

Abstract
Uremic cardiomyopathy, characterized by hypertension, cardiac hypertrophy, and fibrosis, is a complication of chronic kidney disease (CKD). Urea transporter (UT) inhibition increases the excretion of water and urea, but the effect on uremic cardiomyopathy has not been studied. We tested UT inhibition by dimethylthiourea (DMTU) in 5/6 nephrectomy mice. This treatment suppressed CKD-induced hypertension and cardiac hypertrophy. In CKD mice, cardiac fibrosis was associated with upregulation of UT and vimentin abundance. Inhibition of UT suppressed vimentin amount. Left ventricular mass index in DMTU-treated CKD was less compared with non-treated CKD mice as measured by echocardiography. Nephrectomy was performed in UT-A1/A3 knockout (UT-KO) to further confirm our finding. UT-A1/A3 deletion attenuates the CKD-induced increase in cardiac fibrosis and hypertension. The amount of α-smooth muscle actin and tgf-β were significantly less in UT-KO with CKD than WT/CKD mice. To study the possibility that UT inhibition could benefit heart, we measured the mRNA of renin and angiotensin-converting enzyme (ACE), and found both were sharply increased in CKD heart; DMTU treatment and UT-KO significantly abolished these increases. Conclusion: Inhibition of UT reduced hypertension, cardiac fibrosis, and improved heart function. These changes are accompanied by inhibition of renin and ACE.
Keywords: cardiac fibrosis, cardiovascular disease, dimethylthiourea, diuretics, hypertrophy
1 |. BACKGROUND
The prevalence of cardiovascular disease in chronic kidney disease (CKD) patients reaches 65%, compared with 32% in patients without CKD, in the population aged 66 and older.1 Many CKD patients die within 3 years from the time of being diagnosed with uremic cardiomyopathy.2 Uremic cardiomyopathy mainly consists of left ventricular hypertrophy and interstitial fibrosis3 that is associated with hospitalization and the risk of death.4 Cardiac fibrosis is a constant finding in heart biopsies and necropsy studies in CKD patients, which is related to retention of water, sodium, and urea within the body. An increase in cardiac fibrosis is also associated with hypertension and activation of the renin-angiotensin system (RAS).5,6 Activation of the RAS contributes to cardiac hypertrophy, and blocking angiotensin II (AII) receptor type I suppresses left ventricular hypertrophy.7
Urea plays a major role in the urine concentrating mechanism. Urea transporter (UT) is a membrane transport protein, transporting urea. The UT-A (Slc14a2) and UT-B (Slc14a1) genes encode six and two protein isoforms, respectively.8,9 UT-A1 and UT-A3 are expressed in the inner medullary collecting duct (IMCD) and UT-A2 is expressed in thin descending limbs. UT-A4 mRNA is detected in rat kidney medulla. UT-A5 is in testis and UT-A6 is in colon. UT-B is expressed in descending vasa recta, and also in erythrocytes, brain, liver, and colon.8,10 Our previous study found that UT is also located in the heart and is upregulated in CKDs patients and animals.11 In UT-A1/A3−/− mice, urine volume is increased compared with wild-type mice, which indicates these mice have a decreased ability to concentrate their urine.8 Similar effects are seen with inhibitors of UT.12 Therefore, an inhibition of UT may be useful to prevent volume overload and could show positive cardiac effects.
The RAS is important to both cardiovascular and renal physiology and pathology.13 This system is mainly comprised of renin, angiotensinogen (AGT), angiotensin-converting enzyme (ACE), angiotensin I (Ang I), angiotensin II (Ang II), and angiotensin I or II receptors. Upregulation of the RAS is involved with the induction and progression of hypertension, atherosclerosis, cardiac hypertrophy, heart fibrosis, and heart failure.14–16 RAS inhibitors can reduce disease progression in patients with cardiovascular or renal disease by 20% compared with other anti-fibrotic therapies.17
The purpose of this study is to find therapeutic strategies to attenuate uremic cardiomyopathy that may provide a particular cardio-protective benefit in CKD. We used 5/6 nephrectomy (Nx) as a CKD mouse model to determine whether the inhibition of UT may reduce hypertension, cardiac hypertrophy, and fibrosis in these mice. Our studies may provide useful information for the treatment of cardiomyopathy.
2 |. METHODS
2.1 |. Animals and treatments
All animal surgical protocols and procedures were approved by the Emory Institutional Animal Care and Use Committee and adhere to NIH standards for animal use. All mice were maintained at 19-21°C on a 12-h/12-h light-dark cycle, were fed a standard rodent diet and allowed free access to drinking water. Male C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). The animal experimental protocol is shown in Figure S1 CKD mice underwent 5/6Nx as previously described.18,19 Briefly, CKD was induced by removing the right kidney. Mice were rested for 1 week and then a second surgery performed to remove two poles of the left kidney in anesthetized mice (xylazine 12 mg/kg, ketamine 60 mg/kg). Hemostasis was achieved by cautery and pressure. Initially, mice were fed with low protein chow (Harlan Teklad: 14% protein, 3.5% fat, 49% carbohydrate) and special water (0.45% NaCl) after 5/6 nephrectomy. After 7 days, mice were fed with a normal protein diet (23% protein). Two weeks before harvest mice, high-protein diet (40% protein) was provided. Blood urea nitrogen (BUN) is measured the rate of conversion of NADH to NAD monitored at 340 nm using BUN Kinetic Procedure Kit (Thermo Electron, Louisville, CO.). Dimethylthiourea (DMTU, Sigma-Aldrich, MO) is a urea derivative that inhibits urea transport. DMTU was administered at a dose of 100 mg/kgBW/day by osmotic mini pump (DURECT Corporation, Cupertino, CA) beginning 1 week after the second surgery. The total DMTU treatment was 8 weeks. BP was measured by non-invasive photoplethysmography using the tail-cuff method (BP-2000 Blood Pressure Analysis System, Visitech Systems Inc, Apex, NC). BP data were analyzed by BP-2000 analysis software (Visitech Systems Inc).
The details of UT-A1/A3−/− (UT KO) mice are provided in our previous publication.20 Briefly, UT KO mice (the C57BL/6 background) were established by Fenton et al.21,22 Male UT KO mice were a generous gift from Dr Mark Knepper (National Institutes of Health). After these mice were mated with female WT mice (C56BL6), the colony of UT KO mice was re-derived from breeding stock.23 CKD in the UT KO mice was produced by uninephrectomy surgery.24
2.2 |. Urine and serum analysis
Mice were housed in metabolic cages for collection of 24-hour urine samples. Urine and serum osmolality were measured using a Wescor 5520 Vapor Pressure Osmometer (Wescor Inc Logan, UT). Urea levels of urine and serum were measured by enzyme method as previously described (Infinite Reagent, Thermo Fisher Scientific).24
2.3 |. Western blot analysis
Proteins (30 μg/lane) were size separated by SDS-PAGE and western blotting was performed as previously described.25 Primary antibodies against the UT were produced in our lab.26 The UT polyclonal antibody was made by immunizing rabbits against the carboxyl-terminal peptide region—19 amino acids. Synthetic polypeptide was synthesized by the Emory University Microchemical Facility and HPLC-purified. The polypeptide was conjugated to keyhole limpet hemocyanin, dissolved in Freund’s complete adjuvant and injected into rabbits. The rabbits were periodically reinjected. The immunizing polypeptide was linked to a Sulfo-Link column to affinity-purify. The UT is highly conserved, so the antibody recognizes the protein in multiple animals, including rat, mouse, chimp, human, dog, and squirrel. TGF-beta (ab179695) was purchased from Abcam, α-SMA (A2547) was purchased from Sigma-Aldrich, and vimentin (#5741) was purchased from Cell Signaling Technology. Bands were standardized by Ponceau S for total protein and quantitated with ImageJ (NIH).
2.4 |. Immunohistochemistry and histology
Hearts were fixed with 4% paraformaldehyde and immunohistochemistry staining was performed as previously described.25 Masson’s Trichrome staining was performed following the manufacture’s recommended protocol (IMEB Inc, San Marcos, CA). Stained sections were examined with an Olympus inverted microscope IX71. Quantitative evaluation of fibrosis area (blue color) to total area was performed by computerized analysis using Olympus CellSens software.
2.5 |. Quantitative RT-qPCR analysis of mRNA
Total RNA was extracted using Tri-Reagent (Molecular Research). RNA was reverse transcribed to synthesize cDNA using M-MLV Reverse Transcriptase (Invitrogen). Designed primers are listed in Table S1. Expression of individual mRNA was standardized by 18s gene and calculated as the difference between the threshold values of two genes (ΔΔcq).
2.6 |. Echocardiographic evaluations of cardiac function
Echocardiography was performed on a heating pad on lightly anesthetized mice (under 1%-2% isoflurane, in oxygen) using a Vevo 3100 ultrasound system (VisualSonics) as described previously.5 Recording of echocardiographic images was performed in random order with respect to the treatment or control animals. The acquisition of images and evaluation of data were performed by an independent operator, who was blinded to the treatment.
2.7 |. Cell culture and adenovirus transduction
H9c2 cells (CRL-1446; ATCC, Manassas, VA) were cultured in growth media [Dulbecco’s modified Eagle medium plus 10% fetal bovine serum, 100 U/mL of penicillin, 100 μg/mL of streptomycin, and 2 mM L-glutamine] and studied between passages 3 and 9.
Cardiac fibroblasts were isolated from the heart of 1-month old C57BL6 mice. Hearts were minced into a coarse slurry and gently agitated for 1 h at 37°C in DMEM with 25 mM HEPES (pH 7.4) plus 0.1% pronase (Calbiochem, San Diego, CA). The digest was passed through a 100-μm filter and then plated on culture flasks. The primary fibroblasts were cultured with DMEM with 10% fetal bovine serum. Passages two to five were used for experiments.
Viral transduction: the adenovirus, Ad-UT, was produced and amplified in our lab using AdEasy Adenoviral vector system.18 The adenovirus transduction unit was achieved by serial dilutions.
2.8 |. Statistical analysis
All data are presented as mean ± SEM. A two-tailed Student’s t test was used for comparisons of two groups. For a comparison of more than two groups, we used one-way ANOVA with a post hoc analysis by Bonferroni test. To analyze an interaction between two factors, we used two-way ANOVA. Statistical differences with P < .05 were considered significant. N represents the number of animals per condition in an experiment.
3 |. RESULTS
3.1 |. Uremic cardiomyopathy is characterized by hypertension, cardiac hypertrophy, and fibrosis
At 8 weeks after 5/6Nx, the systolic blood pressure (SBP) of the CKD mice significantly increased compared to sham mice (107 mm Hg vs 132 mm Hg, P < .005). The diastolic BP (DBP) showed a trend toward an increase but did not reach a statistically significant difference (Figure 1A). Heart size was increased in the 5/6Nx mice and was accompanied by increased heart weight (Figure 1B). Cardiac fibrosis was strongly identified in the heart of CKD mice by Masson’s Trichrome staining (Figure 1C). In addition, mRNA of collagen type 1a1, type 4a1, and fibronectin were increased in CKD hearts compared to sham-operated mice (Figure 1D). These data suggest that cardiac hypertrophy and fibrosis were induced in this animal model of CKD.
FIGURE 1.
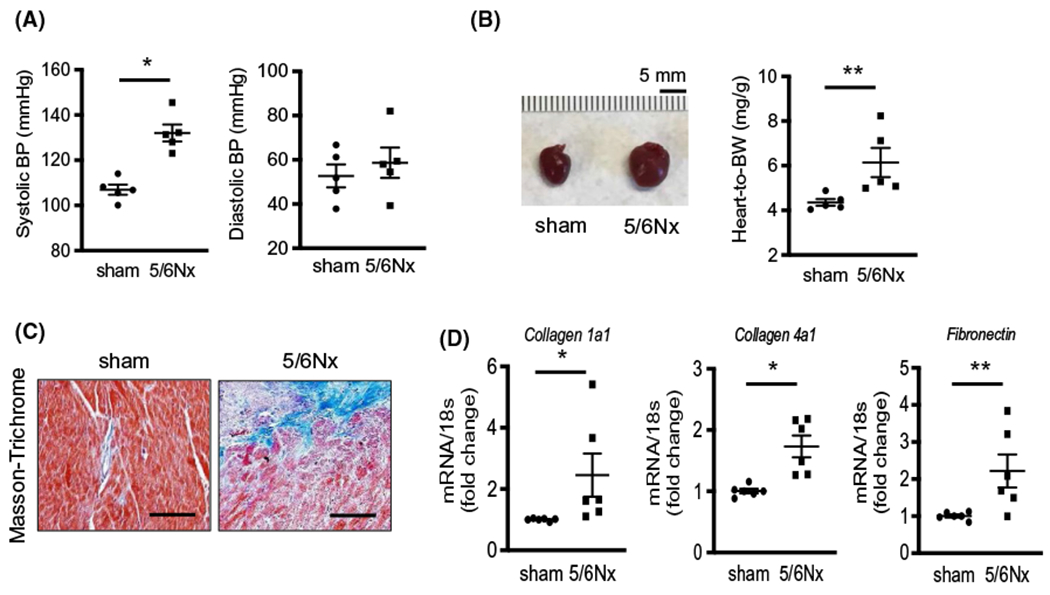
Uremic cardiomyopathy is characterized by hypertension, cardiac hypertrophy, and fibrosis. Sham and 5/6Nx mice were sacrificed and samples were harvested at 8 weeks after CKD was produced. A, Systolic and diastolic blood pressure (BP) were measured by tail-cuff method in sham and 5/6Nx mice. B, Pictures of mouse heart and heart weight (mg) which was normalized by body weight (BW) (g) at sacrifice. C, Masson’s Trichrome staining in mouse heart tissues. Fibrosis is indicated by blue color. Pictures were observed by ×200 magnification. Scale bar is 100 μm. D, Quantitative mRNA expression in mouse hearts performed by real-time PCR. Individual gene expression was calculated by ΔΔcq and standardized by housekeeping gene 18s. All data: mean ± SEM (N = 5). *P < .005, **P < .05 by two-tailed student’s t test
3.2 |. Cardiac fibrosis in uremic heart is associated with upregulation of UT and vimentin expression
An earlier study from our group showed that UT protein levels increase in uremic and hypertensive hearts from human and rat.11 To identify whether the increase in UT associated with the pathologic changes of cardiomyopathy, we measured the amount of UT and vimentin, a pro-fibrotic protein, and found that the UT protein level increased 1.6-fold in the hearts of 5/6Nx mice and was associated with a 1.9-fold increase in the vimentin level (Figure 2A). The increase in vimentin was confirmed by immunohistochemistry (Figure 2B). To explore whether the increased vimentin is induced by UT, we overexpressed UT in cultured H9c2 cells (cardiac myoblasts) by adenovirus-mediated gene transfer (Figure 2C). The intervention of UT significantly increased the amount of vimentin in Ad-UT-treated cells compared with Ad-ctrl-treated cells (Figure 2D). To distinguish whether urea and UT were involved with the increased vimentin levels, we added 40 mmol/L urea into the culture medium and tested vimentin protein amount. We found that urea alone increased vimentin abundance in ad-ctrl-treated cells. However, this increase is much higher in cells overexpressing UT (Figure S3).
FIGURE 2.
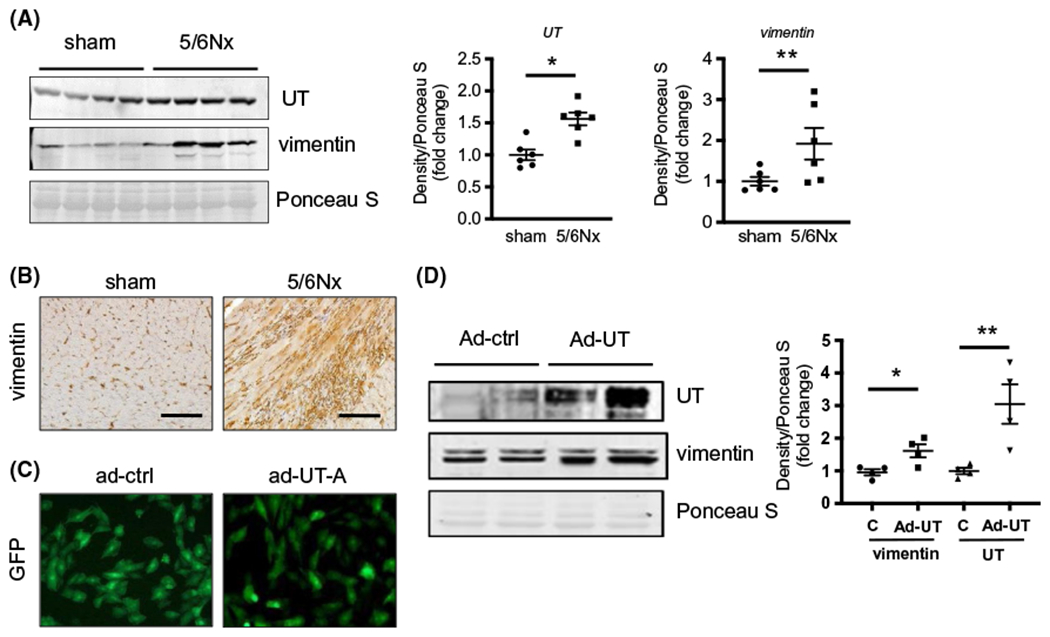
Cardiac fibrosis in uremic heart was associated with upregulation of UT and vimentin expression. A, Western blot of lysates from sham and 5/6Nx mouse hearts probed for UT-A and vimentin proteins. The right point graphs show the levels of each protein in the sham group compared with the level in the 5/6Nx group (designated as one-fold). Data: mean ± SEM (N = 6). *P < .005, **P < .05 by two-tailed student’s t test. B, Immunohistochemistry staining for vimentin (brown) in mouse heart tissues. All pictures were observed by ×200 magnification. Scale bar is 100 μm. C, Immunofluorescence images for GFP in H9c2 cells. Left panel is H9c2 cells transduced with adenovirus (ad)-GFP and right is ad-UT-A/GFP. Pictures of these cells were taken 48 hours after virus transduction. D, Western blot of lysates from H9c2 cells 48 hours after transduction. Data: mean ± SEM (N = 4). *P < .005, **P < .05 vs H9c2 cells with ad-UT-A treatment by one-way ANOVA
3.3 |. Inhibition of UT altered the hemodynamics in CKD mice
Since an increase in UT is associated with an increase in the pro-fibrosis protein vimentin, we investigated whether inhibition of UT by DMTU could prevent fibrosis in the heart of CKD mice. The phenotypic characteristics of mice were determined 8 weeks after completing the 5/6Nx (Table 1). BUN levels in the 5/6Nx mice (both vehicle and DMTU treatment) were significantly higher than in sham mice verifying a functional decline in the kidney. There was no significant difference in BUN values with DMTU treatment compared to vehicle treatment in either sham or CKD mice. Inhibition of UT by DMTU increased urine volume and decreased urine osmolality in sham mice. The abundance of UT was analyzed by Western blot in DMTU-treated sham and CKD mice. We found that UT protein amount had a tendency to decrease in CKD/DMTU-treated mice compared with CKD/vehicle-treated mice, but the differences did not reach statistical significance (Figure S4).
TABLE 1.
Phenotypic characteristics of WT and 5/6Nx mice
| Parameter | Sham | Sham + DMTU | 5/6Nx + vehicle | 5/6Nx + DMTU |
|---|---|---|---|---|
| Initial BW (g) | 25.8 ± 0.4 | 25.0 ± 0.6 | 26.5 ± 0.6 | 26.0 ± 0.5 |
| Final BW (g) | 29.8 ± 0.4 | 25.9 ± 1.1* | 27.6 ± 0.7* | 26.4 ± 0.5* |
| BUN (mg/dL) | 24.9 ± 1.5 | 23.3 ± 2.1 | 50.1 ± 1.6*† | 47.2 ± 3.3*† |
| Urine urea (mmol/L) | 1510 ± 154 | 1555 ± 128 | 761 ± 48*† | 926 ± 95*† |
| 24 hr intake volume/BW (mL/g) | 0.159 ± 0.028 | 0.129 ± 0.006 | 0.324 ± 0.012*† | 0.265 ± 0.028*† |
| 24 hr Urine volume/BW (mL/g) | 0.023 ± 0.004 | 0.058 ± 0.010* | 0.148 ± 0.008*† | 0.170 ± 0.013*†# |
| Water in-out balance/BW (mL/g) | 0.145 ± 0.010 | 0.081 ± 0.010* | 0.185 ± 0.013† | 0.114 ± 0.014‡ |
| 24 hr Urine osmolality (mOsmol/kg H2O) | 3595 ± 559 | 2378 ± 133* | 908 ± 40*† | 1206 ± 116*†# |
| Plasma osmolality (mOsmol/kg H2O) | 322 ± 2 | 322 ± 1 | 330 ± 2 | 329 ± 3 |
Note: Volume in-out balance/BW was calculated by [(24 hr intake volume) − (24hr urine volume)]/BW, All data; mean ± SEM (N = 5-8).
Abbreviations: BW, body weight, BUN, blood urea nitrogen.
P < .05 vs sham + vehicle;
P < .05 vs sham + DMTU;
P < .05 vs 5/6Nx + vehicle by one-way ANOVA with Bonferroni test;
P < .05 for interaction between 5/6Nx and DMTU (P < .0001 in 24 hr urine volume/BW and 24 hr urine osmolality for the factor of 5/6Nx; P < .05 in 24 hr urine volume/BW and 24 hr urine osmolality for the factor of DMTU) by two-way ANOVA.
3.4 |. Inhibition of UT by DMTU ameliorated CKD-induced hypertension, cardiac hypertrophy, and improved heart function
Inhibition of UT suppressed CKD-induced increases in SBP in 5/6Nx mice (DMTU: 119 ± 3 mm Hg vs vehicle: 130 ± 2 mm Hg, P < .05) (Figure 3A). DMTU suppressed the CKD-induced increase in heart size (Figure 3B). The ratio of heart weight to body weight (BW) in 5/6Nx + vehicle (4.50 ± 0.09 mg/g) was significantly increased compared with sham + vehicle mice (3.91 ± 0.06 mg/g, P < .005). DMTU suppressed the CKD-induced increase in heart weight (5/6Nx + DMTU: 3.74 ± 0.12 mg/g vs 5/6Nx + vehicle: 4.50 ± 0.09 mg/g, P < .001) (Figure 3C). We also used dry brain weight to normalize heart weight and there were similar changes in heart weight (1.34 ± 0.09 mg/mg vs 1.04 ± 0.04 mg/mg, P < .01) (Figure 3C, right panel). Dry brain weight was used to normalized heart weight for two reasons: 1-CKD mice have muscle wasting, so the body weight is significantly lower than sham-operated mice; 2-The body weight also could be altered by either edema (in CKD) or dehydration (by DMTU). To identify whether UT inhibition had an impact on heart function, a cardiac echocardiographic analysis was performed. Both intraventricular septum and posterior wall thicknesses were greater in 5/6Nx + vehicle than the other three groups (Table 2). LV end-diastolic volume and LV mass index were significantly increased in 5/6Nx + vehicle compared with sham + vehicle; treatment with DMTU eliminated these increases (Table 2 and Figure 3D). In addition, the percentage of fractional shortening was decreased in CKD heart, which indicates an impaired efficiency of the heart in ejecting blood. In 5/6Nx + vehicle mice, two of five had a pericardial effusion occur, but not in the other three cohorts (Figure 3D). These data suggest that the intervention of DMTU diminished CKD-induced hypertension, suppressed cardiac hypertrophy, and improved heart function in CKD mice.
FIGURE 3.
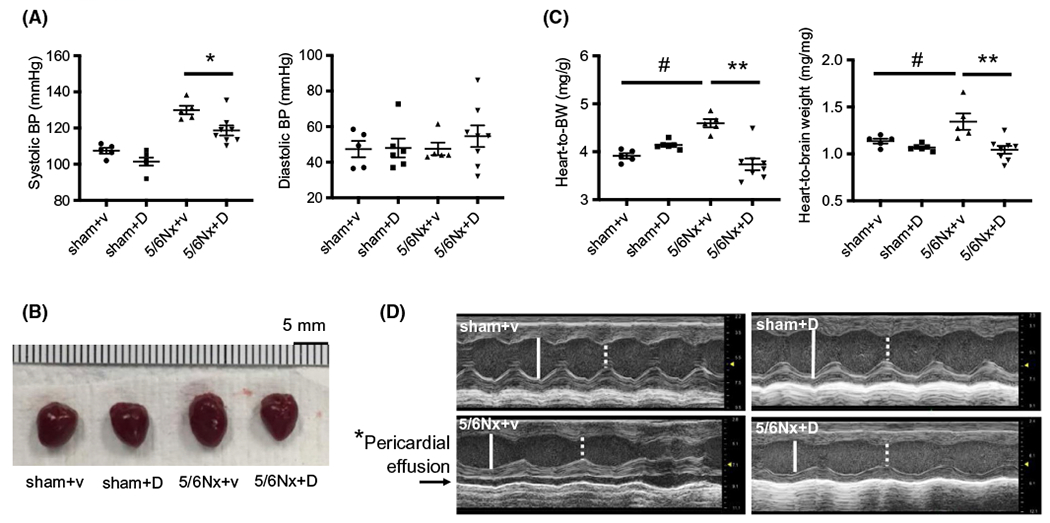
Inhibition of UT-A by dimethylthiourea (DMTU) ameliorated CKD-induced cardiac hypertrophy and improved heart function. A, Systolic and diastolic blood pressure at 8 weeks after completion of the CKD (5/6Nx) surgery. Blood pressure measurement of mice was performed by tail-cuff method. V = vehicle, D = DMTU treatment. B, Pictures of mouse hearts at sacrifice. C, Heart weight (mg) normalized by body weight (g) (left panel) and brain dry weight (mg) (right panel) at sacrifice. All data: mean ± SEM (N = 5-8), *P < .05, **P < .001, #P < .005 by one-way ANOVA. D, Echocardiography was performed on isoflurane anesthetized mice at 8 weeks of CKD. Images in each panel are M-mode images. White solid line—LVEDD: left ventricular endo-diastolic dimension; and white dash line—LVESD: left ventricular end-systolic dimension. Arrow in panel of 5/6Nx + vehicle denotes fluid space indicative of pericardial effusion. *P < .05 between CKD/+vehicle to the other three groups by Chi square analysis
TABLE 2.
Echocardiogram analysis of mice after 5/6 nephrectomy and DMTU treatment
| Sham + vehicle | Sham + DMTU | 5/6Nx + vehicle | 5/6Nx + DMTU | |
|---|---|---|---|---|
| BW (g) | 29.9 ± 0.4 | 25.8 ± 1.2 | 27.1 ± 0.8 | 25.8 ± 0.6 |
| IVSs (mm) | 1.05 ± 0.00 | 1.05 ± 0.00 | 1.17 ± 0.00* | 1.08 ± 0.00†# |
| IVSd (mm) | 0.47 ± 0.02 | 0.44 ± 0.00 | 0.51 ± 0.01* | 0.49 ± 0.01 |
| PWs (mm) | 0.50 ± 0.02 | 0.52 ± 0.03 | 0.53 ± 0.01 | 0.52 ± 0.01 |
| PWd (mm) | 0.54 ± 0.02 | 0.53 ± 0.00 | 0.59 ± 0.01 | 0.55 ± 0.01 |
| LVEDD (mm) | 3.69 ± 0.12 | 3.80 ± 0.18 | 3.43 ± 0.18 | 3.43 ± 0.06 |
| LVESD (mm) | 2.37 ± 0.13 | 2.45 ± 0.14 | 3.34 ± 0.19* | 2.77 ± 0.02†# |
| FS (%) | 32 ± 1.5 | 33 ± 0.5 | 27 ± 0.5* | 30 ± 0.5† |
| LVEDV (μL) | 57.5 ± 1.0 | 57.3 ± 1.8 | 66.6 ± 0.6* | 62.6 ± 0.3†# |
| LVESV (μL) | 22.4 ± 1.4 | 21.8 ± 1.1 | 28.4 ± 0.4* | 23.6 ± 0.3†# |
| LVM (mg) | 89 ± 4 | 97 ± 7 | 113 ± 8* | 89 ± 3† |
| LVMI (mg/g) | 2.97 ± 0.09 | 3.73 ± 0.15 | 4.18 ± 0.27* | 3.44 ± 0.13†# |
Note: All data; mean ± SEM (N = 5-8).
Abbreviations: BW, body weight; FS, percentage of fractional shortening; calculated by 100*(LVEDD − LVESD)/LVEDD; IVSd, interventricular septal thickness at end-diastole; IVSs, interventricular septal thickness at end-systole; LVEDD, left ventricular end-diastolic dimension; LVEDV, left ventricular end-diastolic volume; LVESV, left ventricular end-systolic volume; LVM, left ventricular mass; LVMI, left ventricular mass index (ratio of LVM to BW); PWd, posterior wall thickness at end-diastole; PWs, posterior wall thickness at end-systole.
P < .05 vs sham + vehicle;
P < .05 vs 5/6Nx + vehicle by one-way ANOVA with Bonferroni test;
P < .05 for interaction between 5/6Nx and DMTU (P < .0005 in IVSs, LVESD, LVEDV, and LVESV and P < .05 in LVMI for the factor of 5/6Nx; P < .05 in IVSs, LVESD, LVEDV, and LVESV and P = .95 in LVMI for the factor of DMTU) by two-way ANOVA.
3.5 |. Inhibition of UT attenuated CKD-induced cardiac fibrosis
The impact of DMTU on fibrosis in the hearts of CKD mice was examined by Masson’s staining (Figure 4A). The percentage of fibrotic area in heart was sharply increased in 5/6Nx + vehicle (1.04 ± 0.15%) vs sham + vehicle (0.07 ± 0.03%), but DMTU reduced the increase in fibrotic area in the CKD mouse (0.57 ± 0.07%) (Figure 4A). By immunohistochemistry staining α-smooth muscle actin (α-SMA) proteins increased in 5/6Nx + vehicle, but they were significantly less in 5/6Nx + DMTU group (Figure 4B). The mRNA expression of fibronectin, α-SMA, and collagen 4A1 were limited in the heart from 5/6Nx + DMTU mice compared with 5/6Nx + vehicle mice (Figure 4C–E). These data suggest that the provision of DMTU diminished CKD-induced cardiac fibrosis.
FIGURE 4.
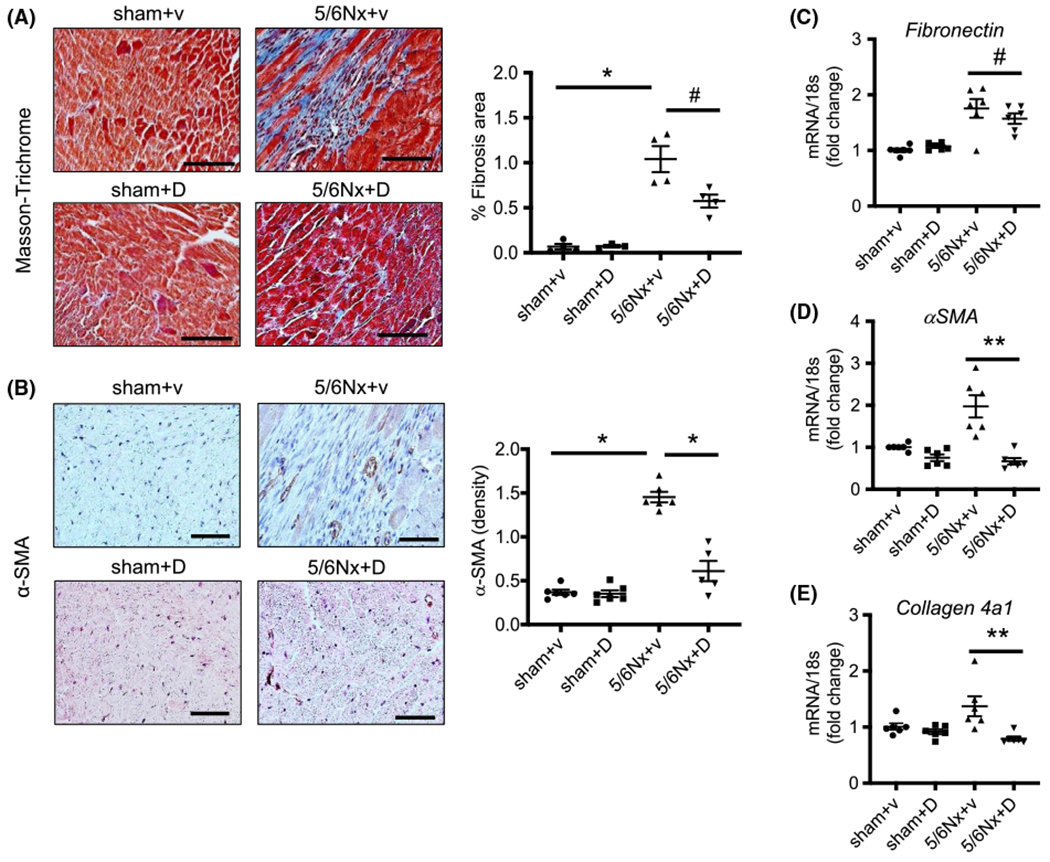
Inhibition of UT-A attenuated CKD-induced cardiac fibrosis. A, Masson-Trichrome staining was performed in mouse heart tissues. Pictures were observed at ×200 magnification, scale bar is 100 μm. Blue color indicated fibrosis. The right point graph shows the percentage of fibrosis area per entire area as calculated using the Olympus CellSens imaging software (N = 4). B, Immunohistochemistry staining for α-SMA in mouse heart tissue with ×400 magnification, scale bar is 50 μm. The right point graph shows that the percentage of α-SMA area per entire area was calculated by Olympus CellSens imaging software (N = 4). C-E, Quantitative mRNA expression of fibronectin, α-SMA, and collagen 4a1 in mouse hearts performed by real-time PCR. Individual gene expression was calculated by ΔΔcq and standardized by housekeeping gene 18s. All data: mean ± SEM (N = 6). *P < .0001, **P < .005, #P < .05 by one-way ANOVA
3.6 |. Deletion of UT decreased the CKD-induced increase in systolic blood pressure, suppressed cardiac fibrosis, and improved heart function
To further explore whether inhibition of UT ameliorates uremic cardiomyopathy, we performed nephrectomy in urea transport knockout (KO) and wild-type (WT) littermate mice. In our initial study, we found that KO mice do not tolerate 5/6 nephrectomy, as they died in 1 week. Therefore, we used uninephrectomy to induce CKD. BUN was significantly increased in both KO/CKD and WT/CKD mice vs no-CKD mice, which provided the evidence for this CKD model (Table 3). KO mice do not have the ability to concentrate urine, so KO/sham mice have low urine osmolality and high urine volume. KO/CKD mice had significantly lower urine osmolality and higher urine volume than KO/sham mice. In both WT and KO mice, CKD increased 24 h urine volume and plasma osmolality (Table 3).
TABLE 3.
Phenotypic characteristics of Wild-type and UT-KO mice
| WT | UT-KO | |||
|---|---|---|---|---|
| WT/sham | WT/Nx | KO/sham | KO/Nx | |
| BW (g) | 29.2 ± 1.2 | 26.7 ± 0.3 | 30.5 ± 1.8 | 26.8 ± 0.6 |
| BUN (mg/dL) | 29.2 ± 1.8 | 52.3 ± 2.0* | 26.3 ± 0.9 | 55.1 ± 1.7# |
| Urine urea (mmol/L) | 1566 ± 114 | 1767 ± 216 | 304 ± 40 | 252 ± 60 |
| 24 hr urea (mmol/day) | 4.767 ± 0.353 | 5.043 ± 0.311 | 3.824 ± 0.120 | 5.092 ± 0.529 |
| 24 hr intake volume/BW (mL/g) | 0.213 ± 0.039 | 0.291 ± 0.021 | 0.621 ± 0.069 | 1.198 ± 0.185## |
| 24 hr Urine volume/BW (mL/g) | 0.110 ± 0.015 | 0.109 ± 0.011 | 0.436 ± 0.055 | 0.970 ± 0.183## |
| Volume in-out balance/BW (mL/g) | 0.103 ± 0.025 | 0.181 ± 0.016** | 0.184 ± 0.026 | 0.229 ± 0.019 |
| 24 hr Urine osmolality (mOsmol/kg H2O) | 2289 ±140 | 2307 ± 288 | 636 ± 11 | 511 ± 52## |
| Plasma osmolality (mOsmol/kg H2O) | 301 ± 2 | 309 ± 1** | 306 ± 2 | 314 ± 3## |
Note: 24 hr urea was calculated by Urine urea (mmol/L)*24 hr Urine volume (mL)/1000, Volume in-out balance/BW was calculated by {(24 hr intake volume) − (24 hr urine volume)} v/BW, All data; mean ± SEM (n = 5-6).
Abbreviations: BW, body weight; BUN, blood urea nitrogen.
P < .0001 vs WT-sham
P < .05 vs WT-sham
P < .0001 vs UT-A1/A3−/−-sham
P < .05 vs UT-A1/A3−/−-sham, by two-way ANOVA.
There is no difference in systolic blood pressure (SBP) in each group at baseline, measured 3 days after nephrectomy surgery. After 8 weeks of CKD, SBP was significantly increased in WT/CKD mice compared to WT/sham mice (125 ± 3 vs 93 ± 2 mm Hg, P = .0001) (Figure 5A). This increase was limited in KO/CKD mice vs KO/sham (110 ± 4 mm Hg vs 102 ± 4 mm Hg P = .12). In CKD mice, deletion of UT significantly decreased SBP (125 ± 3 mm Hg vs 110 ± 4 mm Hg P < .05) (Figure 5A).
FIGURE 5.
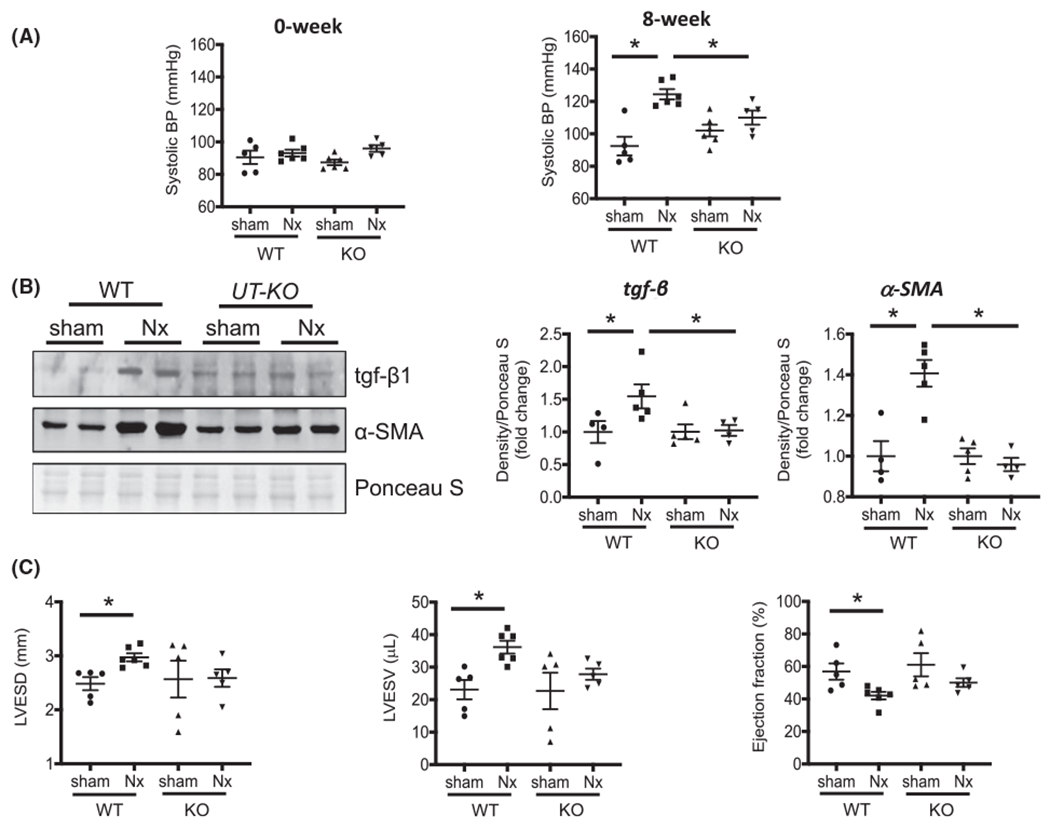
Deletion of UT decreased the CKD-induced increase in systolic blood pressure, suppressed cardiac fibrosis, and improved heart function. A, Systolic blood pressure (BP) were measured by tail-cuff method in WT and KO mice with or without Nx. B, Western blot analysis Tgf-β and α-SMA for lysates of mouse hearts. The right point graphs show the levels of each protein in the sham group compared with the level in the other three groups (designated as one-fold). Protein level normalized by total protein density of Ponceau S staining. Data: mean ± SEM (N = 4). *P < .05 by two-way ANOVA analysis. C, Echocardiography was performed on isoflurane anesthetized mice at 8 weeks after the surgery for mice. LVESD, left ventricular end-systolic dimension; LVESV, left ventricular end-systole volume
To identify whether KO of UT prevented cardiac fibrosis, we measured protein abundance in the heart of WT/sham, WT/CKD, KO/sham, and KO/CKD mice. In WT mice, CKD significantly increased tgf-β levels vs sham mice (Figure 5B). In CKD mice, knockout of UT did not enhance tgf-β compared with KO/sham mice. Deletion of UT abolished the CKD-induced increase in α-SMA, a key protein indicating fibrosis (Figure 5B). To identify whether deletion of UT has an impact on heart function, we performed echocardiography analysis for mice (Figure 5C). In WT/CKD mice, left ventricular end-systolic dimensions (LVESD) were larger, and left ventricular end-systole and -diastole volumes (LVESV and LVEDV) were significantly increased compared with WT/sham. However, LVESD, LVESV, and LVEDV did not have significant changes in KO/CKD mice vs KO/sham mice. In WT mice, the decline of ejection fraction (EF) appeared in CKD, but not in KO/CKD mice, compared with KO/sham mice (Figure 5C). These data suggest that deletion of UT ameliorated hypertension, decreased cardiac fibrosis, and improved heart function.
3.7 |. Inhibition or deletion of UT attenuated CKD-induced upregulation of the RAS
Upregulation of the RAS is related to cardiac hypertrophy and fibrosis.27 In our CKD mice, the mRNA levels of renin and ACE were sharply increased in CKD heart; UT inhibition by DMTU treatment eliminated the increased renin and ACE expression (Figure 6A,B). The mRNA of angiotensin II receptor type1 (Agtr1) did not significantly change in either CKD- or DMTU-treated mouse hearts (Figure 6C). The expression of angiotensin II receptor type2, aldosterone receptor and mas receptor did not change significantly (Figure S2). Similar changes were also found in UT knockout mice. The expression of renin was significantly increased in WT/CKD compared to WT/sham mice; UT deletion removed this increase (Figure 6D). ACE was also increased in WT/CKD heart; UT knockout in CKD mice only slightly decreased ACE compared to KO/sham mice, but did not reach statistical difference (Figure 6E). However, UT knockout significantly decreased CKD-induced upregulation of Agtr1 (Figure 6F). To discover whether the relationship between the UT and the RAS is directly related to each other, we isolated fibroblasts from mouse heart. In cultured cardiac fibroblasts, overexpression of UT increased the expression of renin (Figure 7A), ACE (Figure 7B) and Agtr1 (Figure 7C). UT inhibition by DMTU partially removed these increases. These data suggest that the impact of UT inhibition was to limit cardiac fibrosis and hypertrophy related to the reduced expression of renin, ACE and Agtr1.
FIGURE 6.
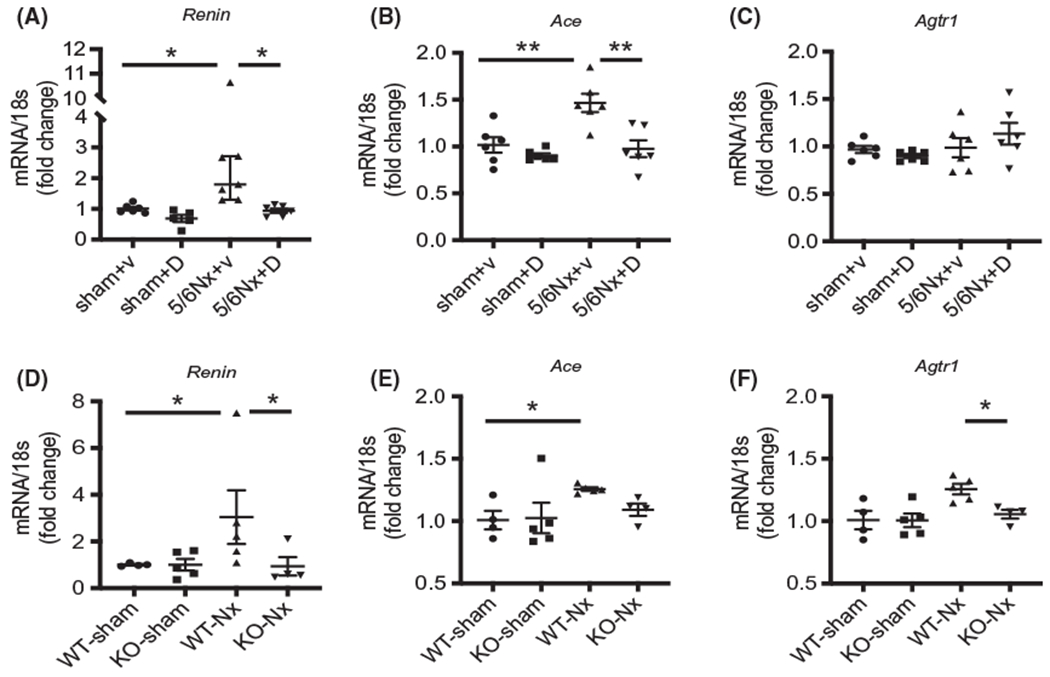
Inhibition of UT-A suppressed CKD-induced upregulation of renin and angiotensin-converting enzyme (ACE). Quantitative mRNA expression in mouse hearts performed by real-time PCR. All gene expressions were calculated by ΔΔcq and standardized by housekeeping gene 18s. A-C, mRNA from mice heart in sham/vehicle, sham/DMTU, 5/6Nx/vehicle, and 5/6Nx/DMTU mice. D-F, mRNA from mice heart in ET/sham, WT/CKD, KO/sham, and KO/CKD mice. A and D are Renin; B and E are ACE (angiotensin-converting enzyme); and C and F are Agtr1: angiotensin II receptor type 1. All data: mean ± SEM (N = 4-6). *P < .05, **P < .005 by one-way ANOVA analysis
FIGURE 7.
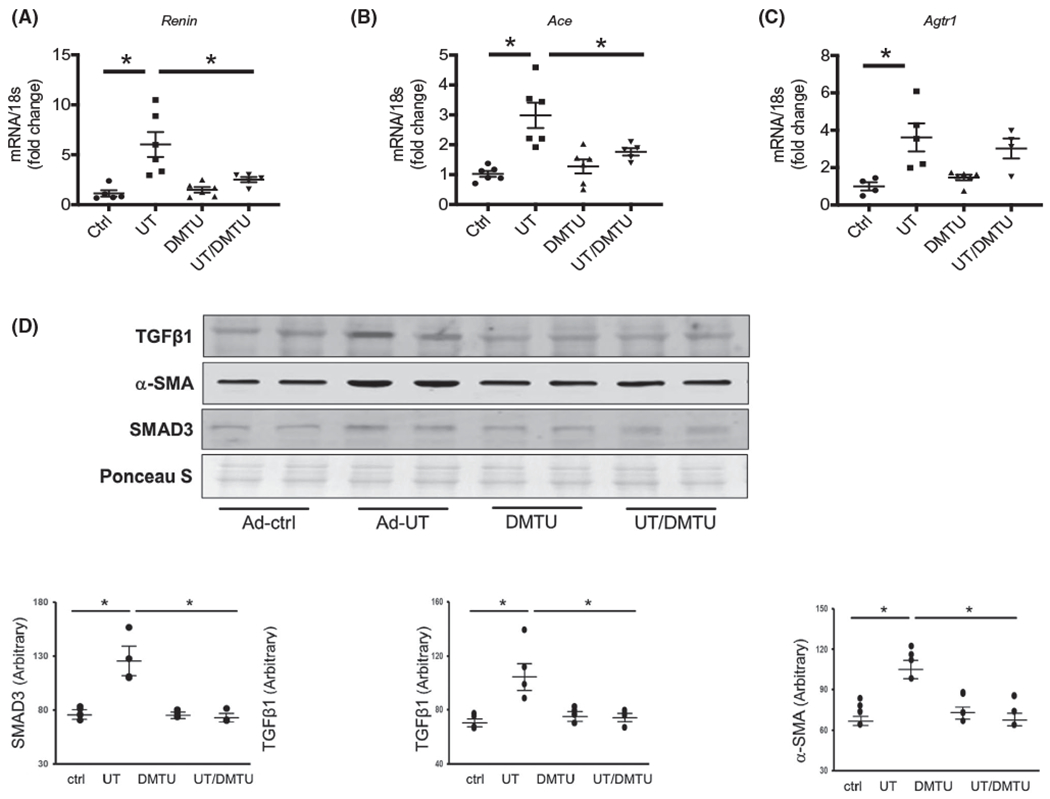
Overexpression UT stimulated fibroblast activation to myofibroblasts in primary cultured cardiac fibroblasts. (A, B & C) cultured fibroblasts treated with adenovirus to overexpress UT. Quantitative mRNA expression in these cells performed by real-time PCR. All gene expressions were calculated by ΔΔcq and standardized by housekeeping gene 18s. A, Renin; B, Ace (angiotensin-converting enzyme); and C, Agtr1: angiotensin II receptor type1. D, Primary cultured cardiac fibroblasts transduced with Ad-UT for 48 hours. Western blot of lysates were performed for α-SMA and TGF. The point graphs show the levels of each protein comparing in arbitrary units. Protein level normalized by total protein density of Ponceau S staining. All data: mean ± SEM (N = 4-6). *P < .05, **P < .005 by one-way ANOVA analysis
3.8 |. Overexpression UT stimulated fibroblast differentiation into myofibroblasts in primary cultured cardiac fibroblasts
In cultured cardiac fibroblastic cells, we found that the myofibroblast marker, αSMA, was significantly increased when overexpressing UT, which suggests that these cells differentiate into myofibroblasts (Figure 7D). In parallel with this change, the protein levels of the tgf-β cascade proteins Smad 3 were also significantly increased (Figure 7D). Inhibition of UT by DMTU partially removed these increases. These data suggest that upregulation of urea transport contributes fibroblast differentiation into myofibroblasts leading to cardiac fibrosis. One of the mechanisms by which UT inhibition prevents or reduces cardiac fibrosis could be through limiting this differentiation.
4 |. DISCUSSION
Uremic cardiomyopathy generally consists of cardiac hypertrophy and fibrosis induced by upregulation of the RAS, hypertension, and volume overload.3,7 To date, the beneficial therapies for cardiomyopathy in CKD patients are antihypertensive drugs and diuretics, such as ACE inhibitors, furosemide, or Ca2+ channel blockers.28,29 However, there are reports of these therapies having insufficient benefit or harmful side effects. Our study indicates that inhibition of UT suppresses the RAS, attenuates hypertension, and decreases volume retention, resulting in prevention of cardiac hypertrophy and fibrosis in mice with CKD (Figure 8).
FIGURE 8.
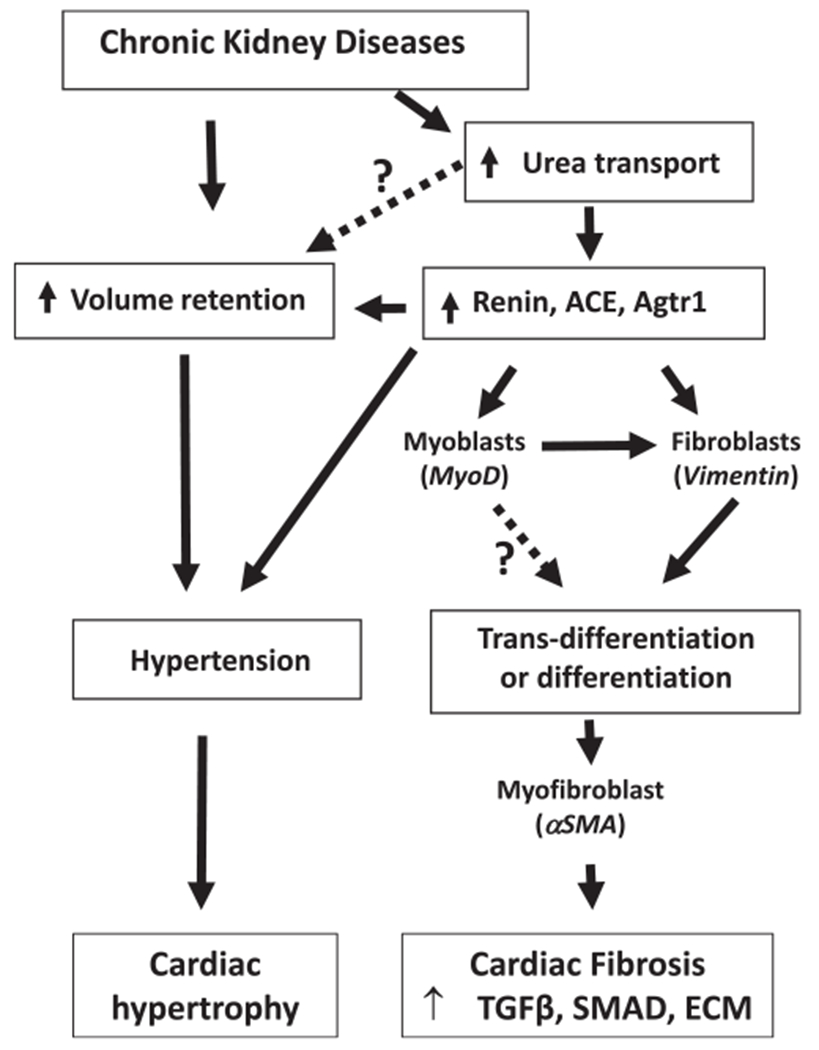
Summary. Chronic kidney disease induces volume retention and upregulation of urea transport. Volume retention cause hypertension leading to cardiac hypertrophy. Upregulation of urea transport increases the RAS and results in hypertension. The increase in the RAS also promotes fibroblast and acquisition of α-SMA expression and results in their becoming myofibroblasts. These myofibroblastic cells synthesize and deposit the extracellular matrix (ECM) components leading to heart fibrosis. In CKD, inhibition of urea transport attenuates hypertension and limits cardiac hypertrophy and fibrosis
A major finding of this study is that inhibition of UT attenuates CKD-induced hypertension and cardiac fibrosis. The majority of UT expression is in kidney, but it can also be found in colon, erythrocytes, brain, and liver.8,10 In our previous study, we found that UT is located in the heart and is upregulated in CKDs patients and animals.11 In the current study, we found that upregulation of UT is related to hypertension and cardiac fibrosis in CKD mice. Inhibition of UT, either by the UT inhibitor DMTU, or by genetic deletion of UT in mice, can decrease CKD-induced cardiac hypertrophy and fibrosis. In addition, the benefits of DMTU include reduced hypertension and improved heart function. In agreement with our study, other investigators reported that inhibition of UT by DMTU plays a protective role for acute myocardial infarction and diabetic cardiac dysfunction.30,31 The obvious mechanism by which DMTU contributes to cardiac protection is by reducing volume retention.
Volume overload is well known as a risk factor for mortality in CKD and end stage renal disease patients.32 Volume overload and hypertension lead to the development of left ventricular hypertrophy to maintain wall stress. Amelioration of hypervolemia and hypertension is a key step in preventing uremic cardiomyopathy. Whether UT directly regulates volume retention is controversial.33 However, our data show that inhibition of UT has a great benefit for reducing volume overload. UT and aquaporin 2 are the major transporters that support increased absorption of urea and water, respectively, in the kidney collecting duct. In knockout mice lacking UT-A1 and UT-A3, urine volumes are increased about three-fold and urine osmolality is decreased to about 35%, even in the presence of apparently normal aquaporin 2 levels.21,34 In a clinical trial, treating volume overload by Tolvaptan improved cardiac congestion and decreased mortality with kidney dysfunction.35
The second major finding of this study is that the inhibition of UT was involved with a decrease in the CKD-induced increase in renin and ACE. There is emerging evidence that upregulation of the RAS plays crucial roles in cardiovascular pathophysiology, including causing hypertension, cardiovascular remodeling, and end organ damage.14 The increase in ACE and angiotensin II is closely related with left ventricular hypertrophy and fibrosis in high-fat feeding mice.36 The local activation of the RAS in heart is associated with cardiac hypertrophy and fibrosis.15,16 Most guidelines for the management of patients with cardiovascular disease recommend ACE inhibitors as first-choice therapy.37 ACE inhibitors and angiotensin-receptor blockers (ARBs) were protective against atrial fibrillation in the patients following cardioversion.38,39
We have three experimental models to support that the upregulation of UT is associated with increased renin and ACE. In primary cultured cardiac fibroblasts cells, overexpression of UT accelerates renin, ACE, and Agtr1 expression. In C57BL6 mice with CKD, the UT inhibitor DMTU abolished the renal failure-induced increase in renin and ACE. In UT-KO mice, enhanced renin was absent in CKD mice. In these mice, UT knockout also eliminated CKD-induced upregulation of Agtr1. Unfortunately, this phenomenon was not seen in CKD mice treated with DMTU. We speculate that this could be due to more UT inhibition in UT knockout mice than in mice treated with a UT inhibitor. In fibroblast culture experiments, we found that overexpression of UT not only increases renin and ACE, but also leads to increased levels of aSMA, tgf-β, and Smad2/3, resulting in the promotion of fibrosis. In support of these cell culture results, we have evidence that inhibition or deletion of UT could attenuate CKD-induced cardiac fibrosis in animals. These beneficial effects are related to downregulation of renin and ACE.
The third major finding of this study is that upregulation of UT promotes fibroblast differentiation into myofibroblasts and has the potential to induce myoblast trans-differentiation to fibroblasts. Laurino et al reported that in cardiac pathologies, over-activation of the RAS response for myoblasts results in trans-differentiation into fibroblasts.40 Myofibroblasts were first described in 1971 and play important roles for maintaining tissue remodeling.41 Their pathologic role has been described in many tissues. Under stress or injury conditions, fibroblasts differentiation into myofibroblasts for tissue repair. Fully differentiated myofibroblasts express αSMA, which is the most reliable marker of the myofibroblast phenotype and fibrosis, while fibroblasts only express vimentin.42 These myofibroblastic cells synthesize and deposit the ECM components for tissue repair. Myofibroblasts will disappear by apoptosis after healing. However, if these cells fail to undergo apoptosis, they will lead to ongoing pathology and will cause fibrosis or scarring.43
In primary cultured cardiac fibroblasts, upregulation of UT increases the αSMA amount, which indicates that UT increased fibroblast differentiation to myofibroblasts. Concomitant with this change, tgf-β, and Smad2/3 also increased, which means that the fibrotic pathway was activated. Additional evidence supports that over-activation of UT promotes fibrosis as seen in cardiac myoblasts, H9C2 cells. Adenovirus-mediated UT transfer increases vimentin abundance. Since vimentin is a fibroblast marker and linked with epithelial mesenchymal transition, we postulate that the increased level of UT promotes myoblast trans-differentiation to fibroblasts.
5 |. CONCLUSIONS
In summary, inhibition or deletion of UT suppress hypertension, and attenuates both cardiac hypertrophy and fibrosis that result from CKD. The mechanism by which inhibition of UT decreases fibrosis, while not fully understood, appears to be associated with suppression of the RAS and decline in volume retention, even when kidney function declines. In addition, UT promotes fibroblast differentiation into myofibroblasts. The inhibition of UT provides multiple benefits to counteract fibrosis. Our study suggests that UT inhibitors may be an attractive therapeutic option for preventing uremic cardiomyopathy and other fibrosis-related disorders.
Supplementary Material
Acknowledgments
Funding information
This work was supported by NIH/NIDDK grant R01 DK41707 to JMS; Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad to AK; the American Heart Association Discover and Innovation Grants supported by Bayer Group (17IBDG33780000) to XHW The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH, or the US Government
Abbreviations:
- ACE
angiotensin-converting enzyme
- AGT
angiotensinogen
- Agtr1
angiotensin II receptor type1
- AII
angiotensin II
- BUN
blood urea nitrogen
- BW
body weight
- CKD
chronic kidney disease
- DMTU
dimethylthiourea
- EF
ejection fraction
- IMCD
inner medullary collecting duct
- LVEDV
left ventricular end-diastole volumes
- LVESD
left ventricular end-systolic dimensions
- LVESV
left ventricular end-systole volumes
- Nx
nephrectomy
- RAS
renin-angiotensin system
- SBP
systolic blood pressure
- UT
urea transporter
- αSMA
α-smooth muscle actin
Footnotes
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists. However, Drs. Sands and Klein are inventors on Patent 9827222: Treating or preventing nephrogenic diabetes insipidus. They are founders of NephroDI Therapeutics. Dr Sands also serves on the board of directors of NephroDI Therapeutics and is a consultant for Third Kidney and Bristol-Myers Squibb.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Institutional Animal Care and Use Committee (IACUC) at Emory University approved all experiments (protocol # 4000152).
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the Supporting Information section.
REFERENCE
- 1.United States Renal Data System. Annual Data Report; 2018;4:79–98. [Google Scholar]
- 2.Trespalacios FC, Taylor AJ, Agodoa LY, Bakris GL, Abbott KC. Heart failure as a cause for hospitalization in chronic dialysis patients. Am J Kidney Dis. 2003;41:1267–1277. [DOI] [PubMed] [Google Scholar]
- 3.London GM. Cardiovascular disease in chronic renal failure: pathophysiologic aspects. Semin Dial. 2003;16:85–94. [DOI] [PubMed] [Google Scholar]
- 4.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 5.Wang B, Zhang A, Wang H, et al. miR-26a limits muscle wasting and cardiac fibrosis through exosome-mediated microRNA transfer in chronic kidney disease. Theranostics. 2019;9:1864–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy DJ, Elkareh J, Shidyak A, et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am J Physiol Renal Physiol. 2008;294:F450–F454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamakawa H, Imamura T, Matsuo T, et al. Diastolic wall stress and ANG II in cardiac hypertrophy and gene expression induced by volume overload. Am J Physiol Heart Circ Physiol. 2000;279:H2939–H2946. [DOI] [PubMed] [Google Scholar]
- 8.Klein JD, Blount MA, Sands JM. Molecular mechanisms of urea transport in health and disease. Pflugers Arch. 2012;464:561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sands JM. Renal urea transporters. Curr Opin Nephrol Hypertens. 2004;13:525–532. [DOI] [PubMed] [Google Scholar]
- 10.Inoue H, Kozlowski SD, Klein JD, Bailey JL, Sands JM, Bagnasco SM. Regulated expression of renal and intestinal UT-B urea transporter in response to varying urea load. Am J Physiol Renal Physiol. 2005;289:F451–F458. [DOI] [PubMed] [Google Scholar]
- 11.Duchesne R, Klein JD, Velotta JB, et al. UT-A urea transporter protein in heart: increased abundance during uremia, hypertension, and heart failure. Circ Res. 2001;89:139–145. [DOI] [PubMed] [Google Scholar]
- 12.Esteva-Font C, Cil O, Phuan PW, et al. Diuresis and reduced urinary osmolality in rats produced by small-molecule UT-A-selective urea transport inhibitors. FASEB J. 2014;28:3878–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gumprecht J, Domek M, Lip GYH, Shantsila A. Invited review: hypertension and atrial fibrillation: epidemiology, pathophysiology, and implications for management. J Hum Hypertens. 2019;33(12):824–836. [DOI] [PubMed] [Google Scholar]
- 14.Forrester SJ, Booz GW, Sigmund CD, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. 2018;98:1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Mello WC, Frohlich ED. Clinical perspectives and fundamental aspects of local cardiovascular and renal renin-angiotensin systems. Front Endocrinol. 2014;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ainscough JF, Drinkhill MJ, Sedo A, et al. Angiotensin II type-1 receptor activation in the adult heart causes blood pressure-independent hypertrophy and cardiac dysfunction. Cardiovasc Res. 2009;81:592–600. [DOI] [PubMed] [Google Scholar]
- 17.Romero CA, Orias M, Weir MR. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol. 2015;11:242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang XH, Hu Z, Klein JD, Zhang L, Fang F, Mitch WE. Decreased miR-29 suppresses myogenesis in CKD. J Am Soc Nephrol. 2011;22:2068–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang XH, Du J, Klein JD, Bailey JL, Mitch WE. Exercise ameliorates chronic kidney disease-induced defects in muscle protein metabolism and progenitor cell function. Kidney Int. 2009;76:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klein JD, Wang Y, Mistry A, et al. Transgenic restoration of urea transporter A1 confers maximal urinary concentration in the absence of urea transporter A3. J Am Soc Nephrol. 2016;27:1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenton RA, Chou CL, Stewart GS, Smith CP, Knepper MA. Urinary concentrating defect in mice with selective deletion of phloretin-sensitive urea transporters in the renal collecting duct. Proc Natl Acad Sci U S A. 2004;101:7469–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fenton RA, Knepper MA. Urea and renal function in the 21st century: insights from knockout mice. J Am Soc Nephrol. 2007;18:679–688. [DOI] [PubMed] [Google Scholar]
- 23.Ilori TO, Blount MA, Martin CF, Sands JM, Klein JD. Urine concentration in the diabetic mouse requires both urea and water transporters. Am J Physiol Renal Physiol. 2013;304:F103–F111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang B, Zhang C, Zhang A, Cai H, Price SR, Wang XH. MicroRNA-23a and microRNA-27a mimic exercise by ameliorating CKD-induced muscle atrophy. J Am Soc Nephrol. 2017;28:2631–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Efe O, Klein JD, LaRocque LM, Ren H, Sands JM. Metformin improves urine concentration in rodents with nephrogenic diabetes insipidus. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doran JJ, Klein JD, Kim YH, et al. Tissue distribution of UT-A and UT-B mRNA and protein in rat. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1446–R1459. [DOI] [PubMed] [Google Scholar]
- 27.von Lueder TG, Wang BH, Kompa AR, et al. Angiotensin receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. 2015;8:71–78. [DOI] [PubMed] [Google Scholar]
- 28.Marquez DF, Ruiz-Hurtado G, Ruilope LM, Segura J. An update of the blockade of the renin angiotensin aldosterone system in clinical practice. Expert Opin Pharmacother. 2015;16:2283–2292. [DOI] [PubMed] [Google Scholar]
- 29.Perkovic V, Ninomiya T, Arima H, et al. Chronic kidney disease, cardiovascular events, and the effects of perindopril-based blood pressure lowering: data from the PROGRESS study. J Am Soc Nephrol. 2007;18:2766–2772. [DOI] [PubMed] [Google Scholar]
- 30.Kinugawa S, Tsutsui H, Hayashidani S, et al. Treatment with dimethylthiourea prevents left ventricular remodeling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res. 2000;87:392–398. [DOI] [PubMed] [Google Scholar]
- 31.Yeih DF, Yeh HI, Hsin HT, et al. Dimethylthiourea normalizes velocity-dependent, but not force-dependent, index of ventricular performance in diabetic rats: role of myosin heavy chain isozyme. Am J Physiol Heart Circ Physiol. 2009;297:H1411–H1420. [DOI] [PubMed] [Google Scholar]
- 32.Zoccali C, Moissl U, Chazot C, et al. Chronic fluid overload and mortality in ESRD. J Am Soc Nephrol. 2017;28:2491–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitada K, Daub S, Zhang Y, et al. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest. 2017;127:1944–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fenton RA, Flynn A, Shodeinde A, Smith CP, Schnermann J, Knepper MA. Renal phenotype of UT-A urea transporter knockout mice. J Am Soc Nephrol. 2005;16:1583–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greene SJ, Gheorghiade M, Vaduganathan M, et al. Haemoconcentration, renal function, and post-discharge outcomes among patients hospitalized for heart failure with reduced ejection fraction: insights from the EVEREST trial. Eur J Heart Fail. 2013;15:1401–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin N, Wang Y, Liu L, Xue F, Jiang T, Xu M. Dysregulation of the renin-angiotensin system and cardiometabolic status in mice fed a long-term high-fat diet. Med Sci Monit. 2019;25:6605–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Messerli FH, Bangalore S, Bavishi C, Rimoldi SF. Angiotensin-converting enzyme inhibitors in hypertension: to use or not to use? J Am Coll Cardiol. 2018;71:1474–1482. [DOI] [PubMed] [Google Scholar]
- 38.Kumagai K, Nakashima H, Urata H, Gondo N, Arakawa K, Saku K. Effects of angiotensin II type 1 receptor antagonist on electrical and structural remodeling in atrial fibrillation. J Am Coll Cardiol. 2003;41:2197–2204. [DOI] [PubMed] [Google Scholar]
- 39.Healey JS, Baranchuk A, Crystal E, et al. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: a meta-analysis. J Am Coll Cardiol. 2005;45:1832–1839. [DOI] [PubMed] [Google Scholar]
- 40.Laurino A, Spinelli V, Gencarelli M, et al. Angiotensin-II drives human satellite cells toward hypertrophy and myofibroblast trans-differentiation by two independent pathways. Int J Mol Sci. 2019;20(19):4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–550. [DOI] [PubMed] [Google Scholar]
- 42.Darby IA, Laverdet B, Bonte F, Desmouliere A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol. 2014;7:301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aarabi S, Bhatt KA, Shi Y, et al. Mechanical load initiates hypertrophic scar formation through decreased cellular apoptosis. FASEB J. 2007;21:3250–3261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.