

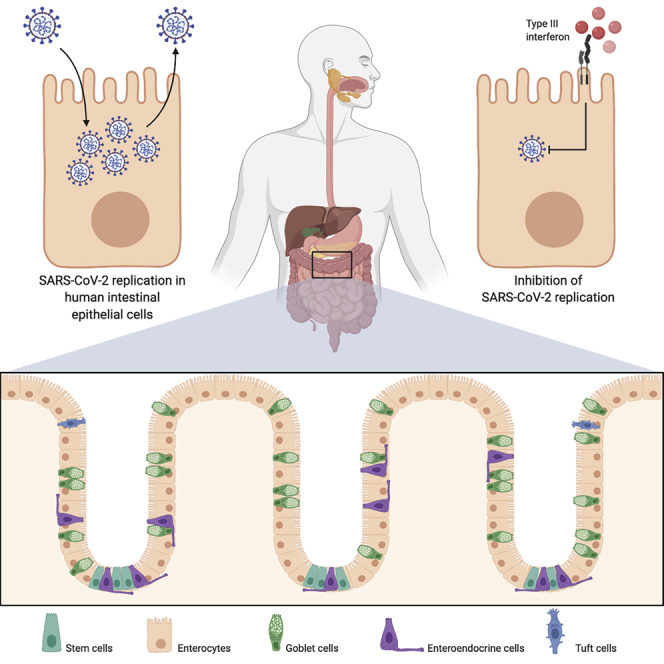
Summary
Severe acute respiratory syndrome-related coronavirus-2 (SARS-CoV-2) is an unprecedented worldwide health problem that requires concerted and global approaches to stop the coronavirus 2019 (COVID-19) pandemic. Although SARS-CoV-2 primarily targets lung epithelium cells, there is growing evidence that the intestinal epithelium is also infected. Here, using both colon-derived cell lines and primary non-transformed colon organoids, we engage in the first comprehensive analysis of the SARS-CoV-2 life cycle in human intestinal epithelial cells (hIECs). Our results demonstrate that hIECs fully support SARS-CoV-2 infection, replication, and production of infectious de novo virus particles. We found that viral infection elicits an extremely robust intrinsic immune response where interferon-mediated responses are efficient at controlling SARS-CoV-2 replication and de novo virus production. Taken together, our data demonstrate that hIECs are a productive site of SARS-CoV-2 replication and suggest that the enteric phase of SARS-CoV-2 may participate in the pathologies observed in COVID-19 patients by contributing to increasing patient viremia and fueling an exacerbated cytokine response.
Keywords: SARS-CoV-2, human intestinal epithelial cells, interferon, IFN, interferon lambda, intrinsic immunity, organoids, interferon stimulted genes, ISGs
Graphical Abstract
Highlights
-
•
Human intestinal epithelium cells (hIECs) can be infected by SARS-CoV-2
-
•
hIECs support SARS-CoV-2 replication and produce de novo viruses
-
•
SARS-CoV-2 infection can be controlled in hIECs by type I and III interferons
Stanifer et al. find that SARS-CoV-2 could infect the human gastrointestinal tract and efficiently produce new viruses. Importantly, they find that the cytokines type I and III interferons, which are naturally made by cells in response to viral infection, are protective and could be used as an antiviral strategy.
Introduction
Coronaviridae is a large family of single-stranded positive-sense enveloped RNA viruses that can infect most animal species (human as well as domestic and wild animals). They are known to have the largest viral RNA genome and are composed of four genera (Cui et al., 2019). Generally, infection by human coronaviruses results in mild respiratory tract symptoms, and they are known to be one of the leading causes of the common cold (Moriyama et al., 2020; Paules et al., 2020). However, in the last 18 years, we have witnessed the emergence of highly pathogenic human coronaviruses, including the severe-acute-respiratory-syndrome-related coronavirus (SARS-CoV-1), the Middle-East-respiratory-syndrome-related coronavirus (MERS-CoV), and, at the end of 2019, the severe-acute-respiratory-syndrome-related coronavirus-2 (SARS-CoV-2) (Lu et al., 2020). SARS-CoV-2 is responsible for the coronavirus-associated acute respiratory disease or coronavirus disease 19 (COVID-19) and represents a major global health threat, and coordinated efforts are urgently needed to treat the viral infection and stop the pandemic.
Although SARS-CoV-2 primarily targets cells of the lung epithelium, causing respiratory infection, there is growing evidence that the intestinal epithelium can also be infected. Multiple studies have reported gastrointestinal symptoms such as diarrhea at the onset of the disease and have detected the prolonged shedding of large amounts of coronavirus genomes in the feces even after the virus is not detectable in oropharyngeal swabs (Wu et al., 2020b; Xiao et al., 2020; Xing et al., 2020; Xu et al., 2020b; Wölfel et al., 2020). Although one study revealed the isolation of infectious virus particles from stool samples (Wang et al., 2020), to date, it remains unclear how many people shed infectious viruses in feces. Most critically, it remains unknown whether or not there is a possibility for fecal transmission of SARS-CoV-2, but multiple health agencies worldwide have highlighted this possibility. The presence of such a large amount of coronavirus genomes in feces is hardly explainable by a swallowing virus replicating in the throat or by a loss of barrier function of the intestinal epithelium, which will allow the release of viruses or genomes from the inside of the body (circulation or lamina propria) to the lumen of the gut. Instead, it is likely due to an active replication in the intestinal epithelium. Recently, intestinal biopsies of SARS-CoV-2 infected patients clearly show the presence of replicating viruses in epithelial cells of the small and large intestine (Xiao et al., 2020). SARS-CoV-2 infection of the gastrointestinal tract is supported by the fact that ACE2, the virus receptor (Hoffmann et al., 2020), is expressed in intestinal epithelial cells (Zhao et al., 2020; Lukassen et al., 2020; Wu et al., 2020a; Venkatakrishnan et al., 2020), and single-cell sequencing analysis suggest that its expression is even higher on intestinal cells compared to lung cells (Xu et al., 2020a). This highlights that SARS-CoV-2 is not restricted to the lung and also infects the gastrointestinal tract. Importantly, many animal coronaviruses are well known to be enteric and are transmitted via the fecal-oral route (Wang et al., 2019; Wang and Zhang, 2016). Additionally, the presence of human pathogenic coronaviruses in the gastrointestinal tract was previously reported for SARS-CoV-1 and MERS-CoV but remained seriously understudied (Leung et al., 2003; Wong et al., 2020; Zhou et al., 2017). Although it is now clear that human coronaviruses, particularly SARS-CoV-2, are found in feces and can infect the gastrointestinal tract, the importance of its enteric phase for viremia, pathogenesis, and patient prognosis remains unknown.
To combat the current pandemic of COVID-19 and prepare for potential future emerging zoonotic coronaviruses, we need to gain a better understanding of the molecular basis of SARS-CoV-2 infection, replication, and de novo infectious virus production in a tissue-specific manner. Here, we engaged in studying SARS-CoV-2 infection of human intestinal cells. For this, we exploited both human intestinal epithelial cell (hIEC) lines and human organoid culture models to characterize how these cells support SARS-CoV-2 replication and de novo infectious virus production and how they respond to viral infection. Direct comparison of both primary and transformed cells shows that hIECs fully support SARS-CoV-2 infection and de novo production of infectious virus particles. Interestingly, viral infection elicited a robust intrinsic immune response where interferon (IFN) mediated responses were efficient at controlling SARS-CoV-2 replication and de novo infectious virus production. Importantly, human primary intestinal epithelial cells responded to SARS-CoV-2 infection by producing only type III IFN. Taken together, our data clearly highlight the importance of the enteric phase of SARS-CoV-2, and this should be taken into consideration when developing hygienic/containment measures and antiviral strategies and when determining patient prognosis.
Results
Efficient Infection of hIECs by SARS-CoV-2
As there is growing evidence that the gastrointestinal tract is infected by SARS-CoV-2, we engaged in studying virus infection in human intestinal epithelial cells (IECs). First, SARS-CoV-2 (strain BavPat1) was propagated in the green monkey cell line Vero. To detect viral infection, we used an antibody directed against a region of the nucleoprotein (N) that is conserved between of SARS-CoV-1 and SARS-CoV-2. Additionally, we used the J2 antibody, which detects double-stranded RNA (dsRNA), which is a hallmark of RNA virus replication (Targett-Adams et al., 2008). Cells positive for N were always positive for dsRNA; the N signal was found to be dispersed within the cytosolic area, whereas dsRNA was found in discrete foci likely corresponding to replication compartments (Harak and Lohmann, 2015) (Figure S1A). Supernatants of infected Vero cells were collected at 48 h post-infection (hpi), and the amount of infectious virus particles present was measured using a TCID50 approach on Vero cells (Figure S1B). The colon-carcinoma-derived lines T84 and Caco-2 cells were then infected with SARS-CoV-2 at a MOI of 0.5 (as determined in Vero cells). These cells expressed both ACE2 and TMPRSS2 (Figures S1C and S1D). At different times post-infection, T84 and Caco-2 cells were fixed and immunostained using the anti-N and anti-dsRNA antibodies (Figure 1A). Results show that SARS-CoV-2-infected Caco-2 cells were readily detected as early as 4 hpi and, by 24 hpi, most of the cells were infected (Figure 1B). Similar results were observed in the T84 cells, but detection of infection was slightly delayed compared to Caco-2 cells, and, although the same amount of SARS-CoV-2 was used to infect these cells, only ∼20% of cells were infected at 24 hpi (Figure 1B). These observations are in agreement with the increase in viral genome copy numbers over time (Figure 1C) and the release of infectious virus particles in the supernatant of infected T84 and Caco-2 cells (Figure 1D). Interestingly, infection of hIECs by SARS-CoV-2 was associated with the generation of an IFN-mediated intrinsic immune response. Concomitant with the differences observed in virus replication and de novo virus production observed between T84 and Caco-2 cells, T84 cells mounted a much stronger immune response than Caco-2 cells (Figures 1E and S2A), although much fewer T84 cells were infected (Figure 1B). Of note, the protein level of secreted type I IFN was under the detection limit (data not shown). Altogether, these results show that IECs are readily infected by SARS-CoV-2 and that infection of Caco-2 cells lead to a weaker intrinsic immune response that is associated with more de novo infectious virus production compared to T84 cells. This observation suggests that the IFN-mediated immune response controls SARS-CoV-2 infection in IECs.
Figure 1.

Human IECs Support SARS-CoV-2 Infection, Replication, and De Novo Infectious Virus Production
T84 and Caco-2 cells were infected with SARS-CoV-2 at a MOI of 0.5.
(A) At indicated time points, cells were fixed and indirect immunofluorescence was performed against the viral N protein (red) and dsRNA (green). Nuclei were stained with DAPI (blue). Representative images are shown. Scale bars, 10 μm.
(B) Same as (A), except the number of SARS-CoV-2-positive cells was quantified in 10 fields of view for each time point.
(C) At indicated time points, RNA was harvested, and q-RT-PCR was used to evaluate the copy number of the SARS-CoV-2 genome.
(D) At indicated time points, supernatants were collected from infected T84 and Caco-2 cells. The amount of de novo virus present in the supernatants was determined using a TCID50 assay.
(E) Same as (C), except the upregulation of IFN-β1 and IFN-λ was evaluated.
Error bars indicate standard deviation. n = 3 biological replicates.
IFN Hinders SARS-CoV-2 Infection in hIECs
To directly test the function of IFNs in controlling/restricting SARS-CoV-2 replication and de novo infectious virus production in hIECs, T84 and Caco-2 cells were mock-treated or pretreated with type I (IFN-β1) or type III (IFN-λ) IFNs. 24 h post-treatment, cells were infected with SARS-CoV-2 at a MOI of 0.5 (as determined in Vero cells), in the presence or absence of IFNs (Figure 2A). 24 hpi, cells were immunostained using both the anti-N and anti-dsRNA antibodies. Results show that pretreatment of cells with either IFN significantly interfered with SARS-CoV-2 infection of both T84 and Caco-2 cells (Figure 2B). This decrease of the number of SARS-CoV-2-infected cells pretreated with either IFN was associated with an inhibition of the increase in viral genome copy numbers (Figure 2C) and a significant decrease in the release of infectious de novo virus particles (Figure 2D). These results show that both IFN-β1 and IFN-λ provided in trans can induce an efficient antiviral state in IECs preventing/controlling SARS-CoV-2 infection. This strongly suggests that the IFN-mediated immune response can control SARS-CoV-2 infection in human IECs.
Figure 2.

IFN-β and IFN-λ Control SARS-CoV-2 Infection of hIECs
(A) Schematic describing the experimental setup.
(B) T84 and Caco-2 cells were pretreated with IFNs 24 h prior to infection. 24 hpi, virus infection was evaluated by indirect immunofluorescence for the viral N protein (red) and dsRNA (green). Nuclei were stained with DAPI (blue). Representative images are shown. The number of SARS-CoV-2-positive cells was quantified in 10 fields of view for each time point. Scale bars, 10 μm.
(C) Same as (B), except RNA was harvested and qRT-PCR was used to evaluate the copy number of SARS-CoV-2 genome.
(D) 24 hpi, supernatants were collected from infected T84 and Caco-2 cells. The amount of de novo virus present in the supernatants was determined using a TCID50 assay.
n = 3 biological replicates. Error bars indicate standard deviation.
IFN-Mediated Intrinsic Immune Response Controls SARS-CoV-2 Infection in HIECs
To address whether the endogenous levels of IFNs generated by IECs controls SARS-CoV-2 replication and de novo infectious virus production, we exploited our previously reported IECs depleted of either the type I IFN receptor (IFNAR1; AR−/−) or the type III IFN receptor (IFNLR1; LR−/−) or depleted of both IFN receptors (double knockout [dKO]). To control that our cells were functionally knocked out for the type I IFN (AR−/−) and/or the type III IFN receptor (LR−/−), T84 cells were treated with either IFN, and the production of the IFN-stimulated gene (ISG) IFIT1 was evaluated. As expected, type I IFN receptor knockout cells (AR−/−) only responded to IFN-λ, whereas type III IFN receptor knockout cells (LR−/−) only responded to IFN-β1 (Figure S3A). IFN receptor dKO cells did not respond to either IFN (Figure S3A). Wild-type (WT) or IFN receptor knockout T84 cells were infected with SARS-CoV-2 at a MOI of 0.1 (as determined in T84 cells). 24 hpi, cells were immunostained using the anti-N antibody, and the number of infected cells was quantified using fluorescent microscopy (Figure 3A). Results showed that depletion of the type I IFN receptor (AR−/−) resulted in a slight increase in the number of infected cells. Importantly, depletion of the type III IFN receptor (LR−/−) resulted in a significant increase of cell infectivity by a factor of approximately seven. Similar results were obtained when both the type I and type III IFN receptors were depleted (dKO) (Figure 3B). Interestingly, this increase in the number of infected cells upon depletion of the type III IFN receptor (LR−/−) was associated with a significant increase in viral genome copy numbers (Figure 3C) and with a three-orders-of-magnitude increase in de novo infectious virus production (Figure 3D). Together, these results suggest that the type-III-IFN-mediated immune response actively participates in controlling SARS-CoV-2 infection in human IECs.
Figure 3.

Type III IFN Receptor Controls SARS-CoV-2 Replication in hIECs
(A–D) WT T84 cells and T84 cells depleted of the type I IFN receptor (AR−/−), the type III IFN receptor (LR−/−), or both (dKO) were infected with SARS-CoV-2 at an MOI of 0.1. (A) 24 hpi, infection was analyzed by indirect immunofluorescence of the viral N protein (red). Nuclei were stained with DAPI (blue). Representative images are shown. Scale bars, 10 μm. (B) Same as (A), except the number of SARS-CoV-2-positive cells was quantified in 10 fields of view for each cell type. (C) 24 hpi, RNA was harvested and the change in the copy number of SARS-CoV-2 genome was evaluated by qRT-PCR. (D) 24 hpi, supernatants were collected from all cell types. The amount of de novo virus present in the supernatants was determined using a TCID50 assay.
(E–G) WT T84 cells were mock-treated or pretreated with pyridine-6, a pan-JAK inhibitor, for 2 h prior to SARS-CoV-2 infection. (E) 24 hpi, infection was analyzed by indirect immunofluorescence of the viral N protein (red). Nuclei were stained with DAPI (blue). Representative images are shown. Scale bars, 10 μm. (F) 24 hpi, RNA was harvested and the change in the copy number of SARS-CoV-2 genome was evaluated by qRT-PCR. (G) 24 hpi, supernatants were collected from mock-treated or pan-JAK-treated cells. The amount of de novo virus present in the supernatants was determined using a TCID50 assay.
n = 3 biological replicates. Error bars indicate standard deviation.
To unambiguously address the importance of the IFN-mediated antiviral response, we used the pan-JAK inhibitor (pyridone-6) to inhibit the STAT1 phosphorylation activation and block the production of ISGs. As we previously reported, treatment of T84 cells with the pan-JAK inhibitor fully inhibits signal transduction downstream both the type I and type III IFN receptors (Pervolaraki et al., 2017; and data not shown). Mock-treated and pyridone-6 pretreated T84 cells were infected with SARS-CoV-2 for 24 h and analyzed using fluorescence microscopy following immunostaining using the anti-N antibody. Results show both a significant increase in the number of infected cells (Figure 3E) and an increase of viral genome copy number in cells treated with the pan-JAK inhibitor (Figure 3F). Importantly, and in agreement with the results observed in cells depleted of the type III IFN receptor, this increase in infectivity was also associated with an increase in infectious de novo virus particle production (Figure 3G). Altogether, these results strongly support a model where type-III-IFN-mediated signaling controls SARS-CoV-2 infection in hIECs.
Human Primary Intestinal Organoids Support SARS-CoV-2 Infection, Replication, and De Novo Infectious Virus Production
To address whether primary hIECs can be infected by SARS-CoV-2 and support de novo virus production, we used human-colon-derived organoids from two distinct donors. Intact ultrastructural organization and differentiation to all cell types (e.g., enterocytes, Goblet cells, enteroendocrine cells, and stem cells) was confirmed using confocal fluorescent microscopy (Figure 4A) and qRT-PCR against cell-type-specific transcripts (Figure 4B). As quantification of the number of infected cells in 3D organoids is very challenging, we exploited previously established protocols to differentiate and infect human intestinal organoids in two dimensions with viruses (Ettayebi et al., 2016; Stanifer et al., 2020). Nondifferentiated organoids were seeded on human-collagen-coated iBIDI chambers. At 24 h post-seeding, differentiation was induced by removal of Wnt3a and reducing the amounts of R-spondin and Noggin for 4 days. Upon full differentiation, organoids were infected with SARS-CoV-2. At 24 hpi, infection was analyzed by immunostaining using the anti-N and anti-dsRNA antibodies and by qRT-PCR (Figure 4C). Results show that independent of the donor, colon organoids were readily infected by SARS-CoV-2, as noted by the presence of cells positive for both N and dsRNA (Figure 4D). Quantification revealed that ∼8%–13% of cells were infected in each donor (Figure 4E). This infection was associated with an increase of viral genome copy number (Figure 4F). Interestingly, infection of organoids led to no type I IFN (IFN-β1) production but an extremely large upregulation of type III IFN (IFN-λ) (Figures 4G, S2B, and S3B). To determine if exogenously added IFNs could prevent SARS-CoV-2 infection, colon organoids were mock-treated or pretreated with IFN-β1 and IFN-λ and then subsequently infected with SARS-CoV-2 for 24 h. Results show that pretreatment of colon organoids with both IFN-β1 and IFN-λ significantly impaired infection (Figure 4H). This was associated with a reduction of SARS-CoV-2 genome copy numbers (Figure 4I) and a decrease in infectious de novo virus particle production (Figure 4J). Altogether, these results show that human colon organoids can support SARS-CoV-2 infection, replication, and de novo infectious virus production and that the type III IFN response plays a critical role in controlling virus replication.
Figure 4.

Human Colon Organoids Support SARS-CoV-2 Infection, Replication, and De Novo Infectious Virus Production
(A) Bright-field and immunofluorescence images showing human colon organoids that express markers for enterocytes (E-cadherin [E-Cad]), goblet cells (mucin [MUC-2]), and enteroendocrine cells (synaptophysin [SYP]). Representative images are shown. Scale bars, 25 μm.
(B) Colon organoids were differentiated prior to infection with SARS-CoV-2. Differentiation was confirmed by qRT-PCR for stem cell markers (OLFM4), enterocytes (sucrase isomaltase [SI]), and goblet cells (MUC-2).
(C) Schematic describing method for infection of 2D colon organoids with SARS-CoV-2.
(D–G) Colon organoids were seeded in 2D and differentiated prior SARS-CoV-2 infection. 24 hpi, cells were analyzed for virus replication and immune response. (D) Infection was analyzed by indirect immunofluorescence of the viral N protein (red) and dsRNA (green). Nuclei were stained with DAPI (blue). Representative images are shown. Scale bars, 10 μm. (E) The number of SARS-CoV-2-positive cells were quantified in 10 fields for each donor. (F) RNA was harvested, and the change in the copy number of SARS-CoV-2 genome was evaluated by qRT-PCR. (G) Same as F, except the upregulation of IFN-β1 and IFN-λ was evaluated.
(H–J) Colon organoids were pretreated with IFNs 24 h prior to infection. (H) 24 hpi, virus infection was evaluated by indirect immunofluorescence for the viral N protein (red) and dsRNA (green). Nuclei were stained with DAPI (blue). Representative images are shown. The number of SARS-CoV-2-positive cells was quantified in 10 fields of view for each time point. Scale bars, 10 μm. (I) RNA was harvested, and qRT-PCR was used to evaluate the copy number of SARS-CoV-2 genome. (J) Supernatants were collected from infected organoids. The amount of de novo viruses present in the supernatants was determined using a TCID50 assay.
n = 3 biological replicates. Error bars indicate standard deviation.
Discussion
Increasing numbers of clinical reports detail that COVID-19 patients not only have severe respiratory symptoms but also show indications of gastrointestinal pathologies. To efficiently combat the pandemic, it is necessary to fully appreciate the complexity of the disease, and this requires that the virus life cycle and its interaction with the host are characterized for each site of infection (e.g., lung versus gut). Here, we report that hIECs, including primary non-transformed epithelial cells, fully support SARS-CoV-2 infection, replication, and de novo infectious virus production. Interestingly, infection of primary hIECs by SARS-CoV-2 induces a robust intrinsic immune response characterized by the production of type III IFNs, but not type I IFN. Pretreatment of hIECs with exogenous IFNs prior to SARS-CoV-2 infection results in a major drop of the number of infected cells, a reduction of viral replication, and a significant decrease in the production of infectious de novo virus particles. Concomitantly, inhibition of type III IFN signaling by genetic ablation of its specific receptor results in a significant increase of SARS-CoV-2 replication and secretion. Together, our results demonstrate that hIECs are a productive site of SARS-CoV-2 replication, and this may have critical implications for the etiology of both SARS-CoV-2 infection and the pathologies observed in COVID-19 patients. Additionally, our work highlights the central role of type III IFN in controlling SARS-CoV-2 at the intestinal epithelium.
Primary hIECs cells support robust replication of SARS-CoV-2 and secretion of infectious de novo virus particles. We found that ∼10% of the cells are infected in a human-colon-derived organoid, leading to a modest but significant increase in viral genome copy number (Figure 4F) and de novo infectious virus particles (Figure 4J) over time. This modest replication of SARS-CoV-2 is explained by the fact that (1) only a small fraction of the cells are infected by SARS-CoV-2, and (2) as shown in this work, IFNs are potent inhibitors of SARS-CoV-2 replication (Figures 2 and 3), and organoids are highly immunoresponsive upon viral infection (Stanifer et al., 2020) (Figure 4G). Our findings are in agreement with recent work from Lamers et al. showing that infection of human intestinal organoids by SARS-CoV-2 induces a strong immune response characterized by a strong expression of ISGs (Lamers et al., 2020). Interestingly, in this work, expression of type III IFN was shown to be very low. This is contrary to our data showing high production of type III IFN; however, we monitored IFN-λ2/3 levels, while the other study monitored IFN-λ1 levels. Further work is necessary to address if different subtypes of type III IFNs are induced by SARS-CoV-2. It is well known that in vivo intestinal epithelium cells are less immunoresponsive because of the gut microenvironment (e.g., microbiota and tissue-specific immune cells) and as such they will very likely show a severely dampened immune response allowing for an even greater SARS-CoV-2 replication.
Analysis of the single-cell RNA sequencing data from the Colon Atlas revealed that only 3.8% of human colon epithelial cells express very low levels of the SARS-CoV-2 receptor ACE-2 (Figures S4A–S4E). This is very different to the small intestine where ACE-2 appears to be more expressed (Qi et al., 2020). This low ACE-2 level could explain why we have a small percentage of infected cells in our colon organoids (Figure 4E). On the contrary, TMPRSS2 seems to be not a limiting factor in the colon (Figures S4C–S4E). Interestingly, q-RT-PCR and western blot analysis do not support single-cell analysis and clearly show that ACE-2 is expressed in both our carcinoma derived lines and our colon organoids (Figures S1C and S1D). The discrepancy between the single-cell RNA sequencing and classical molecular and biochemical approaches is likely the results of (1) the sequencing being not deep enough to detect ACE-2 in individual cells and (2) RNA expression not necessarily matching the protein expression levels. These observations highlight that although analyzing data from single-cell RNA sequencing atlases could be very informative, their findings should be validated in tissues, as they may be misleading or miss important sites of virus replication.
Human colon carcinoma Caco-2 cells produce very large amounts of infectious viral particles. This is higher than the titers obtained in Vero cells, which are commonly used to isolate and propagate SARS-CoV-2 (Harcourt et al., 2020) (Figures S1B and S1C). Similarly, the lung cell line Calu-3 produced much fewer infectious virus particles than Caco-2 cells (Figure S1C). Interestingly, in a study comparing 13 different human cell lines, Caco-2 cells were the only cell type found to support SARS-CoV-1 replication, and, compared to green monkey cells, Caco-2 cells were very efficient in producing infectious de novo virus particles and did not show cytopathic effects (Mossel et al., 2005). These observations that Caco-2 are excellent culture models supporting both SARS-CoV-2 and SARS-CoV-1 further highlight the potentially central role of intestinal epithelial cells in COVID-19 patients. Intriguingly, T84 cells, which are also colon-carcinoma-derived cells, supported SARS-CoV-2 infection, replication, and de novo infectious virus production but to a much lesser extent compared to Caco-2 cells. Analysis of the intrinsic immune response generated upon infection of both cell lines revealed that T84 cells are more immunoresponsive than Caco-2 cells. In light of the results obtained by pretreating cells with exogenous IFNs, we propose that T84 cells, by being able to mount a stronger and faster immune response than Caco-2 cells, can better restrict SARS-CoV-2 infection. This model is fully supported by our IFN receptor knockout T84 cells, in which virus replication and de novo virus production are drastically increased to levels similar to the ones observed in Caco-2 cells.
Exogenously added type I and III IFNs induce an antiviral state in our hIECs, thereby restricting SARS-CoV-2 replication in these cells. Similar observations were made with type I IFN in Vero cells (Mantlo et al., 2020). Interestingly, infection of Calu-3 human lung epithelial cells by SARS-CoV-2 also seems to mount an immune response (Lokugamage et al., 2020), whereas in A549 cells, in ferret models and human lung tissues, SARS-CoV-2 infection leads to the induction of a restricted and muted immune response lacking the induction of type I and type III IFNs (Blanco-Melo et al., 2020; Chu et al., 2020). The reason for the difference in the amount of IFN made upon infection of lung versus intestinal epithelial cells is currently unclear. Like intestinal epithelial cells (Pervolaraki et al., 2017, 2018; Pott et al., 2011), lung epithelial cells are normally highly immunoresponsive and make IFNs upon viral infection (Crotta et al., 2013; Ye et al., 2019). The lack of IFN in lung epithelial cells following SARS-CoV-2 infection might be specific for this virus in this tissue (Blanco-Melo et al., 2020), as SARS-CoV-1 induces IFN production in infected lung tissue (Chu et al., 2020). Importantly, our work shows that compared to the airway epithelium, the intestinal epithelium produces a typical antiviral response. This highlights that host-pathogen interaction should be considered in a tissue-specific manner, as different cellular responses and viral countermeasures might be established among the lung, gut, and other organs.
Interestingly, in human colon organoids, we observed that only type III IFN is made upon SARS-CoV-2 infection, although human intestinal organoids are capable of making both type I and III IFN upon enteric virus infection (Pervolaraki et al., 2018; Stanifer et al., 2020). The lack of type I induction could be due to a kinetic delay of type I IFN production compared to type III IFN but this will need to be carefully addressed in further experiments. Another possibility is that SARS-CoV-2 encodes a specific antagonist that counteracts the production of type I IFN only. However, further studies are necessary to prove this novel concept. Additionally, the observation that the knockout of the type III IFN receptor leads to a much greater increase of SARS-Cov-2 infection, replication, and de novo virus production compared to knockout of the type I receptor (Figures 3A–3D) further highlights the central role of type III IFN in SARS-CoV-2 infection of human intestinal epithelium cells.
We propose that the gut is an active site of replication for SARS-CoV-2, and this could account for the viremia observed in COVID-19 patients and the presence of large amounts of SARS-CoV-2 genomes in the feces. The origin of the replicating SARS-CoV-2 in the intestinal epithelium is still not clear. To date, only one paper reported the isolation of infectious virus from stool samples (Wang et al., 2020). Further characterization of the SARS-CoV-2 enteric life cycle is necessary to determine whether the viral infection observed in the gut is due to fecal/oral transmission or is a manifestation of virus spreading from the lung to the gut. In the context of the gut we foresee that at the onset of SARS-CoV-2 infection, hIECs will mount an antiviral response through the type III IFN signaling pathway. As immune cells participate in mounting an innate immune response, type I IFN will be secreted from these cells and will be able to act on intestinal epithelial cells further reinforcing their antiviral state against SARS-CoV-2. In respect to the severe pathologies observed in the lung, which are believed to be caused by a cytokine storm, the findings that lung epithelial cells mount a muted immune response upon SARS-CoV-2 infection suggests that the cytokines are coming from an alternative source. This source could be from local immune cells or the gastrointestinal mucosa given the large immune response generated by primary hIECs. As a matter of fact, many cytokines are released from epithelial cells toward the lamina propria (tissue side) (Stanifer et al., 2016) and will quickly enter the circulation to potentially fuel and promote the inflammation and pathology observed in the lung.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SARS CoV NP | Sino Biologicals | Cat#MM05 |
| Mouse monoclonal anti-dsRNA (J2) | Scions | Cat#10010200; RRID:AB_2651015 |
| Mouse monoclonal anti-E-cadherin | BD Transductions | Cat#610181 |
| Rabbit polyclonal anti-ACE2 | Abcam | Cat#ab15348 |
| Rabbit polyclonal anti-Mucin-2 | Santa Cruz Biotech | Cat# sc-15334; RRID:AB_2146667 |
| Mouse monoclonal anti-SYP | Santa Cruz Biotech | Cat#sc-17750; RRID:AB_628311 |
| Mouse polyclonal anti-beta actin | Sigma Aldrich | Cat#A5441 |
| Alexa Fluor Goat anti mouse 488 | Thermo Fischer Scientific | Cat#A-11001; RRID:AB_2534069 |
| Alexa Fluor Goat anti rabbit 488 | Thermo Fischer Scientific | Cat#A-11034; RRID:AB_2576217 |
| Alexa Fluor Goat anti mouse 568 | Thermo Fischer Scientific | Cat#A-21124; RRID:AB_2535766 |
| Alexa Fluor Goat anti rabbit 568 | Thermo Fischer Scientific | Cat#A-11011; RRID:AB_143157 |
| Anti-Mouse IgG IRDye CW800 | Li-Cor | Cat#926-32212; RRID:AB_621847 |
| Anti-rabbit IgG IRDye CW800 | Li-Cor | Cat#926-32213; RRID:AB_621848 |
| Anti-mouse IgG IRDye CW680 | Li-Cor | Cat#926-68072; RRID:AB_10953628 |
| Bacterial and Virus Strains | ||
| BavPat1/2020 | European Virology Archives | 026V-03883 |
| Biological Samples | ||
| Human colon resections | University Hospital Heidelberg | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Advanced DMEM/F12 | Thermo Fischer Scientific | Cat# 12634010 |
| HEPES | Thermo Fischer Scientific | Cat3 15630080 |
| Penicillin/Streptomycin | Thermo Fischer Scientific | Cat#15140122 |
| GlutaMAX | Thermo Fischer Scientific | Cat# 35050061 |
| EDTA | Sigma Aldrich | Car#E9884 |
| MatriGel. GFR, LDEV free | Corning | Cat#354230 |
| B27 | Thermo Fischer Scientific | Cat#17504-044 |
| N-acetyl-cysteine | Sigma Aldrich | Cat# A9165 |
| Recombinant mouse EGF | Thermo Fischer Scientific | Cat# PMG8043 |
| [Leu15]-Gastrin I | Sigma-Aldrich | Cat# G9145 |
| A83-01 | Tocris | Cat#2939 |
| Recombinant human IGF-1 | BioLegend | Cat#590904 |
| Recombinant human FGF basic | Peprotech | Cat#100-18B |
| Y-27632 | Caymann Chemicals | Cat#10005583 |
| Mouse recombinant noggin | Peprotech | Cat#250-38 |
| Human recombinant IFN-beta 1 | Biomol | Cat#86421 |
| Recombinant human IFNL1 | Peprotech | Cat#300-02L |
| Recombinant human IFNL2 | Peprotech | Cat#300-02K |
| Recombinant human IFNL3 | Biomol | Cat#179-ML-025 |
| Pyridone 6 | Calbiochem | Cat#420099-500 |
| Collagen from human placenta | Sigma Aldrich | Cat#C5533-5MG |
| 0.05% Trypsin-EDTA | Thermo Fischer Scientific | Cat#25300054 |
| iTaq Universal SYBR green Supermix | BioRad | Cat#1725120 |
| Parafolmaldehyde | Sigma Aldrich | Cat#158127 |
| Triton X-100 | Sigma Aldrich | Cat#X100 |
| DAPI | Sigma Aldrich | Cat#D9542 |
| Fetal Bovine Serum | Capricorn | Cat#FBS-11A |
| DMEM, high glucose | Thermo Fischer Scientific | Cat#11965092 |
| DMEM/F12 | Thermo Fischer Scientific | Cat#11320033 |
| Draq5 | Abcam | Cat#ab108410 |
| Beta propiolactone | Sigma Aldrich | Cat#457604 |
| Critical Commercial Assays | ||
| RNAeasy RNA extraction kit | QIAGEN | Cat#74104 |
| iSCRIPT cDNA synthesis kit | BioRad | Cat#1708890 |
| DIY IFNL2/3 ELISA | PBL Interferon source | Cat#61830-1 |
| Deposited Data | ||
| Single-cell RNA sequencing data of human colon samples. | Smillie et al., 2019 | Single Cell Portal, accession number SCP259 (https://singlecell.broadinstitute.org/single_cell/study/SCP259/intra-and-inter-cellular-rewiring-of-the-human-colon-during-ulcerative-colitis) |
| Experimental Models: Cell Lines | ||
| T84 human colon carcinoma cells | ATCC | CCL-248 |
| T84 IFNLR−/− | Pervolaraki et al., 2018 | N/A |
| T84 IFNAR−/− | Pervolaraki et al., 2018 | N/A |
| T84 IFNLR/IFNAR−/− dKO | Pervolaraki et al., 2018 | N/A |
| Caco-2 human colorectal adenocarcinoma | ATCC | HTB-37 |
| Vero E6 | ATCC | CRL 1586 |
| Human colon organoids | This paper | N/A |
| L-WRN | ATCC | CRL-3276 |
| Oligonucleotides | ||
| TBP for ccactcacagactctcacaac | Eurofins | N/A |
| TBP rev ccactcacagactctcacaac | Eurofins | N/A |
| HPRT1 for cctggcgtcgtgattagtgat | Eurofins | N/A |
| HPRT1 rev agacgttcagtcctgtccataa | Eurofins | N/A |
| IFNL2/3 for gccacatagcccagttcaag | Eurofins | N/A |
| IFNL2/3 rev tgggagaggatatggtgcag | Eurofins | N/A |
| IFNb1 for gccgcattgaccatctat | Eurofins | N/A |
| IFNb1 rev gtctcattccagccagtg | Eurofins | N/A |
| OLFM4 for acctttcccgtggacagagt | Eurofins | N/A |
| OLFM4 rev tggacatattccctcactttgga | Eurofins | N/A |
| SI for aatccttttggcatccagatt | Eurofins | N/A |
| SI rev gcagccaagaatcccaaat | Eurofins | N/A |
| MUC-2 for tgtaggcatcgctcttctca | Eurofins | N/A |
| MUC-2 rev gacaccatctacctcacccg | Eurofins | N/A |
| ACE-2 for tcaaggaggccgagaagttc | Eurofins | N/A |
| ACE-2 rev ttcctgggtccgttagcatg | Eurofins | N/A |
| TMPRSS2 for acctgatcacaccagccatg/ | Eurofins | N/A |
| TMPRSS2 rev cttcgaagtgaccagaggcc | Eurofins | N/A |
| COV1 for gcctcttctgttcctcatcac | Eurofins | N/A |
| COV1 rev agacagcatcaccgccattg | Eurofins | N/A |
| Software and Algorithms | ||
| R (version 3.6.1) | Comprehensive R Archive Network (CRAN) | https://cran.r-project.org/ |
| Seurat R package (version 3.1.4) | R package | https://satijalab.org/seurat/get_started.html (RRID:SCR_016341) |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Megan L. Stanifer (m.stanifer@dkfz.de)
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code. No custom code was used to analyze these data and all methods and packages used are cited in the methods section.
Experimental Model and Subject Details
Cells
T84 human colon carcinoma cells (ATCC CCL-248) and their knock-out clones were maintained in a 50:50 mixture of Dulbecco’s modified Eagle’s medium (DMEM) and F12 (GibCo) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (GIBCO). Caco-2 human colorectal adenocarcinoma (ATCC HTB-37) and Vero E6 (ATCC CRL 1586) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (GibCo) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (GIBCO).
Viruses
SARS-CoV-2 (strain BavPat1) was obtained from Prof. Christian Drosten at the Charité in Berlin, Germany and provided via the European Virology Archive. The virus was amplified in Vero E6 cells.
Human organoid cultures
Human tissue was received from colon resection from the University Hospital Heidelberg. This study was carried out in accordance with the recommendations of the University Hospital Heidelberg with informed written consent from all subjects in accordance with the Declaration of Helsinki. All samples were received and maintained in an anonymized manner. The protocol was approved by the “Ethics commission of the University Hospital Heidelberg” under the protocol S-443/2017. Stem cells containing crypts were isolated following 2 mM EDTA dissociation of tissue sample for 1 h at 4°C. Crypts were spun and washed in ice cold PBS. Fractions enriched in crypts were filtered with 70 μM filters and the fractions were observed under a light microscope. Fractions containing the highest number of crypts were pooled and spun again. The supernatant was removed, and crypts were re-suspended in Matrigel. Crypts were passaged and maintained in basal and differentiation culture media (see Table S1).
Method Details
Antibodies and Inhibitors
Mouse monoclonal antibody against SARS-CoV NP (Sino biologicals MM05), mouse monoclonal against J2 (scions), mouse monoclonal against E-cadherin (BD Transductions #610181), mouse monoclonal anti-SYP (Santa Cruz Biotechnology sc-17750), and rabbit polyclonal anti-Mucin-2 (Santa Cruz Biotechnology# sc-15334) were used at 1:500 for immunofluorescence. Rabbit polyclonal ACE-2 antibody (Abcam) was used at 1:500 and mouse polyclonal actin was used at 1:5000 for western blot. Secondary antibodies were conjugated with AF488 (Molecular Probes), AF568 (Molecular Probes), CW680 (Li-Cor) or CW800 (Li-Cor) directed against the animal source. Human recombinant IFN-beta1a (IFNβ) was obtained from Biomol (#86421) was used at a final concentration of 2,000IU/mL. Recombinant human IFNλ 1 (IL-29) (#300-02L), IFNλ 2 (IL28A) (#300-2K)) were purchased from Peprotech and and IFNλ 3 (IL-28B) was purchased from BioMol. All three lambda interferons were combined and were used at a concentration of 100ng/mL each to make a final concentration of 300ng/mL, Pyridone 6 (Calbiochem #420099-500)was used at a final concentration of 2uM.
Viral infections
All SARS-CoV-2 infections were performed the MOI indicated in the text. Media was removed from cells and virus was added to cells for 1 hour at 37°C. Virus was removed, cells were wash 1x with PBS and media or media containing inhibitors/cytokines was added back to the cells.
2D organoid seeding
8-well iBIDI glass bottom chambers were coated with 2.5% human collagen in water for 1 h prior to organoids seeding. Organoids were collected at a ratio of 100 organoids/transwell. Collected organoids were spun at 450 g for 5 mins and the supernatant was removed. Organoids were washed 1X with cold PBS and spun at 450 g for 5 mins. PBS was removed and organoids were digested with 0.05% Trypsin-EDTA (Life technologies) for 5 mins at 37°C. Digestion was stopped by addition of serum containing medium. Organoids were spun at 450 g for 5 mins and the supernatant was removed and organoids were re-suspended in normal growth media at a ratio of 250 μL media/well. The collagen mixture was removed from the iBIDI chambers and 250 μL of organoids were added to each well.
RNA isolation, cDNA, and qPCR
RNA was harvested from cells using RNAeasy RNA extraction kit (QIAGEN) as per manufactures instructions. cDNA was made using iSCRIPT reverse transcriptase (BioRad) from 250 ng of total RNA as per manufactures instructions. q-RT-PCR was performed using iTaq SYBR green (BioRad) as per manufacturer’s instructions, TBP or HPRT1 were used as normalizing genes. Primer used:
Indirect Immunofluorescence Assay
Cells seeded on iBIDI glass bottom 8-well chamber slides. At indicated times post-infection, cells were fixed in 4% paraformaldehyde (PFA) for 20 mins at room temperature (RT). Cells were washed and permeabilized in 0.5% Triton-X for 15 mins at RT. Primary antibodies were diluted in phosphate-buffered saline (PBS) and incubated for 1h at RT. Cells were washed in 1X PBS three times and incubated with secondary antibodies and DAPI for 45 mins at RT. Cells were washed in 1X PBS three times and maintained in PBS. Cells were imaged by epifluorescence on a Nikon Eclipse Ti-S (Nikon).
In-cell Western
20,000 Vero E6 cells were seeded per well into a 96-well dish 24h prior to infection. 100uL of harvested supernatant was added to the first well. Seven 1:10 dilutions were made (all samples were performed in triplicate). Infections were allowed to proceed for 24h. 24h post-infection cells were fixed in 2% PFA for 20mins at RT. PFA was removed and cells were washed twice in 1X PBS and then permeabilized for 10mins at RT in 0.5% Triton-X. Cells were blocked in a 1:2 dilution of Li-Cor blocking buffer (Li-Cor) for 30mins at RT. Cells were stained with 1/1000 dilution anti-dsRNA (J2) for 1h at RT. Cells were washed three times with 0.1% Tween in PBS. Secondary antibody (anti-mouse CW800) and DNA dye Draq5 (Abcam) were diluted 1/10,000 in blocking buffer and incubated for 1h at RT. Cells were washed three times with 0.1% Tween/PBS. Cells were imaged in 1X PBS on a LICOR (Li-Cor) imager.
Western blot
At time of harvest, media was removed, cells were rinsed once with 1X PBS and lysed with 1X RIPA (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris, pH 8.0 with phosphatase and protease inhibitors (Sigma-Aldrich)) for 5 mins at room temperature (RT). Lysates were collected and equal protein amounts were separated by SDS-PAGE and blotted onto a nitrocellulose membrane by wet-blotting (Bio-Rad). Membranes were blocked with Li-Cor blocking buffer (Li-Cor) for one hour at RT. Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Membranes were washed 3X in TBS-T for 5 mins at RT. Secondary antibodies were diluted in blocking buffer and incubated at RT for 1 hour with rocking. Membranes were washed 3X in TBS-T for 5 mins at RT and scanned on a Li-Cor scanner.
ELISA
Supernatants were collected at indicated time points. Virus in supernatants was inactivated with 0,05% beta propiolactone (BPL) (Sigma-Aldrich) overnight at 4°C. BPL was hydrolyzed at 37°C for 2 hours prior to ELISA. ELISA standards were also diluted in 0.05% BPL overnight following 2h hydrolyzation at 37°C. IFNλ2/3 was evaluated using the IFNλ2/3 DIY ELISA (PBL Interferon source) as per manufacturer’s instructions.
Re-processing of the single-cell RNaseq Human Colon Atlas
The data from the single-cell transcriptome human colon atlas (Smillie et al., 2019) was obtained from the broad institute Single Cell Portal accession number SCP259 (https://singlecell.broadinstitute.org/single_cell/study/SCP259/intra-and-inter-cellular-rewiring-of-the-human-colon-during-ulcerative-colitis). Expression matrices of the epithelial subset of 30 samples were imported and individually analyzed using the Seurat software, version 3.1.4 (https://github.com/satijalab/seurat). Quality filtering was performed, and cells having fewer than 500 genes and more than 30% of UMI count mapped to mitochondrial genes were discarded. Consecutively, the resulting datasets were normalized, scaled and high-variance genes genes were selected. Reciprocal PCA-based data integration was done to merge the samples. Afterward, the resulting batch-corrected counts were used for calculating PCA-based dimensionality reduction and unsupervised Louvain clustering. Furthermore, UMAP visualization was calculated using 50 neighboring points for the local approximation of the manifold structure. Cell type annotation was based on the unsupervised clustering and the metadata provided by the colon atlas (Smillie et al., 2019).
Quantification and Statistical Analysis
Unless otherwise stated, P values were calculated by a two-tailed unpaired t test using the GraphPad Prism software package. All samples were analyzed without blinding or exclusion of samples. N values indicate the number of biological replicates for a sample.
Acknowledgments
This work was supported by a research grant from the Chica and Heinz Schaller Foundation and Deutsche Forschungsgemeinschaft (DFG) project numbers 240245660 (SFB1129), 278001972 (TRR186), 415089553 (Heisenberg), and 272983813 (TRR179) (S.B.) and DFG project number 416072091 (M.L.S.). S.T. was supported by the Darwin Trust of Edinburgh. Work of R.B. was supported by DFG project numbers 272983813 (TRR179), 240245660 (SFB1129), and BA1505/8-1. T.A. is supported by the European Research Council (grant agreement 773089). We would like to acknowledge Vibor Laketa and the Infectious Diseases Imaging Platform (IDIP) in Heidelberg, Germany for support with image acquisition and analysis. We also thank Christian Drosten at the Charité, Berlin and the European Virus Archive (EVAg) for the provision of the SARS-CoV-2 strain BavPat1.
Author Contributions
M.L.S. conceived and carried out experiments, interpreted the results, and wrote the manuscript. C.K., M.M., and C.M.Z. performed experiments. S.T. analyzed the single-cell data. M.C., H.G.K., T.A., and R.B. contributed to the concept of the study and critical discussions. S.B. conceived experiments, interpreted results, and wrote the manuscript. The final version of the manuscript was approved by all authors.
Declaration of Interests
The authors declare no competing interests.
Published: June 19, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107863.
Contributor Information
Megan L. Stanifer, Email: m.stanifer@dkfz.de.
Steeve Boulant, Email: s.boulant@dkfz.de.
Supplemental Information
References
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Møller R., Panis M., Sachs D., Albrecht R.A., tenOever B.R. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. bioRxiv. 2020 doi: 10.1101/2020.03.24.004655. [DOI] [Google Scholar]
- Chu H., Chan J.F.-W., Wang Y., Yuen T.T.-T., Chai Y., Hou Y., Shuai H., Yang D., Hu B., Huang X. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa410. Published online April 9, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotta S., Davidson S., Mahlakoiv T., Desmet C.J., Buckwalter M.R., Albert M.L., Staeheli P., Wack A. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013;9:e1003773. doi: 10.1371/journal.ppat.1003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettayebi K., Crawford S.E., Murakami K., Broughman J.R., Karandikar U., Tenge V.R., Neill F.H., Blutt S.E., Zeng X.-L., Qu L. Replication of human noroviruses in stem cell-derived human enteroids. Science. 2016;353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harak C., Lohmann V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology. 2015;479-480:418–433. doi: 10.1016/j.virol.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt J., Tamin A., Lu X., Kamili S., Sakthivel S., Kumar, Murray J., Queen K., Tao Y., Paden C.R., Zhang J. Isolation and characterization of SARS-CoV-2 from the first US COVID-19 patient. bioRxiv. 2020 doi: 10.1101/2020.03.02.972935. [DOI] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers M.M., Beumer J., van der Vaart J., Knoops K., Puschhof J., Breugem T.I., Ravelli R.B.G., Paul van Schayck J., Mykytyn A.Z., Duimel H.Q. SARS-CoV-2 productively infects human gut enterocytes. Science. 2020 doi: 10.1126/science.abc1669. Published online May 1, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung W.K., To K.F., Chan P.K.S., Chan H.L.Y., Wu A.K.L., Lee N., Yuen K.Y., Sung J.J.Y. Enteric involvement of severe acute respiratory syndrome-associated coronavirus infection. Gastroenterology. 2003;125:1011–1017. doi: 10.1016/j.gastro.2003.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage K.G., Hage A., Schindewolf C., Rajsbaum R., Menachery V.D. SARS-CoV-2 sensitive to type I interferon pretreatment. bioRxiv. 2020 doi: 10.1128/JVI.01410-20. 2020.03.07.982264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Stratton C.W., Tang Y.-W. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J. Med. Virol. 2020;92:401–402. doi: 10.1002/jmv.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukassen S., Lorenz Chua R., Trefzer T., Kahn N.C., Schneider M.A., Muley T., Winter H., Meister M., Veith C., Boots A.W. ). SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020;39:e105114. doi: 10.15252/embj.20105114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantlo E., Bukreyeva N., Maruyama J., Paessler S., Huang C. Potent antiviral activities of type I interferons to SARS-CoV-2 infection. bioRxiv. 2020 doi: 10.1016/j.antiviral.2020.104811. 2020.04.02.022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M., Hugentobler W.J., Iwasaki A. Seasonality of respiratory viral infections. Annu. Rev. Virol. 2020 doi: 10.1146/annurev-virology-012420-022445. Published online March 20, 2020. [DOI] [PubMed] [Google Scholar]
- Mossel E.C., Huang C., Narayanan K., Makino S., Tesh R.B., Peters C.J. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. J. Virol. 2005;79:3846–3850. doi: 10.1128/JVI.79.6.3846-3850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paules C.I., Marston H.D., Fauci A.S. Coronavirus infections: more than just the common cold. JAMA. 2020;323:707–708. doi: 10.1001/jama.2020.0757. [DOI] [PubMed] [Google Scholar]
- Pervolaraki K., Stanifer M.L., Münchau S., Renn L.A., Albrecht D., Kurzhals S., Senís E., Grimm D., Schröder-Braunstein J., Rabin R.L., Boulant S. Type I and type III interferons display different dependency on mitogen-activated protein kinases to mount an antiviral state in the human gut. Front. Immunol. 2017;8:459. doi: 10.3389/fimmu.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervolaraki K., Rastgou Talemi S., Albrecht D., Bormann F., Bamford C., Mendoza J.L., Garcia K.C., McLauchlan J., Höfer T., Stanifer M.L., Boulant S. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 2018;14:e1007420. doi: 10.1371/journal.ppat.1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott J., Mahlakõiv T., Mordstein M., Duerr C.U., Michiels T., Stockinger S., Staeheli P., Hornef M.W. IFN-λ determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA. 2011;108:7944–7949. doi: 10.1073/pnas.1100552108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F., Qian S., Zhang S., Zhang Z. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem. Biophys. Res. Commun. 2020;526:135–140. doi: 10.1016/j.bbrc.2020.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie C.S., Biton M., Ordovas-Montanes J., Sullivan K.M., Burgin G., Graham D.B., Herbst R.H., Rogel N., Slyper M., Waldman J. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell. 2019;178:714–730.e22. doi: 10.1016/j.cell.2019.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanifer M.L., Rippert A., Kazakov A., Willemsen J., Bucher D., Bender S., Bartenschlager R., Binder M., Boulant S. Reovirus intermediate subviral particles constitute a strategy to infect intestinal epithelial cells by exploiting TGF-β dependent pro-survival signaling. Cell. Microbiol. 2016;18:1831–1845. doi: 10.1111/cmi.12626. [DOI] [PubMed] [Google Scholar]
- Stanifer M.L., Mukenhirn M., Muenchau S., Pervolaraki K., Kanaya T., Albrecht D., Odendall C., Hielscher T., Haucke V., Kagan J.C. Asymmetric distribution of TLR3 leads to a polarized immune response in human intestinal epithelial cells. Nat. Microbiol. 2020;5:181–191. doi: 10.1038/s41564-019-0594-3. [DOI] [PubMed] [Google Scholar]
- Targett-Adams P., Boulant S., McLauchlan J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008;82:2182–2195. doi: 10.1128/JVI.01565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan A.J., Puranik A., Anand A., Zemmour D., Yao X., Wu X., Chilaka R., Murakowski D.K., Standish K., Raghunathan B. Knowledge synthesis from 100 million biomedical documents augments the deep expression profiling of coronavirus receptors. BioRxiv. 2020 doi: 10.7554/eLife.58040. 2020.03.24.005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Zhang Y. Animal coronaviruses: a brief introduction. In: Wang L., editor. Animal Coronaviruses. Springer; 2016. pp. 3–11. [Google Scholar]
- Wang Q., Vlasova A.N., Kenney S.P., Saif L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019;34:39–49. doi: 10.1016/j.coviro.2018.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Xu Y., Gao R., Lu R., Han K., Wu G., Tan W. Detection of SARS-CoV-2 in Different Types of Clinical Specimens. JAMA. 2020 doi: 10.1001/jama.2020.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Müller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- Wong S.H., Lui R.N., Sung J.J. Covid-19 and the digestive system . J. Gastroenterol. Hepatol. 2020 doi: 10.1111/jgh.15047. Published online March 25, 2020. [DOI] [PubMed] [Google Scholar]
- Wu C., Zheng S., Chen Y., Zheng M. Single-cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCoV, in the nasal tissue. bioRxiv. 2020 doi: 10.1101/2020.01.26.919985. [DOI] [Google Scholar]
- Wu Y., Guo C., Tang L., Hong Z., Zhou J., Dong X., Yin H., Xiao Q., Tang Y., Qu X. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol. Hepatol. 2020 doi: 10.1016/S2468-1253(20)30083-2. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao F., Tang M., Zheng X., Liu Y., Li X., Shan H. Evidence for gastrointestinal infection of SARS-CoV-2. Gastroenterology. 2020;158:1831–1833.e3. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y.-H., Ni W., Wu Q., Li W.-J., Li G.-J., Wang W.-D., Tong J.-N., Song X.-F., Wing-Kin Wong G., Xing Q.-S. Prolonged viral shedding in feces of pediatric patients with coronavirus disease 2019. J. Microbiol. Immunol. Infect. 2020;53:473–480. doi: 10.1016/j.jmii.2020.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Zhong L., Deng J., Peng J., Dan H., Zeng X., Li T., Chen Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020;12:8. doi: 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Li X., Zhu B., Liang H., Fang C., Gong Y., Guo Q., Sun X., Zhao D., Shen J. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat. Med. 2020;26:502–505. doi: 10.1038/s41591-020-0817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L., Schnepf D., Becker J., Ebert K., Tanriver Y., Bernasconi V., Gad H.H., Hartmann R., Lycke N., Staeheli P. Interferon-λ enhances adaptive mucosal immunity by boosting release of thymic stromal lymphopoietin. Nat. Immunol. 2019;20:593–601. doi: 10.1038/s41590-019-0345-x. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Zhao Z., Wang Y., Zhou Y., Ma Y., Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. bioRxiv. 2020 doi: 10.1101/2020.01.26.919985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Li C., Zhao G., Chu H., Wang D., Yan H.H.-N., Poon V.K.-M., Wen L., Wong B.H.-Y., Zhao X. Human intestinal tract serves as an alternative infection route for Middle East respiratory syndrome coronavirus. Sci. Adv. 2017;3:eaao4966. doi: 10.1126/sciadv.aao4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code. No custom code was used to analyze these data and all methods and packages used are cited in the methods section.