

Abstract
Objectives
VRE are listed, by the WHO, among the leading resistant pathogens causing greatest public concern; hence the spread and transmission of VRE, especially in hospitalized patients, need to be monitored. Despite the advancements in typing methods since the implementation of WGS for outbreak investigations, data interpretation, especially for vancomycin-resistant Enterococcus faecium (VREfm) in an endemic setting, remains challenging. In this study we explored the potential added benefit of incorporating patient movement data and admission screening to accurately estimate the magnitude of an outbreak.
Methods
We sequenced 73 VREfm isolates from patients with bacteraemia (n = 43) and rectal colonization (n = 30/32). Genetic relatedness was determined by SNP distance (≤10) between isolates. Patient movements were visualized in a movement network, along with contact intensity and rectal colonization status prior to infection onset.
Results
ST117, ST80 and ST203 were the predominant STs in our study population. Forty-four percent (18/41) of VREfm bacteraemia cases were of endogenous origin. SNP analysis of infection and colonization isolates revealed nine clonal groups. Eighty-six percent (37/43) of the patients were visualized in a transmission network due to spatiotemporal overlap. Nineteen out of 43 (44%) belonged to five transmission clusters. Incorporation of prior colonization status revealed that transmission was very likely in only 63% (12/19) of patients in these transmission clusters.
Discussion
Although interpretation of WGS data is challenging, incorporation of patient movement data and colonization status by admission screening of high-risk patients may provide additional resolution when interpreting the magnitude of an outbreak in an endemic setting.
Introduction
Enterococcus species are considered one of the leading causes of nosocomial infections in Germany.1 Since their emergence in the mid-1980s in Europe, VRE have become prevalent across Europe and are considered as being major resistant pathogens of public concern.2–4 Moreover, due to multiple intrinsic resistances, especially in Enterococcus faecium, therapeutic options are limited and vancomycin resistance has been linked to increased mortality in patients with VRE bloodstream infections.5,6
Analysis of the prevalence of VRE infections in Germany from 2007 to 2012 revealed a significant increase in VRE surgical site infections and bloodstream infections over those years. This increase is attributable to a high VRE prevalence in four federal states (North Rhine-Westphalia, Hesse, Thuringia and Saxony) in Germany, often referred to as the German ‘VRE belt’.7 The highly clonal population structure of VRE in our region complicates and hampers the analysis and interpretation of molecular typing to investigate patient-to-patient transmission.8
Despite preventive measures, such as admission screening for MDR organisms, and containment measures, by means of isolation, our internal routine surveillance data revealed a significant increase in incidence of VRE bloodstream infections in our hospital over recent years (S. Klein and D. Nurjadi, unpublished data). Therefore, in this study we aimed to investigate whether the abundance of VRE bacteraemia was due to clonal expansion or transmission within an inpatient setting. To this end, we performed WGS on all vancomycin-resistant E. faecium (VREfm) to investigate the clonal relationship of VREfm bacteraemia. Moreover, due to the difficulties in interpretation of WGS data in a VRE endemic area, we wanted to assess the added benefit of combining high-resolution molecular typing with patient movement data and rectal admission screening for VRE. Better integration of typing, epidemiological information and screening of patients at risk may help optimize infection control policy and measures.
Methods
A total of 73 VREfm isolates (43 blood culture and 30 rectal colonization isolates) from 43 patients with VRE bacteraemia between 1 January and 31 December 2016 in our tertiary care hospital were sequenced and analysed for this study. Anonymized baseline characteristics, clinical data and movement data were extracted from routine clinical data. Rectal screening for MDR organisms such as VRE and Gram-negative bacilli was performed routinely for high-risk patients at admission, and weekly in ICUs, as described previously.9
Local infection control policy for VRE
During the study period, VRE-positive patients in intensive and intermediate units, transplant and haemodialysis units and haemato-oncological and paediatric wards, as well as VRE-positive patients with diarrhoea and open wounds, were isolated as an infection prevention measure. Contact isolation precautions consisted of either single-room isolation or organizational bed isolation with a private toilet. Gloves and patient-assigned protective coats were applied for all situations with a risk of (self-) contamination.
Detection of VRE in rectal swabs and blood cultures
The detection of VRE in rectal screening samples and blood culture flasks was performed as part of routine microbiological diagnostic practice. Briefly, rectal swabs were inoculated onto selective agar for VRE (VRE Select, Bio-Rad, Germany) and Columbia blood agar with 5% sheep’s blood (Becton Dickinson, Germany) to check for sampling validity. Species identification was performed using MALDI-TOF MS (Bruker GmbH, Germany). Validation of the first detection of a VRE isolate was performed using PCR, as described previously.10 All VRE isolates were cryopreserved until analysis.
DNA extraction and WGS
Genomic DNA was extracted from an overnight bacterial culture using the DNeasy Blood and Tissue Mini Kit (QIAGEN GmbH, Germany) according to the manufacturer’s protocol. A standard genomic library was prepared from the DNA using the Illumina DNA Flex Kit (Illumina) and sequenced using the Illumina MiSeq platform (2 × 300 bp, paired end). Quality control and assembly were performed as described previously.11
Briefly, raw sequences were trimmed for quality using Sickle 1.33 (parameters: q > 30 and l > 45).12 The cleaned sequences were then assembled using SPAdes 3.13.0 using the option --careful and --only-assembler.13 Contigs obtained from the assembly were curated for length (>1000 bp) and coverage (>10×) to minimize errors and contamination in the draft genome. Annotation was performed using Prokka 1.14.1 (based on Genetic Code Table 11).14 Assembled genome data are available from the NCBI GenBank under the BioProject number PRJNA604888. Raw read data are available upon request. Sequencing statistics are summarized in Table S1 (available as Supplementary data at JAC Online).
Cluster analysis by core-genome MLST (cgMLST)
Classical MLST and cgMLST based on 1779 genes were performed using SeqSphere+ software version 5.0 (Ridom, Münster, Germany) using a standardized cgMLST database for E. faecium.15 For comparisons, core genomes were calculated with Roary using all our isolates in a de novo approach and only genes present in all isolates were considered (1941 genes). Phylogenetic distance was calculated with Gubbins 2.3.4 to take into account recombination events and not overestimate SNPs.16 Gubbins phylogenetic analysis was able to reduce the number of polymorphic sites from 5406 to 1390 sites. SNP profiles identified by Gubbins were used as templates in Phyloviz 2.0 and clonal groups were defined as genomes that were ≤10 SNPs distant in a minimum spanning tree calculated with the eBurst algorithm.17
Cluster/patient movement network
Patient movements were analysed in R 3.6.1 to extract the spatiotemporal overlap between patients using the package Desctools. The resulting matrix was used to perform network analysis using Cytoscape 3.7.1 to represent the spatiotemporal overlap as a connecting edge.
Data analysis
Descriptive statistics were analysed using STATA 13 (StataCorp, USA).
Ethics
Molecular characterization was performed on VRE isolates as part of surveillance and infection control measures at the Heidelberg University Hospital in accordance with the German Infection Protection Act. The Ethics Review Board was consulted regarding the study protocol to ensure conformity to current laws and regulations (S-474/2018).
Results
Between 1 January and 31 December 2016, we identified 43 inpatients with VREfm bacteraemia. There were no vancomycin-resistant Enterococcus faecalis in our VRE blood culture collection. Seventy-four percent (n = 32/43) of these patients screened positive in their rectal swabs for VREfm and 26% (n = 11/43) screened negative. Two-thirds (n = 29/43; 67%) of patients with VREfm bacteraemia were male. The mean age for our study participants was 60.4 years (SD = 13.5 years) with an age range of 20–81 years. All 43 VREfm isolates from blood cultures and 30 of 32 VREfm (2 isolates not recoverable) from rectal swabs were sequenced and analysed for genetic identity.
Molecular background of VRE bacteraemia
The majority of invasive VRE isolates belonged to ST117 (n = 32/43; 74%). Other clonal groups were ST203 (6/43; 14%), ST80 (3/43; 7%), ST17 (1/43; 2%) and ST192 (1/43; 2%). The ST117 clonal group consisted of 12 cgMLST cluster types (CTs; CT17, CT36, CT469, CT17, CT71, CT2558, CT2559, CT2562, CT2565, CT2566, CT2567 and CT3573). The majority of ST117 VREfm in our study population belonged to CT469. All ST203 isolates belonged to CT2561. ST80 isolates belonged to three CTs (CT2560, CT2564 and CT2569). vanB was the predominant resistance mechanism (n = 36/43; 84%) and vanA only accounted for 16% (n = 7/43) of vancomycin resistance in invasive E. faecium in our study population, being associated with the ST203 clonal type (n = 6/6; 100% harboured vanA). The presence and absence of virulence genes associated with biofilm formation (e.g. ecbA) and adhesion genes (e.g. fss3 and scm), as well as resistance genes present in invasive VRE, are displayed in Figure 1.
Figure 1.

Phylogenetic tree of VREfm causing bacteraemia, Heidelberg 2016. The phylogenetic tree was rooted at midpoint. Black squares indicate the presence of virulence and resistance genes, whereas white squares indicate their absence. Red isolate identifiers indicate that both blood culture and rectal isolates were identical (≤10 SNPs). SCGs were defined using an SNP threshold of ≤10 (minimum spanning tree; see Figures S2 and S3).
Several genes conferring resistance to various antibiotics besides vancomycin were detected in our isolate collection. We did not observe any significant associations between clonal groups/clusters and the presence of particular resistance genes. All isolates harboured the aac(6′) gene but only 14% (6/43) harboured the aph(2′′)-Ia gene, conferring high-level gentamicin resistance. The aminoglycoside genes ant(6)-Ia, sat4, aph(2′′)-Ia and aph(3′)-IIIa were absent in all ST203 VREfm isolates. All VREfm isolates in our study harboured the efflux pump transporter genes eat(A) and efmA (Figure 1).
Rectal colonization with VREfm
To better understand the origin and the dynamics of VREfm transmission, rectal and infection isolates were analysed together. Altogether, 32 patients screened positive for VRE colonization in the rectal swabs. The genetic distance was calculated as SNP distance between isolates, as displayed in the phylogenetic tree in Figure 1. Since 2 rectal isolates were not re-culturable, only 30 rectal isolates (out of 32 colonized patients) could be incorporated into the analysis. Sixty percent (18/30) of colonized patients were infected and colonized with identical VREfm strains, i.e. had endogenous infection. The SNP distances between rectal and colonization isolates are displayed in Figure S1. In 18 out of 30 patients (60%), rectal colonization was detected on admission or prior to the onset of VRE bacteraemia.
Putative transmission clusters and patient movement data
SNP distribution within the same MLST clonal complex indicated a threshold of 10 would be reasonable (Figure S2). Using a threshold of 10 SNPs between all sequenced VREfm isolates (n = 73), we could identify nine clonal groups and 14 singletons (groups with one isolate) (Figures 1 and S3). Eight of the SNP-derived clonal groups (SCGs; SCG-1, SCG-2, SCG-3, SCG-4, SCG-5, SCG-6, SCG-7 and SCG-9) belonged to ST117 and isolates in SCG-8 belonged to ST203. SCG-3 and SCG-4 actually consisted of only two isolates (blood culture and rectal) from the same patient in each group. Other SCGs (SCG-1, SCG-2, SCG-5, SCG-6, SCG-7 and SCG-9) were considered as potential transmission clusters (Figure 1). Overall, there was high concordance between the cgMLST and SCG clusters. However, for some highly clonal populations such as ST117/CT71 and ST117/CT469, SCG could deliver a higher resolution, as indicated in Figure 1 (two SCGs belonging to each of ST117/CT71 and ST117/CT469).
Visualization of patient movement data revealed possible epidemiological links between patients, as indicated by the bold connecting lines between nodes and cluster groups. The width of connecting lines corresponds to the duration of contact time between patients. Only 86% (37/43) of the patients shared temporal and spatial overlap, as displayed in Figure 2. Six patients had no spatiotemporal overlap with other patients and so were not included in the network visualization. Nodes with left- and right-hand semicircles of the same colour indicate endogenous infections as the pairwise distance between the blood and rectal isolates was ≤10 SNPs and colonization was detected prior to bacteraemia onset. The movement network suggests five possible transmission networks (TNs), according to epidemiological and SCG overlap. The first transmission cluster, TN1, consisted of P01 and P44 (SCG-1 ST117/CT2565) with three SNPs between the two isolates. The second cluster, TN2, consisting of isolates from SCG-6 (ST117/CT469), was an interconnection of six patients: Patient (P) 08, P17, P19, P25, P41 and P56. The origin of bacteraemia for P08, P17, P19 and P56 was most likely endogenous, as supported by detection of rectal colonization prior to infection onset, whereas P25 and P41 might have acquired the strain from other patients in TN2. Most patients (four out of six patients) in TN2 most likely acquired their colonization with ST117/CT469 VREfm prior to admission, suggesting a low diversity or close clonal relationship in circulating ST117, the predominant clonal group in our region.
Figure 2.
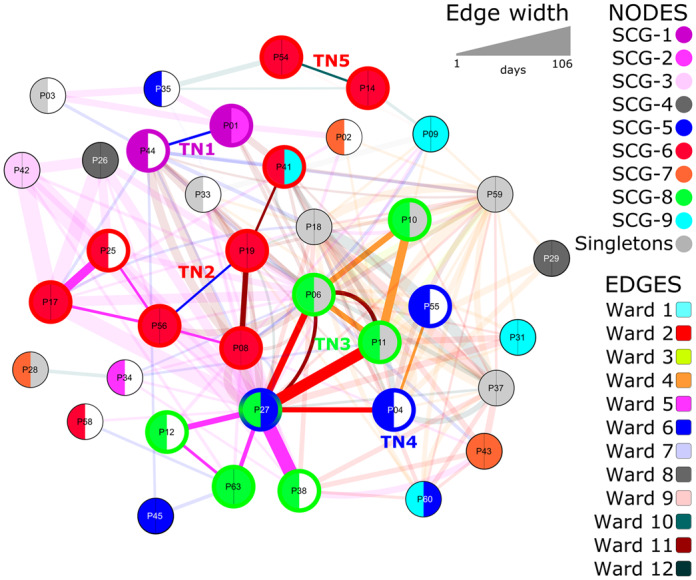
Visualization of patient movement and clonal relationship of VREfm isolates during January–December 2016. VREfm isolates from 37 (out of 43) patients are included in this figure. Edges (lines between nodes) indicate a spatiotemporal (same ward at the same time) overlap in patient movement. Patient isolates within the same clonal group and spatiotemporal overlap are indicated by bold edges, whereas spatiotemporal overlap between non-related strains is indicated by opaque edges. Colours of the edges represent the different wards. Node colours indicate clonal groups. The left-hand semicircle indicates the clonal assignment of the blood culture isolate and the right-hand semicircle indicates that of the colonization isolate. A white right-hand semicircle indicates no colonization or a non-recoverable isolate (n = 12). Clonal group was defined based on an SNP threshold of ≤10. TNs indicate possible transmission clusters as suggested by clonal assignment and epidemiological link. Patients belonging to a TN are indicated by bold borders of the node, corresponding to the SCG colour of their blood culture isolates.
The third network, TN3, contains isolates of P06, P10, P11, P12, P27, P38 and P63, all belonging to SCG-8 ST203/CT2561. Only one patient (P63) in this TN had endogenous bacteraemia. Low frequency of endogenous infections and high connectivity in terms of intensive contact between patients, as indicated by the thickness of connecting lines between the nodes in TN3, suggest patient-to-patient transmission was very likely (Figure 2). In addition, an isolate of P27 in TN3 was connected to both P04 and P55, branching out to TN4 (SCG-5 ST117/CT2558). All patients of TN3 and TN4 were interconnected through Wards 2, 4 and 5. Patient-to-patient transfer in TN5 with P14 and P54 was not very likely, since both patients were colonized prior to infection onset (i.e. endogenous). Altogether, from 19 patients in TN1–5, seven infections were of endogenous origin, therefore only 63% (n = 12/19) of patients were likely to have acquired infections through patient-to-patient transmission.
Discussion
Outbreak investigations are challenging and require a rapid and reliable typing method to determine genetic relatedness between patient isolates, which is essential to demonstrate or contradict nosocomial transmissions. For decades, classical molecular typing of E. faecium has been implemented for outbreak investigations, which has also been the case in our hospital. Although methods such as classical MLST and multi-locus variable-number tandem repeat analysis (MLVA) were once considered as fast and reliable methods for E. faecium outbreak investigations, these methods are obsolete in the era of WGS due to insufficient discriminatory power.18–20 Even with the implementation of WGS, data interpretation can still be challenging, as implied by Zhou et al.,21 who reported that a gene-by-gene-based approach such as using standardized cgMLST nomenclature can be misleading if transfer of a van gene-carrying transposon occurs during an outbreak and that analysis of mobile genetic elements (i.e. transposons) can be a useful tool for outbreak investigations. However, analysis of gene transfer is complex and may require experienced bioinformatics personnel, hence is not always readily available. Therefore, in this study, we explored the possibility of incorporating a patient movement network from routine clinical data for better resolution and interpretation of putative transmission clusters by SNP analysis.
WGS analysis of 73 VREfm isolates from our hospital in the south-west of Germany revealed ST117 as the predominant clonal group circulating in the community and hospital settings, consistent with epidemiological data from neighbouring regions8,22 and surveillance data from the national reference laboratory for staphylococci and enterococci of the Robert Koch Institute, Germany.23vanB-mediated vancomycin resistance was the predominant mechanism, especially in ST117 and ST80 VREfm. Within the ST117 clonal group, we could identify 12 different cgMLST CTs, which suggests diversity within this clonal group. As opposed to a recently published study from a neighbouring Rhine-Main region, in which ST117/CT71 was predominant,8 our data revealed that ST117/CT469 was the main CT of ST117 VREfm in our region. These findings suggest that although ST117 is predominant, the clonal composition of this clonal group may be more diverse than expected, as indicated by the diversity of the cgMLST CTs. Another study from another tertiary hospital in the south-west of Germany also revealed an abundance of ST117;22 unfortunately no cgMLST CT analysis was performed and thus no comparison with our data is possible.
Another prevalent clonal group was ST203 vanA-positive E. faecium, which has been regarded as an emerging clone worldwide. Indeed, ST203 VRE belong to one of the most common clonal groups of VRE causing bacteraemia and outbreaks across the globe.24–26 Even so, unlike the ST203 described from Denmark, our ST203 isolates were cgMLST CT2651 (as opposed to the Danish ST203/CT859). Nevertheless, the epidemiological data and the abundance of this clonal group indicate clinical relevance. Further studies and longitudinal surveillance data are needed to investigate the virulence and transmission potential of this clone.
Our findings suggest that 44% of VREfm bacteraemia in our study population was most likely of endogenous origin, as suggested by WGS. Indeed, the virulence and the origin of Enterococcus species bacteraemia is a highly debated topic. Before the emergence of enterococcal outbreaks in nosocomial settings, colonization was thought to be the most likely origin of these infections. Through the availability and affordability of high-resolution sequencing and numerous outbreak reports on Enterococcus species, outbreaks were later considered as a possible origin of enterococcal bacteraemia. Nevertheless, without prior screening to determine colonization before onset of infection, the designation of infection as endogenous, due to the presence of VREfm in blood cultures and rectal swabs at the same time, is not possible.
Although it is well known that patient-to-patient contact is a major risk factor for the transmission of MDR organisms, visualization of patient movement data in our study gave an important insight into the relevance of patient movement and duration of contact for the acquisition of VREfm. As illustrated in our patient network data in Figure 2, the intensity of contact time in terms of epidemiological overlap in certain wards and the duration of contact is much higher for most patients with non-endogenous VREfm bacteraemia in TN1–5 than those with endogenous bacteraemia, as indicated by the thickness of connecting lines between the nodes. Moreover, we could identify two wards (associated with TN3 and TN4) that were interconnected. This could be explained by the traffic of patients between wards, e.g. P27 was in Wards 2, 5 and 11; and P6 and P11 were in Wards 2, 4 and 11. Upon further inspection, there are certain patient groups that frequently appear in these wards (intensive and intermediate care wards for abdominal surgery and gastroenterology), such as patients with liver transplantation and underlying gastrointestinal conditions. Combining the information from the network visualization and screening data, admission screening may provide additional information to support interpretation of WGS data. We are aware that systematic screening for VRE and isolation is cost-intensive with limited evidence of added benefit. However, our data suggest that, for some patient groups, admission screening may have added benefits to accurately separate transmission from endogenous origin in a highly endemic region.
Our study has limitations. Sampling errors may influence the sensitivity of rectal screening for VRE. Nevertheless, our routine microbiology laboratory performs a validity check of microbial growth on a universal agar medium to minimize false negative results. The lack of consensus threshold for genetic relatedness in terms of SNP distance may have an influence on the cluster classification of WGS data. To minimize this effect, we have chosen a reasonable threshold of ≤10 SNPs for genetic relatedness. On another note, most studies on outbreak investigations of VRE do not include rectal colonization data prior to infection onset. In a non-endemic (i.e. low prevalence) area, this may not be a crucial issue but in a high-prevalence region, where ≥40% of the community may be colonized by VRE, information on the colonization status may avoid overestimation of the magnitude of an outbreak. Current routine microbiological diagnostic practice assumes clonality of bacterial populations growing on agar plates, so that colonization with multiple strains cannot be easily detected. Therefore, there is a possibility that in a case of colonization with more than one VREfm strain, only one strain was picked for further microbiological analysis, which may have led to an underestimation of the total number of endogenous infections.
Taken together, even though WGS is currently the most discriminative typing method available and standardization of typing nomenclature can facilitate comparability between laboratories, great care still needs to be taken when interpreting genetic relatedness for patient-to-patient transmission and outbreak investigation. Incorporation of patient movement data along with admission screening of high-risk patients may provide additional resolution and essential information to avoid overexaggeration of VRE outbreaks and should be taken into account for the implementation of infection control measures.
Supplementary Material
Acknowledgements
We would like to thank Selina Hassel, Nicole Henny and Delal Sahin for excellent technical support.
Funding
This study was supported by internal funding.
Transparency declarations
None to declare.
References
- 1. Behnke M, Hansen S, Leistner R. et al. Nosocomial infection and antibiotic use: a second national prevalence study in Germany. Dtsch Arztebl Int 2013; 110: 627–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uttley AH, Collins CH, Naidoo J. et al. Vancomycin-resistant enterococci. Lancet 1988; 1: 57–8. [DOI] [PubMed] [Google Scholar]
- 3. Werner G, Coque TM, Hammerum AM. et al. Emergence and spread of vancomycin resistance among enterococci in Europe. Euro Surveill 2008; 13: pii=19046. [PubMed] [Google Scholar]
- 4. Tacconelli E, Carrara E, Savoldi A. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 2018; 18: 318–27. [DOI] [PubMed] [Google Scholar]
- 5. DiazGranados CA, Zimmer SM, Klein M. et al. Comparison of mortality associated with vancomycin-resistant and vancomycin-susceptible enterococcal bloodstream infections: a meta-analysis. Clin Infect Dis 2005; 41: 327–33. [DOI] [PubMed] [Google Scholar]
- 6. Chiang HY, Perencevich EN, Nair R. et al. Incidence and outcomes associated with infections caused by vancomycin-resistant enterococci in the United States: systematic literature review and meta-analysis. Infect Control Hosp Epidemiol 2017; 38: 203–15. [DOI] [PubMed] [Google Scholar]
- 7. Gastmeier P, Schroder C, Behnke M. et al. Dramatic increase in vancomycin-resistant enterococci in Germany. J Antimicrob Chemother 2014; 69: 1660–4. [DOI] [PubMed] [Google Scholar]
- 8. Falgenhauer L, Fritzenwanker M, Imirzalioglu C. et al. Near-ubiquitous presence of a vancomycin-resistant Enterococcus faecium ST117/CT71/vanB-clone in the Rhine-Main metropolitan area of Germany. Antimicrob Resist Infect Control 2019; 8: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kocer K, Boutin S, Dalpke AH. et al. Comparative genomic analysis reveals a high prevalence of inter-species in vivo transfer of carbapenem-resistance plasmids in patients with hematological malignancies. Clin Microbiol Infect 2019; doi:10.1016/j.cmi.2019.10.014. [DOI] [PubMed] [Google Scholar]
- 10. Dalpke AH, Hofko M, Zimmermann S.. Development of a real-time PCR protocol requiring minimal handling for detection of vancomycin-resistant enterococci with the fully automated BD Max system. J Clin Microbiol 2016; 54: 2321–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nurjadi D, Boutin S, Dalpke A. et al. Draft genome sequence of Staphylococcus aureus strain HD1410, isolated from a persistent nasal carrier. Genome Announc 2018; 6: e00411–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Joshi NA, Fass JN. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool For FastQ Files (Version 1.33). 2011. https://github.com/najoshi/sickle.
- 13. Bankevich A, Nurk S, Antipov D. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012; 19: 455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014; 30: 2068–9. [DOI] [PubMed] [Google Scholar]
- 15. de Been M, Pinholt M, Top J. et al. Core genome multilocus sequence typing scheme for high-resolution typing of Enterococcus faecium. J Clin Microbiol 2015; 53: 3788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Croucher NJ, Page AJ, Connor TR. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 2015; 43: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Francisco AP, Bugalho M, Ramirez M. et al. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 2009; 10: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Top J, Schouls LM, Bonten MJ. et al. Multiple-locus variable-number tandem repeat analysis, a novel typing scheme to study the genetic relatedness and epidemiology of Enterococcus faecium isolates. J Clin Microbiol 2004; 42: 4503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Homan WL, Tribe D, Poznanski S. et al. Multilocus sequence typing scheme for Enterococcus faecium. J Clin Microbiol 2002; 40: 1963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinholt M, Larner-Svensson H, Littauer P. et al. Multiple hospital outbreaks of vanA Enterococcus faecium in Denmark, 2012-13, investigated by WGS, MLST and PFGE. J Antimicrob Chemother 2015; 70: 2474–82. [DOI] [PubMed] [Google Scholar]
- 21. Zhou X, Chlebowicz MA, Bathoorn E. et al. Elucidating vancomycin-resistant Enterococcus faecium outbreaks: the role of clonal spread and movement of mobile genetic elements. J Antimicrob Chemother 2018; 73: 3259–67. [DOI] [PubMed] [Google Scholar]
- 22. Liese J, Schule L, Oberhettinger P. et al. Expansion of vancomycin-resistant Enterococcus faecium in an academic tertiary hospital in southwest Germany: a large-scale whole-genome-based outbreak investigation. Antimicrob Agents Chemother 2019; 63: e01978–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klare I, Bender JK, Werner G. et al. Eigenschaften, Häufigkeit und Verbreitung von Vancomycin-resistenten Enterokokken in Deutschland—Update 2017/2018. Epid Bull 2019; 35: 365–72. [Google Scholar]
- 24. Johnson PD, Ballard SA, Grabsch EA. et al. A sustained hospital outbreak of vancomycin-resistant Enterococcus faecium bacteremia due to emergence of vanB E. faecium sequence type 203. J Infect Dis 2010; 202: 1278–86. [DOI] [PubMed] [Google Scholar]
- 25. Ryan L, O’Mahony E, Wrenn C. et al. Epidemiology and molecular typing of VRE bloodstream isolates in an Irish tertiary care hospital. J Antimicrob Chemother 2015; 70: 2718–24. [DOI] [PubMed] [Google Scholar]
- 26. Hammerum AM, Baig S, Kamel Y. et al. Emergence of vanA Enterococcus faecium in Denmark, 2005-15. J Antimicrob Chemother 2017; 72: 2184–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.