

Abstract
Epigenetics plays a fundamental role in cancer progression, and developing agents that regulate epigenetics is crucial for cancer management. Among Class I and Class II HDACs, HDAC8 is one of the essential epigenetic players in cancer progression. Therefore, we designed, synthesized, purified, and structurally characterized novel compounds containing N-substituted TZD (P1-P25). Cell viability assay of all compounds on leukemic cell lines (CEM, K-562, and KCL22) showed the cytotoxic potential of P8, P9, P10, P12, P19, and P25. In-vitro screening of different HDACs isoforms revealed that P19 was the most potent and selective inhibitor for HDAC8 (IC50 - 9.3 μM). Thermal shift analysis (TSA) confirmed the binding of P19 to HDAC8. In-vitro screening of all compounds on the transport activity of GLUT1, GLUT4, and GLUT5 indicated that P19 inhibited GLUT1 (IC50 - 28.2 μM). P10 and P19 induced apoptotic cell death in CEM cells (55.19% and 60.97% respectively) and P19 was less cytotoxic on normal WBCs (CC50 - 104.2 μM) and human fibroblasts (HS27) (CC50 - 105.0 μM). Thus, among this novel series of TZD derivatives, compound P19 was most promising HDAC8 inhibitor and cytotoxic on leukemic cells. Thus, P19 could serve as a lead for further development of optimized molecules with enhanced selectivity and potency.
Keywords: N-substituted thiazolidinediones, HDAC, GLUT, Leukemia, Docking
Graphical Abstract
1. Introduction
Epigenetics is the major contributor to cancer initiation, progression, and metastasis [1]. Histone modifications, a central epigenetics mechanism, include acetylation, methylation, phosphorylation, and many others. Histone acetylation unlocks chromatin and promotes gene transcription. Two enzymes control the acetylation of histones: histone acetyltransferases (HATs) and histone deacetylases (HDACs). Acetylation of lysine residues by HATs on histone tails leaves the chromatin structure intact and accessible and is responsible for transcription activation. In contrast, HDAC removes acetyl groups, leading to a more condensed chromatin structure, and, thus, repressing gene transcription [2]. HDACs are also involved in the regulation of cell differentiation, apoptosis, and cell cycle. Malignant cells have higher levels of HDACs compared with healthy cells, which leads to an abnormal histone acetylation pattern.
There are 18 human HDACs classified into four groups based on their structural homology: Class I comprised of HDAC 1–3 and 8, found in the nucleus; Class II comprised of HDAC 4–7, and 10, which are in the nucleus and cytoplasm; Class III comprised of Sirtuins (SIRT) 1–7, found in the nucleus, cytoplasm and/or mitochondria; and Class IV including the newly discovered HDAC 11. HDAC inhibitors approved by FDA for different types of cancer include: Vorinostat, Belinostat, Panobinostat, Romidepsin, and many others are in clinical studies [3–5]. Currently, FDA-approved HDAC inhibitors are generally pan-inhibitors that target more than one HDAC isoform and are associated with severe side effects [6–8]. Hence, isoform-selective HDAC inhibitors with fewer side effects and enhanced potency efforts have become a significant research focus.
HDAC8 is one of the essential epigenetic player in cancer progression and overexpresses in different cancer types, including cancers of the colon, breast, lung, gastric, and pancreas, acute myeloid leukemia (AML), and acute lymphocytic leukemia (ALL). Upregulation of HDAC8 inhibited apoptosis and induced cancer cell proliferation [9–12]. Therefore, targeting HDAC8 could counteract cancer and combat the adverse effects associated with paninhibitors.
There is a big challenge to develop HDAC8-selective inhibitors, since the active site is largely conserved among HDACs in general and class I HDACs including HDAC8 in particular. Therefore, linear hydroxamate inhibitors like SAHA are rather unselective on HDAC isoenzymes. However, HDAC8 contains several structural characteristics that allowed the development of different types of inhibitors with selectivity for this enzyme. First of all, HDAC8 shows significantly higher structural flexibility around the binding pocket, particularly at the outer rim between L1-, L2- and L6-loops as well as at the bottom of the catalytic site at the transition to the acetate release channel. Furthermore, crystal structures of three distinct major conformations of HDAC8-ligand complexes demonstrate the extraordinary malleability of HDAC8 around the active site pocket. There is a transition between the closed (PDB-ID 1T69), a sub-open conformation with a second transient binding pocket (PDB-ID 1T64) and a wide-open protein structure (PDB-ID 1VKG) of HDAC8. These structural features have been exploited by a variety of HDAC-inhibitors with selectivity for HDAC8. Most reported isoenzyme selective HDAC8 inhibitors contain a hydroxamate group as zinc chelating warhead. The selectivity of hydroxamate inhibitors was recently attributed to their L-shape and binding to a subpocket between L1- and L6-loop at the outer rim of the active site pocket that is unique for HDAC8 [14]. The selectivity of other non-hydroxamate inhibitors was explained by the occupation of the transition area between the active site and the acetate release channel at the bottom of the conserved binding pocket [17]. This binding mode is also proposed for recently discovered non-hydroxamate inhibitors with benzothiazine scaffold [18,19]. So called linkerless HDAC8 inhibitors (Fig. 1, compound 2) have been shown to induce the formation of a unique subpocket in HDAC8 by splitting F152 and M274 [20]. Additionally, the transient side pocket, which is observed in crystal structure of PDB-ID 1T64 (trichostatin A) would also be a good candidate for the design of selective HDAC8 inhibitors [21,22]. We have previously reported N-substituted-2,4-thiazolidinedione (TZD) derivative SRR2 as HDAC inhibitor (Fig. 2), wherein, TZD was at the tail portion of the compound, and docking revealed its interaction with zinc [11]. In our recent attempt to develop specific HDAC8 inhibitors, we designed naphthylidene TZD derivatives, with naphthalene as the linker, wherein compound 3h (Fig. 2) inhibited HDAC8 potently (IC50 - 6.7 μM), though it was hypothesized that the TZD moiety would act as zinc binding group (ZBG), the docking analysis shown that the carboxylate group interacted with zinc, and TZD occupied the hydrophobic pocket [23]. Thus, based on our previously reported HDAC inhibitors, we designed novel HDAC8-selective inhibitors. The nature of CAP plays significant role to enhance the potency of compounds, there are variety of functionalities reported which could serve the purpose [24] and thus, we placed variously substituted aryl//heteroaryl/fused heterocyclic aromatic rings at the CAP portion. The next essential moiety is the hydrophobic linker which could be linear or cyclic [25,26], hence, similar to our previous HDAC inhibitor SRR2, we retained linear linker but reduced the length to actually see the difference in the activity (Fig. 2). The next part is ZBG, which is the essential game changer and could exert significant effects on the inhibitory activity of the compounds [27]. Thus, we retained N-substituted TZD moiety in the compound to gain HDAC inhibitory potential and attached it to the linker. Moreover, -CH=CH- functionality attached to ZBG were reported to show promising HDAC8 inhibitory activities, and some of them are currently under clinical investigations [24,28,29]. Therefore, similar to compound 3h, we attached TZD ring to pyridine ring with -CH=CH- which led to a completely novel ZBG, pyridin-2-ylmethylene TZD. Interestingly, the designing located the TZD ring at the end of the linker, which will help us to determine the effects of TZD on the activity. Also, this unique structural design of novel HDAC8 inhibitors with pyridin-2-ylmethylene TZD provided L-shape to the compounds and hence we expect them to show HDAC8 inhibitory potential.
Figure 1.
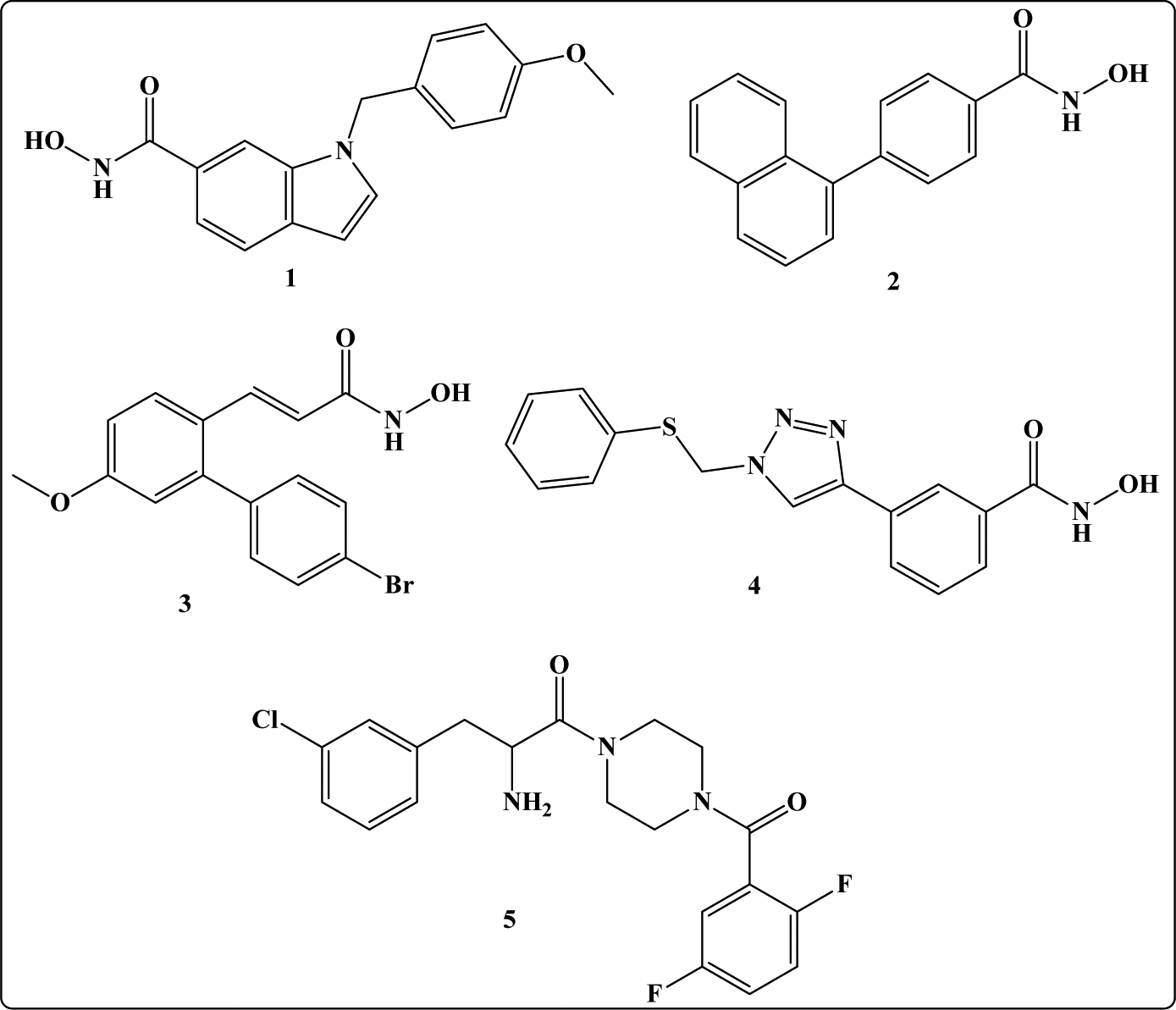
Reported selective HDAC8 inhibitors: PCI34051 (1), linkerless hydroxamic acid (2), ortho-aryl N-hydroxycinnamide (3), NCC149 (4), azetidinone (5) [13–16].
Figure 2.
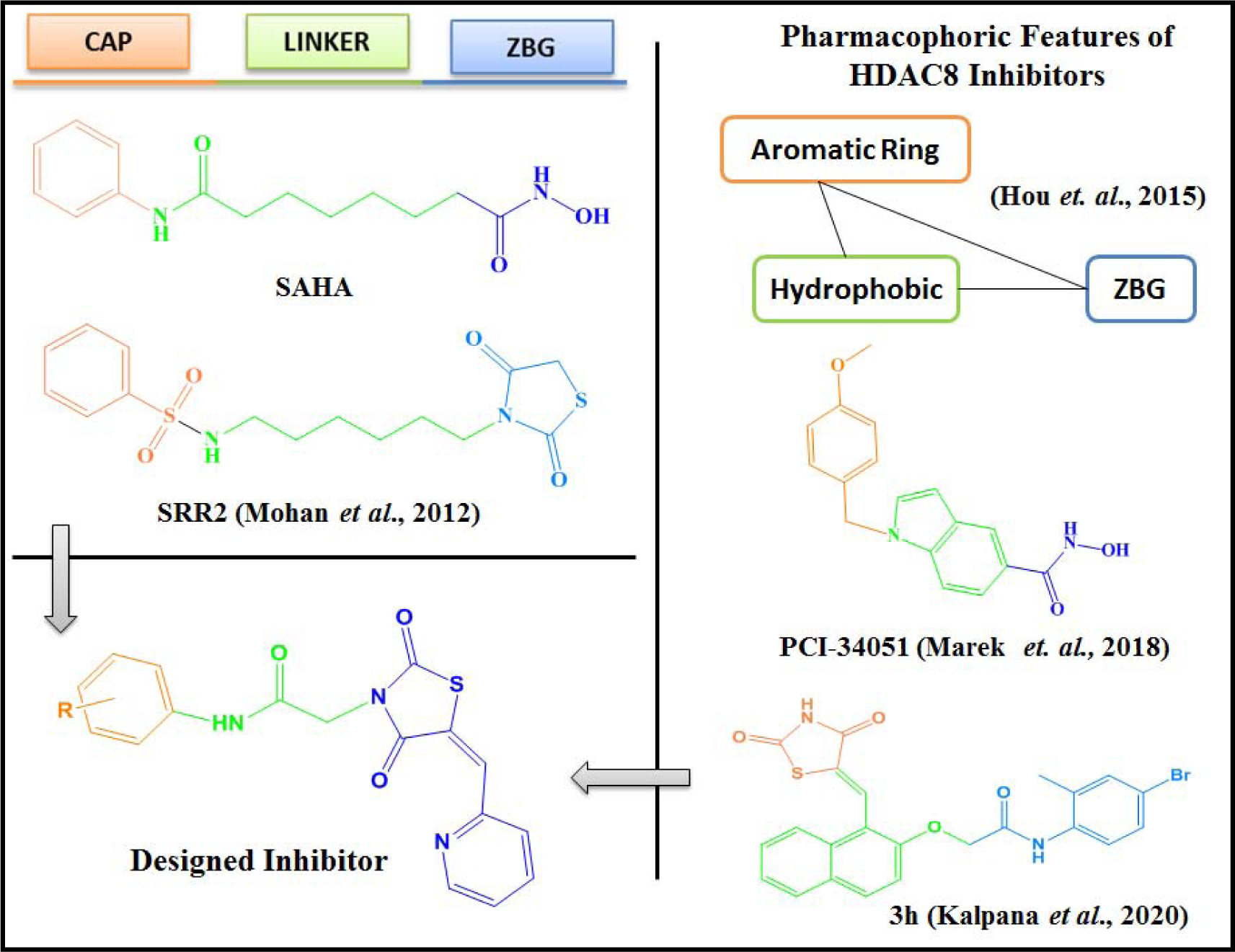
Docking studies of the compounds at the active site of HDAC8 showed zinc binding interactions with the carbonyl group of the TZD ring or the nitrogen atom of the pyridine ring or both (supplementary file). Hence by placing the TZD ring centrally, we expected that these newly designed pyridin-2-ylmethylene TZD derivatives might exhibit potential HDAC8 inhibitory activity. Based on these considerations, in the present work, we designed and synthesized 25 novel compounds (P1-P25) and studied their in-vitro antiproliferative activity.
HDACs are overexpressed in different leukemia subtypes such as acute promyelocytic leukemia (APL) [9,30,31], neuroblastoma [32], and T-cell lymphoma [33]. HDACs overexpression leads to the deacetylation of histones. Increased histone deacetylation causes an increase in the rate of cell cycle and a decrease in the differentiation and apoptosis of leukemic cells. These processes could be important in the identification and treatment of leukemias. Moreover, as our selective HDAC8 inhibitors (naphthylidene TZDs) have proved their in-vitro antiproliferative potential in leukemic cells, we decided to evaluate these newly designed HDAC8 inhibitors in leukemic cell lines.
2. Results and Discussion
2.1. Chemistry
Scheme I illustrates the synthesis of compounds. Synthesis of final compounds P1-P25 proceeded via four steps. The first step in the synthesis of variously substituted amides was as previously reported [23,34–37]. Substituted amines 1a-1y were stirred overnight with chloroacetyl chloride in the presence of potassium carbonate (K2CO3) and dichloromethane (DCM) as a solvent to produce 2a-2y. The second step included knoevenagel condensation of pyridine-2-carboxaldehyde 3 and 2,4-thiazolidinedione 4 in the presence of acetic acid and sodium acetate to yield intermediate 5. By refluxing 5 with 2a-2y in the presence of methanol did not produce P1-P25, so salt of 5 was synthesized in the third step by refluxing 5 with potassium hydroxide in the presence of ethanol to yield knoevenagel salt i.e. 6. In the final step, refluxing vigorously 6 and 2a-2y led to the compounds of interest P1-P25. Knoevenagel salt 6 was common for the synthesis of all the final compounds; therefore, it was synthesized in bulk. The purity of the final compounds P1-P25 was >95% by HPLC. All the synthesized compounds were confirmed structurally by different methods viz. 1H-NMR, 13C-NMR, FTIR, UV Spectroscopy, and Mass Spectrometry (Supplementary file). The detailed synthesis procedure and other spectral observations are in the experimental section.
Scheme 1. Synthetic route of P1-P25.
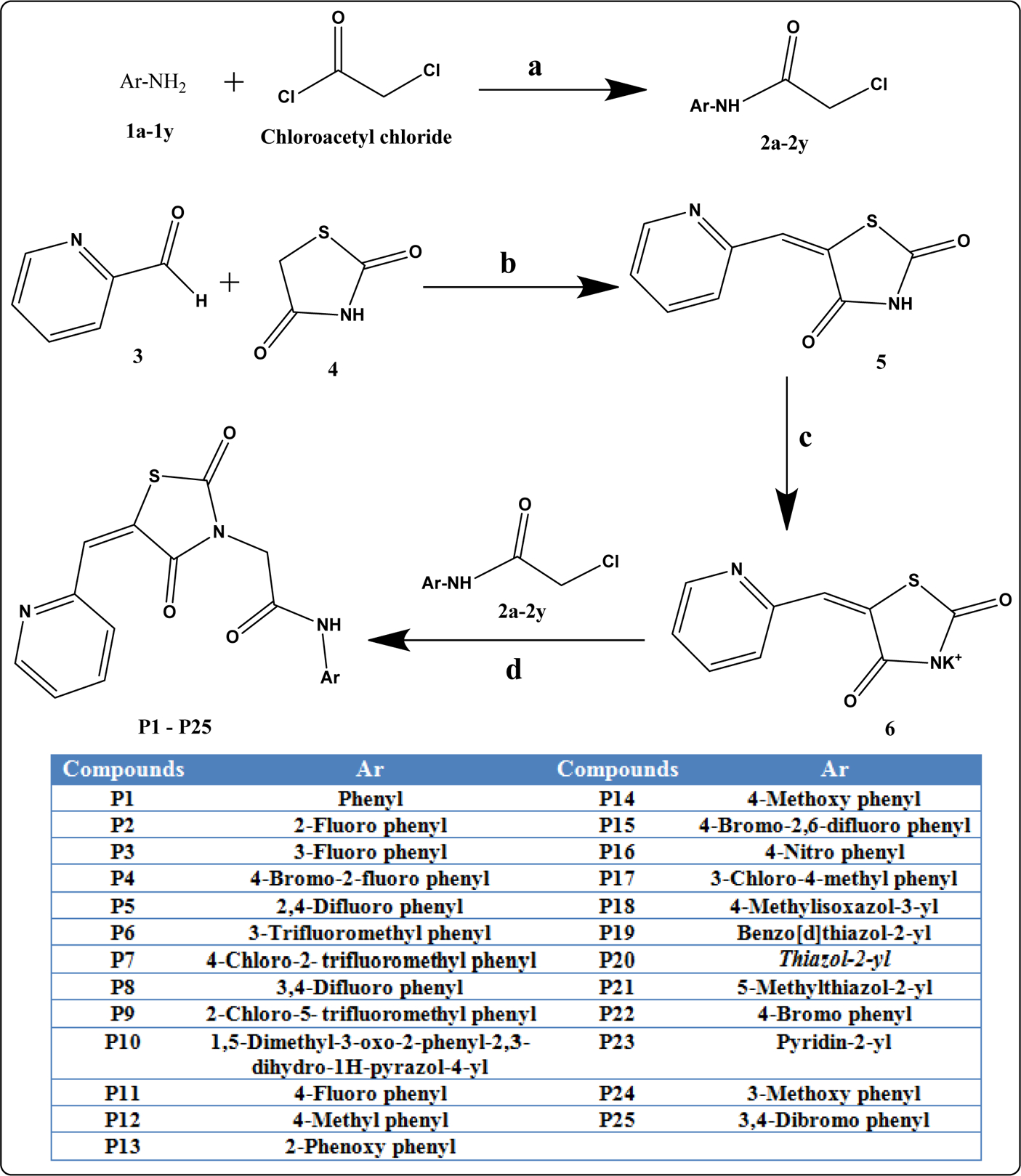
Reaction conditions: a) DCM, K2CO3, 0 0C, stirring; b) AcOH, Sodium acetate, reflux 5 h; c) EtOH, KOH, reflux 3 h; d) Methanol, reflux 6 h.
2.2. Biological evaluation
2.2.1. In-vitro cytotoxicity assay
To evaluate the cytotoxic potential of this series of compounds, we performed the Differential Nuclear Staining (DNS) assay after exposing human cancer cell lines to each compound for 48 h. The DNS assay is typically the first experiment for novel compounds because it provides simple and reliable measure of viability that is quicker/easier to obtain yet robust and comparable to what would be seen if the viability were assessed by flow cytometry. It uses two fluorescent nuclear stains, Hoechst 33342 and Propidium iodide (PI), to selectively label living and dead cells [38]. An initial screening of all 25 compounds, at 50 μM concentration, was conducted on K-562, KCL-22, and CEM leukemia cells. Solutions of each compound were prepared in DMSO, and the desired concentration of each compound was added to cell-containing wells at a final concentration of 1% (v/v) DMSO. Six compounds demonstrated activity in previous biological analysis and considerable cell death in one or more cancer cell lines. These six compounds were selected and assessed with a concentration gradient of 100–0.5 μM via secondary DNS assays on CEM, K-562, and KCL-22 to estimate their CC50 values. Cytotoxicity is reported as CC50 (cytotoxic concentration 50%, concentration of compound needed to kill 50% of the cell population) because in the DNS assay, living and dead cells are differentiated based on their membrane integrity, not on metabolic activity like the popular MTT assay. The CC50 values were estimated by linear interpolation of the two concentrations nearest 50% cell death. Compounds P8, P9, P10, P19, and P25 displayed CC50 values in the mid-micromolar range (Table 1). Out of these six compounds, P12 was the least cytotoxic and did not approach 50% cell death on any cell line at the concentrations used.
Table 1. Cytotoxic concentration 50% (CC50) of compounds P8, P9, P10, P12, P19, and P25 in leukemic cell lines (CEM, K562 and KCL22).
Cells were exposed to different concentrations of compounds for 48 h.
| Compound | Cell Line | CC50 (μM) |
|---|---|---|
| P8 | CEM | 36.9±1.11 |
| K562 | 40.28% cell death at 100 μM | |
| KCL22 | 29.22% cell death at 100 μM | |
| P9 | CEM | 4.80% cell death at 100 μM |
| K562 | 101.8±2.45 | |
| KCL22 | 6.54% cell death at 100 μM | |
| P10 | CEM | 2.39% cell death at 100 μM |
| K562 | 27.06% cell death at 100 μM | |
| KCL22 | 17.4±2.4 | |
| P12 | CEM | 38.67% cell death at 100 μM |
| K562 | 39.42% cell death at 100 μM | |
| KCL22 | 24.81% cell death at 100 μM | |
| P19 | CEM | 79.9±3.74 |
| K562 | 85.4±3.25 | |
| KCL22 | 43.2±0.23 | |
| P25 | CEM | 39±1.69 |
| K562 | 65.6±1.13 | |
| KCL22 | 82.2±0.32 |
2.2.2. In-vitro HDAC enzyme inhibition assay
2.2.2.1. Primary screening on HDAC4 and HDAC8
To determine the HDAC inhibitory potential of the target compounds, we assessed all of them on two isoforms of HDAC viz. HDAC4 and HDAC8. These two isoforms were selected because they possess high deacetylation activity, and belong to two diverse classes of deacetylase: class I (HDAC 8) and class II (HDAC4). The potency and selectivity of the target compounds for the two HDAC isoforms were examined and compared.
The primary screening was done with 50 μM concentration of P1-P25 on HDAC4 and HDAC8. Percent relative activities for all compounds on both HDAC isoforms were determined (Fig. 3A, 3B, and Table 2). Only two compounds, P5 and P12, showed HDAC4 inhibition, with IC50 of 50 μM (Fig. 3C). Interestingly, compounds P10, P12, and P19 displayed moderate to good HDAC8 inhibition, with IC50 values of 23 μM, 47 μM, and 9.3 μM, respectively (Fig. 3D). Among all compounds, P19 and P10 were more selective on HDAC8 over HDAC4, with HDAC8-IC50 in the sub-micromolar range. The presence of the benzothiazole ring may be responsible for the more potent activity of P19 on HDAC8; it was suggested that compounds incorporating the benzothiazole ring had good HDAC8 inhibition [39–43]. Thus, we assume that the benzothiazole ring contributed to the selectivity and activity of P19 against HDAC8. The thermal shift assay (TSA) was performed to examine the effects of P5, P9, P10, P12, and P19 binding to HDAC8 (Supplementary file - Fig. S5). Thermal shift data (ΔT) suggested that the tested compounds did not confer significant stability to the protein; only compound P19 had a weak stabilizing effect of about 0.3 °C.
Figure 3. Screening of P1-P25 on two different classes of HDAC isoforms, HDAC4 and HDAC8.
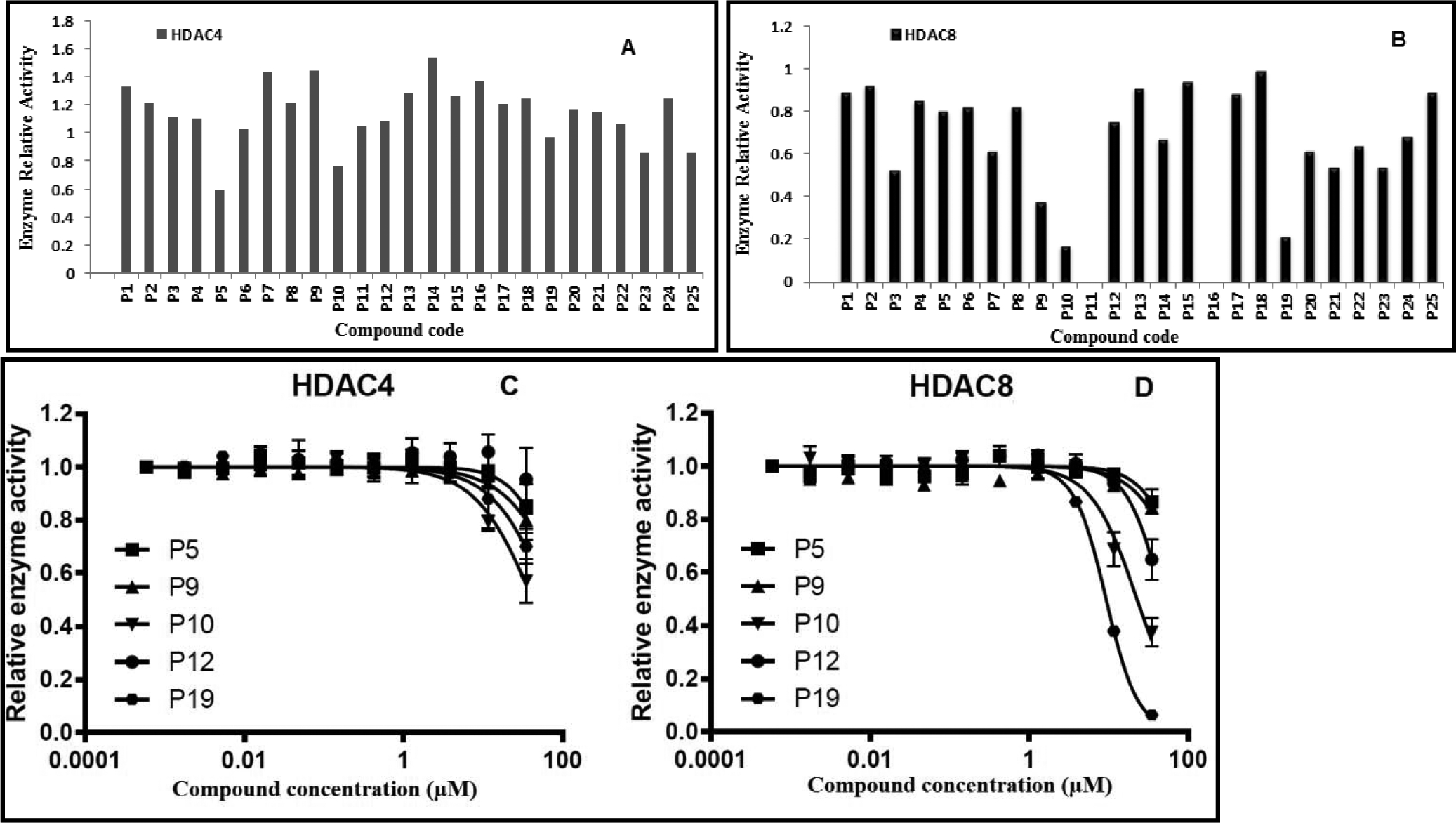
Primary screening of all target compounds at 50 μM against HDAC4 (A) and HDAC8 (B). (C) Dose response curves of P5, P9, P10, P12, and P19 on HDAC4. (D) Dose response curves of P5, P9, P10, P12, and P19 on HDAC8.
Table 2.
Primary screening of P1-P25 on two different classes of HDAC isoforms, HDAC4 and HDAC8.
| Code | HDAC4 IC50 (μM) | HDAC8 IC50 (μM) | Residual HDAC4 activity at 50 μM | Residual HDAC8 activity at 50 μM |
|---|---|---|---|---|
| P1 | >50 | >50 | 100 % | 89 % |
| P2 | >50 | >50 | 100 % | 92 % |
| P3 | >50 | >50 | 100 % | 53 % |
| P4 | >50 | >50 | 100 % | 85 % |
| P5 | 50 | >50 | 59 % | 80 % |
| P6 | >50 | >50 | 100 % | 82 % |
| P7 | >50 | >50 | 100 % | 61 % |
| P8 | >50 | >50 | 100 % | 82 % |
| P9 | >50 | >50 | 100 % | 38 % |
| P10 | >50 | 23±1.1 | 76 % | 17 % |
| P11 | >50 | >50 | 100 % | n.d. |
| P12 | 50 | 47±1.1 | 100 % | 75 % |
| P13 | >50 | >50 | 100 % | 91 % |
| P14 | >50 | >50 | 100 % | 67 % |
| P15 | >50 | >50 | 100 % | 94 % |
| P16 | >50 | >50 | 100 % | n.d. |
| P17 | >50 | >50 | 100 % | 88 % |
| P18 | >50 | >50 | 100 % | 99 % |
| P19 | >50 | 9.3±1.0 | 97 % | 22 % |
| P20 | >50 | >50 | 100 % | 92 % |
| P21 | >50 | >50 | 100 % | 54 % |
| P22 | >50 | >50 | 100 % | 64 % |
| P23 | >50 | >50 | 85 % | 54 % |
| P24 | >50 | >50 | 100 % | 68 % |
| P25 | >50 | >50 | 86 % | 89 % |
IC50-values were determined in triplicates, N=3. Means and standard deviations are provided. n.d. - not determined.
2.2.2.2. HDAC-profiling on a panel of HDACs (HDAC1–8)
In the primary screening on HDAC4 and HDAC8, compounds P10, P12, and P19 were HDAC8 inhibitors (Table 2). P10 and P19, the best HDAC8 inhibitors, were screened on a panel of HDACs (HDAC1–8) to establish if they were selective for HDAC8 (Table 3, Fig. 4). Testing P10 and P19 on a panel of HDACs revealed that P10 inhibited multiple HDAC isoforms such as HDAC2, HDAC5, HDAC6, and HDAC8 in mid-micromolar range. In contrast, P19 displayed HDAC8 inhibition with IC50 in single-digit micromolar range as compared to other HDACs.
Table 3.
IC50 of P10 and P19 on a panel of HDAC.
| Compounds | HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC7 | HDAC8 |
|---|---|---|---|---|---|---|---|---|
| P10 | >50 | 45 | >50 | >50 | 43 | 26 | >50 | 23 |
| P19 | >50 | 41 | >50 | >50 | >50 | 17 | >50 | 9.3 |
| PCI-34051* | 3.0 | 45 | 38 | 10 | >50 | 18 | - | 0.024 |
Kleinschek, A., et al. (2016). “Potent and Selective Non-hydroxamate Histone Deacetylase 8 Inhibitors.” ChemMedChem 11 (23): 2598–2606.
Figure 4.
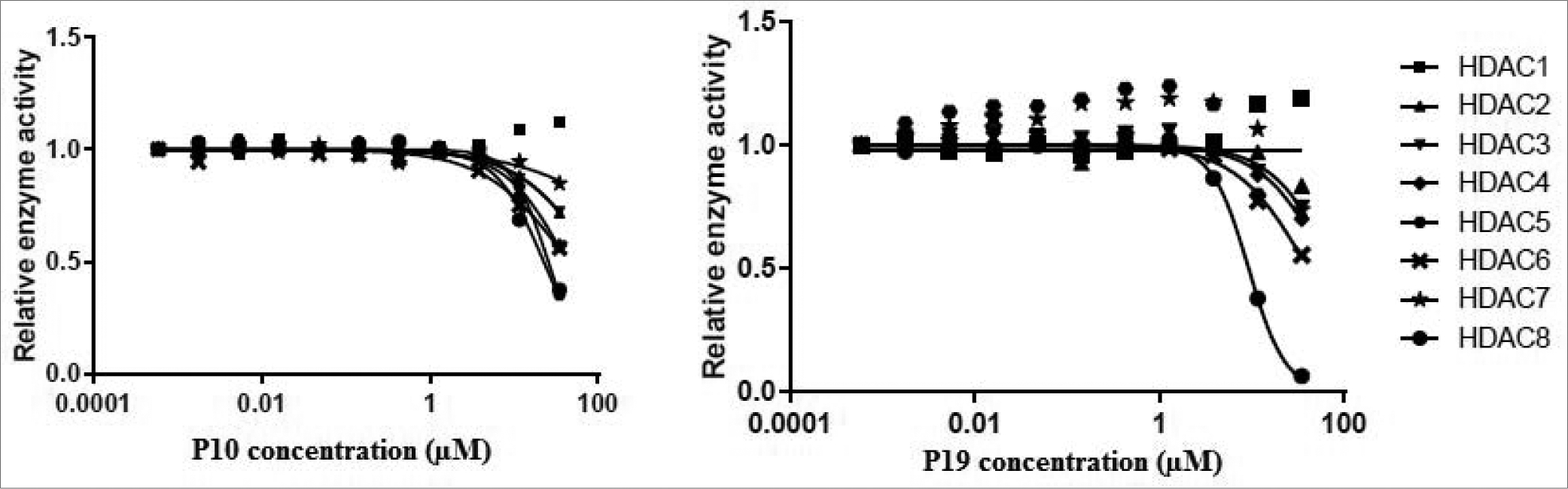
Dose response curve of P10 and P19 on different HDAC isoforms.
2.2.3. GLUT 1, 4, and 5 inhibition assay
The therapeutic effects of HDAC inhibitors stem from their capacity to alter gene expression and acetylation status of histones. Lately, substantial attention has focused on the ability of HDAC inhibitors to modify cellular metabolism; they inhibit glucose transporter 1 (GLUT1)-mediated glucose transport by downregulating GLUT1 expression [44,45]. This downregulation has been linked to a time-dependent decrease in glucose uptake in multiple myeloma cell lines (H929).
Compounds containing N-substituted TZD have been reported as GLUT1 inhibitors [46,47]. Our designed HDAC inhibitors (P1-P25) also hold N-substituted TZD and have exhibited HDAC8 inhibitory activity. Considering that HDAC inhibitors also interfere with GLUT1 expression and to assess their inhibitory potential on GLUTs, we screened all target compounds (P1-P25) for their effect on the transport activity of GLUT 1, 4, and 5 at 50 μM concentration (Fig. 5). For the transport assay, we used the GLUT-specific systems provided by the hexose transport null (hxt0) yeast strains engineered to express a particular human GLUT [48,49]; the only glucose or fructose uptake in these cells is through the recombinant human GLUTs.
Figure 5. Effect of the test compounds on the relative transport activity of GLUT 1, 4, and 5.
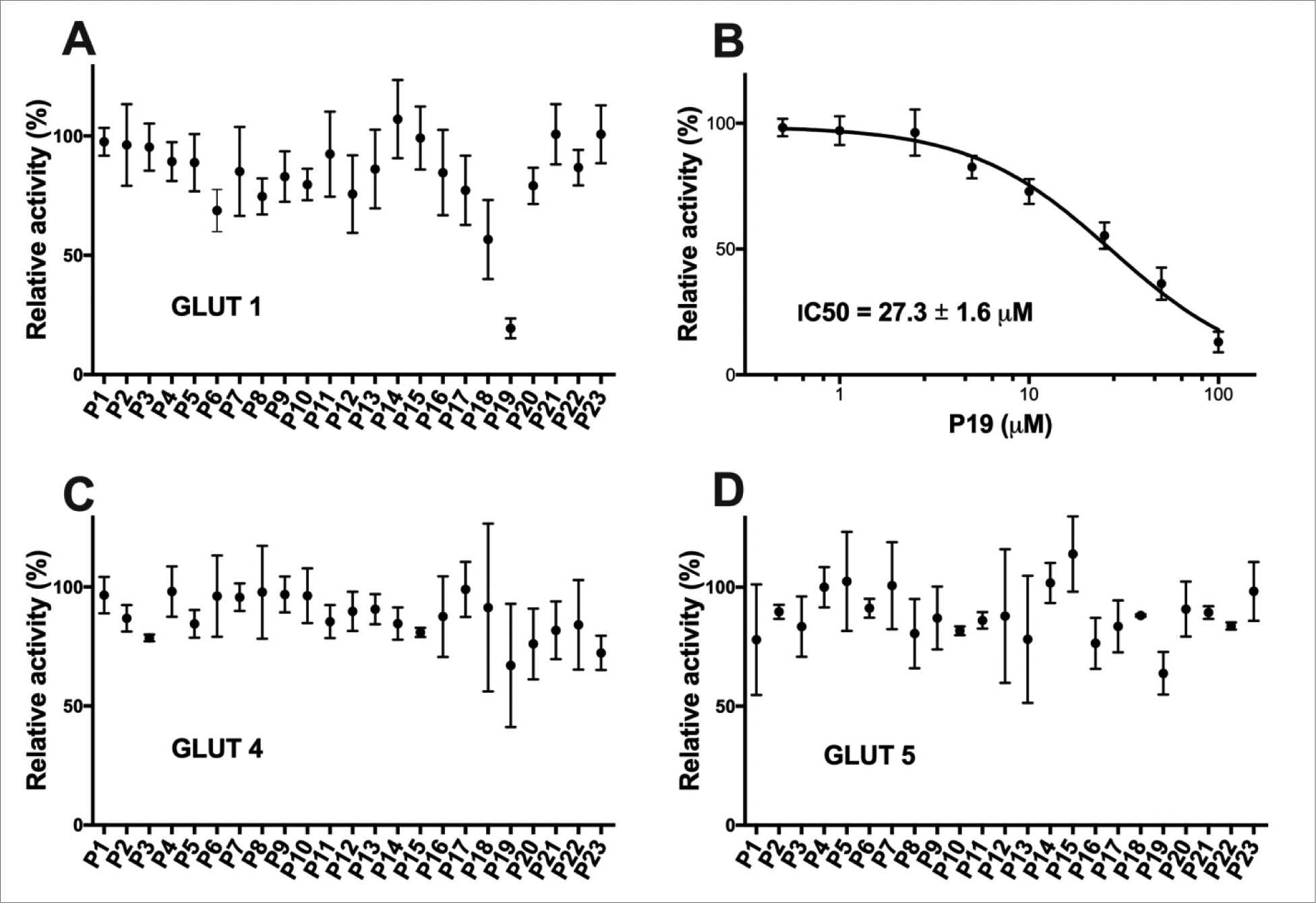
Percent relative activity of GLUT1 (A), GLUT4 (C), and GLUT5 (D) in the presence of 50 μM concentration of test compound. (B) Dose response curve of P19 in GLUT1. Transport assay was initiated by the addition of 5 mM C14-glucose (for GLUT1 and GLUT4) or 10 mM C14- fructose (GLUT5) to hxt0 yeast cells expressing GLUT1, GLUT4, or GLUT5 (see Material and Methods for details). The transport activity was measured in triplicates and means with S.D. are shown.
Among all tested compounds, P19 showed significant inhibition of GLUT1 (IC50 = 28.2 ± 1.8 μM) and remained ineffective on the other two GLUT isoforms (Fig. 5). The rest of the compounds exhibited >50% relative activity at 50 μM concentration on all three GLUT members. Thus, P19 was selective for HDAC8, among other HDAC isoforms, and was also specific for GLU1, compared with GLUT1 and GLUT4. Moreover, our studies also suggest that cancers depending on GLUT1 for the production of energy and glycolysis might be tackled with HDAC inhibitors.
2.2.4. Apoptosis studies by flow cytometry
HDAC8 inhibitors have been found to induce apoptosis in solid and hematological cancer cells through various mechanisms. PCI-34051, a HDAC8 inhibitor have caused apoptosis in T-cell lymphoma cells by caspase activation [7]. In gastric carcinoma cells, HDAC8 inhibition induced apoptosis facilitated by Bcl-2-modifying factor (BMF) and STAT3 [50]. Selective HDAC8 inhibitors have been found to cause dose-dependent selective apoptosis of CD34+ leukemic stem cells and progenitor cells [7]. Our recently reported selective HDAC8 inhibitor has found to induce apoptosis in CEM, lymphoid leukemia CEM cell line [23]. Also, the up-regulation of HDAC8 have been found to inhibit apoptosis in hepatocellular carcinoma, indicating a role of HDAC8 inhibitors in induction of apoptosis [51]. Thus HDAC8 inhibitors have ability to cause apoptotic cell death, and hence to determine if the cytotoxicity was induced by P10 and P19 was apoptosis-related, we stained CEM cells with annexin V-FITC/PI and determined by flow cytometry the percent of cell apoptosis. Incubation of the CEM cells with P10 and P19 at their CC50 concentrations caused an increase in the cell population in the apoptotic region compared with the control. In P10-treated cells, the percent of apoptotic cells was 55.19%, and live cells were only 29.89% when compared with that of control (Fig. 6A, 6C), which was 92.15% (Supplementary file - Fig. S6). P19-treated cells had 60.97% apoptotic cells; the living cells were only 30.86% compared with the control (Fig. 6B, 6C). Thus, as seen with reported HDAC8 inhibitors, compounds P10 and P19 can induce apoptotic death in the CEM cell line.
Figure 6. Analysis of apoptosis induced by compounds P10 and P19 on CEM cells.
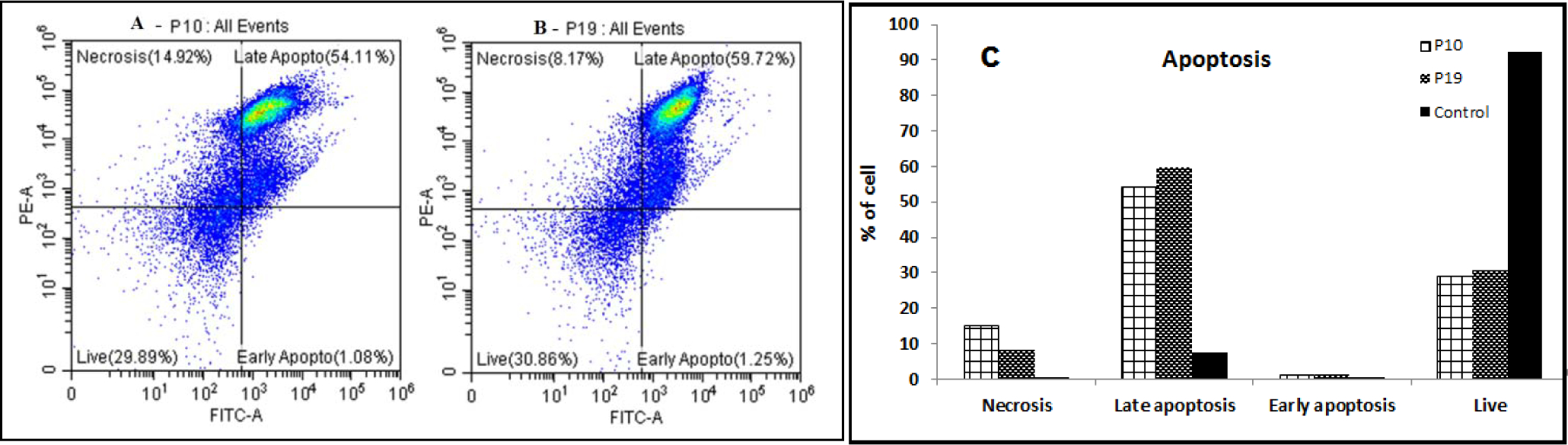
(A-B) Cytograms of test compounds P10 and P19, respectively. CEM cells were treated with IC50 concentration of compounds P10 (A) and compound P19 (B) for 24 h. (C) Graphical representation of apoptotic events of control (1% DMSO), P10-treated and P19-treated cells.
2.2.5. Cell viability of P19-treated non-cancerous cells by MTT and DNS assays
To ascertain the cytotoxicity of P19 on normal and cancerous cells, we performed both MTT and DNS assays to compare their cytotoxicity concentration 50% by two different assays. We determined the cell viability of CEM cell lines and normal white blood cells (WBCs) with the MTT assay and that of CEM cell lines and normal human fibroblast (HS27) with the DNS assay (Supplementary file - Fig. S7). By MTT assay CC50 of P19 in normal WBCs was 104.2 μM, whereas, in CEM cells it was 11.91 μM (Table 4). DNS assay of HS27 showed that CC50 of P19 was 105.0 μM, and of CEM cells was 79.9 μM. Thus, compound P19 could be safe for non-cancerous cells.
Table 4.
Viability of P19-treated cells by MTT and DNS assays.
| P19 | MTT assay (CC50 in μM) | DNS assay (CC50 in μM) |
|---|---|---|
| CEM | 11.91 | 79.9 |
| Non-cancerous cells | 104.2 (WBCs) | 105.0 (HS27) |
2.3. In-silico studies
2.3.1. Molecular docking
Flexible docking was performed with MOE software, using the crystal structures of HDAC8 (PDB ID 1T69) and HDAC4 (PDB ID 2VQJ). To validate the docking protocol, we determined the Root Mean Square Deviation (RMSD) between the crystal pose vs. the docked pose. The corresponding alkyl chains with zinc-chelating hydroxamate warhead of SAHA bound to HDAC8 superpose excellently within the binding pocket of HDAC8 with an RMSD value of 0.9 Å. Similarly, the buried thiophene linker and trifluoromethyl warhead of the redocked ligand in PDB ID 2VQJ showed an excellent RMSD value of 0.6 Å with respect to the crystal structure. The aromatic head groups of SAHA in complex with HDAC8 and the trifluoromethylketone ligand in HDAC4 reach out of the binding pocket were freely rotatable and therefore not considered for the calculation of RMSD. P19 showed the highest experimental activity and the best docking score against HDAC8 (Tables 5). The 2D and 3D ligand-enzyme interaction diagrams illustrating the major interactions of the representative compound P19 with amino acid residue at the active site of HDAC8 are shown in Fig. 7 (Other images in supplementary file). P19 fitted well in the binding pocket of HDAC8 forming hydrogen bonds with the backbone oxygen of G151 and to the side chain of Y306 at the bottom of the active site. Interestingly, P19 interacted with the catalytic zinc ion through carboxamide carbonyl oxygen and not with our hypothesized TZD carbonyl oxygen. The relatively bulky benzothiazole moiety of P19 was inserted between A37 and W141 and protruded into the acetate release channel. The amino acids of the canonical binding tunnel were highly conserved among all classes of zinc dependent HDACs with one major exception: Y306 in class I HDAC8, which was pivotal for the enzyme mechanism, was exchanged against a histidine that was swung away from the catalytic site. However, the amino acids flanking the acetate release channel differed widely, e.g. W141 is unique for HDAC8. Since the bulky benzothiazole group of P19 fitted perfectly into the specific acetate release channel of HDAC8, we hypothesized that this feature together with the hydrogen bond between P19 and Y306, which was not present in HDAC4, could be the reason for the observed inhibitory activity and selectivity towards HDAC8 (Table 2). To visualize the structural differences between HDAC4 and HDAC8, we superposed the best-scoring docking poses of P19 within the crystal structures of both enzymes (Fig. 7C). At first glance, the corresponding conserved amino acids of the binding pockets in HDAC8 and HDAC4 (F208/F227, F152/F168, H142/H158, H143/H159, respectively) showed no perfect overlap but rather significant shifts. Also it was seen that H332, which corresponds to Y306 in HDAC8, was flipped away from the catalytic site opening a selectivity side pocket that is characteristic to all class IIa HDACs. Therefore, H332, unlike Y306 in HDAC8, was not able to form interactions with P19. Most strikingly, HDAC4 did not show an open subpocket at the transition area to the acetate release channel like HDAC8. Consequently, the benzothiazole moiety of P19, which protruded into the acetate release channel of HDAC8, would clash with the surface of HDAC4 in a similar docking pose. The unbeneficial docking pose of P19 at the upper surface of the binding pocket of HDAC4 was in agreement with the observed absent activity against this enzyme (Fig. 7C).
Table 5.
Docking scores target compounds docking to the HDAC8 crystal structure.
| Compound Code | GBVI/WSA dG score | Compound Code | GBVI/WSA dG score |
|---|---|---|---|
| SAHA (redocked) | −11.2 | P13 | −8.0 |
| P1 | −8.0 | P14 | −7.8 |
| P2 | −8.0 | P15 | −7.9 |
| P3 | −7.9 | P16 | −7.3 |
| P4 | −8.2 | P17 | −8.4 |
| P5 | −7.8 | P18 | −8.4 |
| P6 | −8.0 | P19 | −8.6 |
| P7 | −8.4 | P20 | −7.4 |
| P8 | −7.9 | P21 | −7.9 |
| P9 | −7.9 | P22 | −7.5 |
| P10 | −8.4 | P23 | −7.8 |
| P11 | −7.5 | P24 | −8.4 |
| P12 | −8.0 | P25 | −8.1 |
Figure 7.
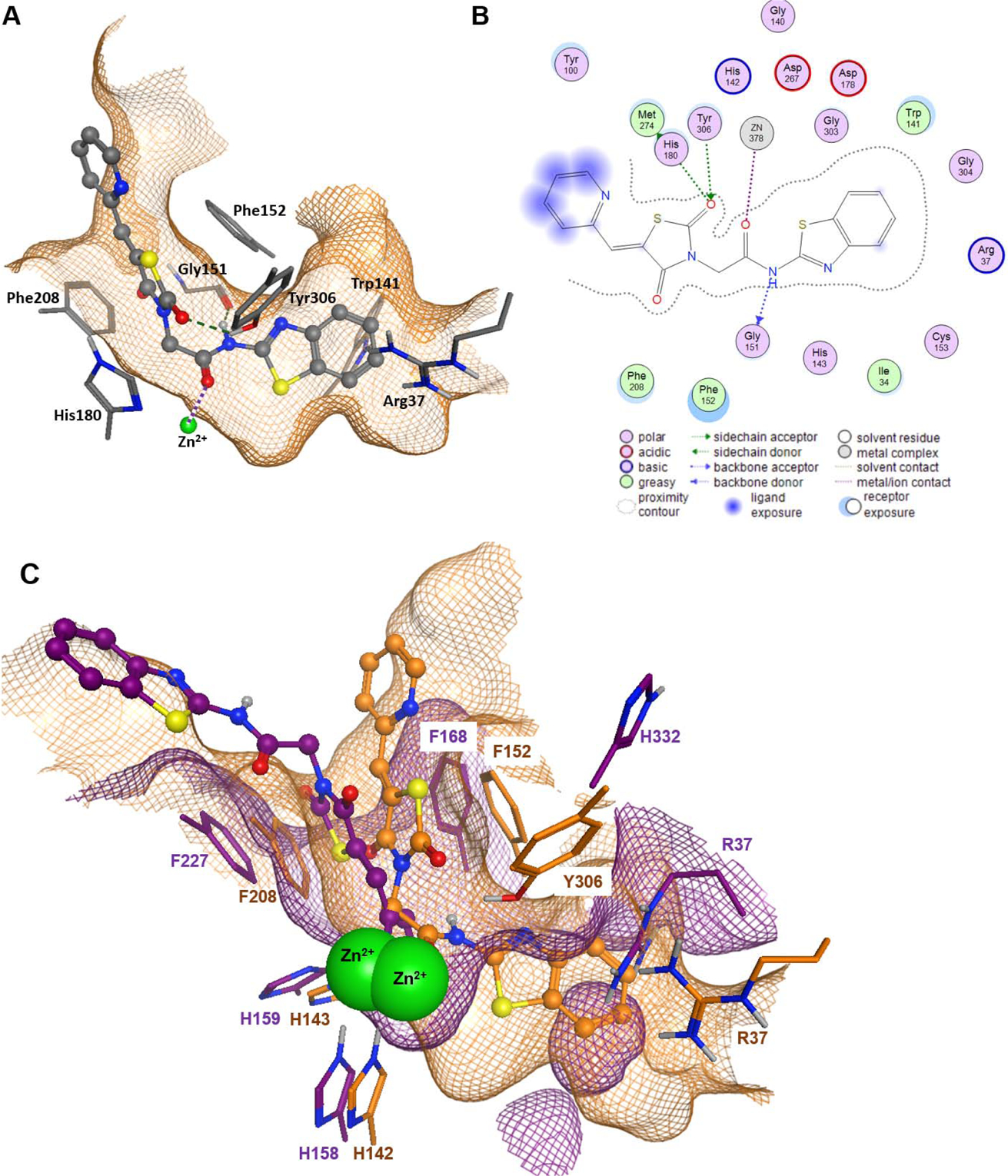
Docking analysis of compound P19 on HDAC8 (PDB ID 1T69) and HDAC4 (PDB ID 2VQJ). A) 3D binding pose of P19 (stick-and-ball model) within the binding pocket of HDAC8. B) 2D ligand interactions between P19 and HDAC8. Hydrogen bonds are indicated by dashed green lines (A) or dotted green or blue arrows (B) and metal ion contacts are shown as dotted magenta lines. C) Superposition of docking poses of P19 in the binding pocket of HDAC8 (PDB ID 1T69, orange) and HDAC4 (PDB ID 2VQJ, magenta). Green spheres represent the catalytic zinc ion of both structures. The amino acids are annotated according to the respective PDB file. The meshed surfaces indicate the respective binding pocket surfaces of HDAC8 (orange) and HDAC4 (magenta).
2.3.2. SwissADME predictions
The therapeutic action of a compound depends on it reaching the desired target site in the body at sufficient concentration described by its ADME (absorption, distribution, metabolism, and excretion) properties. Lipinski’s rule-of-five outlines the probability of a drug to be orally active (i.e., the drug-likeness) and the correlation between the pharmacokinetic and physicochemical parameters. SwissADME has computational models that give fast yet robust predictive data about the physicochemical properties, pharmacokinetics, and drug-likeness along with user-friendly submission and straightforward result interpretation [52]. We used it to predict the physicochemical properties and bioavailability of synthesized compounds P1-P25. All 25 compounds obey Lipinski’s rule of five (Table 6) with no violations, indicating their drug-likeliness. All compounds have an acceptable range of clogP value of 2.00–5.00. Topological polar surface area (TPSA) is a physicochemical descriptor of the surface sum of all the polar atoms in the molecules, and the acceptable region for suitable absorption is between 20 and 130 Å2. Four compounds exhibited higher TPSA than 130 Å2, and the rest fell in the acceptable zone. Compounds solubility ranged from soluble to moderately soluble. The bioavailability score for all compounds was 0.55. In summary, the compounds had drug-likeliness and were good candidates for passive oral absorption.
Table 6.
SwissADME prediction of physicochemical properties and bioavailability of compounds P1-P25.
| Code | TPSAa | Log Pb | Log Sc | ESOLd Class | GIe absorption | Lipinski #violations | Bioavailability Score |
|---|---|---|---|---|---|---|---|
| P1 | 104.67 | 2.08 | −3.4 | Soluble | High | 0 | 0.55 |
| P2 | 104.67 | 2.64 | −3.56 | Soluble | High | 0 | 0.55 |
| P3 | 104.67 | 2.64 | −3.56 | Soluble | High | 0 | 0.55 |
| P4 | 104.67 | 3.4 | −4.47 | Moderately soluble | High | 0 | 0.55 |
| P5 | 104.67 | 3.19 | −3.72 | Soluble | High | 0 | 0.55 |
| P6 | 104.67 | 4.25 | −4.26 | Moderately soluble | High | 0 | 0.55 |
| P5 | 104.67 | 4.9 | −4.86 | Moderately soluble | High | 0 | 0.55 |
| P8 | 104.67 | 3.38 | −4.59 | Moderately soluble | High | 0 | 0.55 |
| P9 | 104.67 | 4.9 | −4.86 | Moderately soluble | High | 0 | 0.55 |
| P10 | 131.6 | 1.87 | −4.21 | Moderately soluble | High | 0 | 0.55 |
| P11 | 104.67 | 2.64 | −3.56 | Soluble | High | 0 | 0.55 |
| P12 | 104.67 | 2.38 | −3.7 | Soluble | High | 0 | 0.55 |
| P13 | 113.9 | 3.87 | −4.86 | Moderately soluble | High | 0 | 0.55 |
| P14 | 113.9 | 2.08 | −3.47 | Soluble | High | 0 | 0.55 |
| P15 | 104.67 | 3.96 | −4.63 | Moderately soluble | High | 0 | 0.55 |
| P16 | 150.49 | 1.98 | −3.47 | Soluble | Low | 0 | 0.55 |
| P17 | 104.67 | 3.04 | −4.3 | Moderately soluble | High | 0 | 0.55 |
| P18 | 145.8 | 2.69 | −4.29 | Moderately soluble | Low | 0 | 0.55 |
| P19 | 130.7 | 1.37 | −2.86 | Soluble | High | 0 | 0.55 |
| P20 | 145.8 | 1.53 | −3.03 | Soluble | Low | 0 | 0.55 |
| P21 | 145.8 | 1.84 | −3.35 | Soluble | Low | 0 | 0.55 |
| P22 | 104.67 | 2.84 | −4.31 | Moderately soluble | High | 0 | 0.55 |
| P23 | 117.56 | 1.47 | −2.94 | Soluble | High | 0 | 0.55 |
| P24 | 113.9 | 2.08 | −3.47 | Soluble | High | 0 | 0.55 |
| P25 | 104.67 | 3.6 | −5.22 | Moderately soluble | High | 0 | 0.55 |
Topological polar surface area;
Log of the partition coefficient (P);
Log solubility;
Estimated aqueous solubility in mg/mL;
Gastrointestinal.
BOILED-Egg is an intrinsic model in SwissADME for predicting brain access or passage of the brain blood barriers (BBB) and passive gastrointestinal absorption (HIA). It is a proficient method that depends on two descriptors WLOGP (lipophilicity) and TPSA (apparent polarity). The white region of BOILED-Egg represents a high probability of passive absorption by the gastrointestinal tract, and the yellow region represents a strong possibility of reaching the brain. Whether the candidate is (PGP+) or not (PGP−) a substrate for P-gp, a multidrug resistance efflux pump responsible for drug clearing, is indicated by points colored red or blue, respectively [53].
BOILED-Egg was plotted for all the 25 compounds (Supplementary file - Fig. S1). None of them were in the yellow region, implying that they may not penetrate the brain. Twenty-one compounds containing the substituted phenyl rings were in the white region, indicating proper absorption. Four compounds (P16, P18, P20, and P21) were outside the egg, suggesting that they neither absorb nor cross the BBB and may have poor bioavailability. These four compounds are substituted five/six-membered aromatic/ heteroaromatic or fused ring viz. nitrophenyl (P16), methylisoxazole (P18), thiazole (P20), or 5-methylthiazole (P21). Accordingly, the biological screening of the compounds showed that these four compounds were inactive or nearly inactive at the receptor site.
2.3.3. Computation of toxic hazards
Checking the alerts for Cramer rules and cytotoxicity by P450-mediated drug metabolism for the compound P19 resulted in high Class III and four sites of metabolism (Supplementary file - Fig. S2 and Fig. S3). Verbose explanation stated that, “Compound has heterocyclic ring with complex substituents (Q7 & 11)”, which could be the reason for being under Class III, and that there are four sites of CYP450-mediated metabolism viz. S-oxidation, N-oxidation, and aromatic hydroxylation. Even though P19 is under Class III and predicted to display toxicity, the possible sites of metabolism by CYP450 could reduce the toxicity by enhancing the compound metabolism.
3. Materials and methods
3.1. In-silico studies
3.1.1. Molecular docking
Molecular docking of the target compounds in the HDAC8 crystal structure was performed with MOE 2019 software (Chemical Computing Group ULC, Canada). The crystal structures of HDAC8 (PDB-ID: 1T69) and HDAC4 (PDB-ID: 2VQJ) were obtained from RCSB Protein Data Bank. The PDB-files were subjected to structure preparation including 3D-protonation for subsequent docking. The partial charges of all protein and ligand atoms were calculated using the implemented Amber14 force field. Molecular docking was performed choosing the triangle matcher for placement of the ligand in the binding site and ranked with the London dG scoring function. The best 30 poses were passed to the refinement and energy minimization in the pocket using the induced fit method and then rescored with the GBVI/WSA dG scoring function.
3.1.2. SwissADME predictions tool
SwissADME is a reliable free online tool [52] that predicts the physicochemical properties of the compounds. The bioavailability and pharmacokinetic parameters of any synthetic compound is obtained by inputting its structure on the website http://www.swissadme.ch/index.php#.
3.1.3. Computation of toxic hazards
Toxtree is a free, user-friendly, and flexible open-source application tool, developed by IDEA Consult Ltd. (Sofia, Bulgaria). It assesses the toxic hazards of a compound by applying a decision tree approach. Toxtree decision tree can be used for applying the Cramer rules, Cramer rules with extensions, Verhaar scheme, Skin irritation and corrosion prediction, eye irritation, and corrosion prediction, Benigni/Bossa rule base (for mutagenicity and carcinogenicity), Michael Acceptors, Skin sensitization alerts, START biodegradability, cytochrome P450-mediated drug metabolism, DNA binding alerts, protein binding alerts etc. It can evaluate the Threshold of Toxicological Concern (TTC) of the material or their potential toxicity. Cramer rules predict toxicological dangers of the molecules when administered orally. In this, molecules are grouped into three classes based on a decision tree. This involves 33 structural rules, and places the assessed molecules into one of three classes: Class I (Low) molecules with low oral toxicity (simple chemical structures with efficient modes of metabolism); Class II (Intermediate) molecules with intermediate toxicity; and Class III (High) molecules that have reactive functional groups and possible significant toxicity. SMARTCyp is a reactivity model that estimates the molecule site(s) labile to metabolism by Cytochromes P450 isoform 3A4 [54,55].
3.2. Chemistry
All reagents, solvents, and chemicals were purchased from commercial sources viz. Sigma Aldrich, S.D. Fine Chem. Ltd., Himedia, and VWR. Reactions were monitored by Thin Layer Chromatography (TLC) using Merck precoated silica gel 60 F-254 plates under short wavelength UV-light (254nm) to detect the UV absorbing spots to ensure the completion of the reaction and also the purity of compounds at each step. All intermediates were purified by recrystallization with suitable solvents such as chloroform, methanol, ethanol, etc. The final compounds were purified by either recrystallization or column chromatography. Column chromatography was performed on silica gel 60 (60 to 120 mesh) with combinations of suitable solvents. The purity of all final products (≥95%) was determined using an Agilent 1200 high-performance liquid chromatography (HPLC) system; software- EZ chrome Elite. The chromatographic column was HemochromIntsil A31 C18 5U 150 mm × 4.6 mm Sn-B180127, with detection at 300 nm. UV-visible detector was used with the flow rate of 1 mL/min. The oven temperature was 30°C; gradient elution with a run time of 10 min using Methanol: Formic Acid (1%) (Formic acid: in 1000 mL double distilled water 1mL formic acid was added) in 80:20 ratio. The melting points of all the intermediates and final compounds were determined with VEEGO, MODEL: VMP-DS Melting Point apparatus.
Structural characterization of the intermediates was done by FTIR and 1H-NMR, and of the final step products by FTIR, 1H-NMR, 13C-NMR, and mass spectrometry. IR was recorded with JASCO FT/IR- 4100 typeA spectrometer using direct sampling technique. 1H-NMR spectra were recorded on Bruker Avance 400 MHz Spectrometer using DMSO-d6 as solvent.13C-NMR was recorded on a Bruker Avance Spectrometer at 100 MHz using DMSO-d6 as solvent. All shifts of 1H NMR are in δ (ppm) units relative to the signals for the solvent DMSO (δ- 2.50 ppm). All coupling constants (J values) are in hertz (Hz). NMR abbreviations are: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and dd, doublet of doublets. The mass spectrum was recorded on LC-MS Agilent Technologies 1260 Infinity instrument. To determine the absorption of synthesized compounds at different wavelengths, we performed UV Spectroscopy (Supplementary file).
The 25 new derivatives of pyridyl containing N-substituted TZDs were synthesized as shown in Scheme 1. Variously substituted chloroacetylated amides (2a-2y) were prepared as previously reported [34–37,56,57]. All chloroacetylated amides were purified by recrystallization with suitable solvents and then used for the next reaction steps. The compound 5-(pyridin-2-ylmethylene)thiazolidine-2,4-dione (5) was synthesized by following the Knoevenegel condensation. Synthesis of potassium salt of (5) was done by refluxing potassium hydroxide in ethanol to produce potassium-2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-ide (6). The substituted chloroacetylated amides (2a-2y) and 6 were added to methanol (10 ml) in round-bottom flask. This solution was then refluxed and after completion the reaction was stopped and the mixture was cooled. The solid precipitated out was collected and purified to produce the final products.
3.2.1. Synthesis of 5-(pyridine-2-ylmethylene)thiazolidine-2,4-dione (5):
2-Pyridinecarboxaldehyde 3 (5 ml, 0.046 mol), thiazolidine-2,4-dione (4) (5 g, 0.0427 mol) and sodium acetate (3 g, 0.036 mol) were added to acetic acid (5 ml) and refluxed for 5 h. The reaction mixture was then cooled to RT and the precipitated solid was collected by filtering under vacuum, washed several times with water, and dried under RT. Crude solid was purified by recrystallization from an appropriate solvent to obtain brown shiny crystals (5). Yield 9 g (90%); Brown shiny crystal; M.P. 232 °C (Charred); IR (cm−1): 3136, 1737, 1614. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 7.41–7.42 (m, 1H), 7.80–7.84 (m, 2H), 7.90–7.94 (m, 1H), 8.73–8.74 (m, 1H), 12.40 (s, 1H).
3.2.2. Synthesis of potassium-2,4-dioxo-5-(pyridine-2-ylmethylene)thiazolidin-3-ide (6):
To the solution of potassium hydroxide (5 g, 0.089 mol) in ethanol (25 ml), (5) (10 g, 0.048 mol) was added with stirring, and the reaction mixture was refluxed for 3 h. The fine solid obtained after cooling the reaction mixture was collected by filtering and washed with cold ethanol to obtain potassium salt 6. Yield 10 g (66%); Light brown color solid; M.P. 292.4 °C (Charred); IR (cm−1): 3037, 1626, 1662. 1H-NMR (400 MHz, DMSO-d6, δ ppm). 7.18–7.21 (m, 1H), 7.26 (s, 1H), 7.52 (d, J=8Hz, 1H), 7.74–7.78 (m, 1H), 8.61–8.62 (m, 1H).
3.2.3. Synthesis of N-(substitutedaryl/heteroaryl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P1-P25):
Intermediate 6 (1 g, 0.004 mol) and variously substituted amides (2a-2y) (0.004 mol) were dispensed together in a round-bottom flask along with 10 mL methanol. This mixture was refluxed and the reaction was monitored for completion by TLC using appropriate mobile phase of Hexane: Ethyl acetate (1:1). After 5–6 h the reaction was stopped and the mixture was cooled. The precipitated solid was collected by filtering under vacuum and purified by recrystallization (chloroform: methanol, 2:1) or column chromatography (hexane: ethyl acetate, 1.5: 0.5) with an appropriate solvent to give the corresponding final products (P1-P25).
3.2.3.1. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-phenylacetamide (P1):
Yield 0.9 g (56%); M.P. 282.8 °C (Charred); buff white color solid; IR (cm−1): 3267, 1739, 1656, 1683, 1620, 1599, 1388. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.50 (s, 2H), 7.08 (s, 1H), 7.32 (s, 2H), 7.46 (s, 1H), 7.55 (s, 2H), 7.91–8.00 (m, 3H), 8.79 (s, 1H), 10.39 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.355, 119.175, 123.714, 124.264, 125.266, 128.002, 128.854, 129.461, 137.635, 138.320, 149.342, 151.035, 163.880, 165.544, 170.979. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 339, LC-MS (m/z, I %): 338 [(M-H)+, 100%]. HPLC Purity: % Area 98.32, RT 2.99 mins.
3.2.3.2. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(2-fluorophenyl)acetamide (P2):
Yield 1 g (60%); M.P. 293.9 °C (Charred); buff white color solid; IR (cm−1): 3263, 1737, 1666, 1686, 1616, 1546, 1386, 1298. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.58 (s, 2H), 7.16–7.18 (m, 2H), 7.25–7.30 (m, 1H), 7.46 (t, J= 5.8 Hz, 1H), 7.85–7.91 (m, 2H), 7.94–8.00 (m, 2H), 8.78 (d, J= 4 Hz, 1H), 10.26 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.192, 115.476, 115.669, 123.862, 124.274, 124.476, 125.250, 128.007, 129.472, 137.645, 149.347, 151.028, 164.503, 165.510, 170.955. UV- Spectrum (10 ppm, λmax - 331.7 nm). Theoretical mass: 357, LC-MS (m/z, I %): 355.9 [(M-2H)+, 100%]. HPLC Purity: % Area 97.54, RT 2.97 mins.
3.2.3.3. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(3-fluorophenyl)acetamide (P3):
Yield 0.85 g (75%); M.P. 250.4 °C (Charred); buff color solid; IR (cm−1): 3306, 1739, 1685, 1666, 1602, 1543, 1388, 1325. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.52 (s, 2H), 6.916 (t, J= 7.6 Hz, 1H), 7.27–7.29 (m, 1H), 7.35 (q, J= 13 Hz, 1H), 7.44–7.53 (m, 2H), 7.89–7.98 (m, 2H), 8.00 (s, 1H), 8.78 (d, J= 3.2 Hz, 1H), 10.63 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.372, 105.891, 106.153, 110.118, 110.326, 114.943, 124.283, 125.186, 128.019, 129.542, 130.519, 130.612, 137.638, 139.928, 140.039, 149.341, 151.006, 160.897, 163.298, 164.251, 164.334, 165.500, 170.962. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 357, LC-MS (m/z, I %): 356.1 [(M-H)+, 100%]. HPLC Purity: % Area 97.28, RT 3.34 mins.
3.2.3.4. N-(4-bromo-2-fluorophenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P4):
Yield 0.6 g (52%); M.P. charred above 300 °C; light gray color solid; IR (cm−1): 3306, 1737, 1672, 1597, 1602, 1543, 1386, 1213, 686. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.58 (s, 2H), 7.38 (d, J= 8.4 Hz, 1H), 7.46 (q, J= 6.6 Hz, 1H), 7.60–7.63 (dd, J= 10.6 Hz, 1H), 7.83.−7.86 (m, 1H), 7.87–7.91 (m, 1H), 7.94–7.96 (m, 1H), 7.98 (s, 1H), 8.78 (d, J= 4.4 Hz, 1H), 10.33 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.214, 118.813, 119.041, 124.287, 125.034, 125.204, 127.583, 128.014, 129.517, 137.649, 149.344, 151.009, 164.661, 165.478, 170.938. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 434, LC-MS (m/z, I %): 433.9 [(M-H)+, 100%], 435.9 [(M-H)+, 90%]. HPLC Purity: % Area 97.72, RT 4.4 mins.
3.2.3.5. N-(2,4-difluorophenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P5):
Yield 0.95 g (77%); M.P. 284.9 °C (Charred); white color solid; IR (cm−1): 3273, 1737, 1672, 1552, 1614, 1505, 1384, 1222. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.55 (s, 2H), 7.06 (t, J= 7.8 Hz, 1H), 7.31 (t, J = 8.8 Hz, 1H), 7.45 (t, J= 5.8 Hz, 1H), 7.77–7.80 (q, J= 11 Hz, 1H), 7.87–7.89 (m, 1H), 7.93–7.95 (m, 1H), 7.98 (s, 1H), 8.77 (d, J= 3.6 Hz, 1H), 10.27 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.056, 104.201, 111.350, 124.296, 125.223, 127.983, 129.481, 137.648, 149.334, 150.968, 164.590, 165.493, 170.981. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 375, LC-MS (m/z, I %): 374 [(M-H)+, 100%]. HPLC Purity: % Area 95.82, RT 3.31 mins.
3.2.3.6. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(3-(trifluoromethyl)phenyl)acetamide (P6):
Yield 1.2 g (80%); M.P. 244.5 °C (Charred); buff white color solid; IR (cm−1): 3273, 1741, 1685, 1666, 1616, 1545, 1390, 1214. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.54 (s, 2H), 7.44 (t, J= 8.8 Hz, 2H), 7.58 (t, J= 8 Hz, 1H), 7.72–7.74 (m, 1H), 7.88–7.90 (m, 1H), 7.94–7.96 (m, 1H), 7.99 (s, 1H), 8.03 (s, 1H), 8.78 (d, J= 4 Hz, 1H), 10.76 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.365, 115.260, 120.123, 122.800, 124.305, 125.176, 128.004, 129.560, 130.177, 137.649, 138.993, 149.338, 150.967, 164.631, 165.506, 170.997. UV- Spectrum (10 ppm, λmax - 332.4 nm). Theoretical mass: 407, LC-MS (m/z, I %): 407 [(M+)+, 100%]. HPLC Purity: % Area 97.59, RT 2.98 mins.
3.2.3.7. N-(4-chloro-2-(trifluoromethyl)phenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P7):
Yield 0.8 g (64%); M.P. charred above 300 °C; cream color solid; IR (cm−1): 3217, 1743, 1620, 1668, 1608, 1537, 1309, 1282, 779. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.52 (s, 2H), 7.45 (t, J= 5.8 Hz, 1H), 7.49–7.51 (m, 1H), 7.76–7.78 (m, 1H), 7.82 (s, 1H), 7.87–7.89 (m, 1H), 7.93–7.95 (m, 1H), 7.98 (s, 1H), 8.77 (d, J= 3.6 Hz, 1H), 10.18 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.416, 113.673, 117.503, 119.575, 119.948, 121.114, 123.837, 126.396, 131.352, 131.797, 133.114, 133.448, 147.955, 149.093, 164.934, 165.442, 167.703. UV- Spectrum (10 ppm, λmax - 332 nm). Theoretical mass: 441, LC-MS (m/z, I %): 439.9 [(M-H)+, 100%]. HPLC Purity: % Area 97.99, RT 3.99 mins.
3.2.3.8. N-(3,4-dichlorophenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P8):
Yield 1.1 g (84%); M.P. 269.4 °C (Charred); white color solid; IR (cm−1): 3336, 1737, 1668, 1620, 1585, 1531, 1325, 779. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.51 (s, 2H), 7.45 (d, J= 6.4 Hz, 2H), 7.55–7.58 (m, 1H), 7.87–7.89 (m, 2H), 7.93–7.97 (m, 2H), 8.77 (s, 1H), 10.72 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.349, 119.285, 120.487, 124.294, 125.143, 125.316, 127.996, 129.569, 130.791, 131.124, 137.629, 138.280, 149.320, 150.946, 164.534, 165.475, 170.982. UV- Spectrum (10 ppm, λmax - 332.2 nm). Theoretical mass: 406, LC-MS (m/z, I %): 405.9 [(M-H)+, 100%]. HPLC Purity: % Area 98.68, RT 3.44 mins.
3.2.3.9. N-(2-chloro-5-(trifluoromethyl)phenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P9):
Yield 0.5 g (45%); M.P. 282.6 °C (Charred); white color solid; IR (cm−1): 3306, 1741, 1676, 1618, 1583, 1548, 1327, 1130, 781. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.65 (s, 2H), 7.46 (t, J= 6 Hz, 1H), 7.57 (d, J= 8.4 Hz, 1H), 7.77 (d, J= 8.4 Hz, 1H), 7.88–7.90 (m, 1H), 7.94–7.98 (m, 1H), 7.99(s, 1H), 8.14 (s, 1H), 8.77 (d, J= 4 Hz, 1H), 10.35 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.222, 121.459, 122.823, 124.317, 125.155, 128.009, 129.566, 130.908, 135.101, 137.658, 149.345, 150.957, 165.307, 165.477, 170.974. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 441, LC-MS (m/z, I %): 439.9 [(M-H)+, 100%]. HPLC Purity: % Area 98.70, RT 4.48 mins.
3.2.3.10. N-(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene) thiazolidin-3-yl)acetamide (P10):
Yield 0.93 g (73%); M.P. 254.6 °C (Charred); light yellow color solid; IR (cm−1): 3304, 3052, 1743, 1658, 1618, 1585, 1550, 1300. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 2.10 (s, 3H), 3.04 (s, 3H), 4.46 (s, 2H), 7.33 (d, J= 7.2 Hz, 3H), 7.34–7.51 (m, 3H), 7.86–7.88 (m, 1H), 7.93–7.84 (m, 1H), 7.97 (s, 1H), 8.76 (d, J= 3.2 Hz, 1H), 9.57 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 11.024, 35.725, 42.835, 106.286, 123.804, 124.246, 125.357, 126.512, 127.948, 129.107, 129.331, 134.759, 137.625, 149.327, 151.012, 152.074, 161.452, 164.916, 165.520, 170.971. UV- Spectrum (10 ppm, λmax - 331.5 nm). Theoretical mass: 449, LC-MS (m/z, I %): 448.1 [(M-H)+, 100%].. HPLC Purity: % Area 97.32, RT 3.01 mins.
3.2.3.11. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(4-fluorophenyl)acetamide (P11):
Yield 1 g (80%); M.P. 274.5 °C (Charred); buff white color solid; IR (cm−1): 3265, 1737, 1681, 16758, 1616, 1548, 1330, 1298. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.49 (s, 2H), 7.15 (t, J= 7.8 Hz, 2H), 7.45 (s, 1H), 7.56 (s, 2H), 7.88 (d, J= 7.2 Hz, 1H), 7.94 (d, J= 7.2 Hz, 1H), 7.98 (s, 1H), 8.78 (s, 1H), 10.46 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.249, 115.319, 115.542, 121.033, 121.110, 124.276, 125.230, 127.981, 129.476, 134.648, 137.630, 149.326, 150.981,157.026, 159.413, 163.871, 165.530, 171.008. UV- Spectrum (10 ppm, λmax - 332 nm). Theoretical mass: 357, LC-MS (m/z, I %): 356 [(M-H)+, 100%]. HPLC Purity: % Area 95.74, RT 3.14 mins.
3.2.3.12. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(p-tolyl)acetamide (P12):
Yield 1.3 g (90%); M.P. 287.3 °C (Charred); white color solid; IR (cm−1): 3250, 3047, 1737, 1681, 1651, 1614, 1548, 1323. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 2.25 (s, 3H), 4.48 (s, 2H), 7.13 (d, J= 8.32 Hz, 2H), 7.42–7.48 (m, 3H), 7.91–7.93 (m, 1H), 7.95–7.99 (m, 1H), 8.01 (s, 1H), 8.79 (d, J= 4.96 Hz, 1H), 10.28 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.803, 113.675, 117.543, 119.165, 119.565, 119.927, 129.214, 132.680, 135.822, 147.948, 149.116, 163.547, 165.083, 167.854. UV- Spectrum (10 ppm, λmax - 332.3 nm). Theoretical mass: 353, LC-MS (m/z, I %): 351.8 [(M-2H)+, 100%]. HPLC Purity: % Area 98.56, RT 4.48 mins.
3.2.3.13. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(2-phenoxyphenyl)acetamide (P13):
Yield 0.75 g (80%); M.P. 212.3 °C (Charred); gray color solid; IR (cm−1): 3265, 1739, 1688, 1668, 1620, 1585, 1107, 1230, 1327. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.53 (s, 2H), 6.68 (s, 1H), 7.03 (d, J= 7.2 Hz, 2H), 7.10–7.18 (m, 3H), 7.39–7.97 (m, 3H), 7.87 (d, J= 6.8 Hz, 1H), 7.97 (s, 3H), 8.77 (s, 1H), 10.05 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.255, 118.463, 118.660, 123.269, 123.561, 123.638, 124.268, 125.224, 125.288, 127.978, 128.952, 129.442, 129.950, 137.629, 147.401, 149.331, 150.989, 156.428, 164.419, 165.515, 170.963. UV- Spectrum (10 ppm, λmax - 331.7 nm). Theoretical mass: 431, LC-MS (m/z, I %): 430.1 [(M-H)+, 100%]. HPLC Purity: % Area 97.97, RT 3.34 mins.
3.2.3.14. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(4-methoxyphenyl)acetamide (P14):
Yield 1.2 g (89%); M.P. 284.8 °C (Charred); buff white color solid; IR (cm−1): 3252, 1739, 1685, 1658, 1618, 1583, 1107, 1257, 1327. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 3.06 (s, 3H), 4.57 (s, 2H), 7.38 (d, J=8 Hz, 2H), 7.44 (d, J=5.6 Hz, 1H), 7.60 (d, J=10 Hz, 1H), 7.81–7.89 (m, 2H), 7.93–7.98 (m, 2H), 8.77 (d, J=3.2 Hz, 1H), 10.39 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.179, 118.811, 119.033, 124.309, 125.023, 125.163, 127.550, 128.010, 129.531, 137.657, 149.341, 150.955, 151.978, 164.664, 165.482, 170.982. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 369, LC-MS (m/z, I %): 368.2 [(M-H)+, 100%]. HPLC Purity: % Area 97.11, RT 2.9 mins.
3.2.3.15. N-(4-bromo-2,6-difluorophenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P15):
Yield 0.8 g (70%); M.P. charred above 300 °C; buff white color solid; IR (cm−1): 3250, 1749, 1541, 1678, 1610, 1583, 1384, 1234, 779. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.52 (s, 2H), 7.44 (s, 2H), 7.57 (d, J= 5.6 Hz, 1H), 7.87–7.98 (m, 3H), 8.77 (s, 1H), 10.22 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 42.668, 116.086, 124.295, 125.343, 127.979, 129.432, 137.665, 149.346, 151.612, 164.704, 165.383, 170.850. UV- Spectrum (10 ppm, λmax - 331.8 nm). Theoretical mass: 452, LC-MS (m/z, I %): 453.8 [(M-H)+, 100%], 452 [(M-H)+, 90%]. HPLC Purity: % Area 99.03, RT 2.94 mins.
3.2.3.16. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(4-nitrophenyl)acetamide (P16):
Yield 0.55 g (40%); M.P. 289.4 °C (Charred); cream color solid; IR (cm−1): 3259, 1741, 1564, 1678, 1614, 1593, 1379, 1317. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.57 (s, 2H), 7.46 (t, J= 6 Hz, 1H), 7.79 (d, J= 8.8 Hz, 2H), 7.88–7.90 (m, 1H), 7.95 (m, 1H), 7.99 (s, 1H), 8.23 (d, J= 8.8 Hz, 2H), 8.78 (d, J= 4 Hz,1H), 11.04 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.500, 119.039, 124.344, 125.040, 125.086, 128.040, 129.649, 137.670, 142.604, 144.318, 149.346, 150.933, 165.019, 165.467, 170.983. UV- Spectrum (10 ppm, λmax - 332.5 nm). Theoretical mass: 384, LC-MS (m/z, I %): 383.1 [(M-H)+, 100%]. HPLC Purity: % Area 99.16, RT 3.81 mins.
3.2.3.17. N-(3-chloro-4-methylphenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P17):
Yield 1 g (83%); M.P. 273.5 °C (Charred); buff white color solid; IR (cm−1): 3246, 2949, 1737, 1581, 1656, 1614, 1579, 1388, 779. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 2.26 (s, 3H), 4.49 (s, 2H), 7.27–7.34 (q, J= 16.8 Hz, 2H), 7.45 (t, J= 6 Hz, 1H), 7.71 (s, 1H), 7.87–7.89 (m, 1H), 7.93–7.95 (m, 1H), 7.98 (s, 1H), 8.77 (d, J= 4 Hz, 1H), 10.52 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 18.863, 43.303, 117.835, 119.192, 124.303, 125.186, 128.004, 129.513, 130.508, 131.320, 133.067, 137.330, 137.651, 149.341, 150.966, 164.114, 165.515, 171.005. UV- Spectrum (10 ppm, λmax - 332 nm). Theoretical mass: 387, LC-MS (m/z, I %): 386 [(M-H)+, 100%]. HPLC Purity: % Area 96.36, RT 4.68 mins.
3.2.3.18. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(4-methylisoxazol-3-yl)acetamide (P18):
Yield 0.68 g (50%); M.P. 272.6 °C (Charred); buff white color solid; IR (cm−1): 3219, 2982, 1743, 1529, 1687, 1616, 1568, 1325. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 2.36 (s, 3H), 4.51 (s, 2H), 6.55 (s, 1H), 7.45 (t, J= 6 Hz, 1H), 7.87–7.88 (m, 1H), 7.93–7.95 (m, 1H), 7.97 (s, 1H), 8.77 (d, J= 4.6 Hz, 1H), 11.40 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 12.034, 43.053, 96.126, 124.300, 125.180, 127.993, 129.537, 137.641, 149.325, 150.944, 157.537, 164.506, 165.431, 169.985, 170.963. UV- Spectrum (10 ppm, λmax - 332 nm). Theoretical mass: 344, LC-MS (m/z, I %): 343 [(M-H)+, 100%]. HPLC Purity: % Area 98.69, RT 2.67 mins.
3.2.3.19. N-(benzo[d]thiazol-2-yl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P19):
Yield 1.3 g (89%); M.P. 263.9 °C (Charred); white color solid; IR (cm−1): 3227, 1735, 1602, 1666, 1614, 1583, 1383. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.68 (s, 2H), 7.32 (t, J= 7.4 Hz, 1H), 7.44–7.48 (m, 2H), 7.77 (d, J= 8 Hz, 1H), 7.89–7.90 (m, 1H), 7.93–7.98 (m, 2H), 8.00 (s, 1H), 8.78 (d, J= 4.4 Hz, 1H), 11.40 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 30.782, 35.806, 42.934, 120.655, 121.781, 123.893, 124.358, 125.134, 126.309, 128.048, 129.693, 131.336, 137.683, 149.357, 150.933, 162.465, 165.420, 165.666, 170.996. UV- Spectrum (10 ppm, λmax - 331.7 nm). Theoretical mass: 396, LC-MS (m/z, I %): 394.9 [(M-2H)+, 100%]. HPLC Purity: % Area 97.45, RT 4.5 mins.
3.2.3.20. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(thiazol-2-yl)acetamide (P20):
Yield 0.85 g (68%); M.P. charred above 300 °C; buff white color solid; IR (cm−1): 3246, 1726, 1602, 1678, 1579, 1473, 1386. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.61 (s, 2H), 7.23 (d, J= 3.6 Hz, 1H), 7.45–7.49 (m, 2H), 7.88–7.90 (m, 1H), 7.94–7.98 (m, 1H), 7.99 (s, 1H), 8.78 (d, J= 4.4 Hz, 1H), 12.59 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 42.691, 114.045, 124.346, 125.162, 128.034, 129.617, 137.684, 149.362, 150.946, 165.435, 170.984. UV- Spectrum (10 ppm, λmax - 332.3 nm). Theoretical mass: 346, LC-MS (m/z, I %): 344.9 [(M-2H)+, 100%]. HPLC Purity: % Area 98.34, RT 2.78 mins.
3.2.3.21. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(5-methylthiazol-2-yl)acetamide (P21):
Yield 0.67 g (60%); M.P. charred above 300 °C; buff white color solid; IR (cm−1): 3338, 2841, 1735, 1500, 1678, 1579, 1465, 1381. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 2.32 (s, 3H), 4.58 (s, 2H), 7.15 (s, 1H), 7.45 (t, J= 6 Hz, 1H), 7.87–7.89 (m, 1H), 7.93–7.95 (m, 1H), 7.98 (s, 1H), 8.77 (d, J= 4.4 Hz, 1H), 12.38 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 11.027, 42.659, 124.322, 125.175, 126.887, 128.013, 129.584, 137.660, 149.341, 150.945, 165.427, 170.975. UV- Spectrum (10 ppm, λmax - 332 nm). Theoretical mass: 360, LC-MS (m/z, I %): 358.9 [(M-2H)+, 100%]. HPLC Purity: % Area 97.49, RT 3.73 mins.
3.2.3.22. N-(4-bromophenyl)-2-(2,4-dioxo-5-(pyridin-2-yl methylene)thiazolidin-3-yl)acetamide (P22):
Yield 0.9 g (88%); M.P. 279.3 °C (Charred); buff color solid; IR (cm−1): 3254, 1734, 1548, 1676, 1581, 1410, 1386, 727. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.50 (s, 2H), 7.43–7.53 (m, 5H), 7.86–7.88 (m, 1H), 7.92–7.94 (m, 1H), 7.97 (s, 1H), 8.76 (d, J= 3.6 Hz, 1H), 10.55 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.337, 115.394, 121.171, 124.280, 125.178, 127.988, 129.518, 131.668, 137.616, 149.319, 150.952, 164.123, 165.507, 171.007. UV- Spectrum (10 ppm, λmax - 332.7 nm). Theoretical mass: 416, LC-MS (m/z, I %): 418 [(M+2H)+, 100%], 416 [(M+)+, 75%]. HPLC Purity: % Area 97.69, RT 4.28 mins.
3.2.3.23. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(pyridin-2-yl)acetamide (P23):
Yield 1.1 g (66%); M.P. 256.3 °C (Charred); buff white color solid; IR (cm−1): 3265, 1739, 1543, 1678, 1620, 1577, 1386. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.57 (s, 2H), 7.13 (s, 1H), 7.45 (s, 1H), 7.78 (s, 1H), 7.87–7.98 (m, 4H), 8.34 (s, 1H), 8.78 (s, 1H), 10.94 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.329, 113.530, 119.898, 124.303, 125.205, 128.002, 129.509, 137.653, 138.417, 148.083, 149.342, 150.972, 151.244, 164.796, 165.519, 171.012. UV- Spectrum (10 ppm, λmax - 331.4 nm). Theoretical mass: 340, LC-MS (m/z, I %): 339 [(M-H)+, 100%]. HPLC Purity: % Area 97.76, RT 2.77 mins.
3.2.3.24. 2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)-N-(3-methoxyphenyl)acetamide (P24):
Yield 0.92 g (85%); M.P. 249.3 °C (Charred); buff white color solid; IR (cm−1): 3252, 1741, 1556, 1681, 1604, 1581, 1386. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 3.71 (s, 3H), 4.49 (s, 2H), 6.65 (d, J= 7.2 Hz, 1H), 7.07 (d, J= 6.8 Hz, 1H), 7.20–7.24 (m, 2H), 7.44 (s, 1H), 7.86–7.97 (m, 3H), 8.77 (s, 1H), 10.42 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.331, 54.953, 104.867, 109.289, 111.396, 124.271, 125.202, 127.980, 129.492, 129.711, 137.617, 139.449, 149.322, 150.965, 159.528, 163.947, 165.532, 171.024. UV- Spectrum (10 ppm, λmax - 332 nm). Theoretical mass: 369, LC-MS (m/z, I %): 368 [(M-H)+, 100%]. HPLC Purity: % Area 97.25, RT 3.09 mins.
3.2.3.25. N-(3,4-dibromophenyl)-2-(2,4-dioxo-5-(pyridin-2-ylmethylene)thiazolidin-3-yl)acetamide (P25):
Yield 1 g (78%); M.P. 272 °C (Charred); buff white color solid; IR (cm−1): 3340, 1737, 1531, 1666, 1618, 1583, 1381. 1H-NMR (400 MHz, DMSO-d6, δ ppm) 4.51 (s, 2H), 7.44–7.46 (m, 2H), 7.56–7.58 (m, 1H), 7.87–7.90 (m, 2H), 7.93–7.95 (m, 1H), 7.98 (s, 1H), 8.77 (s, 1H), 10.73 (s, 1H). 13C-NMR (400 MHz, DMSO-d6): 43.344, 119.277, 120.466, 124.313, 125.123, 125.308, 128.012, 129.578, 130.806, 131.120, 137.646, 138.275, 149.332, 150.937, 164.543, 165.477, 170.991. UV- Spectrum (10 ppm, λmax - 331.9 nm). Theoretical mass: 494, LC-MS (m/z, I %): 496.9 [(M+2H)+, 100%],496 [(M+2H)+, 70%]. HPLC Purity: % Area 95.73, RT 5.61 mins.
3.3. Biological Evaluation
3.3.1. In-vitro cytotoxicity assays
The Differential Nuclear Staining (DNS) assay is a live cell imaging-based assay, which utilizes two nuclear dyes to easily label living and dead cells [38]. An initial assessment of cytotoxicity on this series of 25 compounds was conducted via the DNS assay upon exposure of CEM, K-562 and KCL-22 human cancer cell lines to 50 μM of compounds for 48 h. Compounds were dissolved and diluted in Dimethyl sulfoxide (DMSO) to reach the desired concentration. DMSO, Hydrogen peroxide, and untreated cells were utilized as vehicle, positive, and negative controls, respectively, in all experiments. Cells were seeded in a 96-well plate at a density of 10,000 cells/well in 100 μL of complete culture media. Images of stained cells were collected with the GE Healthcare Life Sciences IN Cell Analyzer 2000. Two h prior to imaging, Hoechst and Propidium iodide (PI) stains were added to each well to distinguish between living and dead/dying cells. Hoechst is a dye able to permeate the membranes of all cells within a sample, whereas PI only penetrates into cells with compromised membranes. Colocalization of Hoechst (blue) and PI (red) signals indicate the dead cell population. In this initial screening, experimental samples were assessed singularly, and control samples in quadruplicate. Subsequent DNS assays on the CEM, K-562, KCL-22 cell lines, were then performed to determine the CC50 values of the compounds that showed cytotoxicity in the initial screening and activity in other biological analyses. Cells were seeded in an identical manner but were treated with a concentration gradient of compound (P8, P9, P10, P12, P19, and P25) ranging from 0.5 μM to 100 μM. Experimental samples and controls were evaluated for cytotoxicity in triplicate after 48 h of incubation.
3.3.1.1. Statistical analysis
For each sample group, the average of replicates and their corresponding standard deviations are displayed. Statistical significance was determined by two-tailed Student’s t-test comparing experimental treatments to vehicle controls (DMSO). P values < 0.05 were deemed significant and graphically displayed using asterisks (*=P<0.05, **=P<0.01, ***=P<0.001).
3.3.2. HDAC enzyme activity assay
Recombinant HDAC4 and 8 were produced as in [58]. The other HDAC isoenzymes were purchased from BPS Bioscience. Serial dilutions of the inhibitor in the assay buffer (25 mM Tris-HCl, pH 8.0, 75 mM KCl, 0.001 % Pluronic F-127) were incubated with HDAC in a black 96-well microtiter half-area plate (Greiner) for 60 min at 30 °C. Subsequently the reaction was started by the addition of 20 μM Boc-Lys(trifluoroacetyl)-AMC (Bachem) as a substrate for HDAC4, 5 and 8, and 50 μM Boc-Lys(acetyl)-AMC as substrate for HDAC1, 2, 3, and 6. After incubation for 60 min at 30 °C, the reaction was stopped by the addition of 1.7 μM SATFMK for HDAC4, 5, and 8, and 4.17 μM suberoylanilide hydroxamic acid (SAHA, Cayman Chemical Company) for HDAC1, 2, 3, and 6. The deacetylated substrate was transformed into a fluorescent product by the addition of 0.4 mg/ml trypsin (Applichem). The release of AMC was followed in a microplate reader (PheraStar Plus, BMG Labtech) at 450 nm (λEx = 350 nm) and correlated to enzyme activity. Dose-response curves were produced with GraphPad Prism and fitted to a four parameters logistic function to obtain IC50 values [59]
Where,
EA is the enzyme activity at a given inhibitor concentration x; Emax and E0 are the enzyme activities determined at zero and complete inhibition, respectively; IC50 represents the inhibitor concentration at which half the enzyme molecules are inhibited; h is the slope of the curve.
3.3.3. GLUT 1, 4 and 5 transport inhibition assay
Synthesized compounds were examined for their effect on the transport activity of GLUT1 [48], GLUT4 [48], and GLUT5 [49] expressed in hexose transporter null yeast cells (hxt0). Yeast cell culturing was done at 30 °C with shaking (180 rpm). VW4000fgy1 yeast cells expressing GLUT1 [48] were cultured for 2–3 days in the synthetic complete media without uracil (SC-uracil) with 2% (w/v) maltose. Cells were washed once in SC-uracil, 2% (w/v) glucose media, transferred in the same media so that OD600nm~0.5, and grown further for 1–2 days. VW4000fgy1erg4 yeast cells expressing GLUT4 [48] were cultured like GLUT1, but in media with lower concentrations of maltose and glucose: SC-uracil, 1% (w/v) maltose media for the initial cell culture, and SC-uracil, 0.2% (w/v) glucose media for the final cell culture. VW4000 yeast cells expressing GLUT5 [49] were cultured for one day in YEP [1% (w/v) yeast extract and 2% (w/v) peptone], 2% (w/v) maltose media, 100 μg/ml geneticin G418. Cells were washed once in YEP, 2% (w/v) fructose media transferred in the same media so that initial OD600nm~0.5, and grown further for 1–2 days. For transport assay, cells in the hexose media were centrifuged (1000 × g, 5 minutes), washed once in PBS buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4), then resuspended in PBS buffer at an OD600nm ~ 10; each assay contained 100 μl of this cell solution.
The transport assay was started by the addition of C14-hexose to a final concentration of 5 mM glucose for GLUT1 or GLUT4, and 10 mM fructose for GLUT5. Transport assay was stopped after 10 minutes by the addition of 3-ml ice-chilled Quench buffer (0.1 M KPi, 0.1 M LiCl, pH 5.5) to the assay, followed by filtration under vacuum on a glass fiber filter (GC50; Advantec, Tokyo, Japan), another wash with 3-ml Quench buffer, and one more filtration. The filtration membranes containing the cells were transferred to scintillation vials, combined with 10 ml of Scintillation buffer (BioSafeII; Research Products International, Mount Prospect, IL, USA), and, after brief vortexing, radioactivity was measured with a scintillation counter (Tri-carb 2900TR, Perkin Elmer, USA). As all synthesized compounds were solubilized in DMSO, controls for calculating the relative activity included 1% (v/v) DMSO -to account for DMSO concentration in the transport assay due to inhibitor addition- and saturating concentrations of known inhibitors for GLUTs [200 μM phloretin for GLUT1 and GLUT4 [60], and 100 μM N-[4-(methylsulfonyl)-2-nitrophenyl]-1,3-benzodioxol-5-amine (MSNBA) for GLUT5 [61]. Initial inhibition screening was done at 100 μM concentration; for the compounds that decreased the relative activity by 50% or more, inhibitor concentration was varied in the transport assay from 0.1 to 100 μM to determine inhibitor IC50. Data were analyzed with GraphPad Prism (San Diego, CA, USA).
3.3.4. Apoptosis studies by flowcytometry
The initiation of apoptosis by compounds P10 and P19 was studied by flow cytometry. Cells were seeded in a 24-well flat-bottom micro-plate and incubated overnight at 37 °C in a CO2 incubator for 24 h. The media was replaced with fresh media, and the cells were incubated with IC50 concentration of P10 and P19 for 24 h. Untreated cells were the negative control. Post incubation, cells were washed with PBS, centrifuged for 5 min at 500 × g at 4 °C, and the supernatant was discarded. Cell pellets were re-suspended in ice-cold 1X Binding Buffer, to which 1 μL of annexin V-FITC solution and 5 μL PI (propidium iodide). Tubes were kept in ice and incubated for 15 min in the dark, 400 μL of ice-cold 1X binding buffer was added. Cell preparations were analyzed by flow cytometry (BD Accuri™ C5 flow cytometer, BD Biosciences, CA, USA). Cytometry data were analyzed by FlowJo software (version 10.1, Ashland, OR, USA) [62].
3.3.5. Cell viability on CEM cell line and normal WBCs by MTT assay
3.3.6.1. WBCs isolation procedure
A volume of 2.5 mL HiSep was transferred to a 15 mL clean centrifuge tube and overlayed with 7.5 ml diluted blood, followed by centrifugation at 400 × g at room temperature for 30 minutes. The supernatant containing most of the plasma and platelet was discarded by aspiration. The mononuclear cells were transferred to a clean centrifuge tube to which 10 ml of isotonic phosphate buffered saline was added and mixed by gentle aspiration. This was followed by centrifugation at 200× g, at room temperature for 10 minutes. The cells were washed again with isotonic phosphate buffered saline and re-suspended in an RBC lysis for 5 mins. The tube was then centrifuged at 300× g for 5 mins, and the supernatant was discarded. Cells were washed with PBS twice and again centrifuged at 300× g for 5 mins. The cells were re-suspended in RPMI 1640 medium, and maintained in a CO2 incubator at 37 °C (95% humidity and 5% CO2) until completion of MTT assay experiments.
3.3.6.2. Cytotoxicity determination by MTT assay
The effect of compound P19 on the viability of CEM cell lines and normal human WBCs were determined by the MTT assay. MTT is a colorimetric assay that measures the reduction of yellow 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) by mitochondrial succinate dehydrogenase. The reduction of MTT can only occur in metabolically active cells. The level of P19 activity was determined by measuring cell viability. The cells were seeded at a density of approximately 5×103 cells/well in a 96-well flat-bottom micro plate, and maintained at 37 °C in 95% humidity and 5% CO2 overnight. The cells were incubated with different concentrations of P19 (100, 75, 50, 25, 10, and 2.5 μM) for another 48 h. The cells in each well were washed twice with phosphate buffer solution, and 20 μL of the MTT staining solution (5 mg/ml in phosphate buffer solution) was added to each well, and the plate was incubated at 37 °C. After 4 h, 100 μL of dimethyl sulfoxide (DMSO) was added to each well to dissolve the formazan crystals, and the absorbance at 570 nm was recorded with a micro-plate reader [63]. Graph Pad Prism Version 5.1 was used to calculate the CC50. The DMSO concentration used for the experiments was less 1.5%. All the concentrations were used in duplicates.
The formula used for determining the cell viability:
4. Conclusion
We synthesized, purified, and characterized the structures of all 25 compounds through various methods (FTIR, 1H-NMR, 13C-NMR, and mass spectrometry). Notably, these compounds do not contain a hydroxamate or trifluoromethylketone zinc-binding group typical for canonical HDAC inhibitors used in clinical therapy, trials or academic research. Two compounds, P10 and P19 showed moderate activity against HDAC8 and a remarkable selectivity against other HDAC isoenzymes. Docking analysis of P19 with best activity (IC50 of 9.3 μM) and docking score against HDAC8 revealed significant binding interactions with the catalytic zinc ion and amino acid residues such as Y306, and G151 in the active site pocket of HDAC8 as well as partial occupation of the specific acetate release channel of HDAC8. This finding is in agreement with the observed experimental isoenzyme selectivity. However, the proposed binding mode has to be confirmed by detailed crystallographic studies. Most interestingly, six compounds (P8, P9, P10, P12, P19, and P25) showed cytotoxicity against leukemic cell lines (K562, CEM, and KCL22) in mid-micromolar range. Moreover, the best active compound P19 was less cytotoxic for non-cancerous cells (WBCs IC50 - 104.2 μM and HS27 IC50 - 105.0 μM). P10 and P19 exhibited apoptotic death in CEM cells. As HDAC inhibitors can affect the expression of GLUT1, we also checked the effect of the compounds on GLUT1, and two other GLUT isoforms GLUT4, and GLUT5. We found that among all 25 compounds only P19 significantly inhibited GLUT1 (IC50 28.2 μM), and it did so without affecting GLUT4 or GLUT5. Thus, P19 is a promising anti-cancer agent, with selectivity and potency against two cancer targets - HDAC8 and GLUT1, offering a starting point for the development of novel non-hydroxamate compounds with enhanced HDAC8 inhibitory potency.
Supplementary Material
HIGHLIGHTS.
N-substituted TZDs, P1-P25 were designed, synthesized & structurally characterized.
Compound P19 was most active against HDAC8 and produced thermal stabilization.
It showed apoptotic cell death and cytotoxicity in leukemic cell lines.
P19 also interfered with glucose transporter 1 - GLUT1.
It was also found less cytotoxic on non-cancerous cells (WBCs and HS27).
Acknowledgments.
This research work was supported by the grant, “Indo-Poland Joint Research Programme” from the Department of Science and Technology (DST), Government of India. Project Reference Number DST/INT/Pol/P-27/2016. In addition, the cytotoxicity work was supported by NIMHD grant 5U54MD007592 to the Border Biomedical Research Center (BBRC) at UTEP.
Abbreviations.
- TZDs
Thiazolidinediones
- HDAC
Histone Deacetylase
- GLUT
Glucose Transporter
- HATs
Histone acetylases
- AML
Acute myeloid leukemia
- ALL
acute lymphocytic leukemia
- APL
Acute promyelocytic leukaemia
- ZBG
Zinc Binding Group
- RMSD
Root Mean Square Deviation
- TPSA
Topological Polar Surface Area
- AcOH
Acetic acid
- EtOH
Ethanol
- DCM
Dichloromethane
- K2CO3
Potassium carbonate
- 1H-NMR
Proton Nuclear Magnetic Resonance
- Hz
Hertz
- J
Coupling Constant
- 13C-NMR
Carbon Nuclear Magnetic Resonance
- FTIR
Fourier-transform infrared spectroscopy
- UV
Ultraviolet Spectroscopy
- HPLC
High Performance Liquid Chromatography
- M.P.
Melting Point
- DNS
Differential Nuclear Staining
- DMSO
Dimethyl sulfoxide
- TSA
Thermal Shift Assay
- MTT
3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Mottamal M, Zheng S, Huang TL, Wang G, Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents, Molecules. 20 (2015) 3898–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Min C, Moore N, Shearstone JR, Quayle SN, Huang P, van Duzer JH, Jarpe MB, Jones SS, Yang M, Selective Inhibitors of Histone Deacetylases 1 and 2 Synergize with Azacitidine in Acute Myeloid Leukemia, PLoS ONE. 12 (2017) e0169128 10.1371/journal.pone.0169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Benedetti R, Conte M, Altucci L, Targeting Histone Deacetylases in Diseases: Where Are We?, Antioxid Redox Signal. 23 (2015) 99–126. 10.1089/ars.2013.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gryder BE, Sodji QH, Oyelere AK, Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed, Future Med Chem. 4 (2012) 505–524. 10.4155/fmc.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Suraweera A, O’Byrne KJ, Richard DJ, Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi, Front. Oncol 8 (2018) 92 10.3389/fonc.2018.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Amin SA, Adhikari N, Jha T, Structure-activity relationships of hydroxamate-based histone deacetylase-8 inhibitors: reality behind anticancer drug discovery, Future Medicinal Chemistry. 9 (2017) 2211–2237. 10.4155/fmc-2017-0130. [DOI] [PubMed] [Google Scholar]
- [7].Balasubramanian S, Ramos J, Luo W, Sirisawad M, Verner E, Buggy JJ, A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas, Leukemia. 22 (2008) 1026–1034. 10.1038/leu.2008.9. [DOI] [PubMed] [Google Scholar]
- [8].Chakrabarti A, Melesina J, Kolbinger FR, Oehme I, Senger J, Witt O, Sippl W, Jung M, Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases, Future Medicinal Chemistry. 8 (2016) 1609–1634. [DOI] [PubMed] [Google Scholar]
- [9].Ceccacci E, Minucci S, Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia, Br J Cancer. 114 (2016) 605–611. 10.1038/bjc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chakrabarti A, Oehme I, Witt O, Oliveira G, Sippl W, Romier C, Pierce RJ, Jung M, HDAC8: a multifaceted target for therapeutic interventions, Trends in Pharmacological Sciences. 36 (2015) 481–492. 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- [11].Mohan R, Sharma AK, Gupta S, Ramaa CS, Design, synthesis, and biological evaluation of novel 2, 4-thiazolidinedione derivatives as histone deacetylase inhibitors targeting liver cancer cell line, Medicinal Chemistry Research. 21 (2012) 1156–1165. [Google Scholar]
- [12].Yang F, Zhao N, Ge D, Chen Y, Next-generation of selective histone deacetylase inhibitors, RSC Adv. 9 (2019) 19571–19583. 10.1039/C9RA02985K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hou X, Du J, Liu R, Zhou Y, Li M, Xu W, Fang H, Enhancing the Sensitivity of Pharmacophore-Based Virtual Screening by Incorporating Customized ZBG Features: A Case Study Using Histone Deacetylase 8, J. Chem. Inf. Model 55 (2015) 861–871. 10.1021/ci500762z. [DOI] [PubMed] [Google Scholar]
- [14].Marek M, Shaik TB, Heimburg T, Chakrabarti A, Lancelot J, Ramos-Morales E, Da Veiga C, Kalinin D, Melesina J, Robaa D, Schmidtkunz K, Suzuki T, Holl R, Ennifar E, Pierce RJ, Jung M, Sippl W, Romier C, Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants, J. Med. Chem 61 (2018) 10000–10016. 10.1021/acs.jmedchem.8b01087. [DOI] [PubMed] [Google Scholar]
- [15].Schweipert M, Jänsch N, Sugiarto WO, Meyer-Almes F-J, Kinetically selective and potent inhibitors of HDAC8, Biological Chemistry. 400 (2019) 733–743. 10.1515/hsz-2018-0363. [DOI] [PubMed] [Google Scholar]
- [16].Suzuki T, Muto N, Bando M, Itoh Y, Masaki A, Ri M, Ota Y, Nakagawa H, Iida S, Shirahige K, Miyata N, Design, Synthesis, and Biological Activity of NCC149 Derivatives as Histone Deacetylase 8-Selective Inhibitors, ChemMedChem. 9 (2014) 657–664. 10.1002/cmdc.201300414. [DOI] [PubMed] [Google Scholar]
- [17].Whitehead L, Dobler MR, Radetich B, Zhu Y, Atadja PW, Claiborne T, Grob JE, McRiner A, Pancost MR, Patnaik A, Shao W, Shultz M, Tichkule R, Tommasi RA, Vash B, Wang P, Stams T, Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors, Bioorganic & Medicinal Chemistry. 19 (2011) 4626–4634. 10.1016/j.bmc.2011.06.030. [DOI] [PubMed] [Google Scholar]
- [18].Muth M, Jänsch N, Kopranovic A, Krämer A, Wössner N, Jung M, Kirschhöfer F, Brenner-Weiß G, Meyer-Almes FJ, Covalent inhibition of histone deacetylase 8 by 3,4-dihydro-2H-pyrimido[1,2-c][1,3]benzothiazin-6-imine, Biochimica et Biophysica Acta (BBA) - General Subjects. 1863 (2019) 577–585. 10.1016/j.bbagen.2019.01.001. [DOI] [PubMed] [Google Scholar]
- [19].Wolff B, Jänsch N, Sugiarto WO, Frühschulz S, Lang M, Altintas R, Oehme I, Meyer-Almes F-J, Synthesis and structure activity relationship of 1, 3-benzo-thiazine-2-thiones as selective HDAC8 inhibitors, European Journal of Medicinal Chemistry. 184 (2019) 111756 10.1016/j.ejmech.2019.111756. [DOI] [PubMed] [Google Scholar]
- [20].Tabackman AA, Frankson R, Marsan ES, Perry K, Cole KE, Structure of ‘linkerless’ hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket, Journal of Structural Biology. 195 (2016) 373–378. 10.1016/j.jsb.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Uba AI, Weako J, Keskin Ö, Gürsoy A, Yelekçi K, Examining the stability of binding modes of the cocrystallized inhibitors of human HDAC8 by molecular dynamics simulation, Journal of Biomolecular Structure and Dynamics. (2019) 1–10. 10.1080/07391102.2019.1615989. [DOI] [PubMed] [Google Scholar]
- [22].Schweipert M, Jänsch N, Sugiarto WO, Meyer-Almes F-J, Kinetically selective and potent inhibitors of HDAC8, Biological Chemistry. 400 (2019) 733–743. 10.1515/hsz-2018-0363. [DOI] [PubMed] [Google Scholar]
- [23].Tilekar K, Upadhyay N, Jänsch N, Schweipert M, Mrowka P, Meyer-Almes FJ, Ramaa CS, Discovery of 5-naphthylidene-2,4-thiazolidinedione derivatives as selective HDAC8 inhibitors and evaluation of their cytotoxic effects in leukemic cell lines, Bioorganic Chemistry. 95 (2020) 103522 10.1016/j.bioorg.2019.103522. [DOI] [PubMed] [Google Scholar]
- [24].Banerjee S, Adhikari N, Amin SA, Jha T, Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview, European Journal of Medicinal Chemistry. 164 (2019) 214–240. 10.1016/j.ejmech.2018.12.039. [DOI] [PubMed] [Google Scholar]
- [25].Qin H-T, Li H-Q, Liu F, Selective histone deacetylase small molecule inhibitors: recent progress and perspectives, Expert Opinion on Therapeutic Patents. 27 (2017) 621–636. 10.1080/13543776.2017.1276565. [DOI] [PubMed] [Google Scholar]
- [26].Miller TA, Witter DJ, Belvedere S, Histone Deacetylase Inhibitors,J. Med. Chem 46 (2003) 5097–5116. 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- [27].Zhang L, Zhang J, Jiang Q, Zhang L, Song W, Zinc binding groups for histone deacetylase inhibitors, Journal of Enzyme Inhibition and Medicinal Chemistry. 33 (2018) 714–721. 10.1080/14756366.2017.1417274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bressi JC, de Jong R, Wu Y, Jennings AJ, Brown JW, O’Connell S, Tari LW, Skene RJ, Vu P, Navre M, Cao X, Gangloff AR, Benzimidazole and imidazole inhibitors of histone deacetylases: Synthesis and biological activity, Bioorganic & Medicinal Chemistry Letters. 20 (2010) 3138–3141. 10.1016/j.bmcl.2010.03.092. [DOI] [PubMed] [Google Scholar]
- [29].Adhikari N, Amin SA, Jha T, Selective and nonselective HDAC8 inhibitors: a therapeutic patent review, Pharmaceutical Patent Analyst. 7 (2018) 259–276. 10.4155/ppa-2018-0019. [DOI] [PubMed] [Google Scholar]
- [30].Mummery A, Narendran A, Lee K-Y, Targeting epigenetics through histone deacetylase inhibitors in acute lymphoblastic leukemia, Current Cancer Drug Targets. 11 (2011) 882–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Quintás-Cardama A, Santos FPS, Garcia-Manero G, Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia, Leukemia. 25 (2011) 226–235. 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- [32].Oehme I, Deubzer HE, Lodrini M, Milde T, Witt O, Targeting of HDAC8 and investigational inhibitors in neuroblastoma, Expert Opinion on Investigational Drugs. 18 (2009) 1605–1617. 10.1517/14728220903241658. [DOI] [PubMed] [Google Scholar]
- [33].Monga V, Swami U, Tanas M, Bossler A, Mott SL, Smith BJ, Milhem M, A Phase I/II Study Targeting Angiogenesis Using Bevacizumab Combined with Chemotherapy and a Histone Deacetylase Inhibitor (Valproic Acid) in Advanced Sarcomas, Cancers. 10 (2018) 53 10.3390/cancers10020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bhanushali U, Rajendran S, Sarma K, Kulkarni P, Chatti K, Chatterjee S, Ramaa CS, 5-Benzylidene-2, 4-thiazolidenedione derivatives: Design, synthesis and evaluation as inhibitors of angiogenesis targeting VEGR-2, Bioorganic Chemistry. 67 (2016) 139–147. [DOI] [PubMed] [Google Scholar]
- [35].Joshi H, Marulkar K, Gota V, S Ramaa C, Hydroxy Cinnamic Acid Derivatives as Partial PPAR γ Agonists: In silico Studies, Synthesis and Biological Characterization Against Chronic Myeloid Leukemia Cell Line (K562), Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 17 (2017) 524–541. [DOI] [PubMed] [Google Scholar]
- [36].Kabir A, Tilekar K, Upadhyay N, Ramaa CS, Novel Anthraquinone Derivatives as Dual Inhibitors of Topoisomerase 2 and Casein Kinase 2: In Silico Studies, Synthesis and Biological Evaluation on Leukemic Cell Lines, Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 18 (2018) 1551–1562. [DOI] [PubMed] [Google Scholar]
- [37].Patil V, Tilekar K, Mehendale-Munj S, Mohan R, Ramaa CS, Synthesis and primary cytotoxicity evaluation of new 5-benzylidene-2, 4-thiazolidinedione derivatives, European Journal of Medicinal Chemistry. 45 (2010) 4539–4544. [DOI] [PubMed] [Google Scholar]
- [38].Lema C, Varela-Ramirez A, Aguilera RJ, Differential nuclear staining assay for high-throughput screening to identify cytotoxic compounds, Curr Cell Biochem. 1 (2011) 1–14. [PMC free article] [PubMed] [Google Scholar]
- [39].Choi MA, Park SY, Chae HY, Song Y, Sharma C, Seo YH, Design, synthesis and biological evaluation of a series of CNS penetrant HDAC inhibitors structurally derived from amyloid- β probes, Sci Rep. 9 (2019) 1–12. 10.1038/s41598-019-49784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huang Y, Zhao J, Song Q, Zheng L, Fan C, Liu T, Bao Y, Sun L, Zhang L, Li Y, Virtual screening and experimental validation of novel histone deacetylase inhibitors, BMC Pharmacol Toxicol. 17 (2016) 32 10.1186/s40360-016-0075-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mehta V, Athar M, Jha PC, Panchal M, Modi K, Jain VK, Efficiently functionalized oxacalix [4] arenes: Synthesis, characterization and exploration of their biological profile as novel HDAC inhibitors, Bioorganic & Medicinal Chemistry Letters. 26 (2016) 1005–1010. [DOI] [PubMed] [Google Scholar]
- [42].Oanh DTK, Van Hai H, Park SH, Kim H-J, Han B-W, Kim H-S, Hong J-T, Han S-B, Nam N-H, Benzothiazole-containing hydroxamic acids as histone deacetylase inhibitors and antitumor agents, Bioorganic & Medicinal Chemistry Letters. 21 (2011) 7509–7512. [DOI] [PubMed] [Google Scholar]
- [43].Thanh Tung T, Thi Kim Oanh D, Thi Phuong Dung P, Van Hue TM, Ho Park S, Woo Han B, Kim Y, Hong J-T, Han S-B, Nam N-H, New benzothiazole/thiazole-containing hydroxamic acids as potent histone deacetylase inhibitors and antitumor agents, Medicinal Chemistry. 9 (2013) 1051–1057. [DOI] [PubMed] [Google Scholar]
- [44].Adekola K, Rosen ST, Shanmugam M, Glucose transporters in cancer metabolism, Curr Opin Oncol. 24 (2012) 650–654. 10.1097/CCO.0b013e328356da72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wardell SE, Ilkayeva OR, Wieman HL, Frigo DE, Rathmell JC, Newgard CB, McDonnell DP, Glucose metabolism as a target of histone deacetylase inhibitors, Molecular Endocrinology. 23 (2009) 388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen Ching-Shih, Wang Dasheng, GLUCOSE TRANSPORTER INHIBITORS, US 9,174.951 B2, 2015. [Google Scholar]
- [47].Wei S, Kulp SK, Chen C-S, Energy Restriction as an Antitumor Target of Thiazolidinediones, J. Biol. Chem 285 (2010) 9780–9791. 10.1074/jbc.M109.065466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wieczorke R, Dlugai S, Krampe S, Boles E, Characterisation of mammalian GLUT glucose transporters in a heterologous yeast expression system, Cell. Physiol. Biochem 13 (2003) 123–134. 10.1159/000071863. [DOI] [PubMed] [Google Scholar]
- [49].Tripp J, Essl C, Iancu CV, Boles E, Choe J-Y, Oreb M, Establishing a yeast-based screening system for discovery of human GLUT5 inhibitors and activators, Sci Rep. 7 (2017) 6197 10.1038/s41598-017-06262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Florio M, Heide M, Pinson A, Brandl H, Albert M, Winkler S, Wimberger P, Huttner WB, Hiller M, Evolution and cell-type specificity of human-specific genes preferentially expressed in progenitors of fetal neocortex, ELife. 7 (2018) e32332 10.7554/eLife.32332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Song S, Wang Y, Xu P, Yang R, Ma Z, Liang S, Zhang G, The inhibition of histone deacetylase 8 suppresses proliferation and inhibits apoptosis in gastric adenocarcinoma, International Journal of Oncology. 47 (2015) 1819–1828. 10.3892/ijo.2015.3182. [DOI] [PubMed] [Google Scholar]
- [52].Daina A, Michielin O, Zoete V, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci Rep. 7 (2017) 1–13. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Daina A, Zoete V, A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules, ChemMedChem. 11 (2016) 1117–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bhatia S, Schultz T, Roberts D, Shen J, Kromidas L, Marie Api A, Comparison of Cramer classification between Toxtree, the OECD QSAR Toolbox and expert judgment, Regulatory Toxicology and Pharmacology. 71 (2015) 52–62. 10.1016/j.yrtph.2014.11.005. [DOI] [PubMed] [Google Scholar]
- [55].Patlewicz G, Jeliazkova N, Safford RJ, Worth AP, Aleksiev B, An evaluation of the implementation of the Cramer classification scheme in the Toxtree software, SAR and QSAR in Environmental Research. 19 (2008) 495–524. 10.1080/10629360802083871. [DOI] [PubMed] [Google Scholar]
- [56].Bhanushali U, Kalekar-Joshi S, Kulkarni-Munshi R, Yellanki S, Medishetty R, Kulkarni P, Subramanian Chelakara R, Design, Synthesis and Evaluation of 5-pyridin-4-yl-2-thioxo-[1, 3, 4] oxadiazol-3-yl Derivatives as Anti-angiogenic Agents Targeting VEGFR-2, Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 17 (2017) 67–74. [PubMed] [Google Scholar]
- [57].Pal T, Joshi H, Ramaa CS, Design and development of oxazol-5-ones as potential partial PPAR-γ agonist against cancer cell lines, Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 14 (2014) 872–883. [DOI] [PubMed] [Google Scholar]
- [58].Jänsch N, Meyners C, Muth M, Kopranovic A, Witt O, Oehme I, Meyer-Almes F-J, The enzyme activity of histone deacetylase 8 is modulated by a redox-switch, Redox Biology. 20 (2019) 60–67. 10.1016/j.redox.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Volund A, Application of the Four-Parameter Logistic Model to Bioassay: Comparison with Slope Ratio and Parallel Line Models, Biometrics. 34 (1978) 357 10.2307/2530598. [DOI] [PubMed] [Google Scholar]
- [60].Kasahara T, Kasahara M, Characterization of rat Glut4 glucose transporter expressed in the yeast Saccharomyces cerevisiae: comparison with Glut1 glucose transporter, Biochim. Biophys. Acta 1324 (1997) 111–119. 10.1016/s0005-2736(96)00217-9. [DOI] [PubMed] [Google Scholar]
- [61].George Thompson AM, Ursu O, Babkin P, Iancu CV, Whang A, Oprea TI, Choe J, Discovery of a specific inhibitor of human GLUT5 by virtual screening and in vitro transport evaluation, Sci Rep. 6 (2016) 24240 10.1038/srep24240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Peram MR, Jalalpure S, Kumbar V, Patil S, Joshi S, Bhat K, Diwan P, Factorial design based curcumin ethosomal nanocarriers for the skin cancer delivery: in vitro evaluation, Journal of Liposome Research. (2019) 1–21. [DOI] [PubMed] [Google Scholar]
- [63].Bhat SS, Revankar VK, Kumbar V, Bhat K, Kawade VA, Synthesis, crystal structure and biological properties of a cis-dichloridobis (diimine) copper (II) complex, Acta Crystallographica Section C: Structural Chemistry. 74 (2018) 146–151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.