

Abstract
Parasympathetic neurons in the airways control bronchomotor tone. Increased activity of cholinergic neurons are mediators of airway hyperresponsiveness (AHR) in asthma, however, mechanisms are not elucidated. We describe remodeling of the cholinergic neuronal network in asthmatic airways driven by brain‐derived neurotrophic factor (BDNF) and Tropomyosin receptor kinase B (TrkB). Human bronchial biopsies were stained for cholinergic marker vesicular acetylcholine transporter (VAChT). Human lung gene expression and single nucleotide polymorphisms (SNP) in neuroplasticity‐related genes were compared between asthma and healthy patients. Wild‐type (WT) and mutated TrkB knock‐in mice (Ntrk2tm1Ddg/J) with impaired BDNF signaling were chronically exposed to ovalbumin (OVA). Neuronal VAChT staining and airway narrowing in response to electrical field stimulation in precision cut lung slices (PCLS) were assessed. Increased cholinergic fibers in asthmatic airway biopsies was found, paralleled by increased TrkB gene expression in human lung tissue, and SNPs in the NTRK2 [TrkB] and BDNF genes linked to asthma. Chronic allergen exposure in mice resulted in increased density of cholinergic nerves, which was prevented by inhibiting TrkB. Increased nerve density resulted in AHR in vivo and in increased nerve‐dependent airway reactivity in lung slices mediated via TrkB. These findings show cholinergic neuroplasticity in asthma driven by TrkB signaling and suggest that the BDNF‐TrkB pathway may be a potential target.
Keywords: asthma, lung innervation, neuroplasticity, neurotrophins
Abbreviations
- AHR
airway hyperresponsiveness
- BDNF
brain‐derived neurotrophic factor
- EFS
electric field stimulation
- OVA
ovalbumin
- PCLS
precision cut lung slices
- TrkB
tropomyosin receptor kinase B
- VAChT
vesicular acetylcholine transporter
- α‐SMA
alpha‐smooth muscle actin
1. INTRODUCTION
Asthma is a highly prevalent chronic inflammatory airway disease that imposes significant healthcare and financial burden. 1 While there has been much focus on the inflammatory aspects of asthma and on the roles of resident airway cells, it is important to recall neuronal control of airway tone and contractility. Indeed, the peripheral and central nervous systems play an important role in the symptoms experienced by patients with allergic asthma, such as airway hyperresponsiveness (AHR), cough, and mucus hypersecretion. 2 , 3 The functional effects of efferent nerves in the airways are mainly mediated by postganglionic parasympathetic cholinergic neurons that contract airway smooth muscle (ASM) while nonadrenergic, noncholinergic (NANC) fibers can induce bronchodilation via NO and VIP. 4 Increased cholinergic tone has been long recognized as a major contributor to persistent bronchoconstriction and AHR in asthmatic patients. 5 This is supported by more recent clinical data of long‐acting anticholinergics as effective bronchodilators in asthma. 6 , 7 ACh can contribute to AHR not only via ASM contraction, but also by inducing airway inflammation and structural remodeling via the muscarinic M3 receptor. 8 , 9 , 10 , 11 While the downstream effects of enhanced neuronal activity on airway structure/function have been explored, the upstream mechanisms by which neuronal plasticity occurs are less known. 12
Acute sensitization of sensory nerves via changes in excitability, activation threshold, or transmission have been described extensively in asthma. 3 Tracheal segments of antigen‐exposed animals display increased ACh release and increased contraction in response to electric field stimulation (EFS). 13 , 14 Recently, increased branching of sensory nerves in the airways has been demonstrated to occur in human asthma and to correlate with disease severity. 15 This suggests that airway neurons not only participate in transient acute exacerbations, but neuronal architecture remodeling accompanies persistent AHR. However, it is entirely unknown whether plasticity of cholinergic nerves occurs in asthma. 12
Neurotrophins (NTs) are growth factors that regulate neuron survival, differentiation, and function. 16 Brain‐derived neurotrophic factor (BDNF) and its high affinity receptor Tropomyosin receptor kinase B (TrkB) are important effectors in asthmatic airways. 12 , 17 The expression of BDNF is increased in the lungs after allergen challenge in murine models. 18 , 19 Also, in humans, the expression of BDNF in sputum and bronchial samples correlates with asthma severity. 20 The TrkB receptor is expressed in airway nerve endings 18 and is crucial for the regulation of airway innervation induced by perinatal inflammation. 19 Moreover, BDNF/TrkB signaling contributes to AHR in animal models of asthma. 21 , 22
We hypothesized that cholinergic airway nerves undergo plasticity in asthma driven by BDNF/TrkB signaling and propose a new mechanism via which ACh release is increased in affected airways. Here, using human bronchial biopsies and mouse lung tissue, we show there is a higher cholinergic nerve density in asthma. In samples from asthma patients, we found increased TrkB gene expression and SNPs in BDNF and NRTK2 (TRKB) genes correlated to asthma susceptibility. Furthermore, using animals carrying mutated TrkB receptors, we demonstrate that increased nerve density and AHR depends on TrkB signaling.
2. MATERIALS AND METHODS
2.1. Human bronchial biopsies
Bronchial biopsies were obtained from subjects visiting the University Medical Center Groningen (UMCG, The Netherlands) and collected from the right or left lower lobe, between generation 3 and 6 of the bronchial tree. The samples were immediately embedded in paraffin and stored at −80°C. All patients originated from cohorts investigated are described earlier. 23 , 24 All patients had a previous doctors' diagnosis of asthma, documented bronchodilator reversibility and AHR to histamine (PC20 = <32 mg/mL). The control cohort of respiratory healthy individuals was derived from the Study to Obtain Normal Values of Inflammatory Variables From Healthy Subjects (NORM; NCT00848406). In this study, current smokers and never smokers older than 18 years were recruited. Patients were considered respiratory healthy if they had no respiratory symptoms, no history of respiratory disease and normal pulmonary function. Normal pulmonary function was defined as a postbronchodilator FEV1/FVC higher than lower limit of normal, absence of AHR to methacholine (PC20 < 16 mg/mL) and absence of reversibility (FEV1% predicted to salbutamol <10%). Patients were excluded if they used inhaled or oral corticosteroids within the last 5 years, or during 5 years of their lives. All patients gave their written informed consent. A table of patient characteristics can be found in the Supporting Information (Table S1).
2.2. Staining of human bronchial biopsies
In total, 68 biopsies were included; 31 current asthma patients and 37 healthy individuals. The biopsies were cut at 12 µm thickness, dried for 30 minutes and fixed with buffered PFA. Next, they were blocked during 2 hours with a solution consisting of 5% of donkey serum, 0.3 M glycine, and 1% of milk. Sections were incubated with primary antibody against vesicular acetylcholine transporter (VAChT) (1:100 dilution, Synaptic Systems) and α‐smooth muscle‐actin (α‐SMA) (1:1000 dilution, Sigma Aldrich) overnight at 4°C. Fluorophore conjugated secondary antibodies were added during 90 minutes at room temperature and slides were mounted with ProLongTMGold antifade reagent (Invitrogen). Fields containing smooth muscle area positively stained for α‐SMA were selected and VAChT + nerve fibers quantified in these regions. Images were acquired using Leica DM4000 B microscope coupled with Leica DFC 345FX camera. VAChT positive area was quantified using ImageJ software (NIH) and normalized by α‐SMA area in each individual image. Both acquisition and quantification of the images were performed blindly.
2.3. Gene expression in human bronchial biopsies
RNA sequencing data were obtained from 173 biopsies; 96 asthma patients and 77 healthy individuals, as described previously. 25 In short, total RNA was extracted using AllPrep DNA/RNA Mini kit (Qiagen) according to the manufacturer's instructions. RNA samples were dissolved in 30 μL RNase‐free water and concentration and quality of RNA were checked using a NanoDrop‐1000. RNA samples were further processed using the TruSeq Stranded Total RNA Sample Preparation Kit (Illumina) and the obtained cDNA fragment libraries were run on an Illumina HiSeq2500 sequencer. A table of patient characteristic can be found in the Supporting Information (Table S1).
A linear model was fitted to neurotrophins (NT‐3, NT‐4, BDNF) and their receptors (TrkA [NRTK1], TrkB [NRTK2], TrkC [NRTK3], and p75NTR) gene expression derived from RNA sequencing in bronchial biopsies (expressed as fragments per kilobase of transcript per million mapped reads, FPKM). Age, gender, and smoking status were used as correction factors. All analyses were conducted using R (version 3.3.2).
2.4. Genetic association
Genetic association studies for NTRK2 and BDNF were performed in the Dutch Asthma GWAS (DAG) cohort; a large cohort of asthma patients, which was described previously. 26 , 27 In short, the cohort consists of in total 920 asthma cases and 980 controls from the Northern Netherlands. The DAG cohort was genotyped in two phases and meta‐analyzed afterwards. All asthmatics had a physician's diagnosis of asthma, asthma symptoms, and BHR to either histamine or methacholine. Control subjects had neither asthma or COPD, nor any evidence of significant airway obstruction. All studies were approved by the medical ethical committee. DNA of subjects from DAG I was genotyped on the Illumina 317 Chip, asthmatics of DAG II with the Illumina 370 Duo Chip and the control subjects of DAG II with Illumina Human Hap 600 (Illumina Inc). SNPs in the BDNF and NTRK2 gene were analyzed using a logistic regression with an additive genetic model, and DAG I and II were meta‐analyzed using CMA software. Values were adjusted for principal components (population stratification‐adjustment) and SNPs with adjusted P‐values P < .05 were considered statistically significant. Lung eQTLs were analyzed per significant SNP using the online tool www.gtexportal.org and the direction of gene expression noted.
2.5. Animals
BALB/c mice were purchased from Charles River. Breeding colonies of Ntrk2tm1Ddg/J mice (TrkB knock‐in mice, stock no. 022363) were purchased from the Jackson Laboratories (Bar Harbor, ME, USA). These animals have a mutated ATP‐binding cassette (TrkBF616A) that can be selectively blocked by the addition of the drug 1NMPP1. The drug does not have effect on normal TrkB tyrosine kinase activity and does not affect its expression. 19 , 22 Female animals from 8 to 12 weeks old were used for all experiments. Animals were housed under 12‐h light:dark cycle and were fed and received water ad libitum.
2.6. Acute and chronic asthma protocols
Animals were sensitized to ovalbumin (OVA) (Sigma‐ Aldrich) on days 1, 14, and 21 by intraperitoneal injection of 10 μg of OVA emulsified in 1.5 mg aluminum hydroxide (Aluminject; Pierce Chemical) and diluted to 200 μL with PBS. Subsequently, mice were challenged with saline or OVA aerosols (1% in PBS) for 20 minutes during 3 consecutive days (OVA acute) or twice weekly on consecutive days for 4 weeks (OVA chronic). The aerosol was delivered to a Perspex exposure chamber (9 liter) by a Pari Boy SX nebulizer driven by an airflow of 40 L/min, providing an aerosol with an output of 0.33 mL/min, as described previously. 28 To block TrkB tyrosine kinase activity, 1NMPP1 (20 μM, Millipore) in 0.2% of DMSO was added to the drinking water one week before the first allergen challenge up until the end of the protocol. Control animals received water supplemented with 0.2% of DMSO.
2.7. In vivo airway hyperresponsiveness
Lung function measurements were collected by using flexiVent (Scireq, Canada) invasive airway mechanics system and data were collected by using FlexiWare software Version 7.6, Service Pack 3. At 24 hours after the last challenge, animals were anesthetized by intraperitoneal injection of ketamine (75 mg/kg, Alphasan Nederland BV.) and metomidine (1 mg/kg, Domitor RT, Orion pharma). Following anesthesia, the animals had their tracheas cannulated and lungs ventilated at a frequency of 300 breaths/min and a tidal volume of 10 mL/kg. To promote muscle relaxation, rocuronium bromide was given subcutaneously. Anesthesia was maintained via subcutaneous administration of ketamine/domitor (1/4 of initial dose). Increasing doses of methacholine (3‐50 mg/mL, Sigma‐Aldrich) were given via a nebulizer to assess airway resistance and compliance.
2.8. Vagotomy
In a separated set of animals, bilateral vagotomy was performed by separating the vagus nerve from the carotid artery and sectioning it at the level of the trachea, prior to lung function measurement.
2.9. Lung harvesting
After euthanasia and exsanguination, lungs were harvested. The left lobe was removed and snap frozen for molecular analysis. The remaining lung was inflated with 600 μL of 4% buffered PFA and fixed overnight immersed in the same solution at 4°C. The lungs were washed by continuously replacing a 0.1 M phosphate buffer solution during 48 hours. The superior right lobe was later embedded in paraffin for immunohistochemistry. The middle and inferior right lobes were cryoprotected overnight in 18% of sucrose solution and snap frozen in liquid nitrogen for immunohistochemistry.
2.10. Eosinophil staining
Cryosections of 5 µm thickness were stained with diaminobenzidine for cyanide‐resistant endogenous peroxidase activity. The number of eosinophils around the airways were counted and adjusted for the area (mm2) of the basement membrane.
2.11. Immunohistochemistry for α‐SMA
Cryosections of 5 µm thickness were stained for α‐SMA (1:1000 dilution. Sigma‐Aldrich, Zwijndrecht, The Netherlands) overnight and visualized by staining with horseradish‐peroxidase‐linked secondary antibody and diaminobenzidine. Airways were photographed and the area positively stained (mm2) was calculated and adjusted by the area (mm2) of the basal membrane.
2.12. Immunofluorescence for PGP 9.5 and VAChT in mouse lung sections
Cryosections of 12 µm thickness were processed as described previously for human biopsies. Primary antibodies used were PGP 9.5 (1:500 dilution. GTX109637; GeneTEx); and VAChT (1:100 dilution, Synaptic Systems). PGP 9.5 is characterized and consolidated in the literature as a specific pan‐neuronal marker in multiple studies, especially useful for the staining of peripheral neuronal fibers. 29 , 30 VAChT is consolidated in literature to assess cholinergic innervation. This specific antibody was validated in several studies in the literature. 31 , 32 , 33
Images were acquired using Leica DM4000 B microscope coupled with Leica DFC 345FX camera. Quantification was made using ImageJ software (NIH), where 5 airways from each animal were analyzed for positive staining area (mm2) and adjusted by basement membrane area (mm2). Both acquisition and quantification of the images were performed blindly.
2.13. Precision cut lung slices preparation
A different set of animals was used to prepare precision cut lung slices (PCLS), as described previously. 34 Briefly, following euthanasia with ketamine/domitor, the trachea was cannulated and the lungs filled with a buffered 1.5% of low melting‐point agarose solution. Subsequently, lungs were placed on ice for 15 minutes, in order to solidify, the agarose and individual lobes were separated for slicing. PCLS were prepared from the lobes by slicing using a tissue slicer (CompresstomeTM VF‐300 microtome, Precisionary Instruments). PCLSs were generated at a thickness of 250 μm. After washing, they were incubated in Dulbecco's Modification of Eagle's Medium (DMEM) supplemented with sodium pyruvate (1 mM), nonessential amino acid mixture (1:100), gentamycin (45 mg/mL), penicillin (100 U/mL), streptomycin (100 mg/mL), and amphotericin B (1.5 mg/mL) at 37°C in a humidified chamber under 5% of CO2/95%, as described previously. 34 , 35
2.14. Ex vivo airway hyperresponsiveness
Twenty four hours after incubation, the PCLS were used for airway narrowing studies using methacholine or EFS. Briefly, methacholine dose response curves (10‐9 M‐10‐2 M) were recorded by placing the slice in 1 mL medium and fixing it with a nylon mesh and metal washer as described previously. 34 , 36 For EFS measurements, the slices were inserted in 2 mL medium between two platinum electrodes, which generate increasing frequencies of electric pulses. Biphasic EFS (200 mA, 2 ms, 4 seconds) was applied and frequency response curves (1‐125 Hz in doubling steps) were recorded by stimulating every 2 minutes. Lung slice images were captured in time‐lapse (1 frame per 2 seconds) using an inverted phase contrast microscope (Eclipse, TS100; Nikon). Airway luminal area was quantified using image acquisition software (NIS‐elements; Nikon). Luminal area is expressed as percent of basal.
2.15. Western blot for neurofilament in lung tissue
Protein homogenates were generated by pulverizing lung slices in liquid nitrogen and subsequent sonication in SDS lysis buffer containing protease inhibitors. Protein concentration was determined by Pierce BCA kit (Thermo Scientific, USA) and equal amounts of protein were submitted to electrophoresis in polyacrylamide gel. Proteins were transferred to nitrocellulose membranes and incubated with primary antibody against neurofilament (1:500 dilution. Merck) overnight and HRP conjugated secondary antibody during 2 hours before visualization in G‐box gel documentation system (Syngene, UK).
2.16. ELISA for BDNF in lung tissue
Protein homogenates generated from lung slices were submitted to ELISA quantification of BDNF following the instructions of the commercial kit (DBD00, R&D systems).
2.17. Ethical statement
Human tissue samples from asthmatic individuals were obtained in studies approved by local ethics committee (METc 2001/074, METc 2004/271, and METc 2009/007) in University Medical Center Groningen (UMCG, The Netherlands). The tissue from control healthy individuals of respiratory healthy individuals was derived from the Study to Obtain Normal Values of Inflammatory Variables From Healthy Subjects (NORM; NCT00848406).
All animal experiments were performed in accordance with the national guidelines and approved by the University of Groningen Committee for Animal Experimentation (CCD number: AVD105002016429).
2.18. Statistics
Data are represented as mean ± SEM. The statistical significance of differences between means of groups were determined by appropriate statistical tests as indicated in the text. Statistical analysis was made in GraphPad Prism version 8.0.0 (GraphPad Software) for all animal experiments. For differential gene expression analysis, DESeq2 package was used. For SNP discovery using GWAS, Plink v1.07 was used and CMA was used to perform the meta‐analysis. Differences were considered significant when P value < .05.
3. RESULTS
3.1. Cholinergic innervation is higher in human asthma
To study cholinergic neuroplasticity in asthma, we used sections of bronchial biopsies derived from patients currently diagnosed with asthma and healthy controls. Sections were stained for the cholinergic marker VAChT and normalized by the area of the smooth muscle marker alpha‐smooth muscle actin (α‐SMA). No difference in α‐SMA area was observed in this data set between healthy controls and asthmatics.
We observed that patients with a current asthma diagnosis display 1.9‐fold higher VAChT+ nerve density innervating ASM as compared to healthy subjects (Figure 1A,B), indicating cholinergic neuroplasticity in asthma. VAChT+ area was not correlated with eosinophil levels in blood or biopsies or with forced expiratory volume in one second (FEV1) (Figure S1).
FIGURE 1.

Human asthma presents higher VAChT+ nerves and TrkB gene expression. A, Representative images of biopsies taken from asthma and healthy controls. Fluorescently labeled α‐SMA is stained green (Alexa Fluor 488) and VAChT is stained red (Cy3). Blue arrow represents a VAChT+ nerve fiber. Scale bar = 50 µm. B, Area of VAChT+ nerve fibers normalized by α‐SMA area in human bronchial samples from current asthma patients (n = 31) and healthy controls (n = 37). C, Total raw counts of TrkB expression in current asthma (n = 96) and healthy controls (n = 77). D, Volcano plot showing the differential expression of neurotrophins (BDNF, NT‐3, NT4) and their receptors (TrkA [NRTK1], TrkB [NRTK2], TrkC [NRTK3], and p75NTR [NGFR]) adjusted by age, smoking status, and gender. Data shown as mean ± SEM. The differences were tested for statistical significance using two‐tailed Student's t test. *P < .05. FC, fold change; VAChT, vesicular acetylcholine transporter; α‐SMA, alpha‐smooth muscle actin
3.2. Asthma is correlated with increased TrkB gene expression
Using differential gene expression analysis from RNAseq data derived from patient biopsies with asthma and healthy controls, we assessed expression of neurotrophins (NGF, BDNF, NT‐3, NT‐4) and their receptors (TrkA [NRTK1], TrkB [NRTK2], TrkC [NRTK3], and p75NTR [NGFR]).
Total raw count for TrkB was higher in asthma patients compared to healthy controls (Figure 1C). After adjusting for age, gender, and smoking status as typical confounders, a volcano plot was generated comparing fold change of gene expression. Also after adjustment, TrkB expression was found higher in asthma compared to healthy controls, with no differences for other neurotrophins or receptors (Figure 1D, Table S2). The expression of BDNF was similar between both groups.
3.3. SNPS in NRTK2 and BDNF correlate with asthma
We searched for associations between SNPs in the BDNF and NRTK2 (TrkB) genes with asthma susceptibility using the Dutch Asthma GWAS (DAG) cohorts. Both DAG I and DAG II were screened separately and a meta‐analysis was performed afterwards.
The meta‐analysis showed 5 SNPs in the BDNF and BDNF‐AS genes and 1 SNP in the NRTK2 gene that were significantly associated with asthma susceptibility (Table 1; Table S3). Interestingly, 5 out of these 6 SNPs were eQTLs, of which 2 were lung‐specific eQTLs. For 4 out of the 5 eQTLs, the risk allele was associated with higher BDNF gene expression or lower expression of the antisense BDNF (BDNF‐AS). 37
TABLE 1.
Significant SNPs found in BDNF/BDNF‐AS and TrkB genes after meta‐analysis of two asthma cohorts GWAS (DAG1 and DAG2) vs healthy individuals
| Human Gene | SNP | Allele | Type | Meta‐analysis GWAS1 + GWAS2 | ||
|---|---|---|---|---|---|---|
| OR | Z | Adjusted P‐value | ||||
| BDNF‐AS | rs10835189 | T > G | Intron variant | 0.873693109 | −2.046 | .04075564 |
| BDNF‐AS | rs7481311 | C > T | Intron variant | 0.818444221 | −2.6575 | .007873283 |
| BDNF‐AS | rs10767652 | A > G | Intron variant | 0.873482556 | −2.0454 | .04081459 |
| BDNF‐AS | rs10835211 | G > A | Intron variant | 0.833161232 | −2.5127 | .011981222 |
| BDNF | rs7934165 | G > A | Intron variant | 1.140697219 | 2.02066 | .043314615 |
| TrkB | rs2034107 | G > T | Intron variant | 0.804784012 | −2.042 | .041149236 |
Abbreviations: OR, odds ratio; Z, z‐score.
3.4. BDNF production is increased in OVA‐exposed mice
Next, we used mouse models to explore mechanistically how allergen exposure can induce cholinergic neuroplasticity. Airway remodeling, persistent inflammation, and increased concentration of neurotrophins are characteristics of asthma. 18 Using Balb/c mice sensitized to OVA, we assessed how acute (OVA acute; 3 times) and chronic (OVA chronic; 8 times) inhalation of OVA impact these parameters (Figure 2A).
FIGURE 2.
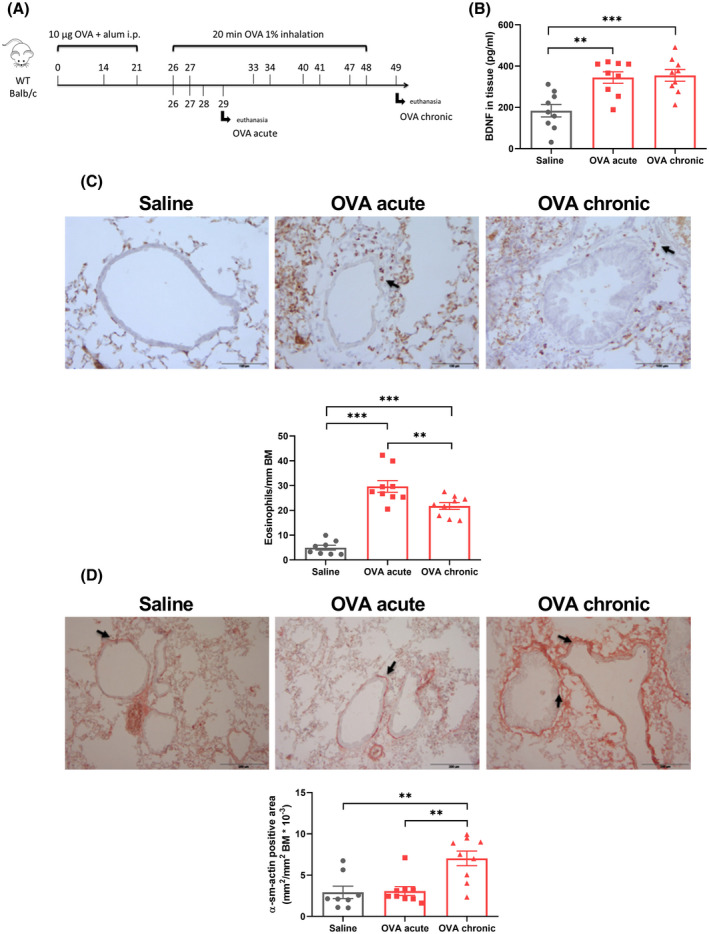
Chronic OVA exposure induces BDNF expression, airway inflammation remodeling and neuronal density. A, WT Balb/c mice were sensitized to OVA via ip injections and challenged via OVA inhalation 3 times (OVA acute) or 8 times (OVA chronic). After 24 hours of the last challenge, the animals were sacrificed and the lungs isolated. B, BDNF concentration in lung tissue measured by ELISA. C, Eosinophil number surrounding the airways normalized by the basement membrane area. Black arrows show positive staining. Scale bar = 100 µm. D, α‐SMA positive area surrounding the airways normalized by basement membrane area. Black arrows show positive staining. Scale bar = 200 µm. Data shown as mean ± SEM. The differences were tested for statistical significance using one‐way analysis of variance (ANOVA) followed by Tukey post hoc test. **P < .01; ***P < .001. BDNF, brain‐derived neurotrophic factor; BM, basement area; OVA, ovalbumin; α‐SMA, alpha‐smooth muscle actin
ELISA showed heightened BDNF protein expression in lung tissue, both after acute and after chronic OVA exposure (Figure 2B). Both acute and chronic OVA exposures‐induced airway eosinophilia (Figure 2C‐D). This increase was greater in the acute model compared to the chronic model, showing that the migration of eosinophils to the lungs is quenched after prolonged inflammatory insult. In contrast, an increased smooth muscle mass was seen in the chronic model but not in the acute model, as assessed by α‐SMA staining (Figure 2E‐F), indicating airway remodeling.
3.5. Airway innervation is higher in chronic OVA‐exposed mice
Based on the increased lung BDNF, we determined whether such increases translate to differences in airway innervation. Using tagged PGP 9.5 (pan‐neuronal marker), we found increased presence of nerve fibers surrounding the airways after chronic OVA exposure, but not after acute OVA exposure (Figure 3A‐B). To confirm this result, we performed Western blot for the neuronal marker neurofilament, which was found to be increased in lung homogenates after chronic OVA exposure (Figure 3C), demonstrating increased neuronal density.
FIGURE 3.
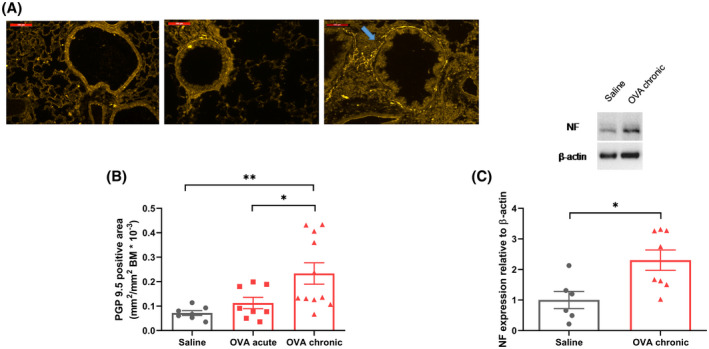
Chronic, but not acute, OVA exposure leads to higher nerve density surrounding the airways. A, Representative image showing fluorescently labeled neuronal marker PGP 9.5 stained with Alexa Fluor 488. Blue arrow indicates a positive fiber. Scale bar = 100 µm. B, Quantification of the neuronal marker PGP 9.5+ area surrounding the airways normalized by the basement membrane area. C, Quantification of neurofilament expression by western blot and representative bands. Data shown as mean ± SEM. The differences were tested for statistical significance in (B) using one‐way analysis of variance (ANOVA) followed by Tukey post hoc test and in (C) using Mann‐Whitney test. *P < .05; **P < .01. BM, basement area; NF, neurofilament; OVA, ovalbumin
3.6. Airway hyperresponsiveness is inhibited by ablation of nerves (vagotomy) in vivo
To assess functional consequences of neuronal remodeling to AHR, we measured airway responsiveness to methacholine in vivo. Acute OVA exposure induced significantly heightened airway resistance and tissue elastance compared to control mice, which was further enhanced after chronic OVA exposure (Figure 4A,B). To verify this difference could be attributed to altered neuronal function, vagotomy was performed prior to methacholine stimulation. Vagotomy prevented the rise in airway resistance and elastance seen after chronic OVA exposure, returning their levels to those observed after acute OVA exposure (Figure 4C,D). Vagotomy did not affect the resistance and elastance values after acute OVA exposure or in the control group, showing that neuronal mediated AHR only occurs after chronic allergen exposure.
FIGURE 4.

Vagotomy prevents chronic OVA exposure‐induced AHR. Animals were cannulated and lung mechanical parameters and dose‐response curves to methacholine were generated using the flexiVent system (A) Airways resistance and (B) tissue elastance in response to increasing doses of nebulized methacholine. (C) Airways resistance and (D) tissue elastance in response to increasing doses of nebulized methacholine after vagotomy is performed. Data shown as mean ± SEM. The differences were tested for statistical significance using two‐way ANOVA followed by Tukey post hoc test. **P < .01; ***P < .001; ****P < .0001 vs saline. # P < .05; ### P < .001 vs OVA acute. AHR, airway hyperresponsiveness; EFS, electric field stimulation; OVA, ovalbumin; PCLS, precision cut lung slices
3.7. Ex vivo electrical, but not methacholine, stimulation reveals AHR in PCLS
To explore the mechanism via which airway neuronal remodeling results in AHR, we applied EFS to PCLS. This model is known to induce airway contraction depending mainly on ACh release via postganglionic vagal parasympathetic neuron depolarization. 38 EFS‐mediated airway contraction was prevented when slices were treated with atropine (1 uM), hexamethonium (5 uM), and lidocaine (1 mM), indicating a specific neuronal response (data not shown). To take into account the effect of direct smooth muscle contraction via M3 receptor activation, methacholine dose‐response curves were also performed in slices from the same animals.
For methacholine, no differences in the concentration‐dependent reduction of airway lumen were found between the groups (Figure 5A). However, when EFS was applied, AHR was observed after OVA exposure. Compared to saline, both acute and chronic OVA groups displayed increased airway narrowing in response to increasing frequencies of EFS. Chronic OVA exposure induced even further increased airway narrowing in comparison to acute OVA exposure (Figure 5B).
FIGURE 5.
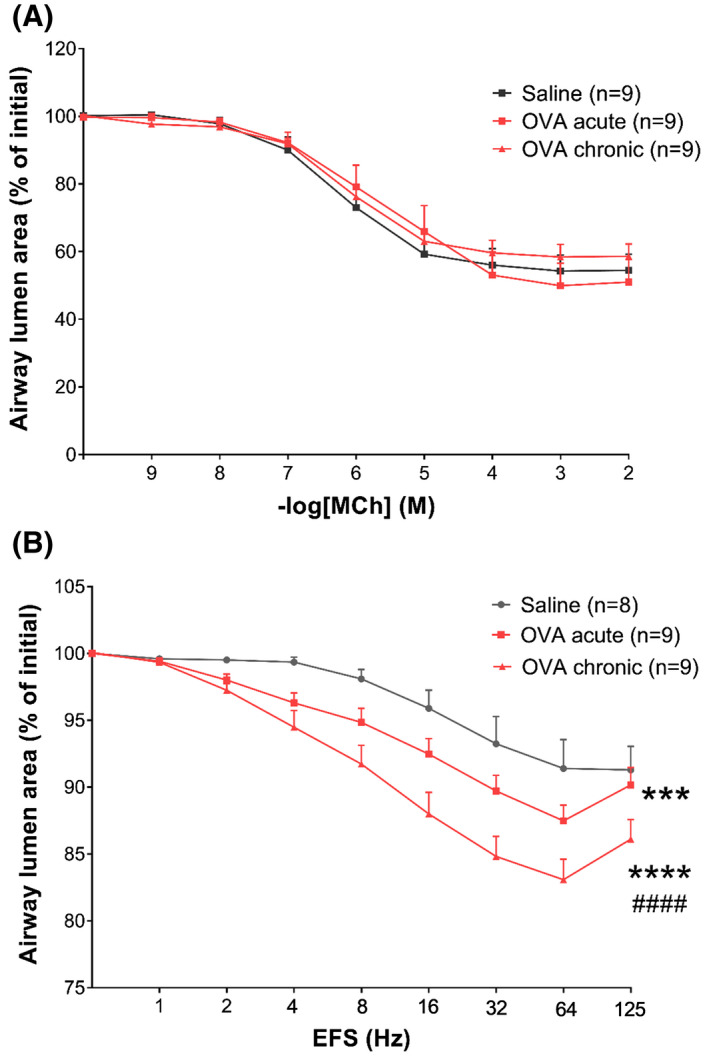
Chronic OVA exposure leads to increased neuronal mediated AHR. A, Airway lumen narrowing in response to increasing doses of methacholine in PCLS. B, Airway lumen narrowing in response to increasing frequencies of EFS in PCLS. Data shown as mean ± SEM. The differences were tested for statistical significance using two‐way analysis of variance (ANOVA) followed by Tukey post hoc test. ***P < .001; ****P < .0001 vs saline. #### P < .0001 vs OVA acute. AHR, airway hyperresponsiveness; EFS, electric field stimulation; OVA, ovalbumin; PCLS, precision cut lung slices
3.8. Disrupting TrkB signaling does not reduce eosinophilic inflammation or structural remodeling
To assess the functional role of BDNF/TrkB signaling in cholinergic neuronal plasticity, a transgenic mouse line carrying a mutated TrkB receptor (TrkBF616A) was used, the tyrosine kinase activity of which can be conditionally and reversibly inhibited by the addition of the drug 1NMPP1. 19 , 22 Animals were exposed to chronic saline (vehicle/saline and 1NMPP1/saline) or OVA (vehicle/OVA and 1NMPP1/OVA) (Figure 6A). 1NMPP1/OVA mice developed similar eosinophilic inflammation (Figure 6B) and smooth muscle mass hypertrophy (Figure 6C) as vehicle/OVA mice.
FIGURE 6.
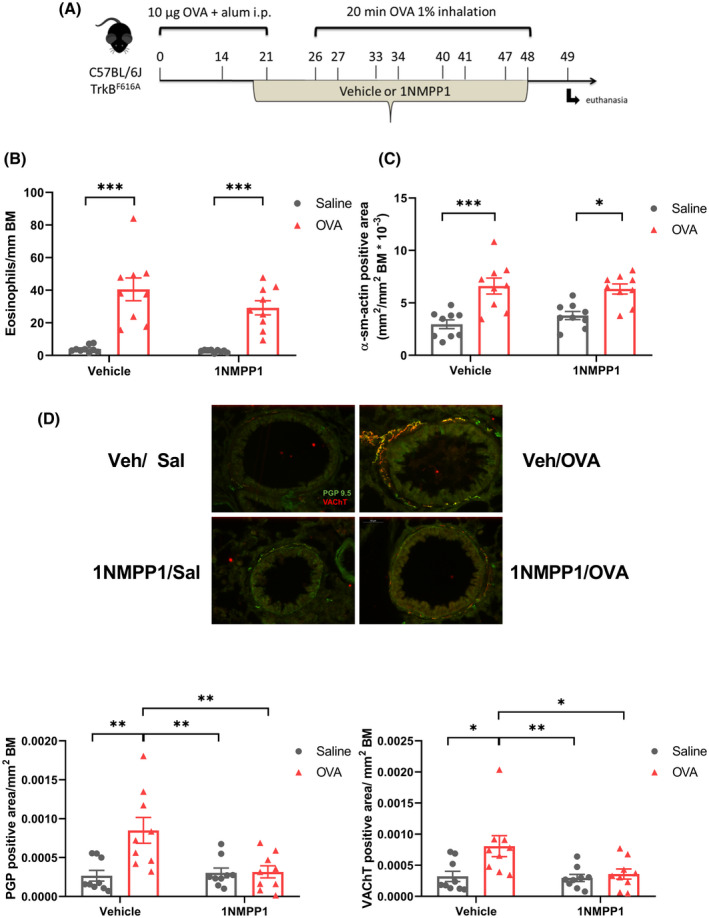
TrkB signaling disruption prevents airway neuronal plasticity but has no effect on inflammation and structural remodeling. A, TrkBF616A mice were submitted to the same protocol described in Figure 2A. A solution of 20 µM of 1NMPP1 was added to the drinking water 1 week before the first challenge with saline (1NMPP1/saline) or OVA (1NMPP1/OVA). Control animals received vehicle (0.2% of DMSO) and were challenged with saline (vehicle/saline) or OVA (vehicle/OVA). B, Eosinophil number surrounding the airways normalized by the basement membrane area. C, α‐SMA positive area surrounding the airways normalized by basement membrane area. D, Quantification of PGP 9.5+ and VAChT+ area surrounding the airways normalized by the basement membrane area. Representative images showing fluorescently labeled PGP 9.5 stained with Alexa Fluor 488 in green and VAChT labeled with Cy3 in red as a merged image. Scale bar 50µm. Data shown as mean ± SEM. The differences were tested for statistical significance using two‐way analysis of variance (ANOVA) followed by Tukey post hoc test. *P < .05; **P < .01; ***P < .001. AHR, airway hyperresponsiveness; OVA, ovalbumin; VAChT, vesicular acetylcholine transporter; α‐SMA, alpha‐smooth muscle actin
3.9. Increased cholinergic innervation in the airways is prevented by disrupting TrkB signaling
Lung sections were prepared and stained for the pan‐neuronal marker PGP 9.5 and the cholinergic marker VAChT. OVA‐exposed animals treated with vehicle (for 1NMPP1, 0.2% of DMSO) displayed increased PGP 9.5 area compared to vehicle/saline and 1NMMP1/saline groups, in line with the data from Balb/c mice (Figure 6D,E). Furthermore, an increase in VAChT+ nerves surrounding the airways, compared to vehicle/saline and 1NMMP1/saline groups was observed, indicative of cholinergic neuroplasticity (Figure 6D,F). Interestingly, this was prevented by disrupting BDNF/TrkB signaling (Figure 6D‐F). Thus, a complete inhibition of PGP+ VAChT+ area compared to vehicle/OVA was found in the 1NMPP1/OVA group. This indicates that TrkB is crucial for the development of increased neuronal branching in response to chronic OVA exposure.
3.10. Disrupting TrkB signaling prevents AHR in lung slices
To assess the functional consequences of preventing neuronal remodeling regarding AHR, slices from TrkBF616A animals submitted to chronic saline and OVA exposure were prepared and analyzed for airway contraction to MCh or EFS. In line with studies from Balb/c mice, no differences in contraction in response to MCh were observed (Figure 7A). Slices from Vehicle/OVA animals responded with greater luminal reduction to increasing electric frequencies compared to Vehicle/Saline or 1NMPP1/Saline, as previously observed. Airways from 1NMPP1/OVA mice displayed similar contraction in response to EFS as the saline exposed groups (Figure 7B), indicating that increased cholinergic nerve density via TrkB signaling is necessary for the AHR seen after chronic OVA exposure.
FIGURE 7.
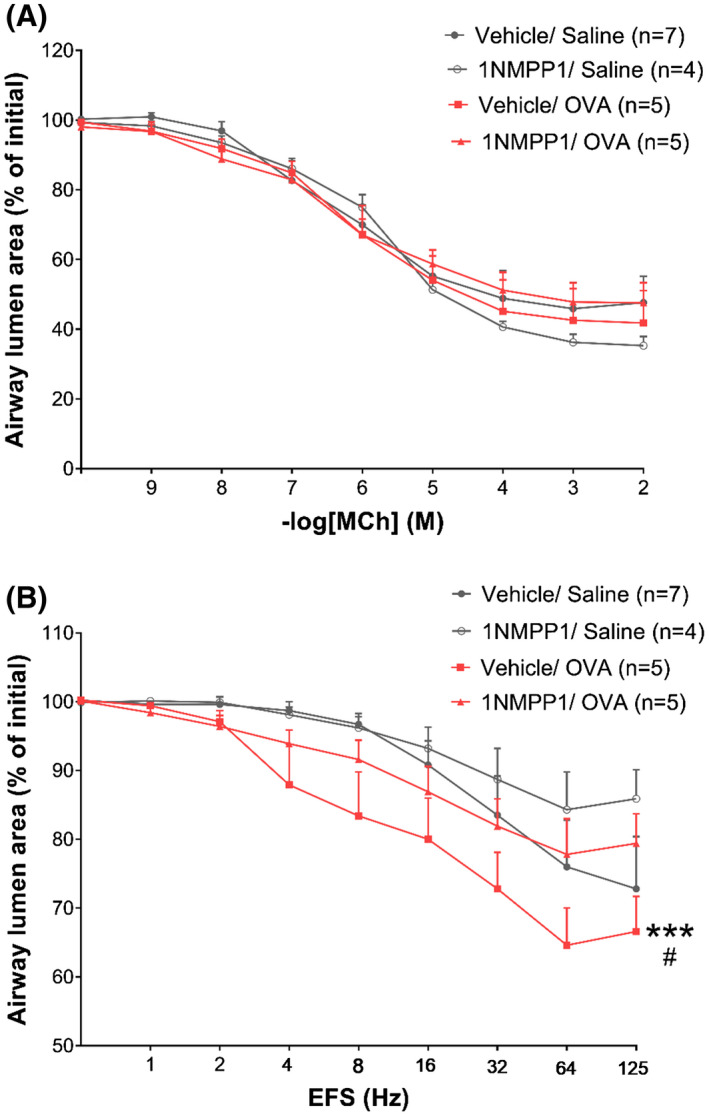
TrkB signaling disruption prevents neuronal mediated AHR. A, Airway lumen narrowing in response to increasing doses of methacholine in PCLS. B, Airway lumen narrowing in response to increasing frequencies of electric field stimulation in PCLS. Data shown as mean ± SEM. The differences were tested for statistical significance using two‐way analysis of variance (ANOVA) followed by Tukey post hoc test. ***P < .001 vs vehicle/saline. # P < .05 vs 1NMMP1/OVA. EFS, electric field stimulation; PCLS, precision cut lung slices
4. DISCUSSION
Allergic asthma induces functional changes in sensory and efferent neurons traveling along the airways that render these hyperreactive to noxious stimuli. 4 The mechanisms underlying this outcome involve enhanced sensory input, central integration, and efferent output, alongside release of inflammatory mediators by sensory neurons. 3 , 39 Recently, it was demonstrated that neuronal remodeling in the airways also occurs in human asthma, as sensory neurons display increased branching and lengthening in bronchial samples from patients with asthma. 15 Here, we report that neuronal plasticity also occurs for the efferent cholinergic system. We show for the first time that bronchial samples from asthma patients display elevated VAChT+ nerve fiber density when compared to biopsies derived from healthy controls. Using an RNAseq data set, we found heightened expression of the TrkB gene in asthma compared to healthy subjects and report SNPs in the NTRK2 (TrkB) and BDNF genes that are associated with asthma susceptibility. We show that TrkB signaling is crucial for the development of increased total and cholinergic nerve density after chronic, but not acute, allergen exposure in mice. This correlated with development of AHR.
We observed that asthma patients have higher cholinergic innervation compared to healthy individuals. Increased cholinergic tone has been previously demonstrated to occur in humans 2 , 40 and in animal models of allergic lung inflammation. 41 , 42 Plasticity of the cholinergic neuronal component in the airways, resulting in decreased depolarization threshold 41 and decreased muscarinic M2 autoreceptor function 42 have been reported following antigen exposure in animals. Here, we suggest a novel mechanism that involves remodeling of cholinergic nerves as one of the contributors to this heightened ACh release and AHR. It would be intriguing to consider the possibility that only some populations of asthmatic patients show increased neuronal Ach. However, in the current study, there was no correlation between increased cholinergic nerve density and clinical parameters. Thus, no correlation was observed between nerve density and FEV1, AHR to histamine or AMP, eosinophilia in blood or biopsies, smoking status, and disease severity. In part, this might be explained by the sample size as for some parameters trends were observed which did not reach statistical significant. Furthermore, it might be affected by the sample population as patients included in the study had relatively mild forms of asthma and it would be of interest to investigate cholinergic innervation in patients with more severe phenotypes. However, in large clinical studies investigating the anticholinergic drug tiotropium as add‐on to ICS plus LABA, it has been shown that tiotropium improves lung function and reduces exacerbation risk irrespective of baseline characteristics including age, gender, disease duration, and onset, frequency of exacerbations, and FEV1 reversibility. 43 , 44 While these clinical data suggest that anticholinergic therapy is effective across a broad asthma population, as currently included in the GINA guidelines as add‐on therapy, 1 they do not provide insights into changes in neuronal aspects of ACh effects per se or of effects on tone vs inflammation. Assessing the mechanisms via which cholinergic neuroplasticity occurs could lead to potential targetable pathways to reduce asthma symptoms.
In mice, we observed that chronic OVA exposure led to increased PGP and VAChT+ cholinergic nerve fibers around the airways, however, acute challenge with OVA did not induce the same effect. This is in line with a study showing increased nerve branching surrounding the airways after acute exposure to OVA early in life, but not in adulthood. 19 It suggests that cholinergic nerve plasticity takes some time to develop and depends on multiple allergen challenges and possibly on profound gene expression changes 45 for sufficient development of a remodeled neuronal network of functional significance in the context of persistent AHR seen in chronic asthma. 46 We did not investigate whether higher cholinergic output is primarily mediated due to phenotypic changes such as isotype switching, 47 or via sprouting of the current neurons, and it would be of interest to further investigate this in future studies.
Interestingly, we could demonstrate that the observed neuronal plasticity after chronic allergen exposure in our study was associated with lung function alterations in mice. An increase in airway resistance and tissue elastance in response to inhaled methacholine was observed after chronic OVA exposure compared to acute OVA or saline exposure. When bilateral vagotomy was performed, the increase in airway resistance observed after chronic OVA exposure was prevented. Thus, the acute component, most likely mediated via airway inflammation, was still present. However, the chronic component was prevented by vagotomy, suggesting that chronic allergen exposure is able to induce alterations in neuronal architecture and functionality, which enhances AHR. Vagotomy was shown previously to reverse methacholine‐induced AHR independently of ASM. 48 This essential role of airway nerves in AHR was confirmed in the current study using lung slices, which allows to disentangle both mechanisms by applying EFS to stimulate nerves as opposed to methacholine to stimulate ASM only. Indeed, AHR was observed only after EFS and not after methacholine stimulation, and more pronounced after chronic OVA exposure. We did not observe increased PCLS airway narrowing in response to methacholine neither after acute nor chronic OVA exposure, despite ASM hyperplasia. Donovan et al 49 previously showed no AHR to methacholine in PCLS obtained from allergen challenged mice either. On the contrary, AHR was clearly visible in response to EFS stimulation in our study. It has been shown that vagal sensory neurons respond to direct bronchoconstrictors, such as methacholine, and cause release of ACh via parasympathetic neurons. 48 Our own data support this contention, as we observed reduced AHR in response to methacholine in OVA‐challenged mice after vagotomy. Therefore, without an intact neuronal reflex, ASM hyperresponsiveness caused by allergen exposure might not be visible in PCLS in vitro, even in response to methacholine, which is considered a direct agonist. 50
Neurotrophins (eg, NGF, BDNF, NT‐3, NT4) and their receptors (TrkA, TrkB, TrkC, and p75) are a family of molecules involved in neuroplasticity. 12 , 16 They are expressed by diverse cell types in the lungs, such as ASM, fibroblasts, epithelium, neurons, and immune cells, 18 , 51 and neurotrophin exposure of isolated ganglia is able to induce cholinergic neurite outgrowth, which can be prevented by treatment with a TrkB antagonist. 52 We observed increased gene expression of TrkB in bronchial biopsies of asthma patients compared to healthy individuals and showed that TrkB signaling is one of the factors responsible for the plasticity of cholinergic airway neurons in mice. When TrkBF616A mice are exposed to allergen, cholinergic neuroplasticity does not occur. Two NTs bind with high affinity to this receptor, BDNF, and NT‐4, and both were shown to be important for neuroplasticity in animal models of asthma. 19 , 22 We found increased BDNF protein expression in OVA exposed mice, however, differential gene expression in human bronchi revealed no elevated expression of either the BDNF or NT‐4 genes. This could be explained by sample heterogeneity, as elevated BDNF expression was found mainly in severe asthma, 20 and patients included in our study had mild asthma. Moreover, an increased neurotrophic signaling in the lungs could be attributed to higher TrkB expression found in asthma samples. Furthermore, we found SNPs in BDNF, BDNF‐AS, and the NTRK2 genes that correlated with asthma susceptibility. This suggests that genetic variation in these genes may contribute to asthma susceptibility. Future studies may further elucidate whether changes in this pathway could predispose individuals for developing asthma.
The main cellular sources for increased BDNF were not investigated in the current study. However, ASM, epithelial cells, fibroblasts, and neurons are all known to secrete BDNF, 18 and TNF‐α and allergens have been shown to increase BDNF release from ASM. 53 In a recent study, chronic exposure to a combination of allergens induced BDNF release by ASM cells, which was found to be crucial for the development of AHR, as ASM‐specific BDNF knockout mice failed to display the same hyperresponsiveness. 22 Silencing of BDNF was not sufficient to reduce eosinophilia, in line with our findings using TrkBF616A mice. A reduction in α‐SMA was not observed in our model and contraction in response to methacholine was comparable for wild‐type (WT) and TrkBF616A mice, suggesting that the changes observed after OVA exposure are confined to altered neuronal innervation and function. Indeed, disrupted TrkB signaling prevented AHR in response to EFS in lung slices after chronic OVA exposure, showing that the increased ACh release is not present in these animals. This is in line with earlier studies demonstrating efficacy of anti‐BDNF in inhibiting AHR in asthma. 21 Furthermore, this suggests that persistent AHR and airway inflammation are not completely dependent on each other, as AHR is inhibited in TrkBF616A mice, whereas airway eosinophilia is not. Accordingly, airway eosinophil‐lowering therapies, such as antibodies against IL‐12 54 and IL‐5 55 and even standard ICSs 56 show only modest reductions of AHR in some patients. Targeting the neuronal compartment might, therefore, be an additional strategy for asthma treatment.
Taken together, we report cholinergic neuronal plasticity in asthmatic human bronchi. Our data support that NTs and their receptors, in particular BDNF and TrkB, represent potential drug targets to reduce neuronal remodeling and to alleviate the symptoms associated with inflammatory lung diseases, which include not only AHR, but also mucus hypersecretion, cough, and dyspnea.
CONFLICT OF INTEREST
GD, MW, SN, SB, JA, AG, EV, CV, BD, JV, GH, MB, NH, WT, CM, and YP have no conflict of interest. LK is the director of Aquilo BV and has received funding for research and lecture fees from Boehringer Ingelheim. RG has received funding for research and lecture fees from Boehringer Ingelheim.
AUTHOR CONTRIBUTIONS
G. Dragunas, G.H. Koppelman, M. van den Berge, N.H.T. ten Hacken, W. Timens, C.D. Munhoz, Y.S. Prakash, R. Gosens, and L.E.M. Kistemaker designed the study; G. Dragunas, M.E. Woest, S. Nijboer, S.T. Bos, J. Asselt, A.P. de Groot, E. Vohlídalová, C.J. Vermeulen, B. Ditz, J.M. Vonk, and L.E.M. Kistemaker performed the experiments; G. Dragunas and L.E.M. Kistemaker drafted the paper. All authors revised and approved the paper.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank M. Weij for technical support with the animal experiments and Prof W. Kummer for technical support with the stainings.
Dragunas G, Woest ME, Nijboer S, et al. Cholinergic neuroplasticity in asthma driven by TrkB signaling. The FASEB Journal. 2020;34:7703–7717. 10.1096/fj.202000170R
Funding information
The authors would like to thank the Lung Foundation Netherlands for financial support (4.2.15.039JO). Additional funding was provided by the US National Institutes of Health (NIHR01HL088029; YP). GD was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (2018/18762‐8). The Dutch Asthma GWAS was supported by Netherlands Asthma Foundation grants AF 95.09, AF 98.48, AF 3.2.02.51, and AF 3.2.07.015 and a grant from the University Medical Center Groningen
REFERENCES
- 1. (GINA), G. I. for A . GINA Report, Global Burden of Asthma; 2018.
- 2. Barnes PJ. Asthma as an axon reflex. Lancet. 1986;327:242‐245. [DOI] [PubMed] [Google Scholar]
- 3. Undem BJ, Taylor‐Clark T. Mechanisms underlying the neuronal‐based symptoms of allergy. J Allergy Clin Immunol. 2014;133:1521‐1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mazzone SB, Canning BJ. Central nervous system control of the airways: pharmacological implications. Curr Opin Pharmacol. 2002;2:220‐228. [DOI] [PubMed] [Google Scholar]
- 5. Fryer AD, Jacoby DB. Muscarinic receptors and control of airway smooth muscle. Am J Respir Crit Care Med. 1998;158:S154‐S160. [DOI] [PubMed] [Google Scholar]
- 6. Moroni‐Zentgraf P, Vandewalker M, Sigmund R, et al. Tiotropium in asthma poorly controlled with standard combination therapy. N Engl J Med. 2012;367:1198‐1207. [DOI] [PubMed] [Google Scholar]
- 7. Casarosa P, Bouyssou T, Germeyer S, Schnapp A, Gantner F, Pieper M. Preclinical evaluation of long‐acting muscarinic antagonists: comparison of tiotropium and investigational drugs. J Pharmacol Exp Ther. 2009;330:660‐668. [DOI] [PubMed] [Google Scholar]
- 8. Kistemaker LEM, Bos ST, Mudde WM, et al. Muscarinic M3 receptors contribute to allergen‐induced airway remodeling in mice. Am J Respir Cell Mol Biol. 2014;50:690‐698. [DOI] [PubMed] [Google Scholar]
- 9. Kistemaker LEM, Slebos DJ, Meurs H, Kerstjens HAM, Gosens R. Anti‐inflammatory effects of targeted lung denervation in patients with COPD. Eur Respir J. 2015;46:1489‐1492. [DOI] [PubMed] [Google Scholar]
- 10. Kistemaker LEM, Hiemstra PS, Bos IST, et al. Tiotropium attenuates IL‐13‐induced goblet cell metaplasia of human airway epithelial cells. Thorax. 2015;70:668‐676. [DOI] [PubMed] [Google Scholar]
- 11. Kistemaker LEM, Bos IST, Menzen MH, Maarsingh H, Meurs H, Gosens R. Combination therapy of tiotropium and ciclesonide attenuates airway inflammation and remodeling in a guinea pig model of chronic asthma. Respir Res. 2016;17:s12931‐016‐0327‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kistemaker LEM, Prakash YS. Airway innervation and plasticity in asthma. Physiology. 2019;34:283‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Larsen GL, Loader JE, Renz H, et al. Increased acetylcholine release in tracheas from allergen‐exposed IgE‐immune mice. Am J Physiol Cell Mol Physiol. 2017;266:L263‐L270. [DOI] [PubMed] [Google Scholar]
- 14. Kageyama N, Igarashi A, Ichinose M, et al. Chronic allergen exposure enhances cholinergic neuro‐transmission in sensitized guinea‐pigs. Eur Respir J. 1995;8:752‐754. [PubMed] [Google Scholar]
- 15. Drake MG, Scott GD, Blum ED, et al. Eosinophils increase airway sensory nerve density in mice and in human asthma. Sci Transl Med. 2018;10:eaar8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nockher WA, Renz H. Neurotrophins and asthma: novel insight into neuroimmune interaction. J Allergy Clin Immunol. 2006;117:67‐71. [DOI] [PubMed] [Google Scholar]
- 17. Barrios J, Xingbin A. Neurotrophins in asthma. Curr Allergy Asthma Rep. 2018;18:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prakash YS, Martin RJ. Brain‐derived neurotrophic factor in the airways. Pharmacol Ther. 2014;143:74‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aven L, Paez‐Cortez J, Achey R, et al. An NT4/TrkB‐dependent increase in innervation links early‐life allergen exposure to persistent airway hyperreactivity. FASEB J. 2014;28:897‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Watanabe T, Fajt ML, Trudeau JB, et al. Brain‐derived neurotrophic factor expression in asthma association with severity and type 2 inflammatory processes. Am J Respir Cell Mol Biol. 2015;53:844‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Braun A, Lommatzsch M, Neuhaus‐Steinmetz U, et al. Brain‐derived neurotrophic factor (BDNF) contributes to neuronal dysfunction in a model of allergic airway inflammation. Br J Pharmacol. 2004;141:431‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Britt RD, Thompson MA, Wicher SA, et al. Smooth muscle brain‐derived neurotrophic factor contributes to airway hyperreactivity in a mouse model of allergic asthma. FASEB J. 2019;33:3024‐3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Broekema M, Volbeda F, Timens W, et al. Airway eosinophilia in remission and progression of asthma: accumulation with a fast decline of FEV1. Respir Med. 2010;104:1254‐1262. [DOI] [PubMed] [Google Scholar]
- 24. Broekema M, Timens W, Vonk JM, et al. Persisting remodeling and less airway wall eosinophil activation in complete remission of asthma. Am J Respir Crit Care Med. 2011;183:310‐316. [DOI] [PubMed] [Google Scholar]
- 25. Wang J, Elliot JG, Ge Q, et al. Unique mechanisms of connective tissue growth factor regulation in airway smooth muscle in asthma: relationship with airway remodelling. J Cell Mol Med. 2018;2826‐2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nieuwenhuis MA, Siedlinski M, van den Berge M, et al. Combining genomewide association study and lung eQTL analysis provides evidence for novel genes associated with asthma. Allergy Eur J Allergy Clin Immunol. 2016;71:1712‐1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nieuwenhuis MAE, Vonk JM, Himes BE, et al. PTTG1IP and MAML3, novel genomewide association study genes for severity of hyperresponsiveness in adult asthma. Allergy Eur J Allergy Clin Immunol. 2017;72:792‐801. [DOI] [PubMed] [Google Scholar]
- 28. Melgert BN, Postma DS, Geerlings M, et al. Short‐term smoke exposure attenuates ovalbumin‐induced airway inflammation in allergic mice. Am J Respir Cell Mol Biol. 2004;30:880‐885. [DOI] [PubMed] [Google Scholar]
- 29. Alberti P, Canta A, Chiorazzi A, et al. Topiramate prevents oxaliplatin‐related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology. 2020;164:107905. [DOI] [PubMed] [Google Scholar]
- 30. Tu NH, Katano T, Matsumura S, et al. Na+/K+‐ATPase coupled to endothelin receptor type B stimulates peripheral nerve regeneration via lactate signalling. Eur J Neurosci. 2017;46:2096‐2107. [DOI] [PubMed] [Google Scholar]
- 31. Salvany S, Casanovas A, Tarabal O, et al. Localization and dynamic changes of neuregulin‐1 at C‐type synaptic boutons in association with motor neuron injury and repair. FASEB J. 2019;33:7833‐7851. [DOI] [PubMed] [Google Scholar]
- 32. Case DT, Burton SD, Gedeon JY, Williams S‐PG, Urban NN, Seal RP. Layer‐ and cell type‐selective co‐transmission by a basal forebrain cholinergic projection to the olfactory bulb. Nat Commun. 2017;8:652 10.1038/s41467-017-00765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gallart‐Palau X, Tarabal O, Casanovas A, et al. Neuregulin‐1 is concentrated in the postsynaptic subsurface cistern of C‐bouton inputs to α‐motoneurons and altered during motoneuron diseases. FASEB J. 2014;28:3618‐3632. [DOI] [PubMed] [Google Scholar]
- 34. Van Dijk EM, Culha S, Menzen MH, Bidan CM, Gosens R. Elastase‐induced parenchymal disruption and airway hyper responsiveness in mouse precision cut lung slices: toward an ex vivo COPD model. Front Physiol. 2017;7:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leus NGJ, Van Der Wouden PE, Van Den Bosch T, et al. HDAC 3‐selective inhibitor RGFP966 demonstrates anti‐inflammatory properties in RAW 264.7 macrophages and mouse precision‐cut lung slices by attenuating NF‐κB p65 transcriptional activity. Biochem Pharmacol. 2016;108:58‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosner SR, Ram‐Mohan S, Paez‐Cortez JR, et al. Airway contractility in the precision‐cut lung slice after cryopreservation. Am J Respir Cell Mol Biol. 2014;50:876‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Modarresi F, van der Brug MP, Fatemi RP, et al. Inhibition of natural antisense transcripts in vivo results in gene‐specific transcriptional upregulation. Nat Biotechnol. 2012;30:453‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schleputz M, Uhlig S, Martin C. Electric field stimulation of precision‐cut lung slices. J Appl Physiol. 2011;110:545‐554. [DOI] [PubMed] [Google Scholar]
- 39. Talbot S, Doyle B, Huang J, et al. Vagal sensory neurons drive mucous cell metaplasia. J Allergy Clin Immunol. 2020. 10.1016/j.jaci.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barnes PJ. Neural control of human airways in health and disease. Am Rev Respir Dis. 1986;134:1289‐1314. [DOI] [PubMed] [Google Scholar]
- 41. Mccaig DJ. Comparison of autonomic responses in the trachea isolated from normal and albumin‐sensitive guinea‐pigs. Br J Pharmacol. 1987;92:809‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fryer AD, Wills‐Karp M. Dysfunction of M2‐muscarinic receptors in pulmonary parasympathetic nerves after antigen challenge. J Appl Physiol. 1991;71:2255‐2261. [DOI] [PubMed] [Google Scholar]
- 43. Kerstjens HAM, Casale TB, Bleecker ER, et al. Tiotropium or salmeterol as add‐on therapy to inhaled corticosteroids for patients with moderate symptomatic asthma: Two replicate, double‐blind, placebo‐controlled, parallel‐group, active‐comparator, randomised trials. Lancet Respir Med. 2015;3:367‐376. [DOI] [PubMed] [Google Scholar]
- 44. Kerstjens HAM, Moroni‐Zentgraf P, Tashkin DP, et al. Tiotropium improves lung function, exacerbation rate, and asthma control, independent of baseline characteristics including age, degree of airway obstruction, and allergic status. Respir Med. 2016;117:198‐206. [DOI] [PubMed] [Google Scholar]
- 45. Leal G, Comprido D, Duarte CB. BDNF‐induced local protein synthesis and synaptic plasticity. Neuropharmacology. 2014;76:639‐656. [DOI] [PubMed] [Google Scholar]
- 46. Postma DS, Kerstjens HAM. Characteristics of airway hyperresponsiveness in asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158:S187‐S192. [DOI] [PubMed] [Google Scholar]
- 47. Pan J, Rhode HK, Undem BJ, Myers AC. Neurotransmitters in airway parasympathetic neurons altered by neurotrophin‐3 and repeated allergen challenge. Am J Respir Cell Mol Biol. 2010;43:452‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McAlexander MA, Gavett SH, Kollarik M, Undem BJ. Vagotomy reverses established allergen‐induced airway hyperreactivity to methacholine in the mouse. Respir Physiol Neurobiol. 2015;212–214:20‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Donovan C, Royce SG, Esposito J, et al. Differential effects of allergen challenge on large and small airway reactivity in mice. PLoS ONE. 2013;8:e74101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chew AD, Hirota JA, Ellis R, Wattie J, Inman MD, Janssen LJ. Effects of allergen on airway narrowing dynamics as assessed by lung‐slice technique. Eur Respir J. 2008;31:532‐538. [DOI] [PubMed] [Google Scholar]
- 51. Ricci A, Felici L, Mariotta S, et al. Neurotrophin and neurotrophin receptor protein expression in the human lung. Am J Respir Cell Mol Biol. 2004;30:12‐19. [DOI] [PubMed] [Google Scholar]
- 52. Ekman M, Zhu B, Swärd K, Uvelius B. Neurite outgrowth in cultured mouse pelvic ganglia—effects of neurotrophins and bladder tissue. Auton Neurosci Basic Clin. 2017;205:41‐49. [DOI] [PubMed] [Google Scholar]
- 53. Vohra PK, Thompson MA, Sathish V, et al. TRPC3 regulates release of brain‐derived neurotrophic factor from human airway smooth muscle. Biochim Biophys Acta ‐ Mol Cell Res. 2013;1833:2953‐2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bryan SA, O'Connor BJ, Matti S, et al. Effects of recombinant human interleukin‐12 on eosinophils, airway hyper‐responsiveness, and the late asthmatic response. Lancet. 2000;356:2149‐2153. [DOI] [PubMed] [Google Scholar]
- 55. Flood‐Page P, Swenson C, Faiferman I, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062‐1071. [DOI] [PubMed] [Google Scholar]
- 56. Szefler S, Weiss S, Tonascia J, et al. Long‐term effects of budesonide or nedocromil in children with asthma. N Engl J Med. 2000;343:1054‐1063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material