

Supplemental Digital Content is available in the text
Keywords: cell-cycle, differential expression, long non-coding RNAs, messenger RNAs, osteoarthritis
Abstract
Background:
A group of differentially expressed long non-coding RNAs (lncRNAs) have been shown to play key roles in osteoarthritis (OA), although they represented only a small proportion of lncRNAs that may be biologically and physiologically relevant. Since our knowledge of regulatory functions of non-coding RNAs is still limited, it is important to gain better understanding of their relation to the pathogenesis of OA.
Methods:
We performed mRNA and lncRNA microarray analysis to detect differentially expressed RNAs in chondrocytes from three OA patients compared with four healthy controls. Then, enrichment analysis of the differentially expressed mRNAs was carried out to define disease molecular networks, pathways and gene ontology (GO) function. Furthermore, target gene prediction based on the co-expression network was performed to reveal the potential relationships between lncRNAs and mRNAs, contributing an exploration of a role of lncRNAs in OA mechanism. Quantitative RT-PCR analyses were used to demonstrate the reliability of the experimental results.
Findings:
Altogether 990 lncRNAs (666 up-regulated and 324 down-regulated) and 546 mRNAs (419 up-regulated and 127 down-regulated) were differentially expressed in OA samples compared with the normal ones. The enrichment analysis revealed a set of genes involved in cell cycle. In total, 854 pairs of mRNA and lncRNA were highly linked, and further target prediction appointed 12 genes specifically for their corresponding lncRNAs. The lncRNAs lncRNA-CTD-2184D3.4, ENST00000564198.1, and ENST00000520562.1 were predicted to regulate SPC24, GALM, and ZNF345 mRNA expressions in OA.
Interpretation:
This study uncovered several novel genes potentially important in pathogenesis of OA, and forecast the potential function of lnc-CTD-2184D3.4, especially for the cell cycle in the chondrocytes. These findings may promote additional aspects in studies of OA.
1. Introduction
Osteoarthritis (OA) is a worldwide musculoskeletal disease, which results in pain, disability and heavy socioeconomic burden.[1] Epidemiologic investigations have shown that the prevalence of OA increases from 40to 60 years with a linear trend, and an obvious gender association exists due to a higher prevalence and severity in females compared with males.[2] The previous studies have demonstrated that cartilage degradation, changes in subchondral bone, proliferation of synoviocytes, and tissue hypertrophy are common features for OA.[3] Despite extensive studies of the complex associations between these risk factors and the pathologic changes, the disease pathogenesis has still remained unclear. Thus, more studies to obtain better understanding on the development and progression of OA are warranted.
As the human genome sequencing has revealed, the human genome contains about 20,000 protein-coding genes, which account for <1% to 2% of the total human genome. It has been estimated that 70% to 90% of our genome is transcribed at some point during development,[4] while such percentage has also been challenged. Long noncoding RNAs (lncRNAs) refer to non-coding transcripts, which are longer than 200 nucleotides in length, including large intergenic noncoding RNAs, natural antisense transcripts, pseudogenes, long intronic noncoding RNAs, and other divergent transcripts.[5] Obviously, focusing on <2% of the coding RNA is insufficient to interpret the complicated mechanism of diseases such as OA. Accumulating evidence suggests that the lncRNAs are functional in several kinds of diseases, including OA. For instance, cartilage injury-related lncRNAs were shown to play a key role in extracellular matrix (ECM) degradation in the pathogenesis of OA.[6] Besides, it has been shown that lncRNA GAS5 regulated the cell survival in the pathogenesis of OA by acting as a negative regulator of miR-21,[7] while lncRNA HOTAIR contributed to the chondrocyte apoptosis in temporomandibular joint OA.[8] Although specific differential lncRNA expressions have been previously found to be related to OA, they represent only a small proportion of the lncRNAs that may be biologically and physiologically relevant. Therefore, it is still necessary to exploit lncRNA and mRNA microarray data and bioinformatics analysis in order to uncover the potential functions of the lncRNAs and their roles in the development of OA, which may promote the pathogenesis of OA.
2. Materials and methods
2.1. Sample collection
This study was approved by the Human Ethics Committee of Xi’an Jiaotong University, and performed according to the principles of the Declaration of Helsinki as revised in 1983. All the participants also gave an informed consent. Human cartilage samples were collected from knees of OA patients undergoing joint replacement surgery and normal donors, who suffered traffic accidents in Shaanxi Han population. The patients were diagnosed as primary knee OA on the basis of clinical evaluation, radiologic imaging, and Western Ontario and McMaster Universities OA Index of pain, stiffness, and function.[9] According to Kellgren–Lawrence grading system, grade III/IV OA patients were selected for this study. The normal control group was diagnosed for not having rheumatoid arthritis, ankylosing spondylitis, or other bone and cartilage diseases. Because of the difficulties in obtaining cartilage biopsies, six OA and seven healthy donors were finally collected for further analyses. After matching according to age and sex (Additional file 1), cartilage from three OA and four normal samples were selected for microarray analysis, and the other three different pairs for quantitative reverse transcription-polymerase chain reaction (qRT-PCR).
2.2. Chondrocyte culture
The chondrocytes from the OA and the healthy donors were isolated from cartilage, which was first digested with 0.25% trypsin for 30 min and then with 0.2% type II collagenase for 4 to 6 h. Then, the chondrocytes were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C and 5% CO2. During the culture period, the medium was replaced every 2 to 3 days.
2.3. Microarray analysis
The differential expression profiles between the OA and the healthy samples were obtained by using the lncRNA+mRNA Human Gene Expression Microarray V4.0, a 4x180K chip (Agilent Technologies). The reverse transcription using CbcScript II reverse transcriptase was adopted after the vitro transcription reactions. The data summarization, normalization and quality control of the lncRNA and mRNA array were conducted by using the GeneSpring software V13.0 (Agilent Technologies). Finally, the genes were considered differentially expressed, when the threshold values were of ≥2 and ≤0.5-fold change and the t test P-value was <.05.
2.4. Arrays and gene set enrichment analysis
The differentially expressed mRNAs were further taken to enrichment analysis using Kyoto Encyclopedia of Genes and Genomes Orthology Based Annotation System, including three types of aspects: disease, pathway, and gene ontology (GO) function. For the GO or pathway terms the significance values had P < .05. Followed by the above enrichment analysis, gene set enrichment analysis (GSEA) was performed to generate gene expression signatures by the GSEA software v2.0 (http://www.broadinstitute.org/gsea).
2.5. Construction of the coding-non-coding gene co-expression network
After the Pearson correlations between the differentially expressed lncRNAs and mRNAs were calculated, the pairs were chosen to construct a coding-non-coding gene co-expression (CNC) network through the open source bioinformatics software Cytoscape (https://cytoscape.org) only when the Pearson correlation coefficients were ≥0.99. In the network analysis, the simplest and most important measures of a gene centrality were defined as the link numbers one node had to the others.
2.6. Target gene prediction
Two kinds of target gene predictions were performed. The cis/trans-prediction was performed by Blat tools (Standalone BLAT v. 35x1 fast sequence search command line tool, downloaded from: http://hgdownload.cse.ucsc.edu/admin/exe/) through the full sequence of the lncRNA and the 3’ UTR of its co-expressed mRNAs.
2.7. Quantitative RT-PCR analyses
Total RNA was isolated from cartilage tissues as described above, and qRT-PCR was performed with RiboTM mRNA/lncRNA qRT-PCR Starter Kit, using the following parameters: 95°C for 5 s, then 60°C for 30 s, and 72°C for 30 s, 40 cycles. The fold change of relative gene expression was calculated with the 2(−ΔΔCt) method, using β-actin as the reference gene. The primer sequences used in this study are shown in Additional file 2.
3. Results
3.1. Differentially expressed lncRNAs
The lncRNA expression profiles of the chondrocytes from cartilage of OA and those of the normal controls were compared on the basis of the microarray data. To be specific, the lncRNA microarray identified 666 up- and 324 down-regulated genes in OA patients compared with the normal subjects (Fig. 1A). Hierarchical clustering analysis showed distinguishable lncRNA expression profiles between the samples and homogeneity within groups (Fig. 1C). There were 45 lncRNAs with fold change value higher than 8, of which 11 down-regulated and 34 up-regulated. Among the 24 chromosomes, chromosome 2 had the most genes compared with the others (Fig. 2A), which also appeared in the top 10 differentially expressed lncRNAs. The most significantly differentially expressed lncRNAs always located at the chromosome 2, including TCONS_00004621 with 39.19 fold change (P-value .0242), ENST00000544821.2 (lnc CTD-2184D3.4) with the highest fold change 27.92 (P-value .0382), hox-HOXD11–34 with 23.10 fold change (P-value .0233). It is noticeable that there were six HOXD10-related lncRNAs among those most significantly down-regulated ones, which also were in the chromosome 2 (Table 1). The classification of the differentially expressed lncRNA included intergenic, intronic, divergent, sense and antisense, and some unknown types. The intergenic lncRNAs were the most common, and the antisense ones covered the second most lncRNAs (Fig. 2C).
Figure 1.
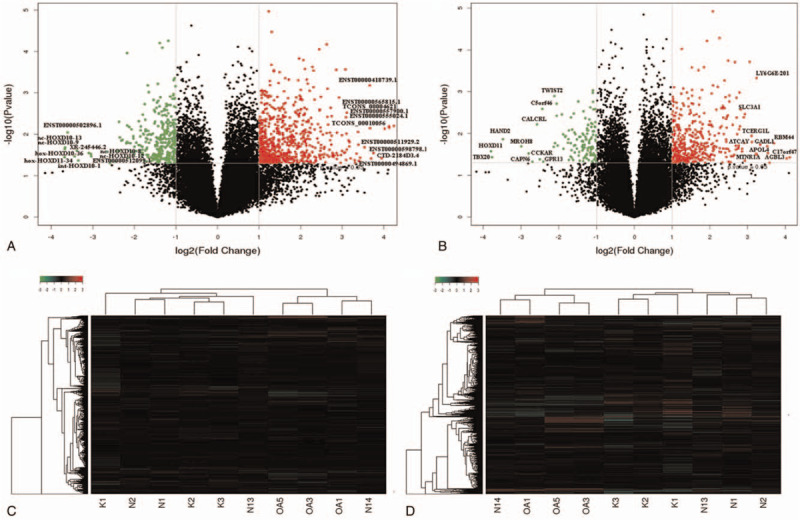
(A) Volcano plot of lncRNA expression. (B) Volcano plot of mRNAs expression. The red and green points respectively indicate up- and down-regulated RNAs with more than 2.0 fold change between OA cartilage and control normal samples. (C) Hierarchical clustering shows a distinguishable lncRNA expression profile between the two groups and homogeneity within groups. (D) Hierarchical clustering shows a distinguishable mRNA expression profile between the two groups and homogeneity within groups.
Figure 2.
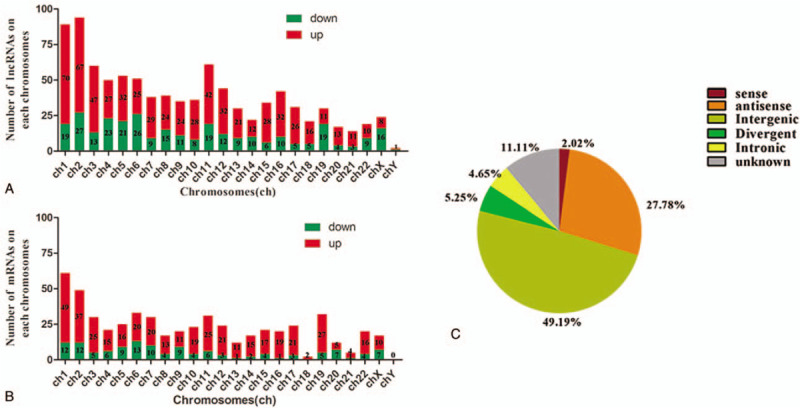
(A) The number of differential lncRNAs on each chromosome. (B) The number of differential mRNAs on each chromosome. (C) Pie-chart of lncRNA classification. The area in each chart assigns to a constructional category by hypergeometric distribution.
Table 1.
Top 10 differentially expressed lncRNAs (OA vs. Normal).

3.2. Differentially expressed mRNAs
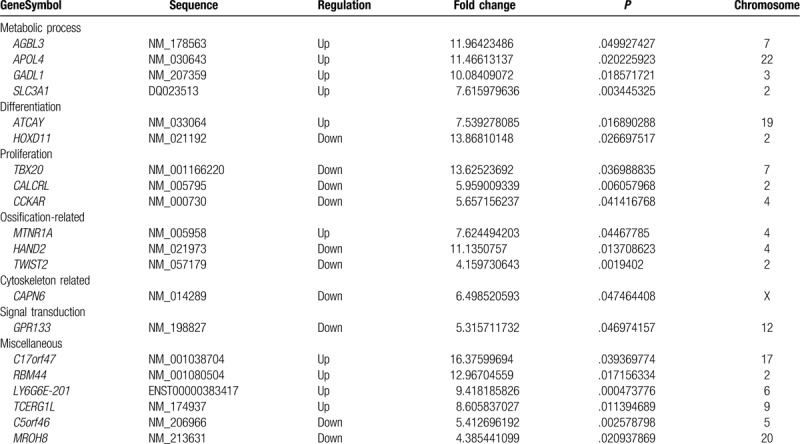
In comparison to the control samples, this study found 546 differentially expressed mRNAs in the OA chondrocytes, and there was a good homogeneity within groups (Fig. 1B and D). On the whole, there were 419 up-regulated and 127 down-regulated mRNAs. Similarly, the number of the up-regulated mRNAs on each chromosome was also higher than down-regulated ones, similarly to lncRNAs (Fig. 2B). The top 10 differentially up- and down-regulations are listed in Table 2, classified according to their GO terms. For the top 10 up-regulated mRNAs, four were involved in metabolic processes, while other up- or down-regulated ones were related to proliferation, differentiation, ossification, cytoskeleton, signal transduction, and miscellaneous functions. The most significantly up-regulated mRNA was C17orf47 with 16.38 fold change (P-value .0394), while the most significantly down-regulated mRNA was HOXD11 with 13.87 fold change (P-value .0267).
Table 2.
Top 10 differentially expressed mRNAs (OA vs normal).
3.3. Arrays and GSEA
Among the top 30 most significantly enriched GO terms (Fig. 3), the most were related to cell cycle, nucleotide metabolism, and lipid metabolic processes. Furthermore, cell cycle and replication accounted for a large proportion in the top 30 most significantly enriched pathway (Fig. 4A). Interestingly and consistent with the above, the GSEA indicated that gene expression sets of nuclear replication were significantly correlated with features of OA versus the normal control (Fig. 4B).
Figure 3.
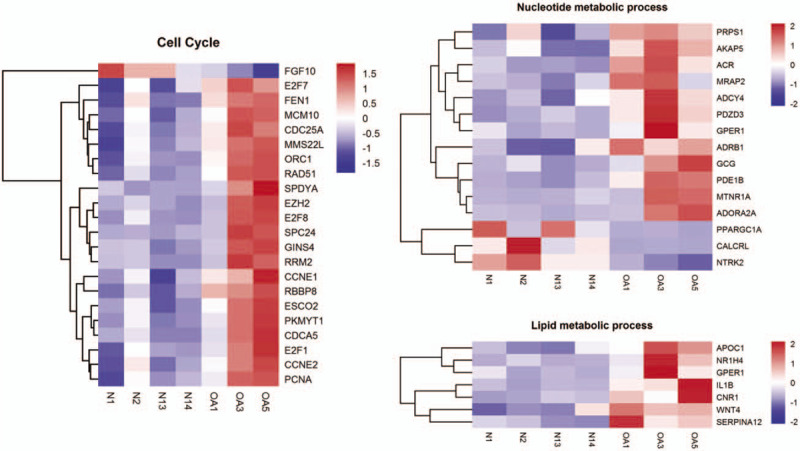
The hierarchical clustering of gene sets related to cell cycle, nucleotide metabolism, as well as lipid metabolic process.
Figure 4.
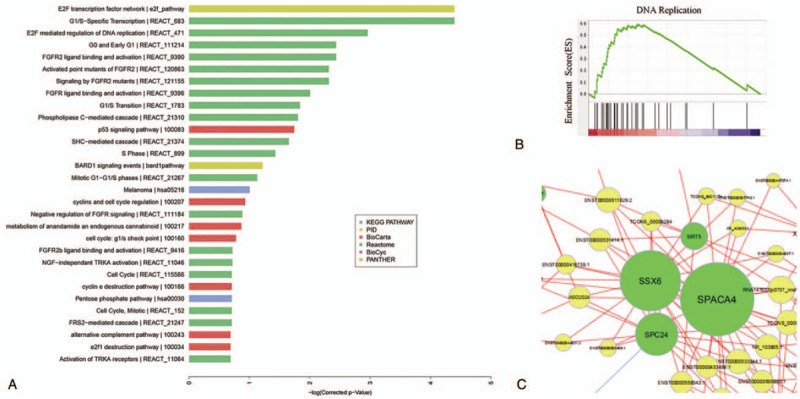
(A) The top 30 significant enriched pathways. Different colours represent different databases. (B) The significant gene expression signature by the GSEA. (C) The center of the co-expression network. The yellow circles represent lncRNAs and green circles represent mRNAs, the size of the circle represents the nodes of the gene in the network (neighbor numbers). The string between 2 genes indicates a tight correlation, the red show positive correlation while the blue show the negative correlation. All pairs were selected with correlation >0.99 or < -0.99 and P value < 0.05.
3.4. Gene co-expression network
The differentially expressed genes of lncRNA/mRNA were selected from the microarray data and then further taken to gene co-expression analysis. Based on the Pearson correlation coefficients, 854 significant correlation pairs composed the co-expression network, in which one lncRNA associated with several mRNAs, and vice versa. Besides, the co-expression pairs were used to draw the network diagram with Cytoscape software. Of these, SPC24, SSX6, SPACA4 were in middle position of network, and connected with most of the lncRNAs (Fig. 4C).
3.5. Target gene prediction
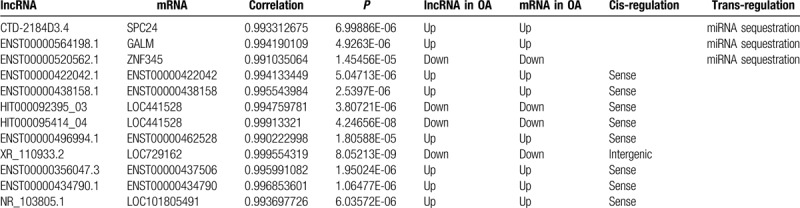
In total, 854 pairs of mRNA and lncRNA were selected for gene co-expression analysis, and were further conducted for target gene prediction. In this process, the target genes of nine pairs were subject to cis-regulation, while the other targets of three pairs were related to trans-regulation. Of these, SPC24, LOC441528, LOC729162, GALM, ZNF345, and LOC101805491 are known as gene symbols (Table 3). Across the enrichment for target genes, SPC24 is a kinetochore complex component, and GALM is related to metabolic pathways, especially galactose metabolism, glycolysis, and gluconeogenesis. Moreover, SPC24 played an important role in co-expression network.
Table 3.
The target genes of lncRNAs and regulation.
3.6. qRT-PCR analyses
In order to confirm the reliability of the expression profiles obtained by the microarray analysis, we randomly selected six differentially expressed lncRNAs (three up- and three down-regulated, fold changes were >3.0) to conduct qRT-PCR with six cartilage samples (three OA and three normal samples). The real-time PCR analysis demonstrated the same trends of up- or down regulation for each lncRNA as the microarray analysis (Fig. 5).
Figure 5.

Figure 5 The heights of the columns in the chart represent the mean fold changes of expression levels of 6 selected genes in OA cartilage to that in normal cartilage, as analyzed by oligonucleotide microarray and PCR. Bars show the mean ± SD. ∗∗=p < 0.01, ∗=p < 0.05, for OA versus normal, by Student's paired t-test.
4. Discussion
At present, there is only a little data available on the lncRNAs related to OA. This study was designed to increase our knowledge of this relation. The study revealed that there were 990 lncRNAs (666 up-regulated and 324 down-regulated) and 546 mRNAs (419 up-regulated and 127 down-regulated) differentially expressed in OA cartilage compared with those of the normal ones. It was observed that the intergenic type of the lncRNAs was the most abundant class of RNAs, and constituted a large proportion of the total lncRNAs. Similar pattern was found in previous studies of the lncRNAs expression profiles in different diseases.[10,11] Compared with the previous findings, HOXD11, TBX20, and TWIST2 were expressed at significantly lower level in OA cartilage, similarly HOXA11, TBX15, and TWIST1 were down-regulated in another OA study.[12] In addition, association between TBX20 and cartilage has not been previously reported. Also HOXD10- and HOXD11-related lncRNAs had lower expression in OA in our study. Under the premise of ensuring strict screening of samples, the data gaps in different studies may be attributed to the biological variety of different populations, such as the physical activity levels and nutritional factors in different populations, which affect occurrence and development of OA,[3,13] particularly in the different geographical areas, such as the northwest and south of China,[10] Asia, and Europe.[12] It is worth mentioning that the result of the qRT-PCR analysis indeed demonstrates the validity of this microarray data.
Among the top 10 differentially expressed RNAs in OA cartilage, HOXD10 and HOXD11 encode a highly conserved family of transcription factors. Besides their role in the regulation of skeletal formation during development,[14] they also participate in tissue remodeling and homeostasis in adults. Thus, their dysregulation can be potentially involved in the development of OA. It has been shown that TWIST1 and TWIST2 interact with the Runx2 to attenuate its function in osteoblast differentiation,[15] and Runx2 and TWIST2 are relevant to osteogenic differentiation capacity of mesenchymal stromal cells.[16] Furthermore, TWIST1 and TWIST2 modulate ATF4-dependent transcriptional activity of osteocalcin promoter in response to parathyroid hormone, which is essential for regulation of endochondral bone formation and calcium homeostasis.[17]
An important finding of this study was also that GSEA revealed numerous significant terms associated with mitotic cell cycle and nucleotide metabolic processes, which are at least partly related to cell cycle. Regarding the cell cycle-related genes cluster, a gene expression profile has indicated HIST1H4C to be involved in DNA replication pathway and cell cycle.[18] AREG is a member of epidermal growth factor receptor ligands, the absence of which can result in reduced content of tibial trabecular bone.[19] The E2F family transcription factors play a functional role in cell cycle control, and the studies have conclusively shown that E2F1 participates in cell cycle as activator, while E2F7 and E2F8 are its repressors.[20] Taken together, several studies have identified many classical cyclin proteins involved in all stages (G1, S, G2, and M) of cell cycle. Genes like CCNE1, CCNE2, PCNA, and SPDYA are all critical regulators of G1-S phase transition, and RRM2 acts on the S phase of cell cycle,[21,22] while CDCA5 causes cell cycle arrest to inhibit cell survival at the G2/M phase.[23] In addition, there is evidence that CENPN may have correlation with mitosis by regulating centromere-kinetochore assembly.[24] Phox2b affects the sub-type specific properties of the neurons by regulating cell cycle exit,[25] so the role of Phox2b in the chondrocytes still remains an issue of further studies. Furthermore, previous study has confirmed that BRSK2 regulated cell-cycle progression through the ubiquitin-proteasome pathway as an important mitotic kinase.[26] As a well-known pro-inflammatory cytokine, interleukin-1β can promote the expression of genes involved in cell survival and proliferation, and it is known to be able to induce chondrocytes apoptosis in an OA model.[27]
The lncRNAs can affect the expression of adjacent coding genes by cis-pattern, and regulate the distant genes even in different chromosomes by trans-pattern. Therefore, the potential functional actions of the differentially expressed lncRNAs are closely relevant to the biological roles of the mRNAs. As the major member of co-expression network, the SPC24 is also a significant target gene of lnc-CTD-2184D3.4. It is a member of NDC80 complex, which is essential for kinetochore integrity and the accurate segregation of chromosomes during mitosis.[28] As important epigenetic regulation factors in the human genome, the lncRNAs regulate DNA methylation, histone modification, chromatin remodeling and gene silencing or activation by epigenetic, transcriptional and post-transcriptional regulation, and other mechanisms.[29] In addition to the direct involvement in gene expression regulation, the lncRNAs compete to bind microRNA with other RNA transcripts because of the same miRNA response element, thereby, achieving mutual exchange and regulation.[30] lnc-CTD-2184D3.4 is predicted for trans-regulation to SPC24 by miRNA sequestration, which may mean that the lncRNAs are mutually regulated with microRNAs and co-participated in the expression of target genes, leading to the microRNA depletion, as competitive endogenous RNA. Therefore, the study may predict the role of lnc-CTD-2184D3.4 in cell cycle regulation in the chondrocytes. However, further knowledge would be needed to understand their role in OA.
The present study aims at the aberrantly expressed RNAs related to cell cycle not mentioned before in OA, either mRNAs or lncRNAs, and furthermore predicts the potential relationships between lncRNAs and mRNAs, contributing to the exploration of roles of lncRNAs in OA mechanism. Based on this primary study, lnc-CTD-2184D3.4 may regulate SPC24 by miRNA sequestration in mitosis process related to cell cycle.
There are a few previous studies, which have somewhat similar focus on the role of RNA regulation in the pathogenesis of OA. A microarray analysis revealed that 121 lncRNAs up- or down-regulated in OA, but the study did not investigate CNC network or target prediction.[31] Whole-transcriptome sequencing was used to identify differentially expressed mRNAs, lncRNAs and circular RNAs in damaged versus undamaged regions of OA knee cartilage.[32] Even the undamaged areas in OA cartilage are inevitably prone to systemic changes the disease inevitably, the problem which our study avoids by using healthy normal controls. Another study used RNA sequencing to identify changed expressions mRNAs and lncRNAs in synovium.[33] However, the results from synovial cells and chondrocytes cannot be directly compared. Whole exome sequencing was also recently used to identify new candidate genes for OA in Finnish families.[34] However, that study focused on rare exonic variants and, therefore, did not concentrate on the differential expressions of lncRNAs. In conclusion, our knowledge of the relationship of coding and non-coding RNAs is still limited, and further functional studies are important to evaluate their roles and interplay in the pathogenesis of OA.
Author contributions
Experiments: Sijia Tan, Peilin Meng, Feng’e Zhang.
Study design: Xiong Guo, Cuiyan Wu.
Writing – original draft: Feng’e Zhang.
Writing – review & editing: Mikko Juhani Lammi.
Supplementary Material
Supplementary Material
Footnotes
Abbreviations: CNC = coding-non-coding gene co-expression, ECM = extracellular matrix, GO = gene ontology, GSEA = gene set enrichment analysis, lncRNA = long non-coding RNA, OA = osteoarthritis, qRT-PCR = quantitative reverse transcription-polymerase chain reaction.
How to cite this article: Zhang F, Lammi MJ, Tan S, Meng P, Wu C, Guo X. Cell cycle-related lncRNAs and mRNAs in osteoarthritis chondrocytes in a Northwest Chinese Han Population. Medicine. 2020;99:24(e19905).
National Natural Science Foundation of China (81502766, 81620108026).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
- [1].Hiligsmann M, Cooper C, Arden N, et al. Health economics in the field of osteoarthritis: an expert's consensus paper from the European Society for Clinical and Economic Aspects of Osteoporosis and Osteoarthritis (ESCEO). Semin Arthritis Rheum 2013;43:303–13. [DOI] [PubMed] [Google Scholar]
- [2].Cooper C, Javaid MK, Arden N. Epidemiology of Osteoarthritis. In Atlas of Osteoarthritis. Tarporley: Springer Healthcare Ltd; 2014. [Google Scholar]
- [3].Glynjones S, Palmer AJ, Agricola R, et al. Osteoarthritis. Lancet 2015;386:376–87. [DOI] [PubMed] [Google Scholar]
- [4].Birney E, Stamatoyannopoulos JA, et al. ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007;447:799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Perkel JM. Visiting “noncodarnia”. Biotechniques 2013;54:301.303–304. [DOI] [PubMed] [Google Scholar]
- [6].Liu Q, Zhang X, Dai L, et al. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol 2014;66:969–78. [DOI] [PubMed] [Google Scholar]
- [7].Song J, Ahn C, Chun CH, et al. A long non-coding RNA, GAS5, plays a critical role in the regulation of miR-21 during osteoarthritis. J Orthop Res 2014;32:1628–35. [DOI] [PubMed] [Google Scholar]
- [8].Zhang C, Wang P, Jiang P, et al. Upregulation of lncRNA HOTAIR contributes to IL-1(-induced MMP overexpression and chondrocytes apoptosis in temporomandibular joint osteoarthritis. Gene 2016;586:248–53. [DOI] [PubMed] [Google Scholar]
- [9].Ehrich EW, Davies GM, Watson DJ, et al. Minimal perceptible clinical improvement with the Western Ontario and McMaster Universities osteoarthritis index questionnaire and global assessments in patients with osteoarthritis. J Rheumatol 2000;27:2635–41. [PubMed] [Google Scholar]
- [10].Fu M, Huang G, Zhang Z, et al. Expression profile of long noncoding RNAs in cartilage from knee osteoarthritis patients. Osteoarthritis Cartilage 2015;23:423–32. [DOI] [PubMed] [Google Scholar]
- [11].Yu Q, Guo W, Shen J, et al. Effect of glucocorticoids on lncRNA and mRNA expression profiles of the bone microcirculatory endothelial cells from femur head of Homo sapiens. Genom Data 2015;4:140–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Karlsson C, Dehne T, Lindahl A, et al. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthritis Cartilage 2010;18:581–92. [DOI] [PubMed] [Google Scholar]
- [13].McAlindon T, Felson DT. Nutrition: risk factors for osteoarthritis. Ann Rheum Dis 1997;56:397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Suzuki M, Kuroiwa A. Transition of Hox expression during limb cartilage development. Mech Dev 2002;118:241–5. [DOI] [PubMed] [Google Scholar]
- [15].Bialek P, Kern B, Yang X, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell 2004;6:423–35. [DOI] [PubMed] [Google Scholar]
- [16].Ulrich C, Rolauffs B, Abele H, et al. Low osteogenic differentiation potential of placenta-derived mesenchymal stromal cells correlates with low expression of the transcription factors Runx2 and Twist2. Stem Cells Dev 2013;22:2859–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Danciu TE, Li Y, Koh A, et al. The basic helix loop helix transcription factor Twist1 is a novel regulator of ATF4 in osteoblasts. J Cell Biochem 2012;113:70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wissing M, Sonne S, Westergaard D, et al. The transcriptome of corona radiata cells from individual MII oocytes that after ICSI developed to embryos selected for transfer: PCOS women compared to healthy women. J Ovarian Res 2014;7:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Qin L, Tamasi J, Raggatt L, et al. Amphiregulin is a novel growth factor involved in normal bone development and in the cellular response to parathyroid hormone stimulation. J Biol Chem 2005;280:3974–81. [DOI] [PubMed] [Google Scholar]
- [20].Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009;9:785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Whitfield ML, Sherlock G, Saldanha AJ, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell 2002;13:1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Porter LA, Kong-Beltran M, Donoghue DJ. Spy1 interacts with p27Kip1 to allow G1/S progression. Mol Biol Cell 2003;14:3664–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shen Z, Yu X, Zheng Y, et al. CDCA5 regulates proliferation in hepatocellular carcinoma and has potential as a negative prognostic marker. Onco Targets Ther 2018;11:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dutertre M, Chakrama FZ, Combe E, et al. A recently evolved class of alternative 3′-terminal exons involved in cell cycle regulation by topoisomerase inhibitors. Nat Commun 2014;5:3395. [DOI] [PubMed] [Google Scholar]
- [25].Dubreuil V, Hirsch MR, Pattyn A, et al. The Phox2b transcription factor coordinately regulates neuronal cell cycle exit and identity. Development 2000;127:5191–201. [DOI] [PubMed] [Google Scholar]
- [26].Li R, Wan B, Zhou J, et al. APC/CCdh1 targets brain-specific kinase 2 (BRSK2) for degradation via the ubiquitin-proteasome pathway. PLoS ONE 2012; 7: e45932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhou PH, Liu SQ, Peng H. The effect of hyaluronic acid on IL-1β-induced chondrocyte apoptosis in a rat model of osteoarthritis. J Orthop Res 2008;26:1643–8. [DOI] [PubMed] [Google Scholar]
- [28].McCleland ML, Kallio MJ, Barrettwilt GA, et al. The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochore-microtubule attachment. Curr Biol 2004;14:131–7. [DOI] [PubMed] [Google Scholar]
- [29].Chen LL, Carmichael GG. Decoding the function of nuclear long non-coding RNAs. Curr Opin Cell Biol 2010;22:357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell 2011;146:353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Xing D, Liang JQ, Li Y, et al. Identification of long noncoding RNA associated with osteoarthritis in humans. Orthop Surg 2014;6:288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li H, Yang HH, Sun ZG, et al. Whole-transcriptome sequencing of knee joint cartilage from osteoarthritis patients. Bone Joint Res 2019;8:288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Xiang S, Li Z, Bian Y, et al. Identification of canged expression of mRNAs and lncRNAs in osteoarthritic synovium by RNA-sequencing. Gene 2019;15:55–61. [DOI] [PubMed] [Google Scholar]
- [34].Skarp S, Kämäräinen OP, Wei GH, et al. Whole exome sequencing in Finnish families identifies new candidate genes for osteoarthritis. PLoS One 2018;13:e0203313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.