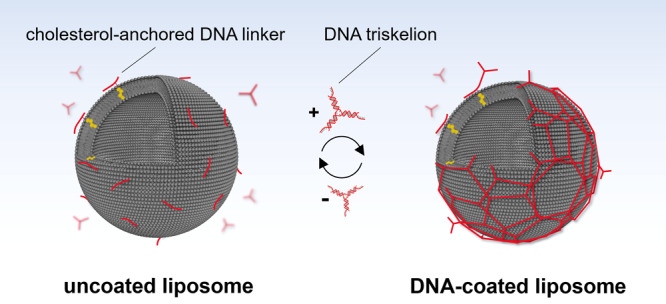
The self-assembly of the protein clathrin on biological membranes facilitates essential processes of endocytosis and has provided a source of inspiration for materials design by the highly ordered structural appearance. By mimicking the architecture of the protein building blocks and clathrin self-assemblies to coat liposomes with biomaterials, advanced hybrid carriers can be derived. Here, we present a method for fabricating DNA-coated liposomes by hydrophobically anchoring and subsequently connecting DNA-based triskelion structures on the liposome surface inspired by the assembly of the protein clathrin. Dynamic light scattering, ζ-potential, confocal microscopy, and cryo-electron microscopy measurements independently demonstrate successful DNA coating. Nanomechanical measurements conducted with atomic force microscopy show that the DNA coating enhances the mechanical stability of the liposomes relative to uncoated ones. Furthermore, we provide the possibility to reverse the coating process by triggering the disassembly of the DNA coats through a toehold-mediated displacement reaction. Our results describe a straightforward, versatile, and reversible approach for coating and stabilizing lipid vesicles through the assembly of rationally designed DNA structures. This method has potential for further development toward the ordered arrangement of tailored functionalities on the surface of liposomes and for applications as hybrid nanocarriers.
Keywords: DNA nanotechnology, biomimetics, liposome, clathrin, atomic force microscopy, cryo-electron microscopy
The protein clathrin plays a major role in cell trafficking by mediating endocytosis.1 In its monomeric form, clathrin has the shape of a triskelion. Upon an external stimulus, it forms spherical complexes surrounding a membrane vesicle with an alternating pattern of hexagons and pentagons.2,3 In addition to its biological significance, the structural elegance of clathrin has inspired the development of artificial functional materials that mimic clathrin lattices and their self-assembly.4−6
DNA is increasingly exploited as a building material for predictable and precise assembly of various structures on the nanoscale.7−10 DNA nanostructures can be conceived by specifically designing the individual nucleotide sequences, which then form unique shapes aided by the Watson–Crick base-pairing interactions. Furthermore, the choice of DNA as a material allows for multiple biomedical applications due to essential characteristics, such as water solubility and biodegradability.11,12 The conjugation of DNA and liposomes has enabled their use as carriers13,14 or artificial systems imitating the structure or functionality of certain membrane proteins.15−21 Many of the so far presented approaches of DNA structures anchored to the liposome surface use spatially uncontrolled attachment.20,22,23 The arrangement of DNA structures to supramolecular DNA lattices is still mainly restricted to 2D supported lipid bilayers.4,24−27 Larger arrangements of DNA assemblies from spatially defined subunits on the surface of a liposome have predominantly employed DNA origami structures.4,28−30 For instance, DNA origami-based rectangles4 as well as curved DNA origami nanoconstructs29,30 have been interconnected on lipid vesicles. Yet, such DNA origami networks were shown to produce liposome deformation or even destruction, which potentially limits the applicability for purposes such as the transport of cargo in vesicles. Moreover, the use of DNA origami constructs in order to achieve dense vesicle coating is a more complex synthetic approach compared to other DNA assemblies based on DNA tiles or junctions.
This study presents a straightforward method of coating and stabilizing nanoscale lipid vesicles with a semirigid DNA network, based on the connection of three-arm branched DNA junctions inspired by the structure of the clathrin triskelion.
Results and Discussion
Large unilamellar vesicles (LUVs) were prepared by extrusion of a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid suspension in phosphate-buffered saline (PBS) solution through a 200 nm pore size membrane.31 To facilitate coating of the vesicles while reducing DNA aggregation in bulk, a two-step annealing process was applied (Figure 1a). First, LUVs (from now on defined as V) were incubated overnight at room temperature with DNA linkers labeled with a cholesterol tag through a triethylene glycol spacer (Chol-TEG) to render structures referred to as VL (Figure 1a). These linkers were composed of an 18 nucleotide (nt) long DNA duplex containing 18 nt long overhangs on both sides (Figure 1b; Supporting Information Table S1). In the second step, a DNA triskelion (Figure 1c,d) was added to the VL structures to achieve the final construct VLT (Figure 1a) through complementary hybridization with the overhangs of the linker via incubation at 4 °C for 50 min. The triskelion consisted of three DNA strands forming a three-arm branched junction (Figure 1c). Three types of triskelion nanostructures (T1, T2, and T3) were prepared, which differed by the number of unpaired bases (1, 2, or 3 thymine bases, respectively) forming the hinge in the center of the three branches (Supporting Information Table S2). These variations were introduced to study the influence of different mechanical flexibilities on the coating and rigidity of the resulting hybrid structures (VLT1, VLT2, VLT3). Specifically, the 1 thymine hinge (T1 design) was expected to generate the triskelion with the highest rigidity, and the 3 thymine base hinge (T3 design) to generate the most flexible triskelion. This assumption was based on the difference of the persistence lengths and stiffnesses of single-stranded DNA (ssDNA) and double-stranded DNA (dsDNA).32,33 Their correct folding was confirmed by gel electrophoresis (Supporting Information Figure S1) and atomic force microscopy (AFM) (Figure 1d; Supporting Information Figure S2).
Figure 1.

(a) Schematic of the two-step assembly process where the linker attaches first to the vesicles and the triskelion hybridizes subsequently to form an interconnected coating on the vesicle surface (not to scale). (b) Representation of the linker attached to a liposome through a Chol-TEG tag at the 3′-end of one of the oligonucleotides composing the duplex. The 18 nt long overhangs were included on each side of the linker to anneal with the triskelion. (c) Representation of the triskelion with a complementary 18 nt overhang per arm to hybridize with the linker. The three configurations of T1, T2, and T3 differ by the number of unpaired thymine bases in the hinge. (d) AFM micrograph of the T1 triskelion in liquid. Lateral scale bar: 10 nm. (e) Hydrodynamic diameters of the different hybrid structures obtained by DLS with V = 195 ± 10 nm, VL = 190 ± 9 nm, VLT1 = 247 ± 6 nm, VLT2 = 248 ± 8 nm, and VLT3 = 245 ± 7 nm (from counts in intensity). The stated values represent the averages and the standard deviations of five measurements of three independent sample preparations each.
For each step of the assembly process (V, VL, VLT), the changes in size and surface charge were tracked by dynamic light scattering (DLS) and ζ-potential measurements. The size of the liposomes remained in the same range upon linker incubation (VL, Figure 1e). However, after either of the three triskelion designs were added, an increase of the hydrodynamic diameter of ∼45 nm was observed for VLT1, VLT2, and VLT3 (Figure 1e; Supporting Information Figure S5 and Table S6). No size increase was observed when the triskelion was added to V incubated with a linker lacking the Chol-TEG tag (VL-T) or when a triskelion was incubated with VL where the sequences were not complementary to each other (VLTno-match; Supporting Information Figure S6). Further, the incubation of the T1 triskelion with V, even at enhanced concentrations (up to 7 times larger than used for VLT), did not result into a size increase either (Supporting Information Figure S7). These experiments demonstrate that the hydrophobic anchor instead of unspecific DNA adsorption enables the coating process. Alternatively, when the cholesterol-modified linker was assembled with the triskelion in the absence of LUVs (LT), aggregation was observed, evidencing the role of the vesicles in reducing the formation of DNA clusters and templating the hybrid structure assembly (Supporting Information Figure S6). ζ-Potential measurements additionally evidenced the stages of the coating process (Supporting Information Table S6): upon addition of the linker, a decrease in the ζ-potential was observed from −4 ± 1 mV (for V) to −19 ± 2 mV (for VL). Further incubation with the triskelion (VLT) resulted in an additional decrease to −31 ± 2 mV for VLT1, −31 ± 2 mV for VLT2, and −32 ± 2 mV for VLT3. The ζ-potential of VL-T (obtained by incubating V with a linker without the Chol-TEG tag and the T1 triskelion) was −5 ± 1 mV, similar to the value obtained for V (−4 ± 1 mV), further highlighting the necessity of the Chol-TEG modification to enable DNA coating.
Cryo-electron microscopy (cryo-EM) was used to directly visualize the successful coating process. In contrast to V (Figure 2a), the completely assembled hybrid structures (VLT) were observed as lipid vesicles surrounded by a corona of filamentous structures (Figure 2b–d), with the three types of triskelia rendering a similar appearance (see also Supporting Information Figure S8). The appearance of VL resembled unmodified liposomes but with the presence of short spikes and dark spots on the vesicle surface (Supporting Information Figure S8), which can be assigned to the linkers on the surface of the liposomes.
Figure 2.

Cryo-EM images of V (a), VLT1 (b), VLT2 (c), and VLT3 (d). Upon the addition of the DNA structures to the vesicles, a filamentous coat can be seen surrounding the vesicles. Scale bars: 50 nm.
In order to confirm that our DNA self-assembly approach was able to successfully lead to a large-extent DNA network coating the liposome, the mobility of the linker–triskelion assemblies was investigated on the surface of giant unilamellar vesicles (GUVs) through fluorescence recovery after photobleaching (FRAP) measurements.4,34 To this end, the linker was labeled with an ATTO647N fluorophore to assess the different triskelion designs separately. Prior to these experiments, we confirmed that the linker was anchored to the bilayer (Supporting Information Figure S14). Furthermore, to exclude the possibility that the presence of Mg2+ ions (details are in the Experimental Section) could enhance the interactions with the membrane by unspecific adsorption, we added the T1 triskelion labeled with ATTO647N on its own to the GUVs without further attachment functionalities (Supporting Information Figure S14). No considerable adsorption was evident.
After having established the correct anchoring process, we investigated potential differences in the mobility of the DNA coating provided by the three triskelion designs with FRAP measurements. Thus, a circular area was bleached into the DNA coats surrounding the GUVs (Figure 3) at the bottom of the hybrid vesicles. Independently of the triskelion design, the level of fluorescence recovery after approximately 30 s remained at a value around 30% of the prebleaching intensity, indicating similarly hindered mobility (Figure 3 and Supporting Information Figure S15). This can be assigned to no observable differences in the connection capabilities when coating a GUV between VLT1, VLT2, and VLT3. To corroborate this result, we intentionally restricted the complete linker–triskelion hybridization by modifying the sequence of one or two arms of the T1 triskelion (rendering the structures VLT1-1 and VLT1-2, respectively) to prevent complete hybridization with the linker on the liposomes (Supporting Information Table S7 and Figure S16). For these configurations, evident fluorescence recovery to approximately 50% of the prebleaching intensity in the case of VLT1-1 and 70% in the case of VLT1-2 was observed (Figure 3). This correlates to a higher mobility of the DNA coat fragments on the GUV surface compared to that of VLT1, VLT2, or VLT3.
Figure 3.

Fluorescence recovery traces upon photobleaching of the DNA coats and confocal micrographs comparing VLT1 and VLT1-1. The stages prebleaching (1), after bleaching (2), and after approximately 30 s recovery time (3) are shown for the ATTO647N label attached to the linker. Scale bars: 5 μm.
After the coating process was visually confirmed, AFM in solution was conducted to investigate the mechanical properties of the hybrid structures. We expected the rigidity provided by the triskelion to follow T1 > T2 > T3, in agreement with the number of unpaired thymine bases in the hinge.32,33 The hybrid structures were first compared to V and VL by incubating the samples on mica substrates for 1 h under the same conditions (details are in the Experimental Section). By contrast to plain POPC LUVs, which spontaneously rupture upon contact with the substrate due to the specific environmental conditions35 (Figure 4a), VLT conferred significant vesicle stabilization, preventing the coated liposomes from bursting, independently of the triskelion design (VLT1 is shown in Figure 4c, VLT2 and VLT3 in the Supporting Information Figure S10). V and VL by contrast form a roughly 4 nm thick layer on the substrate (Figure 4a,b), matching typical dimensions of lipid bilayers.36 Topographic measurements by AFM performed on 10 different hybrid vesicles for each design (see bottom panel of Figure 4c for a representative profile) yield average vesicle heights of approximately 54.6 ± 12.6 nm (VLT1), 41.6 ± 9.8 nm (VLT2), and 59.3 ± 14.7 nm (VLT3). These values are lower than those of the in-bulk solution diameter of the vesicles, likely due to some deformation and flattening of the vesicles by the measuring AFM tip. Note that given the expected tip compression of the vesicles and tip convolution effects, the spatial dimensions of the hybrid vesicles obtained from AFM cannot be used for precise quantification of the size distribution of LUVs.
Figure 4.

AFM images obtained in solution show a stabilizing influence of the DNA coating on the LUVs. Images and selected height profiles were acquired for (a) V (lateral scale bar: 2.5 μm), (b) VL (lateral scale bar: 2.5 μm), and (c) VLT1 (lateral scale bar: 1 μm). The white lines in the images indicate the positions of the line profiles shown in the panels underneath. (d) Young’s moduli of the three configurations of coated vesicles in dependence of the triskelion design (VLT1: 11.1 ± 0.5 kPa, VLT2: 8.2 ± 0.5 kPa, VLT3: 6.0 ± 0.6 kPa). The error bars correspond to the standard error of the mean of 10 measurements per hybrid vesicle design.
Strikingly, AFM reveals a dependence of the mechanical rigidity of the hybrid structures on the employed triskelion. The Young’s moduli of the hybrid structures follow the trend VLT1 > VLT2 > VLT3 (Figure 4d). For a given VLT, the larger the Young’s modulus is, the lower the flexibility of the triskelion hinge is. As DLS, FRAP, cryo-EM, and AFM topography show no significant difference between the three designs in terms of vesicle coating efficiency, we assign the observed differences in mechanical rigidity to differences in the triskelion design.
The response of the hybrid liposomes to detergent addition was investigated by treating the samples with 0.5% Triton X-100 (TX100). The addition of TX100 completely destroyed V and VL, as denoted by the disappearance of the peaks corresponding to the assembled structures obtained from DLS (Figure 5a,b, respectively). The peak centered at approximately 12 nm is observed in all cases and is assigned to the presence of TX100 micelles in solution (Supporting Information Figure S21). By contrast, after supplementing TX100 to VLT, a second peak in agreement with the size of VLT originally is observed by DLS (Figure 5c). This peak could originate from DNA-coated LUVs capable of resisting detergent solubilization, as similarly reported previously for LUVs coated with enzymatically linked DNA.37 In order to clarify the nature of the remaining particles of VLT size, we imaged V and VLT samples by cryo-EM and by negative staining electron microscopy before and after treatment with 0.5% TX100. Both negative staining and cryo-EM micrographs confirmed the solubilization of V and the presence of micelles after TX100 addition (Supporting Information Figures S18 and S19). However, larger particles were found in the case of the VLT1 sample after TX100 was added (Figure 5d; Supporting Information Figure S20). The morphology of these particles substantially differs from that of untreated VLT1, suggesting that these are composed by DNA assemblies that remain mostly interconnected after detergent addition. Interestingly, TX100 treatment of LUVs coated with triskelion designs with modified sequences to restrict hybridization with the linker, or with unbalanced triskelion and linker concentrations, did not result in remaining interconnected DNA assemblies as only the micelles’ peak could be detected by DLS (Supporting Information Figures S22–S24). This observation further supports the importance of the complete interaction and controlled ratio between triskelion and linker to produce an efficient DNA coating.
Figure 5.

Effect of Triton X-100 addition to (a) V, (b) VL, and (c) VLT1 measured by DLS. (d) Connected DNA assemblies are left of VLT1 after detergent treatment, as shown by cryo-EM. Scale bar: 50 nm.
We next explored the possibility of making the hybrid structures responsive. Aiming to mimic the function of proteins such as Hsc70 in mediating clathrin disassembly and dissociation from liposomes,38,39 the T1 DNA triskelion was engineered to allow for a toehold-displacement-triggered disintegration of the DNA mesh on the lipid vesicle surface (Figure 6a). For this purpose, the number of bases where the linker and triskelion hybridized was decreased to 12, and a 10 nt toehold was introduced (Figure 6a; Supporting Information Table S4). The resulting hybrid structure, referred to as VLT1s, showed a hydrodynamic diameter of 246 ± 13 nm, agreeing with the previously described VLT1, VLT2, and VLT3 structures. Upon addition of a displacement strand (S), the average size decreased to 195 ± 5 nm, a value in the range of VL. This observation, together with the ζ-potential increase from −31 ± 2 to −22 ± 1 mV, following the addition of S, indicates the successful disintegration of the DNA coating.
Figure 6.

Reversibility of the vesicle coating process. (a) Schematic representation of the VLT1s’ disassembly through a toehold-mediated displacement reaction. A 10 nt toehold was included in the T1 triskelion (referred to as T1s) hybridizing with a displacement strand S. (b) Time-resolved and relative fluorescence quenching after addition of S to VLT1s in comparison to a nonhybridizing Scontrol (Sc-), and the effect presented by dilution with PBS (the data represent averages of three sample preparations; error bars show the standard deviation). (c) Cryo-EM images of VLT1s before (left) and after addition of S (right). Scale bars: 50 nm. (d) Top panels: size (hydrodynamic diameters by DLS from counts in intensity) of VLT1s before and after the addition of S, and the subsequent addition of the original T1. The bottom panels collect the DLS data of each sample after the addition of 0.5% TX100.
Further evidence of the disassembly was provided by tracking the fluorescence quenching upon the hybridization of S labeled with a Black Hole II (BHQ-2) quencher and the triskelion labeled with an ATTO550N fluorophore (Figure 6b). Indeed, the addition of S to the VLT1s sample caused virtually complete quenching of the fluorescence signal (Figure 6b, dark red data). On the other hand, the addition of a noncomplementary strand Scontrol (equally labeled with BHQ-2) did not lead to the same degree of fluorescence quenching (Figure 6b, light red data). The fluorescence signal drop observed in this case was similar to the effect caused by simple dilution with the buffer (Figure 6b, gray data).
Cryo-EM was able to verify the success of the disassembly process by visualizing the coated (VLT1s) and uncoated state (VLT1s + S). Indeed, whereas VLT1s contained a DNA mesh as observed before (Figure 2b–d) on the vesicle surface (Figure 6c, left image; Supporting Information Figure S9), the addition of S resulted into the reduction of the DNA corona (Figure 6c, right image; Supporting Information Figure S9).
Sequentially to the dissociation induced by incubation with S, the coating could be reinstated by adding the original T1 triskelion to the beforehand uncoated vesicles, as denoted by a size increase back to 238.0 ± 7.0 nm observed by DLS (Figure 6d). In agreement with the response of VLT1, VLT2, and VLT3 to the presence of TX100, large particles were also detected for VLT1s upon TX100 addition, as indicated by a peak in DLS positioned at the original size (Figure 6d, bottom row). On the contrary, VLT1s vesicles incubated with S were mostly destroyed by the detergent. After further addition of T1 to the formerly uncoated VLT1s (in absence of TX100, Figure 6d), the response to TX100 was the same as that in the original VLT1s, and the characteristic peak centered at the position of the initial size was reinstated.
Conclusions
In conclusion, we have described a straightforward approach to generate DNA-coated lipid vesicles inspired by clathrin self-assembly with enhanced mechanical stability. We demonstrate that the DNA coating can be designed to be disassembled through a toehold-mediated displacement reaction and further reassembled by the subsequent addition of a DNA triskelion as proposed. The DNA coating could be further derivatized to equip liposomes with a variety of functionalities. This possibility is of interest for several purposes, including drug delivery, as these hybrid structures possess the carrier capabilities presented by liposomes as well as the functionality provided by the surrounding DNA coat. The changes in LUV rigidity given by our hybrid system could be used to influence cellular uptake,40 and endogenous nucleic acid molecules can be used to trigger the toehold-mediated disassembly.41 These properties could be exploited to drive targeted delivery approaches.
Experimental Section
Liposome Preparation
LUVs were prepared as previously described31 by extruding a solution of 2 mM POPC in PBS through 200 nm extrusion membranes purchased from Avanti Lipids. POPC was bought as powder from Sigma-Aldrich and diluted to a 200 mM stock in chloroform.
DNA Triskelion Folding and Characterization
DNA oligonucleotides were purchased from Integrated DNA Technologies Inc. (IDT). Linker and triskelion were assembled in PBS and subjected to the thermal programs described in section S1 of the Supporting Information. The linker was assembled at 4 μM concentration (by the equimolar mixture of the two strands) and the triskelion at a 6 μM concentration (by the equimolar mixture of the three strands). Successful folding was assessed by polyacrylamide gel electrophoresis (PAGE, details in Section S2 of the Supporting Information S2) and AFM.
Assembly and Disassembly of the DNA coating on liposomes
The hybrid structures were prepared by first incubating the linker (added at 3 μM) overnight at room temperature with the vesicle solution (2 mM POPC lipids), resulting in a linker concentration of 1.1 μM and POPC concentration of 1.2 mM. Subsequently, the triskelion was added to the mixture (at 4 μM) and incubated for 50 min at 4 °C. The final mixture contained 1 mM POPC, 930 nM linker, and 620 nM triskelion. A molar ratio of 3:2 linker/triskelion was used due to the number of available hybridization sites (2 in case of the linker and 3 in case of the triskelion) to maximize the polymerization degree.
For the toehold-mediated coating disassembly, a T1s triskelion was employed. T1s was designed with shorter complementary domains (12 bp) and a 10 nt long toehold to allow for displacement by an added fully complementary 22 nt long displacement strand (S). Liposome coating was performed as described above to yield VLT1s. S was added to the VLT1s solution at twice the concentration of toeholds present in T1s and incubated for 10 min at 4 °C to achieve the disassembly of the DNA coating (VLT1sS). The reassembly process was carried out with the T1 triskelion following the same protocol as described above for the respective incubation step.
Fluorescent measurements were performed using a Varian Cary Eclipse fluorescence spectrophotometer. To this end, the T1s design was assembled with an ATTO550N fluorophore per strand and the displacement strand with a Black Hole II (BHQ-2) quencher at the matching end. To account for a dilution effect provided by the addition of S, the same volume of PBS was added to the VLT1s sample. Additionally, a displacement strand Scontrol labeled with BHQ-2 likewise was used with an altered sequence interacting with neither the linker nor T1s to examine whether the observed quenching resulted from the intended DNA hybridization or from stochastic quenching only (assessed with NUPACK).42
Sequences of all DNA strands can be reviewed in the Supporting Information Tables S1–S5 and S7).
Hydrodynamic Diameter and ζ-Potential Measurement
Hydrodynamic diameters and ζ-potential were measured with a ZetaSizer Nano ZSP by Malvern Panalytics. All liposome-containing samples were measured at a final lipid concentration of 1 mM in PBS. The untreated vesicles (V) were measured after being diluted 1:1 in PBS (total volume 100 μL) to match the concentration of the hybrid structures. The vesicles with linkers (VL) were measured by topping up the aliquots to 100 μL with PBS for the same reason. The final hybrid vesicles (VLT) were measured directly. The reported results show average values of three preparations for each sample (V, VL, VLT). Controls involved LT1 (incubation without vesicles), VL-T (linker without Chol-TEG), VLTno-match (involving a T1-based triskelion having nonhybridizing sticky ends), and VT (liposomes and the T1 triskelion without the linker).
Cryo-Electron Microscopy
Cryo-EM grids were prepared by applying 3 μL of sample (at 1 nM concentration) on glow discharged holey grids (Quantifoil Cu 1.2/1.3 400 mesh). Excess sample was removed by blotting with filter paper for 4 s prior to plunge-freezing in liquid ethane using a FEI Vitrobot Mark IV at 100% humidity and 4 °C. Data were collected on a FEI Tecnai F20 FEG microscope at 200 kV using a Falcon II (or Falcon III) direct electron detector (always in linear mode). Images were collected at a dose rate of 20 e–/pixel/s, with a total dose of 40 e–/Å2. Magnification was set to 50 000×, yielding a pixel size of 2.08 Å/pixel at the specimen level.
Confocal Microscopy
GUVs were prepared by electroformation using a Nanion Vesicle Prep Pro setup. 2-Oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine (POPC, Sigma-Aldrich) and 1-palmitoyl-2-{6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine (NBD-PC, Avanti Polar Lipids) were dissolved in chloroform to 25 and 1 mg/mL concentrations, respectively, and mixed at the weight ratio of 200:1 (POPC/NBD-PC). Eighty microliters of the lipid mixture at the final concentration of 5 mg/mL were spin-coated on the conducting surface of an indium tin oxide (ITO)-coated glass slide (Nanion/Vision-Tek). After the remaining solvent was evaporated for 1 h in a desiccator, 600 μL of the buffer (1 M sorbitol, 200 mM sucrose) was deposited on the spin-coated layer of lipids within the O-ring chamber, which was subsequently sealed with another ITO-coated slide (conducting surface facing the other). The electroformation chamber was then connected to the Nanion Vesicle Prep Pro, and the electroformation protocol proceeded in three steps: (1) the ac voltage increased linearly from 0 to 3.2 V peak-to-peak (p–p) at 10 Hz over 1 h; (2) the voltage stayed at 3.2 Vpp and 10 Hz for 50 min; (3) the frequency decreased linearly to 4 Hz over 10 min and was maintained for another 20 min. Electroformation was carried out at 37 °C, and vesicles were stored at room temperature and protected from light. Twenty microliters of the GUV solution was pipetted into wells coated with bovine serum albumin. Afterward, the DNA was added at 200 nM final concentration, diluted in an osmotically balanced, glucose-based solution containing 5 mM MgCl2 buffered with 40 mM HEPES buffer (pH 7.4). Hereby, the linker strand (which was not modified by a Chol-TEG) was labeled with an ATTO647N fluorophore. First, 50 μL of the linker solution was added to 20 μL of GUVs and incubated for 2 h at room temperature. Next, 2 μL of 6 μM triskelion was added and incubated for another 50 min at room temperature. The measurements were performed before and after the addition of the triskelion in the same chambers to ensure linker attachment. Imaging was conducted using a Leica TCS SP5 II confocal microscope and a 60× oil immersion objective. The samples were excited by a 488 nm and a 635 nm wavelength laser.
Atomic Force Microscopy and Force Spectroscopy
Samples were prepared placing 20 μL of 25 mM MgCl2 (final concentration 5 mM due to dilution by the samples) onto a freshly cleaved mica followed by 50 μL of the specific sample solution (V, VL, VLT1, VLT2, VLT3) together with 30 μL of 10 mM Tris-HCl (pH 7.4, final concentration 3 mM). All samples were incubated at room temperature for 1 h, followed by the addition of another 20 μL of the 25 mM MgCl2 solution to further promote sample adhesion to the substrate.
Imaging the three triskelion configurations (T1, T2, T3), as well as the combination of linker and T2 triskelion (LT2), was achieved by diluting the samples 10 times in 1× TAEM buffer (1× TAE plus 25 mM MgCl2, pH 7.4) and placing 2 μL of the solution on a mica substrate. Subsequently, 10 μL of 1× TAEM buffer and 2 μL of a 100 mM NiCl2 solution were added. After a minute equilibration time, an additional 60 μL of the 1× TAEM buffer was supplemented.
A commercial Cypher ES AFM (Asylum Research) equipped with direct laser excitation (blueDrive) and temperature control was used for all experiments. Imaging was carried out in amplitude modulation (AM) with the cantilever fully immersed in liquid and at a controlled temperature of 25 ± 0.1 °C. As in standard AM operation, the cantilever was driven at its fundamental resonance frequency and the amplitude kept constant while scanning. The cantilever used for the investigation of V, VL, and VLT (OMCL RC800-PSA, Olympus, Tokyo, Japan) had a nominal spring constant of 0.05 N/m. During imaging, the set-point ratio S = A/A0 between the imaging amplitude A and free amplitude A0 was kept as high as possible (typically S > 0.8) to limit the force applied to the vesicles. For imaging the linker and triskelion, we used a cantilever with a nominal spring constant of 0.38 N/m (ORC8, Bruker, Camarillo, US), keeping a set-point ratio of S ∼ 0.7. All of the images were corrected for tilt (line or plane flattening) and lightly low-pass filtered to remove grainy noise using the WSxM software (Nanotec Electronica, Madrid, Spain).43
For the quantification of the Young’s moduli from spectroscopy measurements, averaged data from at least 10 force curves (FCs) for each sample (VLT1, VLT2, VLT3) were analyzed using custom routines programmed in Igor Pro (WaveMetrics, Lake Oswego, USA). The Young’s modulus was calculated by fitting a suitable region of the FCs with a previously described equation for analyzing vesicular structures.44
Acknowledgments
The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) through the ERC grant PhysProt (Agreement No. 337969). K.N.B. and T.P.J.K. are grateful for financial support from the Biotechnology and Biological Sciences Research Council (BBSRC), the Newman Foundation, the Wellcome Trust and the Cambridge Centre for Misfolding Diseases. K.V. and L.P. acknowledge funding from the BBSRC (Grant BB/M024830/1). J.G.N. received support from Heptares Therapeutics, the Ministerio de Ciencia, Innovación y Universidades (RTI2018-095629-J-I00), and the Fondo Europeo de Desarrollo Regional (FEDER). D.S. is supported by Winton Programme for the Physics of Sustainability, as well as the Engineering and Physical Sciences Research Council (EPSRC). S.H.A. acknowledges funding by the University of Zaragoza (UZ2018-CIE-04 and JIUZ-2018-CIE-04) and by the Gobierno de Aragón-FSE (Research Group E47_17R). The authors would like to thank Michael Schaich, Magdalena A. Czekalska, Dr. Jinbo Zhu, Prof. Ulrich Keyser, and Dr. Lorenzo Di Michele for insightful discussions. The authors would also like to acknowledge the use of Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b09453.
DNA sequence information and folding procedure; supplementary gel electrophoresis, ζ-potential and DLS results; additional AFM images and Young’s modulus analysis; supplementary negative staining and cryo-electron transmission electron microscopy images; confocal microscopy and fluorescence recovery after photobleaching analysis of coated giant unilamellar vesicles (PDF)
Author Present Address
△ (L.C.) Micron School of Materials Science & Engineering, Boise State University, Boise, ID 83725, USA.
Author Present Address
○ (J.G.-N.) Institute of Biocomputation and Physics of Complex Systems (BIFI), University of Zaragoza, BIFI-IQFR (CSIC), 50018, Zaragoza, Spain.
Author Present Address
□ (J.G.-N.) Laboratorio de Microscopías Avanzadas, University of Zaragoza, 50018, Zaragoza, Spain.
The authors declare no competing financial interest.
Notes
A preprint version of this manuscript is available on ChemRxiv: Baumann, K. N.; Piantanida, L.; García-Nafría, J.; Sobota, D.; Voïtchovsky, K.; Knowles, T. P. J.; Hernández-Ainsa, S. Clathrin-Inspired Coating and Stabilization of Liposomes by DNA Self-Assembly; https://doi.org/10.26434/chemrxiv.8859017.v1.
Supplementary Material
References
- McMahon H. T.; Boucrot E. Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Heuser J. E.; Keen J. H.; Amende L. M.; Lippoldt R. E.; Prasad K. Deep-Etch Visualization of 27S Clathrin: A Tetrahedral Tetramer. J. Cell Biol. 1987, 105, 1999–2009. 10.1083/jcb.105.5.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen M.; Roux A. Mechanisms of Clathrin-Mediated Endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- Kocabey S.; Kempter S.; List J.; Xing Y.; Bae W.; Schiffels D.; Shih W. M.; Simmel F. C.; Liedl T. Membrane-Assisted Growth of DNA Origami Nanostructure Arrays. ACS Nano 2015, 9, 3530–3539. 10.1021/acsnano.5b00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaliano G. D.; Vitaliano F.; Rios J. D.; Renshaw P. F.; Teicher M. H. New Clathrin-Based Nanoplatforms for Magnetic Resonance Imaging. PLoS One 2012, 7, e35821. 10.1371/journal.pone.0035821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y.; Hanna M. E. S.; Zhuo D.; Chang K. G.; Bozorg-Grayeli T.; Melosh N. A. Self-Assembly of Mesoscale Artificial Clathrin Mimics. ACS Nano 2017, 11, 9889–9897. 10.1021/acsnano.7b03739. [DOI] [PubMed] [Google Scholar]
- Bell N. A. W.; Keyser U. F. Nanopores Formed by DNA Origami: A Review. FEBS Lett. 2014, 588, 3564–3570. 10.1016/j.febslet.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Benizri S.; Ferey L.; Alies B.; Mebarek N.; Vacher G.; Appavoo A.; Staedel C.; Gaudin K.; Barthélémy P. Nucleoside-Lipid-Based Nanocarriers for Sorafenib Delivery. Nanoscale Res. Lett. 2018, 13, 17. 10.1186/s11671-017-2420-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen U.; Simonsen A. C.; Vogel S. DNA-Controlled Assembly of Soft Nanoparticles. J. Am. Chem. Soc. 2008, 130, 10462–10463. 10.1021/ja8030054. [DOI] [PubMed] [Google Scholar]
- Börjesson K.; Lundberg E. P.; Woller J. G.; Nordén B.; Albinsson B. Soft-Surface DNA Nanotechnology: DNA Constructs Anchored and Aligned to Lipid Membrane. Angew. Chem., Int. Ed. 2011, 50, 8312–8315. 10.1002/anie.201103338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V.; Palazzolo S.; Bayda S.; Corona G.; Toffoli G.; Rizzolio F. DNA Nanotechnology for Cancer Therapy. Theranostics 2016, 6, 710–725. 10.7150/thno.14203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q.; Zhao S.; Liu J.; Song L.; Wang Z. G.; Ding B. Rationally Designed DNA-Based Nanocarriers. Adv. Drug Delivery Rev. 2019, 147, 2–21. 10.1016/j.addr.2019.02.003. [DOI] [PubMed] [Google Scholar]
- Lopez A.; Liu J. DNA Oligonucleotide-Functionalized Liposomes: Bioconjugate Chemistry, Biointerfaces, and Applications. Langmuir 2018, 34, 15000–15013. 10.1021/acs.langmuir.8b01368. [DOI] [PubMed] [Google Scholar]
- Banga R. J.; Chernyak N.; Narayan S. P.; Nguyen S. T.; Mirkin C. A. Liposomal Spherical Nucleic Acids. J. Am. Chem. Soc. 2014, 136, 9866–9869. 10.1021/ja504845f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkholz O.; Burns J. R.; Richter C. P.; Psathaki O. E.; Howorka S.; Piehler J. Multi-Functional DNA Nanostructures That Puncture and Remodel Lipid Membranes into Hybrid Materials. Nat. Commun. 2018, 9, 1521. 10.1038/s41467-018-02905-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göpfrich K.; Zettl T.; Meijering A. E. C.; Hernández-Ainsa S.; Kocabey S.; Liedl T.; Keyser U. F. DNA-Tile Structures Induce Ionic Currents through Lipid Membranes. Nano Lett. 2015, 15, 3134–3138. 10.1021/acs.nanolett.5b00189. [DOI] [PubMed] [Google Scholar]
- Göpfrich K.; Li C.-Y.; Ricci M.; Bhamidimarri S. P.; Yoo J.; Gyenes B.; Ohmann A.; Winterhalter M.; Aksimentiev A.; Keyser U. F. Large-Conductance Transmembrane Porin Made from DNA Origami. ACS Nano 2016, 10, 8207–8214. 10.1021/acsnano.6b03759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langecker M.; Arnaut V.; Martin T. G.; List J.; Renner S.; Mayer M.; Dietz H.; Simmel F. C. Synthetic Lipid Membrane Channels Formed by Designed DNA Nanostructures. Science 2012, 338, 932–936. 10.1126/science.1225624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Wang J.; Rothman J. E.; Pincet F. Accelerating SNARE-Mediated Membrane Fusion by DNA-Lipid Tethers. Angew. Chem., Int. Ed. 2015, 54, 14388–14392. 10.1002/anie.201506844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmann A.; Li C.-Y.; Maffeo C.; Al Nahas K.; Baumann K. N.; Göpfrich K.; Yoo J.; Keyser U. F.; Aksimentiev A. A Synthetic Enzyme Built from DNA Flips 107 Lipids per Second in Biological Membranes. Nat. Commun. 2018, 9, 2426. 10.1038/s41467-018-04821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Journot C. M. A.; Ramakrishna V.; Wallace M. I.; Turberfield A. J. Modifying Membrane Morphology and Interactions with DNA Origami Clathrin-Mimic Networks. ACS Nano 2019, 13, 9973–9979. 10.1021/acsnano.8b07734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langecker M.; Arnaut V.; List J.; Simmel F. C. DNA Nanostructures Interacting with Lipid Bilayer Membranes. Acc. Chem. Res. 2014, 47, 1807–1815. 10.1021/ar500051r. [DOI] [PubMed] [Google Scholar]
- Wu N.; Chen F.; Zhao Y.; Yu X.; Wei J.; Zhao Y. Functional and Biomimetic DNA Nanostructures on Lipid Membranes. Langmuir 2018, 34, 14721–14730. 10.1021/acs.langmuir.8b01818. [DOI] [PubMed] [Google Scholar]
- Avakyan N.; Conway J. W.; Sleiman H. F. Long-Range Ordering of Blunt-Ended DNA Tiles on Supported Lipid Bilayers. J. Am. Chem. Soc. 2017, 139, 12027–12034. 10.1021/jacs.7b06572. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Endo M.; Sugiyama H. Lipid-Bilayer-Assisted Two-Dimensional Self-Assembly of DNA Origami Nanostructures. Nat. Commun. 2015, 6, 8052. 10.1038/ncomms9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Buck A.; Jiang S.; Yan H.; Walter N. G. DNA-Cholesterol Barges as Programmable Membrane-Exploring Agents. ACS Nano 2014, 8, 5641–5649. 10.1021/nn500108k. [DOI] [PubMed] [Google Scholar]
- Suzuki Y.; Endo M.; Yang Y.; Sugiyama H. Dynamic Assembly/Disassembly Processes of Photoresponsive DNA Origami Nanostructures Directly Visualized on a Lipid Membrane Surface. J. Am. Chem. Soc. 2014, 136, 1714–1717. 10.1021/ja4109819. [DOI] [PubMed] [Google Scholar]
- Czogalla A.; Kauert D. J.; Franquelim H. G.; Uzunova V.; Zhang Y.; Seidel R.; Schwille P. Amphipathic DNA Origami Nanoparticles to Scaffold and Deform Lipid Membrane Vesicles. Angew. Chem., Int. Ed. 2015, 54, 6501–6505. 10.1002/anie.201501173. [DOI] [PubMed] [Google Scholar]
- Franquelim H. G.; Khmelinskaia A.; Sobczak J.-P.; Dietz H.; Schwille P. Membrane Sculpting by Curved DNA Origami Scaffolds. Nat. Commun. 2018, 9, 811. 10.1038/s41467-018-03198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grome M. W.; Zhang Z.; Pincet F.; Lin C. Vesicle Tubulation with Self-Assembling DNA Nanosprings. Angew. Chem., Int. Ed. 2018, 57, 5330–5334. 10.1002/anie.201800141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ainsa S.; Ricci M.; Hilton L.; Aviñó A.; Eritja R.; Keyser U. F. Controlling the Reversible Assembly of Liposomes through a Multistimuli Responsive Anchored DNA. Nano Lett. 2016, 16, 4462–4466. 10.1021/acs.nanolett.6b01618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Q.; Wang G.; Jiang J. The Persistence Length and Length per Base of Single-Stranded DNA Obtained from Fluorescence Correlation Spectroscopy Measurements Using Mean Field Theory. Phys. A 2013, 392, 1072–1079. 10.1016/j.physa.2012.09.022. [DOI] [Google Scholar]
- Brunet A.; Tardin C.; Salomé L.; Rousseau P.; Destainville N.; Manghi M. Dependence of DNA Persistence Length on Ionic Strength of Solutions with Monovalent and Divalent Salts: A Joint Theory–Experiment Study. Macromolecules 2015, 48, 3641–3652. 10.1021/acs.macromol.5b00735. [DOI] [Google Scholar]
- Jahnke K.; Weiss M.; Frey C.; Antona S.; Janiesch J.-W.; Platzman I.; Göpfrich K.; Spatz J. P. Programmable Functionalization of Surfactant-Stabilized Microfluidic Droplets via DNA-Tags. Adv. Funct. Mater. 2019, 29, 1808647. 10.1002/adfm.201808647. [DOI] [Google Scholar]
- Hardy G. J.; Nayak R.; Zauscher S. Model Cell Membranes: Techniques to Form Complex Biomimetic Supported Lipid Bilayers via Vesicle Fusion. Curr. Opin. Colloid Interface Sci. 2013, 18, 448–458. 10.1016/j.cocis.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B. A.; Engelman D. M. Lipid Bilayer Thickness Varies Linearly with Acyl Chain Length in Fluid Phosphatidylcholine Vesicles. J. Mol. Biol. 1983, 166, 211–217. 10.1016/S0022-2836(83)80007-2. [DOI] [PubMed] [Google Scholar]
- Ruysschaert T.; Paquereau L.; Winterhalter M.; Fournier D. Stabilization of Liposomes through Enzymatic Polymerization of DNA. Nano Lett. 2006, 6, 2755–2757. 10.1021/nl061724x. [DOI] [PubMed] [Google Scholar]
- Schlossman D. M.; Schmid S. L.; Braell W. A.; Rothman J. E. An Enzyme That Removes Clathrin Coats: Purification of an Uncoating ATPase. J. Cell Biol. 1984, 99, 723–733. 10.1083/jcb.99.2.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E.; Ungewickell H.; Holstein S. E. H.; Lindner R.; Prasad K.; Barouch W.; Martini B.; Greene L. E.; Eisenberg E. Role of Auxilin in Uncoating Clathrin-Coated Vesicles. Nature 1995, 378, 632–635. 10.1038/378632a0. [DOI] [PubMed] [Google Scholar]
- Chen L.; Li X.; Zhang Y.; Chen T.; Xiao S.; Liang H. Morphological and Mechanical Determinants of Cellular Uptake of Deformable Nanoparticles. Nanoscale 2018, 10, 11969–11979. 10.1039/C8NR01521J. [DOI] [PubMed] [Google Scholar]
- Xue H.; Ding F.; Zhang J.; Guo Y.; Gao X.; Feng J.; Zhu X.; Zhang C. DNA Tetrahedron-Based Nanogels for SiRNA Delivery and Gene Silencing. Chem. Commun. 2019, 55, 4222–4225. 10.1039/C9CC00175A. [DOI] [PubMed] [Google Scholar]
- Zadeh J. N.; Steenberg C. D.; Bois J. S.; Wolfe B. R.; Pierce M. B.; Khan A. R.; Dirks R. M.; Pierce N. A. NUPACK : Analysis and Design of Nucleic Acid Systems. J. Comput. Chem. 2011, 32, 170–173. 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- Horcas I.; Fernández R.; Gómez-Rodríguez J. M.; Colchero J.; Gómez-Herrero J.; Baro A. M. WSXM: A Software for Scanning Probe Microscopy and a Tool for Nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. 10.1063/1.2432410. [DOI] [PubMed] [Google Scholar]
- Laney D. E.; Garcia R. A.; Parsons S. M.; Hansma H. G. Changes in the Elastic Properties of Cholinergic Synaptic Vesicles as Measured by Atomic Force Microscopy. Biophys. J. 1997, 72, 806–813. 10.1016/S0006-3495(97)78714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.