

Summary
With more than 450 members, the solute carrier (SLC) group of proteins represents the largest class of transporters encoded in the human genome. Their several-pass transmembrane domain structure and hydrophobicity contribute to the orphan status of many SLCs, devoid of known cargos or chemical inhibitors. We report that SLC proteins belonging to different families and subcellular compartments are amenable to induced degradation by heterobifunctional ligands. Engineering endogenous alleles via the degradation tag (dTAG) technology enabled chemical control of abundance of the transporter protein, SLC38A2. Moreover, we report the design of d9A-2, a chimeric compound engaging several members of the SLC9 family and leading to their degradation. d9A-2 impairs cellular pH homeostasis and promotes cell death in a range of cancer cell lines. These findings open the era of SLC-targeting chimeric degraders and demonstrate potential access of multi-pass transmembrane proteins of different subcellular localizations to the chemically exploitable degradation machinery.
Keywords: transporter, solute carrier, multi-pass transmembrane protein, targeted degradation, proteolysis targeting chimera (PROTAC), degrader, SLC9A1, SLC38A2, E3 ligase, proteasome
Graphical Abstract
Highlights
-
•
SLC transporters are amenable to rapid, proteasome-dependent targeted degradation
-
•
13 different SLCs at different subcellular locations are shown to be degradable
-
•
The first SLC PROTAC d9A-2 targets SLC9A1 and other SLC9 family members
-
•
d9A-2 effectively impairs pH homeostasis and differentially kills cancer cell lines
Bensimon et al. demonstrate that solute carriers (SLCs), multi-TM transporter proteins, are amenable to targeted protein degradation by using the dTAG system and developing the first SLC PROTAC, d9A-2. The compound leads to degradation of SLC9A1, resulting in impaired pH homeostasis and cytotoxicity.
Introduction
To maintain cell viability and support the requirements of proliferation, the molecular building blocks of cells are constantly generated, utilized, and exchanged between the extracellular environment and intracellular compartments. Several families of transmembrane transporters, including solute carriers (SLCs), ion channels, water channels, and ATP-driven pumps, enable the exchange of water, nutrients, ions, and metabolic products across cellular membranes (Hediger et al., 2013). The SLC family, which includes more than 450 genes, is the second-largest family of membrane proteins in the human genome (Lin et al., 2015). The most thoroughly studied members are SLCs mediating the uptake of certain molecules, such as glucose and serotonin, SLCs linked to Mendelian diseases, SLCs involved in drug pharmacokinetics, and SLCs targeted by Food and Drug Administration-approved drugs (César-Razquin et al., 2015, Lin et al., 2015). While most SLCs remain poorly characterized (César-Razquin et al., 2015), our understanding of these transporters has clearly suggested that research into SLCs will unravel fundamental relationships between cell metabolism and pathophysiology, benefit patients with a wide range of indications, and expedite drug development.
The increased metabolic rate exhibited by cancer cells or activated lymphocytes, for example, requires metabolic reprogramming tuned to increase the uptake of the necessary nutrients, support energy and pH homeostasis, supply the building blocks for macromolecular synthesis, complete replication of the genome, and respond to extracellular and intracellular stresses (Pavlova and Thompson, 2016, Sinclair et al., 2013). In recent years, evidence of critical roles for some SLCs in tumorigenic processes has been accumulating, demonstrating that SLCs may be attractive targets for drug development in cancer (El-gebali et al., 2013, Koltai, 2016). However, successful efforts describing the intricate relationships between the signaling networks that drive proliferation and the metabolic networks that enable it have lagged behind in determining the role of SLCs in the context of such networks (Vander Heiden and Deberardinis, 2017).
Generating a gene loss-of-function state in cells is a valuable approach in biology to connect the corresponding encoding proteins to their cellular functions. Mechanistic and functional studies of proteins are inherently limited by the challenges associated with the lengthy process of deriving stable and viable knockout clones for such proteins. These limitations are more pronounced in studies attempting to connect proteins to their functions in dynamic and rapid cellular processes, such as metabolism. Due to the great plasticity and robustness of biological systems (Barkai and Leibler, 1997), long-term perturbations, such as gene loss can lead to considerable adaptation, masking direct consequences of the absence of a specific biological component and revealing the outcome of global effects. Rapid loss of function at the protein level is thus a desirable experimental strategy to circumvent these problems and allow the monitoring of effects that are temporally close to the perturbation being investigated.
Several techniques have been developed in recent years to control the targeted degradation of specific proteins (Mayor-Ruiz and Winter, 2019). A new generation of heterobifunctional small-molecule degraders or PROTACs (proteolysis-targeting chimeras) enabled the rational design of small molecules that induce the selective and rapid degradation of target proteins (Bondeson et al., 2015, Winter et al., 2015). PROTACs operate by inducing molecular proximity between the protein of interest (POI) and a cellular E3 ligase substrate receptor by binding simultaneously to both proteins. This induced proximity leads to ubiquitination and proteasome-mediated degradation of the POI. The modular design, consisting of a warhead binding to the POI, a flexible linker, and a defined E3 ligase ligand, renders a variety of PROTAC development opportunities. Building on phthalimide conjugation as means for the development of PROTACs, tag-based technologies have been introduced, in which a POI is tagged with a domain (also known as “degron”), making it amenable to degradation by specific chemical molecules (Bondeson and Crews, 2017, Winter et al., 2015). In one such method, the degradation tag (dTAG) system, mutated FKBP12 is utilized as the tag that enables the phthalimide-mediated degradation of the POI by a variety of degraders published recently (Erb et al., 2017, Nabet et al., 2018).
The growing list of cellular proteins permissive to targeted degradation includes a number of therapeutically relevant nuclear and cytoplasmic proteins, such as BET proteins, the oncogenic fusion protein BCR-ABL1, and kinases implicated in cell-cycle regulation (Brand et al., 2019, Burslem et al., 2019, Winter et al., 2015). More recently, also single-pass transmembrane kinases (Burslem et al., 2018, Lai and Crews, 2017, Zou et al., 2019), expressed on the plasma membrane, have been shown to be amenable to chemically induced degradation. In all cases, functional degraders are based on published inhibitors binding the cytoplasmic kinase domain of these receptors.
However, several of the top drug targets and disease-associated genes have a multi-pass transmembrane domain topology and are located at different subcellular locations. Prominent examples are G-protein-coupled receptors (GPCRs), ABC transporters, such as the cystic fibrosis transmembrane conductance regulator, and SLCs, prominently represented by the targets of the serotonin-uptake inhibitors and glifozins (Faillie, 2017, Kristensen et al., 2011). Would these proteins be amenable to chemically induced degradation despite their extensive embedding in biological membranes? Moreover, would the required degradation machinery find access to these classes of proteins even when located at different subcellular sites, such as the ER or lysosomes? We set out to comprehensively address these questions by studying several members of the large superfamily of SLCs, which vary in transmembrane topology, post-translational modifications, and subcellular localizations.
Given that published work has focused solely on individual examples of single-pass transmembrane proteins, the issue whether SLCs, or any other several-pass transmembrane protein, would in principle be possible subjects of PROTAC-induced degradation, has been open. This study systematically investigates the possibility that SLCs, despite their complex membrane-embedded topology and heterogeneous subcellular localization, will be amenable to chemical degradation. We first address the question by ectopically expressing SLC-dTAG fusion proteins (Nabet et al., 2018). We also test whether an SLC expressed from the natural chromosomal locus and therefore exposed to more physiological regulation, can be made degradable through dTAG knockin. Finally, we attempt the development of a directly acting degrader of an untagged endogenous SLC. Development of this first-in-class SLC chimeric degrader inaugurates the era of SLC PROTACs, suitable both as cellular tools for elucidating SLC functions but also as a new class of potential drugs.
Results
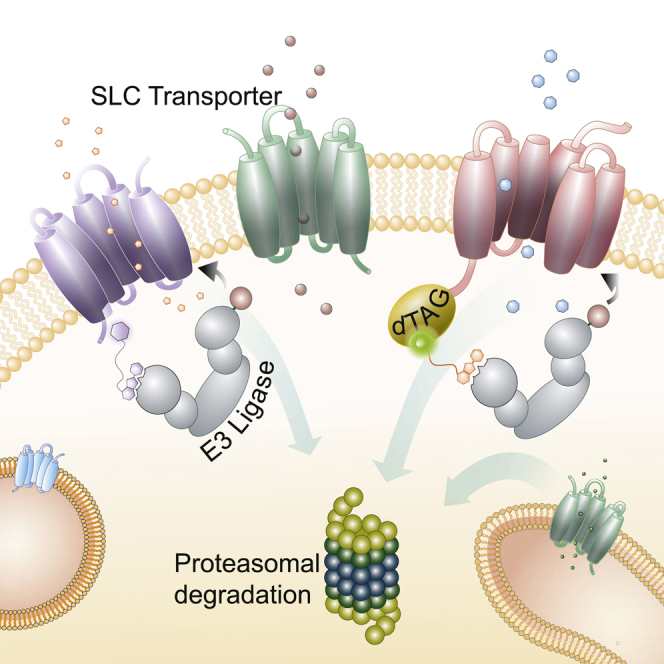
To test whether SLCs are amenable to phthalimide-mediated degradation, we ectopically expressed SLCs as fusions to a mutated FKBP domain, also known as the dTAG system (Erb et al., 2017, Nabet et al., 2018). In brief, a dTAGed protein is subject to degradation by specific chimeric degrader molecules (e.g., dTAG7/dTAG13) that simultaneously bind to the dTAG and the CRL4CRBN E3 ligase, inducing molecular proximity and ensuing degradation by the proteasome. Given that SLCs are expressed in all cellular compartments, we first tested the accessibility of differentially located SLCs to targeted proteolysis (Figure 1A). Each of the tested SLCs was stably expressed in HAP1 cells and subcellular localization was assessed by immunofluorescence microscopy (Figures 1B and S1). In detail, we assayed SLCs located at the plasma membrane (PM: SLC1A5, SLC38A1, SLC2A1, SLC2A3, SLC16A1, and SLC9A1), plasma membrane and vesicles (SLC38A2), endoplasmic reticulum (ER: SLC39A7 and SLC30A9), lysosome (SLC38A9), mitochondria (SLC25A26, SLC25A1, SLC25A19, and MTCH2), and Golgi (SLC35B2 and SLC33A1). Cellular treatment with a specific degrader molecule (dTAG7 or dTAG13) led us to identify that SLCs expressed at plasma membrane, ER, Golgi, and lysosome were amenable to targeted degradation (Figure 1C). As exemplified by SLC2A3, the dTAG could be placed on either the N or C terminus of the SLC if both face the cytoplasm (Figure S2A). The outer mitochondrial protein MTCH2 (SLC25A50) was amenable to degradation, while the inner mitochondrial SLCs tested could not be degraded within the assayed time frame (Figures 1C and S2A). Although some SLCs, such as SLC38A2 or SLC2A3 could be completely degraded after treatment, other SLCs, such as SLC35B2 or SLC39A7 were not degraded to completion (see Table 1). Complete degradation did not appear to correlate with the stable expression level of the dTAG protein or with the expression of the endogenous protein (Figure S2B). Targeted degradation was also validated via protein-specific antibodies, which generally showed good consistency with detection via the HA epitope of the dTAG (Figure S2C). Of note, temporal control of targeted SLC degradation could be influenced by the choice of the respective heterobifunctional molecule: degradation with the PROTAC dTAG7 led to reversible degradation, while dTAG13 maintained the target degradation for at least 48 h (Figure S2D). Collectively, these data confirm that an intracellular target-engagement is sufficient to recruit the CRLCRBN E3 ligase complex and prompt SLC degradation.
Figure 1.

Amenability of SLCs to Targeted Degradation
Exogenous expression of solute carriers (SLCs) tagged by FKBP12F36V (“dTAG”) demonstrates amenability to targeted degradation in different cellular compartments:
(A) Illustration representing SLC proteins from different subcellular locations (plasma membrane, lysosome, mitochondria, Golgi, endoplasmic reticulum) were selected for testing their amenability to targeted degradation. See also Table 1.
(B) Representative images of immunofluorescence (αHA) imaging of the indicated dTAG-HA SLCs confirmed subcellular localization. Scale bar, 10 μm. Co-localization for all SLCs with established cellular markers is detailed in Figure S1.
(C) HAP1 cells expressing dTAG-HA SLCs (as indicated) were treated for 24 h with 0.5 μM dTAG7 or dTAG13. All but three mitochondrial SLCs were amenable to targeted degradation at varying efficiency. See also Figure S2.
Table 1.
SLC Amenability to Targeted Protein Degradation
| SLC | Predicted TMs | Subcellular Localization | Amenable to Targeted Degradation |
|---|---|---|---|
| SLC1A5 | 8 | PM | +++ |
| SLC2A1 | 12 | PM | + |
| SLC2A3 | 12 | PM | +++ |
| SLC16A1 | 12 | PM | ++ |
| SLC38A1 | 11 | PM | +++ |
| SLC9A1 | 12 | PM | +++ |
| SLC38A2 | 11 | PM | +++ |
| SLC38A9 | 11 | Lysosome | +++ |
| SLC30A9 | 5 | ER | ++ |
| SLC39A7 | 6 | ER | + |
| SLC33A1 | 11 | Golgi | ++ |
| SLC35B2 | 9 | Golgi | + |
| SLC25A1 | 6 | Mitochondria (inner) | – |
| SLC25A19 | 6 | Mitochondria (inner) | – |
| SLC25A26 | 6 | Mitochondria (inner) | – |
| MTCH2 | 3 | Mitochondria (outer) | + |
Summarizing the list of SLC proteins tested as targets for degradation, as well as the number of transmembrane (TM) segments predicted by PROTTER (Omasits et al., 2014), their subcellular localization, and an estimate of how amenable to degradation these proteins were. PM, plasma membrane; ER, endoplasmic reticulum.
Related to Figure 1.
Near-complete degradation could be achieved in the low nanomolar range for some SLCs, such as SLC38A2 or SLC9A1, although the dose of degrader required for maximal target degradation varied based on the studied SLC (Figures 2A and S3A). Targeted degradation of SLCs was typically initiated within a few hours depending on the SLC (Figures 2B and S3B). Although SLC38A2 and SLC9A1 stood out as amenable to rapid degradation, other SLCs, such as SLC1A5 or SLC2A3, required 6 h or more. Degradation of ectopically expressed dTAG-SLCs was not unique to the HAP1 cell line, but could also be achieved in other cancer cell lines, such as HCT15, LS180, and others, as exemplified by SLC2A3 and SLC1A5 (Figures 2C and S3C). To validate that the observed SLC destabilization is dependent on an active CRL4CRBN complex, we next set up chemical competition experiments. As expected, dTAG7-mediated degradation was blocked by inhibiting cullin neddylation, and thus CRL activity, by co-treatment with the NAE1 inhibitor MLN4924 (Figures 2D and S3D). Similarly, saturating the CRBN binding site by treatment with excess concentration of pomalidomide prevented measurable SLC degradation (Figures 2D and S2E). Finally, treatment with proteasome inhibitors (bortezomib or MG132) rescued degradation of the SLC. Degradation appeared unaffected upon co-treatment with the lysosomal modulator bafilomycin A1 (Figures 2D and S3D). Thus, targeted degradation of SLCs appears to utilize the proteasome but not the lysosome machinery.
Figure 2.

Characteristics of Targeted Degradation of SLCs by dTAG System
(A) A range of dTAG13 concentrations was tested in cell lines expressing dTAG-HA SLC38A2, SLC16A1, or SLC2A1 for 48 h. The dose required for close to complete degradation varies for these example SLCs. Additional examples are in Figure S3A.
(B) A time course of dTAG-driven SLC degradation. HAP1 cell lines expressing dTAG-HA SLC38A2, SLC9A1, or SLC1A5 were treated with 0.5 μM dTAG7 or dTAG13, and samples were harvested at several time points. The glycosylated form of SLC38A2 (upper band) appeared to be degraded slightly faster than the unglycosylated form. SLC9A1 and SLC1A5 provide additional examples of variation in time required for degradation. Additional SLCs are in Figure S2D.
(C) dTAG-HA SLC2A3 was stably expressed in HAP1, LS180, and HCT15 cells. Following 72 h of treatment with 0.5 μM dTAG13, SLC2A3 was completely degraded.
(D) Chemical “rescue” of dTAG-driven degradation of SLCs. HAP1 cell lines expressing dTAG-HA SLC1A5 or SLC38A9 were treated with 0.5 μM dTAG7 for 12 or 18 h, respectively. These cells were also treated with chloroquine (CQ) (50 μM), bortezomib (bort.) (1 μM), MG-132 (MG) (1 μM), MLN4924 (MLN) (1 μM), pomalidomide (poma.) (10 μM), or bafilomycin A1 (bafi.) (2.5 μM). SLC degradation was rescued by inhibiting CRL activity or the proteasome, but not by inhibiting the lysosome machinery.
See also Figures S3D and S3E.
Next, we aimed to knock in the dTAG domain at a genomic locus to demonstrate the feasibility of chemically controlling an endogenous SLC protein. We selected SLC38A2 for endogenous tagging at the N terminus (Figure 3A). SLC38A2 protein levels have been shown to be kept low under normal culture conditions and strongly induced after amino acid (aa) starvation (Nardi et al., 2015). Indeed, in our cell line model, the endogenously tagged SLC38A2 protein was expressed and localized to cell membranes upon aa starvation (Figure 3A), in proportion to the level of aa depletion. The endogenously tagged SLC38A2 protein was induced rapidly and became visible within 4 h of aa depletion (Figure S4A). Despite the strong induction of expression, the transporter remained amenable to targeted degradation. We found that, even under this strong stimulus, SLC38A2 protein expression was completely ablated by 2 h of pre-treatment with dTAG13 or dTAG7 (Figure 3B). On the other hand, co-treatment with dTAG13 or dTAG7 led to near-complete disappearance of SLC38A2 protein expression, exposing a slight difference between the two degraders, as a function of time, dose, and medium (Figures 3B, S4B, and S4C). By immunofluorescence, a stronger signal was noticed in the Golgi compartment of cells treated with dTAG13, likely related to a polypeptide not having reached maturation (Figure S4D). Since SLC38A2 is rapidly induced by aa starvation, it could be halted during maturation in the ER or the Golgi by treatment with brefeldin A or monensin, respectively. In both cases, SLC38A2 was amenable to near-complete degradation (Figures 3C and S4E), highlighting that targeted degradation is feasible either in these compartments and/or en route between these compartments. It also revealed again a slight difference between dTAG13 and dTAG7 in Golgi-localized SLC38A2 (Figure 3C). To further ascertain that SLC38A2 can be targeted for degradation from the PM, we monitored degradation kinetics after aa starvation. Following the induction and localization of SLC38A2 to PM, the protein was degraded within 3 h post-treatment with dTAG7/13, in line with data for the overexpressed protein (Figure 3D). Finally, SLC38A2 dTAG-mediated degradation was notably more rapid than the natural process by which SLC38A2 protein levels decay to basal levels in response to full medium supplement (Figures 3E and S4G). Altogether, the data for exogenous and endogenous expression of dTAGed SLCs indicate that multi-pass transmembrane proteins, such as SLCs, are within reach of the recruited proteolytic machinery and amenable to proteasome-mediated targeted degradation.
Figure 3.

dTAG Knockin Are Equally Amenable to Targeted Degradation
(A) dTAG knockin: SLC38A2 was tagged at the N terminus with HA-dTAG. Blasticidin was used as the selection marker. The expression of dTAG-HA SLC38A2 was induced by replacing the normal culture medium with DMEM medium deprived of both FBS and amino acids, or supplemented with FBS and only with 5% of non-essential amino acids. Representative immunofluorescence images of HA-dTAG-SLC38A2 expression after 10 h of induction are shown. Scale bar, 50 μm. A time course of this induction can be found in Figure S4A.
(B) The expression of HA-dTAG SLC38A2 was induced by replacing the normal culture medium with medium deprived of amino acids and FBS. Cells were treated with medium only (“none”) or treated with medium and dTAG7/13 in one of two regimes: 2-h pre-treatment (left boxplot) or co-treatment (right boxplot). Expression of the endogenous SLC is induced rapidly and was monitored by immunofluorescence (α-HA) imaging and quantified by automated image analysis. The mean HA intensity was plotted for each time point in each regime separately, with the condition none plotted in both graphs as a shared reference. Two-hour pre-treatment with dTAG7 or dTAG13 leads to complete degradation of SLC38A2. Co-treatment with dTAG13, but not dTAG7, leads to accumulation of a signal corresponding to undegraded polypeptide at the later time points of induction, suggesting a difference in kinetics between the two PROTACs.
(C) The expression of HA-dTAG SLC38A2 was induced by replacing the normal culture medium with medium deprived of amino acids and FBS. Cells were co-treated with dTAG7 (0.5 μM) or dTAG13 (0.5 μM) for 16 h. In addition, cells were treated with brefeldin A (5 μg/mL) or monensin (2 μM), halting the protein in the ER or Golgi compartment, respectively. SLC38A2 is amenable to degradation in and/or en route to both compartments. In the Golgi, a slight fraction of the SLC is not degraded under dTAG13 co-treatment. See also Figure S4E.
(D) Expression, re-localization, and degradation of HA-dTAG SLC38A2 were monitored by immunofluorescence and quantified by automated image analysis. Representative images are presented, and quantification of the data is presented in Figure S4E. HA-dTAG SLC38A2 was induced by replacing the normal culture medium with medium deprived of amino acids and FBS for 10 h. Cells were co-treated, as indicated, with brefeldin A (5 μg/mL) or monensin (2 μM), dTAG7 (0.5 μM), or dTAG13 (0.5 μM). Scale bar, 50 μm.
(E) Cells were treated for 10 h in medium lacking amino acids, leading to the induction of HA-dTAG SLC38A2. dTAG7 (0.5 μM) or dTAG13 (0.5 μM) were then added for the indicated hours, to monitor degradation of the glycosylated protein from the plasma membrane. Near-complete degradation is achievable within 3 h, and is maintained for at least 9 h. As a reference for the natural removal of SLC38A2, cells were refed by a change to full medium for 9 h.
(F) Cells were treated for 18 h in medium supplemented with 5% amino acids, leading to the induction of HA-dTAG SLC38A2. To closely compare the PROTAC-mediated degradation to the natural removal of the protein, cells were treated with dTAG7 (0.5 μM) or refed with full medium for the indicated time points. dTAG7-mediated degradation was initiated within 1 h and nearly completed within 2 h. Removal of the protein after refeeding was noticeably slower from 4 h onward.
See also Figure S4F.
The use of genetically encoded degradation systems, such as the dTAG approach, limits its use to established and genetically tractable cell culture systems. To investigate whether SLCs can be degraded by the CRL4CRBN E3 ligase complex through a chemical entity that engages the endogenous SLC directly, we set out to design a directly acting first-in-class, SLC degrader. SLC9A1, also known as NHE1, is an electroneutral and reversible ion transporter that exchanges one Na+ ion for one H+ ion. It contributes to both cytoplasmic alkalization (pHi), and acidification of the microenvironment (Parks et al., 2013). It has been previously shown to be involved in cancer and therefore is an attractive model target for our purpose (Stock and Pedersen, 2017). Design of the warhead (in short, “w9A”) binding to SLC9A1 was made possible by a modification of a previously reported ligand binding to SLC9A1, intended as an inhibitor (Atwal et al., 2006). We synthesized a focused library of putative SLC9A1 PROTACs (in short, d9A-1…5) by systematically varying linker length, composition and attachment chemistry to the phthalimide-based CRBN binder (Figures 4A and S6A). Degradation of SLC9A1 was assessed in two cell lines, HAP1 and KBM7. The most potent chimeric compound, denoted here as d9A-2, led to degradation of endogenous SLC9A1 at sub-micromolar concentration within 8 h post-treatment (Figures 4B and S5B). Two additional compounds of the same series, denoted as d9A-1 and d9A-3, also led to degradation of the target, albeit at lower potency (Figures S5B and S5C). Interestingly, d9A-5 led to slight accumulation of SLC9A1 (Figure S5C). By inspecting GFP-tagged SLC9A1 in HAP1 cells, it became apparent that the protein accumulated in an unknown intracellular compartment and/or vesicles (Figure S5D). These nuances stress the importance of linker design in generating an efficient chemical degrader, and subsequently we focused on d9A-2 for further characterization.
Figure 4.

SLC9 PROTAC Series
(A) The chemical structure of the SLC degrader d9A-2. See Figure S5A for structures of the related molecules.
(B) HAP1 and KBM7 cells were treated with different concentrations of d9A-2. Within 8 h, degradation of SLC9A1 was observed in both cell lines.
(C) Both WT and CRBN knockout KBM7 cell lines were treated with indicated concentrations of d9A-2 for 12 h. SLC9A1 degradation is observed in WT but not CRBN knockout.
(D) Chemical “rescue” of d9A-2-driven degradation. WT KBM7 cell lines were treated with 0.5 μM d9A-2 for 16 h. These cells were also treated with bortezomib (bort.) (0.25 μM), MG-132 (MG) (1 μM), MLN4924 (MLN) (1 μM), pomalidomide (poma.) (1 μM), or bafilomycin A1 (bafi.) (10 μM). All molecules, apart from bafilomycin, could rescue SLC9A1 from degradation.
(E) Selectivity of d9A-2 was tested across HAP1 cell lines expressing Strep/HA-SLC9A1, SLC9A2, SLC9A3, SLC9A4, SLC9A5, SLC9A6, SLC9A7, SLC9A8, SLC9A9, SLC9B1, and SLC9B2. Cells were treated with varying concentrations of d9A-2 for 16 h, after which degradation of the exogenous SLCs was monitored. At 0.25 μM, SLC9A1 is the only SLC9 member that is completely degraded.
(F) Kinetics of d9A-2-induced degradation tested in HAP1 cell lines expressing Strep/HA-SLC9A1, SLC9A2, SLC9A4, and SLC9A6. d9A-2 (0.75 μM) was added to HAP1 cell lines for the indicated length of time. SLC9A1 is the only protein that is mostly degraded after 6 h.
As expected, genetic ablation of the E3 ligase adaptor CRBN prevented the degradation of SLC9A1 (Figures 4C and S5E). Again in line with a mechanism of controlled proteolysis, SLC9A1 destabilization was abrogated by blocking CRL activity via pharmacologic inhibition of NAE1 via MLN4924. Similarly, competition with excess amounts of pomalidomide blocked d9A-2-induced degradation of SLC9A1 (Figures 4D and S5E). Treatment with proteasome inhibitors (bortezomib or MG132), but not bafilomycin A1, rescued SLC degradation (Figure 4D).
Next, we wanted to assess the selectivity for the degradation of SLC9A1 over other SLC9 family members. To render this survey independent of endogenous transporter expression levels in HAP1 cells, we ectopically overexpressed tagged proteins, corresponding to 11 members of the SLC9 family (A1, A2, A3, A4, A5, A6, A7, A8, A9, B1, and B2). At 16 h post-treatment with d9A-2 at 250 or 750 nM, SLC9A1 demonstrated the most efficient degradation. d9A-2 prompted degradation of the closely related SLC9A2 and SLC9A4, which are not endogenously expressed in HAP1, and also prompted degradation of the more distant SLC9A7 and SLC9B1, which are endogenously expressed in HAP1 (Figure 4E). Comparatively, abundance of the other assayed SLCs was less significantly affected at these experimental conditions. SLC9A1, SLC9A2, and SLC9A4 (but not SLC9A6) degraded at similar time kinetics (Figure 4F) and were at least partially amenable to degradation by d9A-3 (Figure S5F). In sum, while not exclusively selective for SLC9A1, d9A-2 features some level of intra-family selectivity. These experiments further attest to the degradability of SLCs by heterobifunctional inducers of proteolysis.
To assess the consequences of d9A-2 treatment, we tested the ability of cells to recover from acid load, a well-established assay for SLC9 function (Atwal et al., 2006, Loiselle and Casey, 2010, Rotte et al., 2010). We compared d9A-2, at 8 h post-treatment, to the warhead w9A, or to ethylisopropyl amiloride (EIPA), an SLC9 inhibitor of a different chemotype, as control (Pedersen et al., 2007, Harguindey et al., 2013). In HAP1 cells, we found that, at ~10 min post-recovery, d9A-2-, w9A-, or EIPA-treated cells exhibited a pronounced difference in pHi recovery compared with untreated cells. Interestingly, while at this time point the difference in pH was most acute for cells treated with 50 μM of either w9A or EIPA, these differences subsided rapidly. Within 30 min post-recovery, treatment with 1 μM d9A-2 demonstrated a defect in pHi recovery as strong as that observed in treatment with 25 or 50 μM w9A or 50 μM EIPA (Figure 5A), attesting to the potency of d9A-2. Next, we assessed the effects of these molecules on cell viability at 72 h post-treatment. In KBM7 wild-type (WT) versus KBM7 −/− CRBN we found similar sensitivity to w9A, but a significant difference in sensitivity to d9A-2 (Figure 5B). Furthermore, the cytotoxicity profile in KBM7 WT was in agreement with the biochemical evidence for degradation (Figure 5C). Given the cytotoxicity in KBM7 WT, we expanded these investigations and characterized the cytotoxicity of the PROTACs d9A-1…5 in a panel of 43 cancer cell lines, with EIPA and bortezomib as controls. We found that, while cytotoxicity varied between cell lines, d9A-2 is most cytotoxic (half maximal effective concentration [EC50] < 0.1 μM) in cell lines of leukemic origin (Figure 5D). When comparing the activity of all molecules in the series, we also found that, across the tested cell lines, cytotoxicity correlated with the efficacy of SLC9A1 degradation observed by immunoblots (Figure S6A): Activity areas above the dose response were highest for d9A-2, followed by d9A-3 and d9A-1. Importantly cell line sensitivity to d9A-2 correlated with sensitivity to EIPA (Figure S6B), attesting to the involvement of SLC9A1 in cytotoxicity. In sum, d9A-2 treatment leads to impaired pHi recovery and is toxic in multiple cancer cell lines.
Figure 5.

Effect of SLC9A1 Degradation on Cell Proton Transport and Viability
(A) Effect of indicated molecules on pHi recovery was assessed in the acid load assay. To achieve SLC9 degradation, cells were pre-treated for 8 h with d9A-2 (1 or 2.5 μM). As reference, cells were treated (25 or 50 μM) with the warhead w9A from which d9A-2 was developed or EIPA, a molecule known to inhibit SLC9A1, as a control. Following an acid load perturbation, recovery was compared between untreated and each of the treatments. The difference in recovery, compared with untreated cells (ΔpHi), was calculated at indicated time points after recovery. Each condition was assayed in six replicates. The corresponding raw plots are presented in Figure S6C. In comparison with untreated, a significant change in pH recovery was observed for almost all treatments (∗p < 0.05, ∗∗∗p < 0.0005; one-tailed t test). In comparison with 1 μM d9A-2, the change in pH recovery was higher only in 50 μM w9A (5 min, p < 0.05; 10 min, p < 0.0005; 15 min, p < 0.05) or 50 μM EIPA (10 min, p < 0.05). Data are represented as mean ± SD. See also Figure S6C.
(B) Viability WT and CRBN knockout KBM7 was assessed at 72 h post-treatment with d9A-2 or w9A (mean ± SD). The two cell lines display similar sensitivity to the warhead w9A but marked difference in sensitivity to d9A-2
(C) Viability of WT KBM7 was also assessed at 72 h post-treatment with d9A-2, d9A-3, d9A-1, or w9A (mean ± SD), to relate between the effect of these compounds on degradation and cytotoxicity. See also Figures S5 and S6.
(D) EC50 values for d9A-2 were estimated for 43 cancer cell lines tested at 72 h post-treatment. The area above the dose response was calculated to capture and compare dose curves for each molecule of the PROTAC series (Figure S6A). Cell lines of leukemic origin display a marked sensitivity to d9A-2 with EC50 < 0.1 μM.
Discussion
The advent of targeted protein degradation has been one of the most important novelties in pharmacology and drug development of the last decade (Pettersson and Crews, 2019). Paradoxically, the largest group of drug targets and, arguably the most important, has so far not been affected by these new developments: proteins with multi-pass transmembrane domains, such as GPCRs, channels, and transporters. Through the use of the dTAG system and the development of an SLC degrader, we demonstrate that multiple SLCs are amenable to targeted protein degradation by chimeric degrader molecules. As an approach to study the functions of SLCs in rapid processes, such as cellular metabolism, targeted SLC degradation offers several advantages: the benefit of removing a protein completely through chemical matter at rapid timescale, without the limitations of generating knockouts or the scarcity of existing chemical probes against SLCs. While most of SLC-related small-molecule development has been oriented toward target inhibition (Lin et al., 2015), this work opens a new avenue to generating chemical matter to target SLCs by degradation. We expect that, as the development of such compounds is accelerated, these will become valuable tools in de-orphanizing the functions of SLCs, raise the attractiveness of SLCs as a drug target class, and ultimately result in many new pharmacological agents. Moreover, targeting SLCs by degradation, rather than inhibition, would offer the possibility to modulate transport-independent roles, such as acting as receptors for viruses (Sainz et al., 2012, Yan et al., 2012) or modulators of tumor progression (Coothankandaswamy et al., 2013, Payen et al., 2017). As an example for such a possibility, SLC9A1 has been shown to carry out numerous structural functions in the actin cytoskeleton and scaffolding of protein complexes, beyond its activity in proton transport, which may be modulated by one of the compounds in our PROTAC series (Amith and Fliegel, 2013, Baumgartner et al., 2004, Meima et al., 2007).
By characterizing targeted SLC degradation via chemical competition experiments and genetic loss-of-function approaches, we found this process to be E3 ligase and proteasome dependent, as has also been shown for single-pass transmembrane proteins (Burslem et al., 2018, Zhang et al., 2018). The number of transmembrane domains in the SLCs tested was in the range of 3–12 transmembrane domains, but appeared to be inconsequential for degradability. Based on the tested examples, amenability to degradation appears to be related to localization rather than topology. SLCs located at mitochondria and Golgi compartments were less likely to be completely removed compared with SLCs at the plasma membrane, although not all plasma membrane SLCs were equally amenable. In accordance, by chemically halting endogenous dTAG-SLC38A2 en route to the plasma membrane, we showed that this SLC is accessible for complete degradation not only at the plasma membrane but, in principle, also in both the Golgi and ER.
Although both dTAG molecules and d9A series of PROTACs are based on hijacking the CRL4CRBN ligase, we assume that other E3 ligases can also be recruited to SLCs. In fact, we can foresee the development of degraders that harness E3 ligases that are thought to be involved in membrane protein proteostasis, such as NEDD4/NEDD4L (Goel et al., 2015, Lamothe and Zhang, 2016, MacGurn et al., 2012). To exploit targeted protein degradation, a binding event that per se may not lead to any change in protein function can be the basis for a degrader and even utilized to engineer selectivity. This is particularly relevant as we anticipate that SLC structures are enriched in surfaces that may enable small-molecule binding, but not lead to inhibition (Bai et al., 2017, Colas et al., 2016). d9A-2, a first-in-class SLC PROTAC, induced efficient degradation of its cognate target SLC9A1 along with degradation of other SLC9 members, such as the transporters SLC9A2 and SLC9A4. Such selectivity may in fact be beneficial when redundant functions, such as pH homeostasis, are necessary to tackle in diseases, such as cancer (Counillon et al., 2016, Stock and Pedersen, 2017). Alkalization of intracellular pH (pHi) by SLC9A1 has been linked to early malignant transformation, cell migration, and metastases (Harguindey et al., 2013, Parks et al., 2013, Reshkin et al., 2000, Reshkin et al., 2014, Schwab et al., 2012). Despite the promise of targeting pHi as a therapeutic approach in cancer, molecules inhibiting SLC9A1 have not yet been clinically assessed in cancer patients (Harguindey et al., 2013). We show here that d9A-2 is a potent cytotoxic compound, justifying exploration of its potential for further development as a pre-clinical compound. We envision that, in the future, selectivity could be engineered strategically toward any selected SLC9 member targeted by d9A-2 by further modifying the ligand and/or the linker.
Based on the broad range of transporters tested in this study, we anticipate that amenability to targeted degradation will also be true for other multi-pass membrane proteins, such as GPCRs and ion channels. In a recent report, Li et al. (2020) demonstrated the targeted degradation of the α1-adrenergic receptor, and we anticipate more such efforts to follow. Overall, we have shown here that SLCs from a variety of different families, at different cellular localizations, with different transmembrane structures, are suitable for chemically induced degradation. To the best of our knowledge, d9A-2 is the first chemical degrader targeting multi-pass transmembrane domain proteins as large as SLC9s.
Significance
Using the dTAG system, we report that SLC proteins belonging to different families and subcellular compartments are amenable to induced degradation by PROTACs. Amenability to degradation appears to be related to subcellular localization rather than transmembrane topology and is proteasome dependent. As an example, endogenous dTAG-SLC38A2 is rapidly expressed upon amino acid starvation, and amenable to targeted degradation at the plasma membrane and in every subcellular compartment en route. Furthermore, we report the synthesis of d9A-2, a chimeric compound that leads to the degradation of SLC9A1, as well as several other members of the SLC9 family. We show that d9A-2 impairs cellular pH homeostasis, in accordance with the roles of SLC9A1 in alkalization of intracellular pH. d9A-2 is a potent cytotoxic compound in a range of cancer cell lines, attesting to its potential candidacy for pre-clinical development. We anticipate that targeted SLC degradation will afford new opportunities to study the functions of SLCs in rapid cellular processes and to generate new chemical matter for pharmacological modulation of SLC functions.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GAPDH (G-9) Mouse monoclonal antibody (mAb) | Santa Cruz Biotechnology | sc-365062; RRID: AB_10847862 |
| Anti-pan ACTIN Rabbit polyclonal | Cytoskeleton, Inc. | Cat# AAN01; RRID: AB_10708070 |
| Anti-HA.11 Mouse mAb | BioLegend | Cat# 901516; RRID: AB_2820200 |
| Anti-HA (C29F4) Rabbit mAb | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Anti-HA Mouse mAb, Horseradish Peroxidase Conjugated, Clone HA-7 | Sigma Aldrich | Cat# H6533; RRID: AB_439705 |
| Anti-HA High Affinity Rat mAb | Roche | Cat# 11867423001; RRID: AB_390918 |
| Anti-NHE-1/SLC9A1 Mouse mAb, Clone 54 | Santa Cruz Biotechnology | Cat# sc-136239; RRID: AB_2191254 |
| Anti-GLUT1/SLC2A1 Rabbit mAb (D3J3A) | Cell Signaling Technology | Cat# 12939; RRID: AB_2687899 |
| Anti-SNAT1/SLC38A1 Rabbit mAb (D9L2P) | Cell Signaling Technology | Cat# 36057; RRID: AB_2799092 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11006, RRID: AB_2534074 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11012, RRID: AB_2534079 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11005, RRID: AB_2534073 |
| Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) antibody | Jackson ImmunoResearch Labs | Cat# 111-035-003, RRID: AB_2313567 |
| Peroxidase-AffiniPure Goat Anti-Mouse IgG (H + L) antibody | Jackson ImmunoResearch Labs | Cat# 115-035-003, RRID: AB_10015289 |
| Anti-ASCT2/SLC1A5 Rabbit mAb (D7C12) | Cell Signaling Technology | Cat# 8057; RRID: AB_10891440 |
| Anti-Calreticulin Rabbit mAb (EPR3924) | Abcam | Cat# ab92516; RRID: AB_10562796 |
| Anti-ERp72 XP Rabbit mAb (D70D12) | Cell Signaling Technology | Cat# 5033; RRID: AB_10622112 |
| Anti-GM130 XP Rabbit mAb (D6B1) | Cell Signaling Technology | Cat# 12480; RRID: AB_2797933 |
| Anti-LAMP1 XP Rabbit mAb (D2D11) | Cell Signaling Technology | Cat# 9091; RRID: AB_2687579 |
| Anti-AIF XP Rabbit mAb (D39D2) | Cell Signaling Technology | Cat# 5318; RRID: AB_10634755 |
| Bacterial and Virus Strains | ||
| One Shot™ Stbl3™ Chemically Competent E. coli | Invitrogen | C737303 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BCECF, AM | Invitrogen | B1170 |
| MG132 | Selleckchem | Catalog No.S2619 |
| Bortezomib | Selleckchem | Catalog No.S1013 |
| Pomalidomide | Selleckchem | Catalog No.S1567 |
| MLN4924 | MedchemExpress | HY-70062 |
| Bafilomycin A1 | Enzo Life Sciences | BML-CM110-0100 |
| Brefeldin A | BioLegend | 420601 |
| Monensin | BioLegend | 420701 |
| dTAG13 | Gift from N. Gray’s lab | N/A |
| dTAG7 | Synthesized in house | N/A |
| d9A-1 | This study | N/A |
| d9A-2 | This study | N/A |
| d9A-3 | This study | N/A |
| d9A-4 | This study | N/A |
| d9A-5 | This study | N/A |
| w9A | This study | N/A |
| DAPI | Thermo Scientific | Catalog No. D1306 |
| Hoechst 33342 | Thermo Scientific | Catalog No. 62249 |
| Chloroquine | InvivoGen | tlrl-chq |
| EIPA | Sigma Aldrich | A3085 |
| Critical Commercial Assays | ||
| Intracellular pH Calibration Buffer Kit | Invitrogen | P35379 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | G7572 |
| Experimental Models: Cell Lines | ||
| A375 | Gift from S. Wagner’s lab | N/A |
| A549 | ATCC | CCL-185 |
| BT474 | Gift from S. Nijman’s lab | N/A |
| BV173 | DSMZ | ACC 20 |
| CAKI | MD Anderson | N/A |
| DLD1 | Gift from W. Berger’s lab | N/A |
| DU145 | ATCC | HTB-81 |
| HAP1 | Horizon Genomics | N/A |
| HAP1 -/- SLC38A2 | Horizon Genomics | HZGHC001975c003 |
| HAP1 -/- SLC38A9 | Horizon Genomics | HZGHC000777c011 |
| HCT15 | Gift from C. Gasche’s lab | N/A |
| HCT116 | ATCC | CCL-247 |
| HEK293T | ATCC | CRL-3216 |
| HEL | DSMZ | ACC 11 |
| HELA | Gift from M. Hentze’s lab | N/A |
| HL60 | ATCC | CCL-240 |
| HS578T | Gift from S. Nijman’s lab | N/A |
| HUH7 | JCRB | JCRB0403 |
| K562 | ATCC | CCL-240 |
| KBM7 | Gift from T. Brummelkamp’s lab | N/A |
| KBM7 -/- CRBN | Mayor-Ruiz and Winter, 2019 | N/A |
| KCL22 | DSMZ | KCL22 |
| KG1 | DSMZ | ACC 14 |
| KU812 | DSMZ | ACC 378 |
| LAMA84 | DSMZ | ACC 168 |
| LNCaP | Gift from S. Nijman’s lab | N/A |
| LOVO | Gift from C. Gasche’s lab | N/A |
| LOXIMVI | Gift from S. Wagner’s lab | N/A |
| LS180 | ATCC | ACC 168 |
| MCF10A | Gift from S. Kubicek’s lab | N/A |
| MCF7 | ATCC | HTB-22 |
| MDAMB231 | Gift from W. Berger’s lab | N/A |
| ML2 | DSMZ | ACC 15 |
| MOLM13 | DSMZ | ACC 554 |
| MONOMAC6 | DSMZ | ACC 124 |
| MV411 | DSMZ | ACC 102 |
| NCIH2228 | Gift from E. Haura’s lab | N/A |
| NCIH446 | Gift from J. Bradner’s lab | N/A |
| NCIH460 | ATCC | HTB-177 |
| NOMO1 | DSMZ | ACC 542 |
| PC3 | ATCC | CRL-1435 |
| PC9 | Gift from S. Nijman’s lab | N/A |
| SHI1 | DSMZ | ACC 645 |
| SW480 | Gift from W. Berger’s lab | N/A |
| SW620 | Gift from W. Berger’s lab | N/A |
| T47D | ATCC | HTB-133 |
| T98G | gift from T. Decker’s lab | N/A |
| THP1 | ATCC | TIB-202 |
| Oligonucleotides | ||
| SLC38A2_5-PITCH (CGCGTTACATAGCATCGTACGCGT ACGTGTTTGGTAGCTTGAAGAAGGCCGAAACCAT GGCCAAGCCTTTGTCTCAAGAAGAATCC) |
Sigma Aldrich | N/A |
| SLC38A2_3-PITCH (CATCAGCATCCTAGAGCATCGTACG CGTACGTGTTTGGGGGGAAATACTGAATCGTCCCATCTC TGCTTTCTTCATAGATCCGCCGCCACCCGAC) |
Sigma Aldrich | N/A |
| SLC38A2_gRNA_S (CACCGAATACTGAATCGTCCCATTT) | Sigma Aldrich | N/A |
| SLC38A2_gRNA_AS (AAACAAATGGGACGATCCAGTATCC) | Sigma Aldrich | N/A |
| SLC38A2_FWD (CTGGTACTTTTCCACTCGCCT) | Sigma Aldrich | N/A |
| SLC38A2_REV (AGGAGTTGACTTTCACACCAGC) | Sigma Aldrich | N/A |
| Recombinant DNA | ||
| pcDNA3.1_SLC1A5 N-Flag | Gift from F. Bassermann’s lab | N/A |
| pENTR223_SLC2A1_Fusion | Harvard PlasmID | HsCD00378964 |
| pDONR221_SLC2A3_Closed | Harvard PlasmID | HsCD00043135 |
| pDONR221_SLC2A3_Fusion | Harvard PlasmID | HsCD00039983 |
| pcDNA3.1_SLC16A1 N-term Flag | Gift from F. Bassermann’s lab | N/A |
| pDONR221_SLC38A1_Closed | Harvard PlasmID | HsCD00043034 |
| pDONR221_SLC38A2_Closed | Harvard PlasmID | HsCD00043884 |
| pOTB7_hSLC39A7 | Dharmacon | 3345970 |
| pDONR221_SLC38A9 | RESOLUTE consortium | Addgene #132070 |
| pDONR221_SLC25A1 | RESOLUTE consortium | Addgene #132299 |
| pDONR221_SLC9A1 | RESOLUTE consortium | Addgene #132210 |
| pDONR221_SLC9A2 | RESOLUTE consortium | Addgene #132222 |
| pDONR221_SLC9A3 | RESOLUTE consortium | Addgene #132234 |
| pDONR221_SLC9A4 | RESOLUTE consortium | Addgene #132246 |
| pDONR221_SLC9A5 | RESOLUTE consortium | Addgene #132163 |
| pDONR221_SLC9A6 | RESOLUTE consortium | Addgene #132175 |
| pDONR221_SLC9A7 | RESOLUTE consortium | Addgene #132187 |
| pDONR221_SLC9A8 | RESOLUTE consortium | Addgene #132199 |
| pDONR221_SLC9A9 | RESOLUTE consortium | Addgene #132211 |
| pDONR221_SLC9B1 | RESOLUTE consortium | Addgene #132223 |
| pDONR221_SLC9B2 | RESOLUTE consortium | Addgene #132235 |
| pDONR221_SLC30A9 | RESOLUTE consortium | Addgene #132287 |
| pDONR221_SLC33A1 | RESOLUTE consortium | Addgene #132264 |
| pDONR221_SLC35B2 | RESOLUTE consortium | Addgene #132251 |
| pDONR221_SLC25A26 | RESOLUTE consortium | Addgene #132259 |
| pDONR221_SLC25A19 | RESOLUTE consortium | Addgene #132263 |
| pDONR221_MTCH2 | RESOLUTE consortium | Addgene #131954 |
| pLX305-N-dTAG | Nabet et al., 2018 | Addgene #91797 |
| pLX305-C-dTAG | Nabet et al., 2018 | Addgene #91798 |
| psPAX2 | Gift from D. Trono lab | Addgene #12260 |
| pMD2.G | Gift from D. Trono lab | Addgene #12259 |
| pCRIS-PITCHv2-dTAG-BSD | Nabet et al., 2018 | Addgene #91792 |
| px330A | Sakuma et al., 2016 | Addgene #58766 |
| pRRL strepHA | Bigenzahn et al., 2018 | N/A |
| pRRL GFP | Bigenzahn et al., 2018 | N/A |
| Software and Algorithms | ||
| CellProfiler 3.1.8 | McQuin et al., 2018 | https://cellprofiler.org/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| R version 3.4.4 | R Core Team, 2018 | https://www.R-project.org/ |
| SoftMax® Pro | Molecular Devices | Version 7.0 |
| Python (Version 3) | Python Software Foundation | http://www.python.org |
| SciKit-Learn | Pedregosa et al., 2011 |
http://scikitlearn.org |
| Seaborn | https://seaborn.pydata.org/ |
https://github.com/m waskom/seaborn/tre e/v0.9.0 |
| PRISM v8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
| DMEM w 4.5 g/L Glucose w/o Amino Acids w 2.0 g/L NaHCO3 | PAN BIOTECH | P04-01546S1 |
| MEM Non-essential Amino Acid Solution (100×) | Sigma Aldrich | M7145 |
| TurboFectin 8.0 | OriGene | TF81001 |
| DirectPCR® DNA Extraction System | Viagen Biotech (VWR) | Cat No.: 732-3255 |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Giulio Superti-Furga (GSuperti@cemm.oeaw.ac.at).
Materials Availability
All plasmids and compounds generated in this study will be made available on request but we may require a payment and/or a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or any unique software code
Experimental Model and Subject Details
Cell Lines
KBM7 WT (donor sex: male), HAP1 WT (donor sex: male), HAP1 -/-SLC38A2, HAP1 -/-SLC38A9 and KBM7 -/- CRBN (described in (Mayor-Ruiz et al., 2019)) were maintained in IMDM medium (Gibco). HEK293T (donor sex: female) was maintained in DMEM medium. A375 (donor sex: female), A549 (donor sex: male), BT474 (donor sex: female), BV173 (donor sex: male), CAKI (donor sex: male), DLD1 (donor sex: male), DU145 (donor sex: male), NCIH2228 (donor sex: female), HCT116 (donor sex: male), HCT15 (donor sex: male), HEL (donor sex: male), HELA (donor sex: female), HL60 (donor sex: female), HS578T (donor sex: female), HUH7 (donor sex: male), K562 (donor sex: female), KCL22 (donor sex: female), KG1 (donor sex: male), KU812 (donor sex: male), LAMA84 (donor sex: female), LNCaP (donor sex: male), LOVO (donor sex: male), LOXIMVI (donor sex: male), LS180 (donor sex: female), MCF10A (donor sex: female), MCF7 (donor sex: female), MDAMB231 (donor sex: female), ML2 (donor sex: male), MOLM13 (donor sex: male), NOMO1 (donor sex: female), MONOMAC6 (donor sex: male), MV411 (donor sex: male), NCIH446 (donor sex: male), NCIH460 (donor sex: male), PC3 (donor sex: male), PC9 (donor sex: male), SHI1 (donor sex: male), SW480 (donor sex: male), SW620 (donor sex: male), T47D (donor sex: female), T98G (donor sex: male), and THP1 (donor sex: male) were maintained in RPMI medium.
All media were supplemented with 10% (v/v) FBS and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin). Knock-in single cell clones of HA-dTAG SLC38A2 HAP1 cell line were generated as detailed below and maintained in IMDM medium. For starvation, culture media was replaced with DMEM media without amino acids (Pan Biotech). Where indicated, 5% (v/v) of non essential amino acid mixture (Sigma) was added. Information on endogenous SLC9 family member RNA expression levels was retrieved from (Brockmann et al., 2017). Information regarding tissue of origin was obtained from the Cancer Cell Line Encyclopedia (Barretina et al., 2012).
Plasmids and Stable Cell Line Generation
SLC1A5, SLC2A1, SLC2A3, SLC16A1, SLC38A1, SLC38A2 were obtained or sub-cloned as gateway-compatible pENTR/pDONR vectors (Harvard PlasmID Repository). SLC39A7 was obtained from Dharmacon, GE Healthcare. The following vectors were a gift from the RESOLUTE consortium: SLC38A9, SLC25A1, SLC9A1, SLC9A2, SLC9A3, SLC9A4, SLC9A5, SLC9A6, SLC9A7, SLC9A8, SLC9A9, SLC9B1, SLC9B2, SLC30A9, SLC33A1, SLC35B2, SLC25A19, SLC25A26, and MTCH2. cDNAs were transferred into gateway-compatible lentiviral expression vectors: pLX305 dTAG vectors (Addgene #91797/8), pRRL Strep-HA or pRRL mGFP (described previously (Bigenzahn et al., 2018), EF1a promotor driven expression) using LR recombination (ThermoFisher Scientific). Plasmid purification, using QIAprep spin miniprep kit (Qiagen), was performed from E.coli stbl3 cultures that were chemically transformed and grown overnight with the respective selection antibiotic. For the generation of lentiviral stable overexpression cells, HEK293T cells were transfected with psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) and expression vectors using polyethylenimine (PEI). 12 hours post transfection medium was replaced with fresh medium. The medium, containing the virus, was harvested 48 hours later, filtered (0.45 μm), supplemented with 5μg/ml Polybrene (Hexadimethrine bromide, Sigma) and added to target cells. 48 hours after transduction the medium was supplemented with the respective selection antibiotics (puromycin, blasticidin), to derive stably expressing cell populations. HA-dTAG SLC38A2 knock-in single clone cell line was generated by microhomology-mediated end joining with the PITCh system that has been previously described(Nabet et al., 2018, Sakuma et al., 2016). The two vectors pCRIS-PITCHv2-dTAG-BSD (Addgene #91792) and pX330A (Addgene#58766), were adapted to our target by sub-cloning with the following primers, designed in PITCh designer tool (Nakamae et al., 2017):
| SLC38A2_5-PITCH | CGCGTTACATAGCATCGTACGCGTACGTGTTTGGTAGCTTGAA GAAGGCCGAAACCATGGCCAAGCCTTTGTCTCAAGAAGAATCC |
| SLC38A2_3-PITCH |
CATCAGCATTCTAGAGCATCGTACGCGTACGTGTTTGGGGGG AAATACTGAATCGTCCCATCTCTGCTTTCTTCATAGATCC GCCGCCACCCGAC |
| SLC38A2_gRNA_S | CACCGAATACTGAATCGTCCCATTT |
| SLC38A2_gRNA_AS | AAACAAATGGGACGATTCAGTATTC |
500k HAP1 cells, seeded in a 10cm plate, were transiently co-transfected with the pX330A-sgSLC38A2-SpCas9 vector and the pCRIS-PITChv2-BlastR-P2A-2∗HA-dTAG-3∗(GGGGS)-SLC38A2 repair template vector using TurboFectin 8.0 (OriGene). To enrich for clones, cell medium was supplemented with 10ug/ml blasticidin for ten days. Single cell clones were seeded in 384-well plates by limiting dilution and grown to confluence. To verify clones, genomic DNA was extracted using DirectPCR DNA extraction system (Viagen Biotech), followed by PCR and Sanger sequencing, as well as immunoblotting. Primer sequences for verification were as follows:
| SLC38A2_FWD | CTGGTACTTTTCCACTCGCCT |
| SLC38A2_REV | AGGAGTTGACTTTCACACCAGC |
Method Details
Antibodies and Immunoblotting
For immunoblotting, whole cell extracts were prepared using RIPA lysis buffer (25mM Tris/HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, EDTA-free protease inhibitor (Roche) and phosphatase inhibitors 2+3 (Sigma-Aldrich)). Cell extracts were incubated for 15 min on ice in 50 μL RIPA buffer. Lysates were cleared by centrifugation (20,000 g, 10 min, 4°C). Protein extracts were quantified and normalized using the BCA assay (Thermo Scientific). Where indicated, cleared lysates where treated with the enzyme PNGase (NEB) to deglycosylate the proteins. Lämmli Sample Buffer 4x was added to protein extract samples without boiling. Cell lysates were run on SDS-polyacrylamide gel in tris-glycine running buffer and transferred to nitrocellulose membranes Amersham Protran 0.45 μm (GE Healthcare), according to standard methods. The membranes were incubated with the antibodies indicated below and visualized with horseradish peroxidase–conjugated secondary antibodies (described below) using the ECL Western blotting system (Thermo Scientific). The following antibodies were used: GAPDH (Santa Cruz, sc-365062), Actin (Cyoskeleton Inc., #AAN01), HA (Covance, cat #901516), HA (Cell Signaling, #3724), HA7-HRP (Sigma, H6533), HA (Roche, #11867423001), SLC9A1 (Santa Cruz, sc-136239), SLC2A1 (Cell Signaling, #12939), SLC38A1 (Cell Signaling, #36057), SLC1A5 (Cell Signaling, #8057), Calreticulin (Abcam, #ab92516), ERp72 (Cell Signaling, #5033), GM130 (Cell Signaling, #12480), LAMP1 (Cell Signaling, #9091), AIF (Cell Signaling, #5318). Secondary antibody for imaging were goat anti-Rat IgG Alexa Fluor 488 (ThermoFisher, A11006), goat anti-Rabbit IgG Alexa Fluor 594 (ThermoFisher, A11012), goat anti-Mouse IgG Alexa Fluor 594 (ThermoFisher, A11005). The following secondary antibodies were used for immunoblotting: goat anti-mouse HRP (115-035-003, Jackson ImmunoResearch) and goat anti-rabbit HRP (111-035-003, Jackson ImmunoResearch). The full details of antibodies are described in the Key Resources Table.
Immunofluorescence Staining and Imaging
For immunofluorescence detection of the expressed SLCs in HAP1 cells, cells were seeded onto poly-L-lysine hydrobromide (P6282, Sigma-Aldrich)-coated 96-well CellCarrier Ultra plates (PerkinElmer) or onto 13mm No. 1.5H cover glasses (Paul Marienfeld). Cells were incubated and thereafter fixed with 4% formaldehyde in PBS 1x or with ice cold methanol. After fixation, cells were incubated in blocking buffer (0.3% Saponin (47036, Sigma-Aldrich), 10% FCS in PBS 1x) for one hour at room temperature (RT), rocking. Primary antibody staining was performed for 2 hours at RT in blocking solution and fluorophore conjugated secondary antibody staining was applied for one hour after 3 washes in blocking solution. A full list of antibodies is described in the Key Resources Table. Final washing was performed three times and counterstaining was done for 15 min at RT with DAPI (1:1000 in PBS 1x) for nuclei. Cover glasses were mounted using ProLong Gold antifade reagent (Thermo Scientific) and imaged using a Zeiss LSM-780 confocal microscope. Images were prepared using ImageJ(Schneider et al., 2012) Imaging of CellCarrier Ultra plates was performed on an Opera Phenix High Content Screening System (Perkin Elmer).
Automated Image Analysis
For the quantification of cellular HA-tag intensities, CellProfiler v3.1.8 was used(McQuin et al., 2018). In brief, nuclei were identified from DAPI staining and cells as secondary objects from CellMask images. HA-tag intensities were quantified on a single cell level and consecutive plotting of the data was performed in R 3.4.4.
Chemicals
MG132, Bortezomib, Pomalidomide, were obtained from Selleckchem. Ethylisopropyl amiloride (EIPA) was obtained from Sigma. MLN4924 was obtained from MedchemExpress. Bafilomycin A1 was obtained from Enzo Life Sciences and Chloroquine from Inivogen. Brefeldin A and Monensin were obtained as solutions from BioLegend. dTAG13 was a generous gift from the Nathanael Gray laboratory, Dana Farber Cancer Institute. dTAG7 was synthesized in house. All chemicals were dissolved in DMSO and utilized at the concertation described in respective figures.
Synthesis of the Series d9A-1 – d9A-5
The carboxy-warhead (4'-fluoro-3'-methyl-2-(4-(4-methyl-1H-imidazol-5-yl)piperidin-1-yl)-[1,1'-biphenyl]-4-carboxylic acid) and ensuing d9A PROTAC series (d9A-1, d9A-2. d9A-3, d9A-4, d9A-5) were synthesized by Wuxi AppTec as in the scheme:

Intermediate 3 (2.6 g, 8.74 mmol) was synthesized by the reaction of 1-benzyloxycarbonyl-4-piperidone (15 g, 64.31 mmol) with 4-methyl-1H-imidazole (26.4 g, 321.53 mmol) using 2.5 eq KOtBu as base at 140°C in 6.8% yield. Intermediate 3 (1.2 g, 4.04 mmol) was subjected to hydrogenation in EtOH with Pd/C at 15 psi for 12 h, thereby both reducing the tetrahydropyridine ring and deprotecting the CBz group to piperidine 4 (0.66 g crude) in quantitative yield. Compound 4 easily underwent nucleophilic aromatic substitution with ethyl 3-fluoro-4-nitrobenzoate (0.77 g, 3.59 mmol) in MeCN at 20°C using iPr2NEt as base. The obtained nitroarene 7 (0.57 g, 1.60 mmol) was reduced to the aromatic amine 7G (0.37 g, 1.13 mmol) with iron and acetic acid in 70% yield. This amine was diazotized with 2 eq t-BuONO in MeCN and the diazo intermediate was reacted with CuBr2 at 20°C for 12 h to obtain 28% of bromoarene 7H (130 mg, 0.33 mmol). Bromide 7H (50 mg, 0.12 mmol) underwent Suzuki coupling with 4-fluoro-3-methylphenylboronic acid (59 mg, 0.38 mmol) using 10 mol% Pd(dppf)Cl2 as the catalyst, 2 eq Boc2O for the in situ protection of the substrate, and 2 eq. K2CO3 as the base in dioxane with 10% H2O under MW irradiation at 140°C for 1.5 h. The product of the Suzuki coupling, intermediate 7F (70 mg, 0.17 mmol), was obtained in 70% yield, the Boc group having undergone deprotection in situ. It was then hydrolysed with LiOH in THF / EtOH / H2O to the carboxylic acid, d9A warhead. The compounds d9A-1 to d9A-5 were synthesized from the d9A warhead and the aminoalkyl substituted Cereblon ligands (Shanghai Haoyuan Chemexpress) by amide couplings using HATU and iPr2NEt in DMF(Zhang et al., 2018). Final compounds were delivered at LC purity of 94-99%.
Intermediate 3: MS (ESI) m/z 298.2 [M+H]+
Intermediate 4: 1H NMR (400 MHz, D2O) δ ppm 8.50 (s, 1H), 3.58 (br d, J = 13.0 Hz, 2H), 3.28 - 3.13 (m, 3 H), 2.32 (s, 3H), 2.15 (br d, J = 14.1 Hz, 2H), 1.98 (br dd, J = 3.3, 13.5 Hz, 2H). MS (ESI) m/z 166.2 [M+H]+
Intermediate 7: 1H NMR (400 MHz, DMSO-d6) δ ppm 11.60 (br s, 1 H), 7.92 (d, J = 8.4 Hz, 1 H), 7.80 (s, 1 H), 7.60 (d, J = 8.3 Hz, 1 H), 7.36 (s, 1 H), 4.36 (q, J = 7.1 Hz, 2 H), 3.28 (br d, J = 12.1 Hz, 2 H), 2.99 (br t, J = 11.4 Hz, 2 H), 2.73 (br d, J = 7.8 Hz, 1 H), 2.11 (s, 3 H), 1.77 - 1.91 (m, 2 H), 1.70 (br d, J = 11.5 Hz, 2 H), 1.35 (t, J = 7.1 Hz, 3 H). MS (ESI) m/z 359.3 [M+H]+
Intermediate 7G: MS (ESI) m/z 329.3 [M+H]+
Intermediate 7H: 1H NMR (400 MHz, DMSO-d6) δ ppm 11.71 - 11.49 (m, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.67 (d, J = 1.8 Hz, 1H), 7.53 (dd, J = 1.9, 8.3 Hz, 1H), 7.44 - 7.32 (m, 1H), 4.32 (q, J = 7.1 Hz, 2H), 2.79 (br t, J = 11.2 Hz, 2H), 2.74 - 2.66 (m, 1H), 2.12 (br s, 3H), 1.98 - 1.87 (m, 2H), 1.74 (br d, J = 12.3 Hz, 2H), 1.38 - 1.29 (m, 3H). MS (ESI) m/z 392.1 [M+H]+
Intermediate 7F: MS (ESI) m/z 422.2 [M+H]+
Carboxy-warhead w9A (TFA Salt): 1H NMR (400 MHz, DMSO-d6) δ ppm 14.07 (s, 2H), 12.98 (s, 1H), 8.91 (s, 1H), 7.64 (d, J = 6.6 Hz, 3H), 7.58 (d, J = 6.4 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.21 (t, J = 9.1 Hz, 1H), 3.14 (d, J = 11.6 Hz, 2H), 2.81 (s, 1H), 2.66 (s, 2H), 2.31 (s, 3H), 2.24 (s, 3H), 1.66 (s, 4H). MS (ESI) m/z 394.1 [M+H]+)
13C NMR (100 MHz, d6-DMSO) δ 167.8, 160.5 (d, J = 243 Hz), 150.9, 137.8, 136.2 (d, J = 3.58 Hz), 132.7, 131.9 (d, J = 4.07 Hz), 131.7, 131.0, 128.0 (d, J = 8.00 Hz), 124.7 (d, J = 17.4 Hz), 124.1, 124.0, 119.8, 115.4 (d, J = 22.0 Hz), 51.5, 31.7, 31.1, 14.6, 9.31.
d9A-1 (FA salt): 1H NMR (400MHz, CDCl3-d) δ ppm 7.74 - 7.53 (m, 3H), 7.51 - 7.39 (m, 3H), 7.37 - 7.29 (m, 2H), 7.25 - 7.21 (m, 1H), 7.09 (br dd, J = 8.2, 17.6 Hz, 2H), 6.95 (br t, J = 9.1 Hz, 1H), 4.88 - 4.79 (m, 1H), 4.55 - 4.45 (m, 2H), 3.71 - 3.37 (m, 18H), 3.14 (br s, 2H), 2.83 - 2.47 (m, 6H), 2.24 (s, 3H), 2.15 (br s, 3H), 2.08 - 1.99 (m, 1H), 1.75 - 1.48 (m, 4H). MS (ESI) m/z 882.3 [M+H]+)
d9A-2 (TFA salt): 1H NMR (400 MHz, DMSO-d6) δ ppm 14.14 (d, J = 13.2 Hz, 1H), 11.11 (s, 1H), 8.92 (s, 1H), 8.57 (t, J = 5.6 Hz, 1H), 7.61 (dd, J = 11.2, 6.8 Hz, 1H), 7.56 (t, J = 5.6 Hz, 3H), 7.32 – 7.26 (m, 1H), 7.23 – 7.16 (m, 1H), 7.13 (d, J = 8.8 Hz, 1H), 7.04 (d, J = 7.2 Hz, 1H), 6.60 (s, 1H), 5.06 (dd, J = 12.8, 5.2 Hz, 1H), 3.60 (t, J = 5.2 Hz, 2H), 3.55 – 3.51 (m, 9H), 3.50 (s, 4H), 3.43 (dt, J = 11.2, 5.2 Hz, 4H), 3.32 (s, 1H), 3.15 (d, J = 10.8 Hz, 2H), 2.94 – 2.79 (m, 3H), 2.71 – 2.58 (m, 3H), 2.30 (s, 3H), 2.24 (s, 3H), 2.12 (s, 1H), 2.00 (dd, J = 13.6, 8.0 Hz, 1H), 1.61 (dd, J = 48.0, 10.8 Hz, 4H). MS (ESI) m/z 869 [M+H]+)
d9A-3 (FA salt): 1H NMR (400MHz, DMSO-d6) δ ppm 11.14 (br s, 1H), 8.54 (br t, J = 5.2 Hz, 1H), 8.03 - 7.95 (m, 1H), 7.80 (t, J = 7.9 Hz, 1H), 7.64 - 7.45 (m, 6H), 7.42 - 7.34 (m, 2H), 7.27 (d, J = 7.7 Hz, 1H), 7.20 (br t, J = 9.0 Hz, 1H), 5.12 (dd, J = 5.5, 13.0 Hz, 1H), 4.79 (s, 2H), 3.56 - 3.41 (m, 16H), 3.12 (br d, J = 10.4 Hz, 2H), 2.98 - 2.76 (m, 1H), 2.66 - 2.58 (m, 5H), 2.35 - 2.29 (m, 3H), 2.08 (s, 3H), 2.06 - 1.99 (m, 1H), 1.75 - 1.63 (m, 6H), 1.58 - 1.50 (m, 2H). MS (ESI) m/z 910.2 [M+H]+)
d9A-4 : 1H NMR (400MHz, DMSO-d6) δ ppm 11.54 (br s, 1H), 11.16 (br s, 1H), 8.56 (br t, J = 5.1 Hz, 1H), 8.05 (br t, J = 5.4 Hz, 1H), 7.86 - 7.79 (m, 1H), 7.61 (br s, 1H), 7.57 (br s, 2H), 7.55 - 7.48 (m, 2H), 7.42 (d, J = 8.4 Hz, 1H), 7.37 (s, 1H), 7.30 (d, J = 7.8 Hz, 1H), 7.22 (t, J = 9.2 Hz, 1H), 5.15 (dd, J = 5.5, 13.1 Hz, 1H), 4.82 (s, 2H), 3.62 - 3.55 (m, 6H), 3.54 - 3.42 (m, 5H), 3.14 (br d, J = 11.1 Hz, 2H), 2.98 - 2.87 (m, 1H), 2.73 - 2.61 (m, 6H), 2.33 (s, 3H), 2.16 - 2.02 (m, 4H), 1.74 - 1.50 (m, 4H). MS (ESI) m/z 838.3 [M+H]+)
d9A-5 : 1H NMR (400MHz, DMSO-d6) δ ppm 11.55 (br s, 1H), 11.16 (br s, 1H), 8.48 (br s, 1H), 7.99 (br s, 1H), 7.85 (br t, J = 7.8 Hz, 1H), 7.53 (br s, 5H), 7.43 (br d, J = 8.6 Hz, 1H), 7.37 (s, 1H), 7.29 (br d, J = 7.5 Hz, 1H), 7.23 (br t, J = 9.2 Hz, 1H), 5.16 (br dd, J = 5.1, 13.0 Hz, 1H), 4.81 (s, 2H), 3.29 (br d, J = 5.5 Hz, 2H), 3.23 - 3.10 (m, 4H), 2.99 - 2.87 (m, 1H), 2.72 - 2.60 (m, 4H), 2.34 (s, 3H), 2.11 (br s, 3H), 2.06 (br s, 1H), 1.68 (br d, J = 10.8 Hz, 2H), 1.62 - 1.45 (m, 7H), 1.36 (br s, 4H). MS (ESI) m/z 806.3 [M+H]+)
Synthesis of the Warhead (w9A), Related to d9A
The SLC9A1 warhead (1-(4'-fluoro-3'-methyl-[1,1'-biphenyl]-2-yl)-4-(4-methyl-1H-imidazol-5-yl)piperidine) was synthesized by Chempartner, as in the scheme:

To a mixture of 1-fluoro-2-nitrobenzene (0.6 g, 4.25 mmol) and 4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidine (1.01 g, 3.40 mmol) in CH3CN (15 mL) was added DIEA (1.1 g, 8.5 mmol). The mixture was stirred at 60oC for 12h. The mixture was concentrated in vacuo. The residue was purified by silica gel chromatography on silica (Petroleum ether/Ethyl acetate = 1:1, v/v) to afford 4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)-1-(2-nitrophenyl)piperidine (1.3 g, 91.3%) as a yellow oil (Mass: find peak 417.1 [M+H]+).
To a solution of 4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)-1-(2-nitrophenyl)piperidine (1.3 g, 3.12 mmol) in MeOH (20 mL) was added Pd/C (0.5 g). The mixture was stirred at 20~25oC for 12h under H2 (15 psi). The mixture was filtered and the mother liquid was concentrated in vacuo to give desired product 2-(4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidin-1-yl)aniline (1.1 g, 85.4% yield) as a yellow oil (Mass: find peak 387.1 [M+1]+).
To a solution of 2-(4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidin-1-yl)aniline (1.1 g, 2.85 mmol) in CH3CN (20 mL) was added t-BuNO2 (0.59 g, 5.70 mmol) and CuBr2 (0.64 g, 2.85 mmol) under ice-bath. The mixture was stirred at 20~25oC for 6h. The reaction was monitored by LCMS and after completion the mixture was extracted with EtOAc (50 mL x 3). The combined organic layers were concentrated in vacuo. The residue was purified by chromatography (Petroleum ether: Ethyl acetate = 1:1, v/v) to afford 1-(2-bromophenyl)-4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidine (0.6 g, 46.8%) as a a yellow solid (Mass: find peak 450.0 [M+1]+).
To a solution of 1-(2-bromophenyl)-4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidine (0.6 g, 1.33 mmol) and (4-fluoro-3-methylphenyl)boronic acid (0.25 g, 1.60 mmol) in dioxane/H2O (6 mL/ 2 mL) was added K2CO3 (0.37 g, 2.66 mmol) and Pd(dppf)Cl2 (95 mg, 0.13 mmol). The mixture was stirred at 100oC under N2 for 5h. The reaction was monitored by LCMS and after completion the mixture was concentrated in vacuo. The residue was purified by chromatography (Petroleum ether: Ethyl acetate = 1:1, v/v) to afford1-(4'-fluoro-3'-methyl-[1,1'-biphenyl]-2-yl)-4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidine (0.35 g, 54.8%) as a yellow solid (Mass: find peak 480.1 [M+1]+).
To a solution of 1-(4'-fluoro-3'-methyl-[1,1'-biphenyl]-2-yl)-4-(4-methyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-imidazol-5-yl)piperidine (0.6 g, 0.42 mmol) in THF (6 mL) was added TBAF (2.1 mL, 2.1 mmol, 1N in THF). The mixture was stirred at 60oC for 12 h. The reaction was monitored by LCMS and after completion the mixture was extracted with EtOAc (20 mL x 3). The combined organic layers were concentrated in vacuo. The residue was purified by Prep-HPLC to afford 1-(4'-fluoro-3'-methyl-[1,1'-biphenyl]-2-yl)-4-(4-methyl-1H-imidazol-5-yl)piperidine (50 mg, 34.3%) as a white solid.(Mass: find peak 350.2 [M+1]+).
Warhead w9A: 1H NMR (500 MHz, MeOD) δ 7.57-7.51 (m, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.42 (s, 1H), 7.31 – 7.23 (m, 1H), 7.20 (dd, J = 7.5, 1.6 Hz, 1H), 7.13 (d, J = 7.9 Hz, 1H), 7.10-7.04 (m, 2H), 3.20 (d, J = 11.8 Hz, 2H), 2.64 (t, J = 11.9 Hz, 3H), 2.34 (d, J = 1.5 Hz, 3H), 2.17 (s, 3H), 1.75 (qd, J = 12.5, 3.4 Hz, 2H), 1.63 (d, J = 10.7 Hz, 2H).
13C NMR (100 MHz, MeOD) δ 160.2 (d, J = 243 Hz), 151.0, 137.3 (d, J = 3.74 Hz), 134.3, 132.4, 131.5 (d, J = 4.72 Hz), 130.8, 127.9, 127.7 (d, J = 7.88 Hz), 124.0 (d, J = 16.9 Hz), 122.2, 118.3, 114.2 (d, J = 22.5 Hz), 52.1, 33.5, 31.9, 13.2, 9.26.
pHi Measurements
HAP1 cells were plated on 96 black clear-well dishes coated with poly-L Lysine (Sigma), at 40,000 cells per well, in IMDM medium supplemented with 10% (v/v) FBS. On the next day, intracellular pH (pHi) was determined by the following procedure: Where indicated, cells were pre-treated for eight hours with d9A-2. Next, cells were washed once with IMDM medium without FBS. Cells were stained for 30 minutes with 3 μM BCECF, AM (2',7'-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein, Acetoxymethyl Ester, Invitrogen), followed by two washes with RPMI media (phenol red free). Cells were then incubated in RFH media: RPMI media (phenol red free) supplemented with 10% (v/v) FBS and 25 mM HEPES (pH = 7.4).
To determine steady-state pHi, fluorescence of BCECF at Ex490/Em535 and Ex440/Em535 was acquired every 60 seconds using a plate reader (SpectraMax i3x; Molecular Dynamics). Following 5 minutes in steady-state, cells were loaded by treatment for 15 minutes with NH4Cl (15mM) in RFH media. To examine the recovery from acid load, media was then replaced with fresh RFH media. Where indicated, w9A or EIPA were added with the recovery media. Each condition was measured in six replicates. The fluorescence ratios were converted to pHi by calibrating the fluorescence in each well, at each time point, with an intracellular pH calibration kit (pH range 5.5-7.5, Invitrogen), measured simultaneously on the same plate. Cells treated with 1 μM d9A-2 were calibrated with a matched calibration curve, while all other samples were calibrated based on the untreated cells’ calibration curve. For pHi calculations and consecutive plotting, Python 3.7.3 was used, with SciKit-Learn(Pedregosa et al., 2011) and Seaborn. In brief, a linear regression model, based on the conducted pH calibration curves was used to calculate pHi of samples; pHi differences (ΔpHi) represent the mean calculated pH of treated cells subtracted from the mean calculated pHi of untreated cells at 5, 10, 15 and 30 min after the recorded pHi-minimum of each sample.
Cellular Viability Assay
For comparison of different cancer cell lines, compounds were transferred on black 384-well plates using an acoustic liquid handler (Echo, Labcyte). Cells were seeded at densities of 1,000 cells per well for adherent cell lines and 3,000 cells for suspension cell lines. Viability was measured after 72 hours using the CellTiter-Glo assay (Promega) on a plate reader (SpectraMax i3x, Molecular Probes). All measurements were done in technical quadruplicates. Data were normalized to DMSO treated-controls, four-parameter dose response fitting curves were obtained using the R package drc(Ritz et al., 2015) and area above the curve was calculated with R package PharmacoGx(Smirnov et al., 2016). Correlations between d9A-2 toxicity and EIPA or bortezomib toxicity were done using R package ggpubr (spearman correlation method). For comparisons made specifically for KBM7 cells (Figures 5B and 5C), cells were seeded in 96-well plates, at a density of 15,000 cells per well in triplicates. Viability was measured and quantified as described above, and plotted using GraphPad Prism V8. Data is presented as mean +/- SD.
Quantification and Statistical Analysis
Statistical analysis was performed in Microsoft Excel Software by one-tailed t-test, and the normality of distributions was tested using the Shapiro–Wilk test. Data are represented as mean ± SD. The statistical parameters are found in the respective text and figure legends.
Acknowledgments
We are thankful to all the members of the Superti-Furga and Winter laboratories for discussions, feedback and reagents. We are grateful to Stefan Kubicek for critical input and to Bojan Villagos, Adrián César-Razquin, Manuele Rebsamen, Cristina Mayor-Ruiz, Tea Pemovska, Enrico Girardi, and Andrea Casiraghi for their valuable input. CeMM, the Superti-Furga and Winter laboratories are supported by the Austrian Academy of Sciences. We acknowledge receipt of third-party funds from the Austrian Science Fund (FWF F4711-B20 Myeloid Neoplasms. F.K.), Vienna Science and Technology Fund (WWTF LS17-051 Cellular Color Chart V.D.), the European Research Council (ERC AdG 695214 GameofGates, to A.B., M.D.P., and P.E.). M.D.P. is a recipient of a Fulbright Study/Research Grant.
Author Contributions
A.B., G.E.W., and G.S.-F. conceived the study. A.B., M.D.P., P.E., V.D., and F.K. performed the experiments, analyzed the data, and prepared the figures. A.B., M.D.P., G.E.W., and G.S.-F. wrote the manuscript.
Declaration of Interests
A.B., G.E.W., and G.S.-F. are co-authors of a patent application and co-founders of a company related to SLCs. G.S.-F. is the Academic Project Coordinator of the IMI grant RESOLUTE in partnership with Pfizer, Novartis, Bayer, Sanofi, Boehringer Ingelheim, and Vifor Pharma. The G.S.-F. laboratory receives funds from Pfizer and Boehringer Ingelheim.
Published: May 7, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chembiol.2020.04.003.
Supplemental Information
References
- Amith S.R., Fliegel L. Regulation of the Na+/H+ exchanger (NHE1) in breast cancer metastasis. Cancer Res. 2013;73:1259–1264. doi: 10.1158/0008-5472.CAN-12-4031. [DOI] [PubMed] [Google Scholar]
- Atwal K.S., O’Neil S.V., Ahmad S., Doweyko L., Kirby M., Dorso C.R., Chandrasena G., Chen B.-C., Zhao R., Zahler R. Synthesis and biological activity of 5-aryl-4-(4-(5-methyl-1H-imidazol-4-yl)piperidin-1-yl)pyrimidine analogs as potent, highly selective, and orally bioavailable NHE-1 inhibitors. Bioorg. Med. Chem. Lett. 2006;16:4796–4799. doi: 10.1016/j.bmcl.2006.06.077. [DOI] [PubMed] [Google Scholar]
- Bai X., Moraes T.F., Reithmeier R.A.F. Structural biology of solute carrier (SLC) membrane transport proteins. Mol. Membr. Biol. 2017;34:1–32. doi: 10.1080/09687688.2018.1448123. [DOI] [PubMed] [Google Scholar]
- Barkai N., Leibler S. Robustness in simple biochemical networks. Nature. 1997;387:913–917. doi: 10.1038/43199. [DOI] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner M., Patel H., Barber D.L. Na(+)/H(+) exchanger NHE1 as plasma membrane scaffold in the assembly of signaling complexes. Am. J. Physiol. Cell Physiol. 2004;287:C844–C850. doi: 10.1152/ajpcell.00094.2004. [DOI] [PubMed] [Google Scholar]
- Bigenzahn J.W., Collu G.M., Kartnig F., Pieraks M., Vladimer G.I., Heinz L.X., Sedlyarov V., Schischlik F., Fauster A., Rebsamen M. LZTR1 is a regulator of RAS ubiquitination and signaling. Science. 2018;362:1171–1177. doi: 10.1126/science.aap8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D.P., Crews C.M. Targeted protein degradation by small molecules. Annu. Rev. Pharmacol. Toxicol. 2017;57 doi: 10.1146/annurev-pharmtox-010715-103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D.P., Mares A., Smith I.E.D., Ko E., Campos S., Miah A.H., Mulholland K.E., Routly N., Buckley D.L., Gustafson J.L. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M., Jiang B., Bauer S., Donovan K.A., Liang Y., Wang E.S., Nowak R.P., Yuan J.C., Zhang T., Kwiatkowski N. Homolog-selective degradation as a strategy to probe the function of CDK6 in AML. Cell Chem. Biol. 2019;26:300–306.e9. doi: 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann M., Blomen V.A., Nieuwenhuis J., Stickel E., Raaben M., Bleijerveld O.B., Altelaar A.F.M., Jae L.T., Brummelkamp T.R. Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling. Nature. 2017;546:307–311. doi: 10.1038/nature22376. [DOI] [PubMed] [Google Scholar]
- Burslem G.M., Smith B.E., Lai A.C., Jaime-Figueroa S., McQuaid D.C., Bondeson D.P., Toure M., Dong H., Qian Y., Wang J. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol. 2018;25:67–77.e3. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem G.M., Schultz A.R., Bondeson D.P., Eide C.A., Savage Stevens S.L., Druker B.J., Crews C.M. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 2019;79:4744–4753. doi: 10.1158/0008-5472.CAN-19-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- César-Razquin A., Snijder B., Frappier-Brinton T., Isserlin R., Gyimesi G., Bai X., Reithmeier R.A., Hepworth D., Hediger M.A., Edwards A.M. A call for systematic research on solute carriers. Cell. 2015;162:478–487. doi: 10.1016/j.cell.2015.07.022. [DOI] [PubMed] [Google Scholar]
- Colas C., Ung P.M.-U., Schlessinger A. SLC transporters: structure, function, and drug discovery. Medchemcomm. 2016;7:1069–1081. doi: 10.1039/C6MD00005C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coothankandaswamy V., Elangovan S., Singh N., Prasad P.D., Thangaraju M., Ganapathy V. The plasma membrane transporter SLC5A8 suppresses tumour progression through depletion of survivin without involving its transport function. Biochem. J. 2013;450:169–178. doi: 10.1042/BJ20121248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counillon L., Bouret Y., Marchiq I., Pouysségur J. Na+/H+ antiporter (NHE1) and lactate/H+ symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta. 2016;1863:2465–2480. doi: 10.1016/j.bbamcr.2016.02.018. [DOI] [PubMed] [Google Scholar]
- El-gebali S., Bentz S., Hediger M.A., Anderle P. Molecular aspects of medicine solute carriers ( SLCs ) in cancer q. Mol. Aspects Med. 2013;34:719–734. doi: 10.1016/j.mam.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Erb M.A., Scott T.G., Li B.E., Xie H., Paulk J., Seo H.-S., Souza A., Roberts J.M., Dastjerdi S., Buckley D.L. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 2017;543:270–274. doi: 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faillie J.-L. Pharmacological aspects of the safety of gliflozins. Pharmacol. Res. 2017;118:71–81. doi: 10.1016/j.phrs.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Goel P., Manning J.A., Kumar S. NEDD4-2 (NEDD4L): the ubiquitin ligase for multiple membrane proteins. Gene. 2015;557:1–10. doi: 10.1016/j.gene.2014.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harguindey S., Arranz J.L., Polo Orozco J.D., Rauch C., Fais S., Cardone R.A., Reshkin S.J. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs—an integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J. Transl. Med. 2013;11:282. doi: 10.1186/1479-5876-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger M.A., Clémençon B., Burrier R.E., Bruford E.A. The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol. Aspects Med. 2013;34:95–107. doi: 10.1016/j.mam.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltai T. Cancer: fundamentals behind pH targeting and the double-edged approach. Onco Targets Ther. 2016;9:6343–6360. doi: 10.2147/OTT.S115438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen A.S., Andersen J., Jørgensen T.N., Sørensen L., Eriksen J., Loland C.J., Strømgaard K., Gether U. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol. Rev. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamothe S.M., Zhang S. Chapter five. Ubiquitination of ion channels and transporters. Prog. Mol. Biol. Transl Sci. 2016;141:161–223. doi: 10.1016/bs.pmbts.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Li Z., Lin Y., Song H., Qin X., Yu Z., Zhang Z., Dong G., Li X., Shi X., Du L. First small-molecule PROTACs for G protein-coupled receptors: inducing α1A-adrenergic receptor degradation. Acta Pharm. Sin. B. 2020 doi: 10.1016/j.apsb.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L., Yee S.W., Kim R.B., Giacomini K.M. SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 2015;14:543–560. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiselle F.B., Casey J.R. Measurement of intracellular pH. Methods Mol. Biol. 2010;637:311–331. doi: 10.1007/978-1-60761-700-6_17. [DOI] [PubMed] [Google Scholar]
- MacGurn J.A., Hsu P.-C., Emr S.D. Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 2012;81:231–259. doi: 10.1146/annurev-biochem-060210-093619. [DOI] [PubMed] [Google Scholar]
- Mayor-Ruiz C., Winter G.E. Identification and characterization of cancer vulnerabilities via targeted protein degradation. Drug Discov. Today Technol. 2019;31:81–90. doi: 10.1016/j.ddtec.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Mayor-Ruiz C., Jaeger M.G., Bauer S., Brand M., Sin C., Hanzl A., Mueller A.C., Menche J., Winter G.E. Plasticity of the cullin-RING ligase repertoire shapes sensitivity to ligand-induced protein degradation. Mol. Cell. 2019;75:849–858.e8. doi: 10.1016/j.molcel.2019.07.013. [DOI] [PubMed] [Google Scholar]
- McQuin C., Goodman A., Chernyshev V., Kamentsky L., Cimini B.A., Karhohs K.W., Doan M., Ding L., Rafelski S.M., Thirstrup D. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 2018;16:e2005970. doi: 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meima M.E., Mackley J.R., Barber D.L. Beyond ion translocation: structural functions of the sodium-hydrogen exchanger isoform-1. Curr. Opin. Nephrol. Hypertens. 2007;16:365–372. doi: 10.1097/MNH.0b013e3281bd888d. [DOI] [PubMed] [Google Scholar]
- Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamae K., Nishimura Y., Takenaga M., Nakade S., Sakamoto N., Ide H., Sakuma T., Yamamoto T. Establishment of expanded and streamlined pipeline of PITCh knock-in—a web-based design tool for MMEJ-mediated gene knock-in, PITCh designer, and the variations of PITCh, PITCh-TG and PITCh-KIKO. Bioengineered. 2017;8:302–308. doi: 10.1080/21655979.2017.1313645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardi F., Hoffmann T.M., Stretton C., Cwiklinski E., Taylor P.M., Hundal H.S. Proteasomal modulation of cellular SNAT2 (SLC38A2) abundance and function by unsaturated fatty acid availability. J. Biol. Chem. 2015;290:8173–8184. doi: 10.1074/jbc.M114.625137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omasits U., Ahrens C.H., Müller S., Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30:884–886. doi: 10.1093/bioinformatics/btt607. [DOI] [PubMed] [Google Scholar]
- Parks S.K., Chiche J., Pouysségur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer. 2013;13:611–623. doi: 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- Pavlova N.N., Thompson C.B. Perspective the emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen V.L., Hsu M.Y., Rädecke K.S., Wyart E., Vazeille T., Bouzin C., Porporato P.E., Sonveaux P. Monocarboxylate transporter MCT1 promotes tumor metastasis independently of its activity as a lactate transporter. Cancer Res. 2017;77:5591–5601. doi: 10.1158/0008-5472.CAN-17-0764. [DOI] [PubMed] [Google Scholar]
- Pedersen S.F., King S.A., Nygaard E.B., Rigor R.R., Cala P.M. NHE1 inhibition by amiloride- and benzoylguanidine-type compounds inhibitor binding loci deduced from chimeras of NHE1 homologues with endogenous differences in inhibitor sensitivity. J. Biol. Chem. 2007;282:19716–19727. doi: 10.1074/jbc.M701637200. [DOI] [PubMed] [Google Scholar]
- Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. Scikit-learn: machine learning in Python. Machine Learn. Python. 2011;6:2825–2830. [Google Scholar]
- Pettersson M., Crews C.M. PROteolysis TArgeting Chimeras (PROTACs)—past, present and future. Drug Discov. Today Technol. 2019;31:15–27. doi: 10.1016/j.ddtec.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshkin S.J., Bellizzi A., Caldeira S., Albarani V., Malanchi I., Poignee M., Alunni-Fabbroni M., Casavola V., Tommasino M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000;14:2185–2197. doi: 10.1096/fj.00-0029com. [DOI] [PubMed] [Google Scholar]
- Reshkin S.J., Greco M.R., Cardone R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. B Biol. Sci. 2014;369:20130100. doi: 10.1098/rstb.2013.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz C., Baty F., Streibig J.C., Gerhard D. Dose-response analysis using R. PLoS One. 2015;10:e0146021. doi: 10.1371/journal.pone.0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotte A., Pasham V., Eichenmüller M., Bhandaru M., Föller M., Lang F. Upregulation of Na+/H+ exchanger by the AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 2010;398:677–682. doi: 10.1016/j.bbrc.2010.06.135. [DOI] [PubMed] [Google Scholar]
- Sainz B., Barretto N., Martin D.N., Hiraga N., Imamura M., Hussain S., Marsh K.A., Yu X., Chayama K., Alrefai W.A. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 2012;18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma T., Nakade S., Sakane Y., Suzuki K.-I.T., Yamamoto T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016;11:118–133. doi: 10.1038/nprot.2015.140. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab A., Fabian A., Hanley P.J., Stock C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012;92:1865–1913. doi: 10.1152/physrev.00018.2011. [DOI] [PubMed] [Google Scholar]
- Sinclair L.V., Rolf J., Emslie E., Shi Y.-B., Taylor P.M., Cantrell D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013;14:500–508. doi: 10.1038/ni.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov P., Safikhani Z., El-Hachem N., Wang D., She A., Olsen C., Freeman M., Selby H., Gendoo D.M.A., Grossmann P. PharmacoGx: an R package for analysis of large pharmacogenomic datasets. Bioinformatics. 2016;32:1244–1246. doi: 10.1093/bioinformatics/btv723. [DOI] [PubMed] [Google Scholar]
- Stock C., Pedersen S.F. Roles of pH and the Na+/H+ exchanger NHE1 in cancer: from cell biology and animal models to an emerging translational perspective? Semin. Cancer Biol. 2017;43:5–16. doi: 10.1016/j.semcancer.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Deberardinis R.J. Review understanding the intersections between metabolism and cancer biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.E., Buckley D.L., Paulk J., Roberts J.M., Souza A., Dhe-Paganon S., Bradner J.E. Drug development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1 doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Han X.-R., Yang X., Jiang B., Liu J., Xiong Y., Jin J. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK) Eur. J. Med. Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Ma D., Wang Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019;37:21–30. doi: 10.1002/cbf.3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or any unique software code