

Abstract
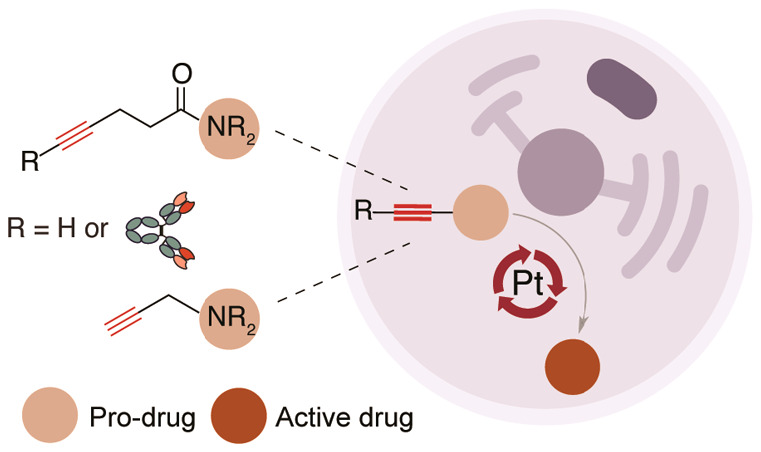
The ability to create ways to control drug activation at specific tissues while sparing healthy tissues remains a major challenge. The administration of exogenous target-specific triggers offers the potential for traceless release of active drugs on tumor sites from antibody–drug conjugates (ADCs) and caged prodrugs. We have developed a metal-mediated bond-cleavage reaction that uses platinum complexes [K2PtCl4 or Cisplatin (CisPt)] for drug activation. Key to the success of the reaction is a water-promoted activation process that triggers the reactivity of the platinum complexes. Under these conditions, the decaging of pentynoyl tertiary amides and N-propargyls occurs rapidly in aqueous systems. In cells, the protected analogues of cytotoxic drugs 5-fluorouracil (5-FU) and monomethyl auristatin E (MMAE) are partially activated by nontoxic amounts of platinum salts. Additionally, a noninternalizing ADC built with a pentynoyl traceless linker that features a tertiary amide protected MMAE was also decaged in the presence of platinum salts for extracellular drug release in cancer cells. Finally, CisPt-mediated prodrug activation of a propargyl derivative of 5-FU was shown in a colorectal zebrafish xenograft model that led to significant reductions in tumor size. Overall, our results reveal a new metal-based cleavable reaction that expands the application of platinum complexes beyond those in catalysis and cancer therapy.
Introduction
The targeting of potent drugs with tumor-specific ligands is an essential feature of drug delivery and cancer therapy.1 Notable in this field are antibody–drug conjugates (ADCs) that use an antibody to transport a drug to cancerous cells and endogenously release it by hydrolysis (low pH, reduction of disulfide bonds) or by proteolysis (e.g., cathepsin B protease).2,3 Although the cleavage of ADC linkers with endogenous triggers is the simplest method for drug release, external small-molecule triggers for extracellular drug release may be more advantageous because they avoid any disparity in cleavage rates caused by variable biology across subjects, and drug release is not dependent on the concentrations of cellular triggers.4−7 In fact, ADCs built with protease cleavable linkers for drug release have been shown recently to not depend on the cathepsin B protease function for efficient and targeted cancer-cell killing.4 The promise of controlled prodrug activation has fueled research into new triggers that enable bond-cleavage reactions to unleash bioorthogonal protecting groups, which deactivate otherwise potent drugs.8 Robillard and co-workers pioneered the development of tetrazine-triggered drug delivery from ADCs.9 By using a noninternalizing ADC consisting of a diabody conjugated to trans-cyclooctene-linked drug monomethyl auristatin E (MMAE),10 the allylic carbamate-containing linker can rapidly react with a tetrazine through an inverse-electron-demand Diels–Alder reaction.10,11 The drug is released within the extracellular tumor environment and showed efficacy in delaying tumor growth in xenograft mice models.10 Other chemical- or light-mediated decaging reactions have also been developed with an array of applications that range from in situ activation of prodrugs to the gain-of-function study on proteins.12−15
Although the recent results with noninternalizing ADCs for click-triggered drug release show promise, there are several issues that remain to be improved, such as the lack of tumor-selectivity of the chemical triggers or their short in vivo retention times that may result in reduced release of the cytotoxic payloads and thus lower efficacy. Additionally, in such applications the tumor payload concentration is determined by the cell-surface antigen expression, which in some cases may be too low to achieve a useful therapeutic response.7,16 This contrasts with the use of internalizing ADCs that have an accumulative effect inside the tumor cells.7,16,17
Metal-mediated decaging of prodrugs has been more extensively reported than small-molecule-mediated decaging.18 Unlike chemical triggers, transition metals can be catalytic, which allows their use in substoichiometric amounts. In these cases, only very small amounts of catalytic metal are required to achieve the desired pharmacologic effect, thereby reducing toxicity and side reactions.19,20 This feature was recently demonstrated by Weissleder and co-workers using palladium nanoparticles that accumulate in tumor cells and serve as cellular catalysts for the activation of different model prodrugs and resulted in tumor growth inhibition.21
Palladium-mediated decaging is indeed the most studied method for prodrug activation, which relies on the cleavage of terminal propargylic and allylic carbamates moieties introduced into small molecule drugs.21−26 Recently, our group developed an internal bifunctional thioether propargyl carbamate linker with a conjugating unit for protein modification and MMAE for palladium-mediated drug release from a nanobody-drug conjugate in cellular systems.27 Other metals, such as ruthenium28−30 and gold,31,32 have been also explored for cleavage and drug release. One of the latest additions to this field was the recent report by Peng Chen and co-workers33 on a copper-releasable reaction for protein gain-of-function and drug activation. Together, these examples highlight the potential of metal-mediated cleavage as a means to achieve controlled and chemically defined drug release.
Whereas the utility of the above-mentioned metals for decaging applications has been extensively demonstrated, other metals have not yet been sufficiently explored. For instance, platinum is widely used in catalysis,34 but has found few applications in chemical biology, possibly as a result of its intrinsic cytotoxicity. However, in the context of cancer therapy, we hypothesized that the use of platinum complexes [e.g., Cisplatin (CisPt) used in the clinic]35,36 as catalysts for cleavage reactions could be propitious for bioorthogonal activation of prodrugs in tumor cells.
“Bioorthogonal” is perhaps unfitting terminology for a compound known to react with water, nucleic acids, amino acids, and proteins.37,38 However, CisPt is one of the most commonly used chemotherapy drugs, being used to treat up to 20% of cancer patients.39,40 CisPt was deemed a suitable reagent for the development of a drug decaging reaction because it is highly reactive (half-life in humans of ∼30 min),41,42 accumulates in the tumor, and most importantly, is not present in human biology.43,44 In this way, the activation of prodrugs at the tumor site when the chemical trigger has already accumulated may represent a major achievement. It may be conceivable for metal concentrations to reach 0.25 to 3.7 μgs per g of tumor.41,45,46 For a 1 cm3 tumor (approximately 1 g wet weight), the concentration of CisPt is estimated to be 0.83–12.3 μM.47
Therefore, we were interested in investigating new biorthogonal cleavage reactions catalyzed by platinum for applications in prodrug activation. Here, we demonstrate that pentynoyl tertiary amide and N-propargyl handles introduced into small-molecule drugs are successfully decaged in aqueous solution and cell media using nontoxic amounts of platinum salts (Scheme 1). This strategy was successfully applied to small molecule prodrug activation (MMAE and 5-FU) and further extended to drug release from a noninternalizing ADC in cancer cells. Finally, we show that CisPt-mediated bond cleavage can be used to activate a 5-FU prodrug in a zebrafish xenograft model for treatment of colorectal cancer.
Scheme 1. Platinum-Mediated Bioorthogonal Bond Cleavage.
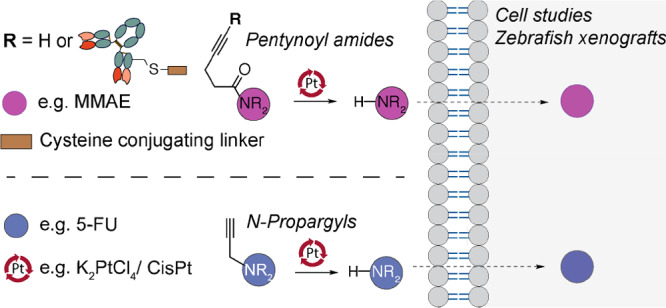
Secondary amines protected in the form of a tertiary pentynoyl amide (top) or N-propargyl (bottom) can be selectively deprotected by platinum reagents like the chemotherapeutic drug CisPt. This strategy was explored for drug activation of the protected MMAE and 5-FU drugs and extended for drug release from an ADC in cancer cells. Ultimately, CisPt-mediated activation of a “5-FU-propargyl prodrug” was evaluated in a zebrafish xenograft model for treatment of colorectal cancer.
Results and Discussion
Engineering of a Platinum-Mediated Decaging Reaction
From studies on the reactivity of platinum complexes, it is apparent that platinum shares many of its reactions with similar complexes of gold.48 We therefore searched the literature for reactions with Au and Pt that would function at room temperature, in aqueous media, and with likely fast kinetics to adapt for CisPt-mediated bioorthogonal decaging reactions. The cyclization of 4-pentynoic acid is well-known to proceed quickly in aqueous media with reaction times ranging from minutes to a few hours49,50 and has even been demonstrated with platinum (II and IV) anticancer complexes (Figure 1a).51 Given the previous studies, a metal-catalyzed mechanism was devised whereby a carbamate carbonyl could be used as an internal nucleophile to cause carbocyclization followed by release of a secondary amine (Figure 1b). Working on this hypothesis, we synthesized the terminal propargyl carbamate 3a (Figure 1c) to verify if the carbonyl could act as a nucleophile and attack the alkyne to subsequently release morpholine 6a in the presence of K2PtCl4. We observed conversions of 20% and 61% for reactions carried out in D2O/CD3OD (3:1) with 0.1 and 2 equiv of metal salt, respectively (Figure 1d, Entries 1 and 2; Figures S1 and S2 of the Supporting Information, SI). Similar yields were found for the reaction with NaAuCl4 (Figure S3). In contrast, if an aliphatic carbamate with no propargyl handle is used (compound SI S1) under the same decaging conditions the free amine is not released (Figures S4 and S5, respectively).
Figure 1.

Platinum-mediated decaging reaction engineering. a. The cyclization of 4-pentynoic acid is known to proceed rapidly in aqueous media with gold and platinum complexes. b. The proposed reaction uses a carboxamide as an internal nucleophile that cyclizes and displaces the secondary amine leaving group, which could be a drug or a fluorophore. c. Model compounds with alkyne amide or carbamate were used to survey the decaging reaction. d. Efficiency of the cleavage reaction under different conditions was assessed by 1H NMR spectroscopy. e. 1H NMR spectroscopy of the decaging of the tertiary amide 4a in the presence of a catalytic amount of K2PtCl4. The reaction generates a cyclized intermediate that undergoes hydrolysis to release morpholine 6a. General procedure for determining decaging conversion by 1H NMR spectroscopy: carbamate and amide compounds (10 mgs) were dissolved in MeOD (0.2 mL) and metal complexes (0.1 or 2 equiv) were added in D2O (0.6 mL) at room temperature in an open vessel for 14 h. The reactions were transferred to an NMR spectroscopic tube and sealed. Conversion was calculated based on the relative ratios of methylene peaks resulting from the starting material and the released amine product. Numerical data are the mean of 2 or 3 replicates.
According to these observations, it should also be possible to decage tertiary amides to release secondary amines (Figure 1c). This was an attractive prospect because amides are often much more stable than their corresponding carbamates.52 Decaging of pentynoyl tertiary amide 4a was monitored by NMR spectroscopy over time and was proven to have comparable rates and yields to the corresponding carbamate 3a (Figure 1d, Entry 4; Figure 1e). Importantly, the reaction proceeds with substoichiometric amounts of the metal complex (Figure 1d, Entry 3; Figure S6). The reactions were also successfully trialed with K2PtCl6 as a representative Pt(IV) species (Figure 1d, Entries 5 and 6; Figure S7) and NaAuCl4 (Figure S8) with good yields.
Overall, these results are important because they demonstrate a decaging reaction of stable protected tertiary amides by using substoichiometric amounts of platinum complexes that could function in water, open air, and without need of extreme temperatures or complex ligands. It is important to note that even after all of the starting material has been consumed (as evidenced by loss of terminal alkyne proton), not all of it has decomposed to release the amine (Figure 1e). This lack of conversion is likely due to side reactions, and nucleophilic attack on the alkyne, which seems the most plausible mechanism. To elucidate this a pentynoyl secondary amide (compound S2) was reacted with K2PtCl4 and NaAuCl4 under similar conditions (Figures S9 and S10). We found that the reaction proceeds with much lower extents of decaging, likely due to the amide nitrogen competing as a nucleophile to yield a stable cyclized product, and thus a smaller yield of released amine.
Mechanistic and Kinetic Studies of the Platinum-Mediated Decaging Reaction
To further study the platinum decaging reaction, the pentynoyl tertiary amide was conjugated to a naphthalimide-based fluorophore to generate fluorescent quenched probe 7 (Figure 2a, see SI for synthetic details).53,54 The reaction was then monitored by the increase in fluorescence upon removal of the protecting group to form fluorescent probe 8. With 50 equiv of K2PtCl4 or CisPt, we found that the fluorescence was restored over a period of 200 min for K2PtCl4 and 300 min for CisPt (Figure 2b), with complete consumption of 7 and formation of corresponding “turned ON fluorophore”, as indicated by LC–MS analysis (Figure S11). For both metals the conversion was accompanied by an initial steady state followed by a marked increase of the fluorescence, which suggested the formation of an activated intermediate. Indeed, it is known that platinum complexes form a series of reactive intermediates by successive replacement of the chloro ligands by water or hydroxyl groups.55−57 We hypothesized that formation of such an aqua intermediate early on could be responsible for the activation of the platinum complexes. This hypothesis was verified using LC–MS studies to follow formation of K2PtCl4– and CisPt–aqua complexes over time, which occurred within 6 h (Figures S12 and S13). Consistent with this hypothesis, platinum salts failed to form the aqua complexes when incubated in the presence of N,N-dimethylformamide (DMF). On the basis of these observations, we further studied the kinetics of the releasing reaction after formation of the aqua complexes (6 h in water/DMF at 37 °C). As expected, activation of the platinum salts significantly accelerated the turn-on half-time from t1/2 = 171 min to t1/2 = 30 min for K2PtCl4 and from t1/2 = 276 min to t1/2 = 60 min for “CisPt” (Figure 2c and Table S1). Accordingly, if the reaction was performed in pure DMF, then formation of the decaged probe was not observed (50 equiv of K2PtCl4 or CisPt for 14 h at 37 °C). This result is in agreement with previous LC–MS studies that suggested the requirement of water to generate the active catalyst. The activation of metal chloride in aqueous solvents has few precedents but has been reported for gold complexes.58 This effect is explained by facilitated ionization of the M–Cl bonds in water.58
Figure 2.

Examination of the platinum-catalyzed bioorthogonal cleavage reaction. a. Naphthalimide-based fluorogenic probes were used to study the cleavage efficiency of the platinum reaction for decaging alkyne-containing molecules. The caged naphthalimide derivatives exhibited high stability in solution and cell media and their quenched fluorescence could be reactivated upon removal of the caging group (λex = 445 nm, λem = 545 nm). b. Changes in fluorescence intensity during the time course of the decaging reaction between fluorogenic probe 7 and platinum salts (K2PtCl4/CisPt). c. Determined half-time for the reaction of 7 with activated and nonactivated platinum salts. d. Decaging kinetics for the pentynoyl amide fluorophore. Rate constants were determined under pseudo first order conditions with a 50 μM final concentration of probe 7 and 10–50 equiv of aqua platinum metals. e. Kinetics profiles of the decaging reaction in the presence of the metal poisons CS2 and EDTA. Error bars represent ± s.d. (n = 3). All experiments were repeated 3 independent times. f. Calculated mechanism for the depropargylation reaction catalyzed by Pt with model substrate 4a. Calculations were performed with an implicit solvent model for water. Geometries and frequencies were calculated with the functional revPBE and, to obtain very accurate energetics, single point energy calculations with DLPNO–CCSD(T) and counterpoise corrections were employed to suppress basis set superposition errors.
In terms of catalytic activity, the reaction of 7 with 0.3 equiv of activated K2PtCl4 complex yielded decaged probe 8 in 98% yield after 72 h at 37 °C (catalyst turnover number 3.3). Upon moving to 2 equiv of the metal complex, the decaged product was obtained in quantitative yield after 4 h at 37 °C (Figure S14). As a comparison, the same study was performed with Pd(OAc)2, a standard palladium complex for N-depropargylation.59 Interestingly, the Pd-reaction proceeded with comparable efficiency, although with slightly better rates of conversion (>98% yield in 1 h, LC–MS analysis). Of relevance, palladium decaging of alkyne amides has never been reported before. Finally, the compatibility and efficiency of the reactions were tested under physiological conditions. The activated aqua complexes were first shown to persist in complete DMEM cell media for at least 16 h at 37 °C, as assessed by LC–MS analysis, although a significant decrease in their concentration was observed over time (Figure S15). Later, the reactions were shown to proceed in cell media with conversions of 69% for K2PtCl4 (50 equiv) and 17% for CisPt (150 equiv) after 14 h at 37 °C (Figures S16 and S17). Similarly, the reaction was also trialed in high salt concentration buffers with high efficiency (t1/2 = 36 min for 50 equiv of K2PtCl4 and t1/2 = 105 min for 100 equiv of CisPt, 37 °C in E3 medium, Figures S18 and S19).
Having found an efficient platinum complex for decaging pentynoyl tertiary amides, we turned our attention to the determination of the rate constant of the reaction (Figure 2d). By fitting the appearance of 8 in the presence of increasing amounts of metal complexes and by using pseudo-first order conditions, the reactions were found to have second order rate constants of 0.230 ± 0.004 M–1 s–1 for K2PtCl4 and 0.080 ± 0.002 M–1 s–1 for CisPt (Figure 2d). These reaction rates are similar to those reported for other metal-assisted decaging reactions.27
To determine the nature of active species involved in the decaging reaction, we performed kinetic experiments with carbon disulfide (Figure 2e, Table S2). CS2 acts as a catalyst poison for homogeneous and heterogeneous Pt(0) reactions, although Pt(II) species are unaffected. As seen in Figure 2e, the reaction rates are similar to those with or without CS2. This result can be attributed to the noninvolvement of Pt(0) species in the reaction. However, the reaction rates were significantly affected by the addition of ethylenediamine tetraacetic acid (EDTA; Figure 2e), possibly due the participation of Pt(II) in the reaction.
We also performed computational studies to help understand the reaction mechanism (Figure 2f). These studies suggest that the most probable operating reaction pathway of substrate 4a is a stepwise process involving the coordination of substrate molecule to Pt(II), followed by an intramolecular attack of the carbonyl oxygen of the Pt-coordinated substrate (CS0) to the pentynoyl moiety, which gives five-membered ring intermediate CP. Different pathways to decomposition of CP were explored (Figure S20); the lowest energy one was the hydration of CP leading to formation of intermediate Q1, which readily decomposes to liberate free amine 6a. The metal complex is then recovered in a subsequent step by hydrolysis of 5 (Figure 2f). The complete calculation for the first reaction turnover of the main mechanism is depicted in Figure S21 and SI Movie. This mechanism is further supported by the identification of intermediate species CS0 by LC–MS (Figures S22–S26). The main difference observed for the reaction with substrate 3a relative to 4a was the higher free energy of activation (ΔΔG# = 2.75 kcal mol–1) for the intramolecular attack of the carbonyl oxygen at the pentynoyl moiety. However, both substrates share the same energy barrier for hydration of CP and release of 6a (Figure S21).
Extending the Decaging Reaction to N-Propargyl Group
Following the discovery of a platinum-cleavable group, we hoped to extend the scope of handles that could be used for decaging. Metal-mediated decaging of N-propargyl handles has been widely explored to modulate the cytotoxic activity of antineoplastic drugs in a controlled manner.22,25 On this basis, we investigated the possibility of using N-propargyl groups introduced on drugs of interest for prodrug activation using platinum triggers. First, and similarly to the pentynoyl amide reaction, an N-propargyl group was used to protect a secondary amine on a naphthalimide derivative to generate fluorogenic probe 9 (Figure 2a). As described above, we tested the reactivity of K2PtCl4 and CisPt before and after formation of the aqua complexes (6 h incubation at 37 °C in DMF/water). Once again, dissociation of the chloride anions in water was found to be crucial for triggering the reactivity of platinum complexes. Indeed, we found that reactions with aqua complexes are faster according to the calculated half-time for the “fluorescent reactions” (from t1/2 = 200 ± 3 min to t1/2 = 27 ± 3 min for K2PtCl4 and from t1/2 = 628 ± 51 min to t1/2 = 303 ± 34 min for CisPt; Table S3). The fluorescence-based assay was also employed to calculate the second-order rate constant for the reaction. Accordingly, the calculated rate constant was 0.120 ± 0.001 M–1 s–1 for K2PtCl4 and 0.0160 ± 0.0004 M–1 s–1 for CisPt (Figure S27). These results show that N-propargyls decage slower than pentynoyl amides. As a reference, the same study was performed with palladium complex Pd(OAc)2, which behaved slightly better than the platinum salts, to promote formation of 10 with a second-order rate constant of 0.39 ± 0.015 M–1 s–1 (Figure S28). The reaction was also subjected to CS2 and EDTA poisoning. CS2 had no effect but EDTA completely inhibited the reaction, which indicates the participation of Pt(II) species (Figure S29 and Table S4). On the basis of these results and LC–MS analysis after 2 h of reaction between K2PtCl4 and probe 9 (Figure S30), we propose that the first turnover of the reaction proceeds as recently disclosed for palladium depropargylation,59 i.e., (i) co-ordination of Pt(II) to alkyne moiety, (ii) attack of a H2O molecule at the propargyl terminal carbon to form an enol, (iii) tautomerization to a more stable Pt-aldehyde complex, and (iv) C–N bond cleavage by either hydrolysis or β-N elimination followed by hydration of Pt-complex (Figure S31). Finally, we investigated the ability of platinum salts to remove the propargyl protecting group in cells (DMEM) and zebrafish (E3) media. The reaction with the fluorogenic probe was monitored for K2PtCl4 and CisPt for 14 h at 37 °C. Efficiencies in E3 media were generally high with the reaction complete in 60 and 150 min for K2PtCl4 and CisPt, respectively (Figure S32). In DMEM, cleavage was less efficient with conversion yields of 67% for K2PtCl4 (50 equiv) and 30% for CisPt (150 equiv) after 14 h at 37 °C (Figure S33).
Platinum-Mediated Decaging in Living Cells
To verify whether platinum-mediated depropargylation would function in cell culture, a pentynoyl amide derivative of antineoplastic drug MMAE was synthesized. MMAE is the drug present in the ADC brentuximab vedotin that is in clinical use to treat patients with relapsed Hodgkin lymphoma and systemic anaplastic large-cell lymphoma,60 and remains the drug of choice for antibody-targeted therapies. In addition, a N-propargyl 5-fluorouracil (pFU) derivative was also tested, which was found to be efficiently decaged and activated with gold nanoparticles31 and palladium complexes.25 When MMAE-am was treated in DMF/water (1:1) for 4 h with 10 equiv of K2PtCl4, complete consumption of MMAE-am was seen by LC–MS with 37% release of MMAE along with the formation of the intermediate Q1s (Figures S20 and S34). In a similar fashion, decaging of pFU proceeds with yields of 46% ± 2 and 72% ± 2 for K2PtCl4 and CisPt, after 14 h reaction with 2 equiv, at room temperature and 37 °C, respectively (Figures S35–S37). These prodrugs (MMAE-am 11 and pFU 12, see the SI for synthetic details) were reacted with platinum salts in cell culture in the hope of observing a “turn-on” of toxicity. Unfortunately, the chemotherapeutic CisPt has a narrow window of nontoxic concentrations for efficient decaging in cells.61 Indeed, CisPt was demonstrated to be toxic in HeLa cells at concentrations as low as 2.5 μM (Figure S38). On the contrary, platinum salts K2PtCl4 and K2PtCl6 did not significantly influence the viability of HeLa cells at concentrations below 50 μM (Figure S38). With both prodrugs, an increase of about 2-fold in toxicity could be observed for some of the tested concentrations when reacted with K2PtCl4 over 3 days in cell culture (Figure 3a and 3b; e.g., 1 nM of MMAE-am and 50 μM of pFU). In contrast, no decrease of cytotoxicity was observed in cells treated independently with cFU 13, a non-decaging control derivative, or in combination with K2PtCl4 (Figure 3b). These control studies indicate that 5-FU was not generated because the alkyl handle does not undergo decaging by K2PtCl4.
Figure 3.
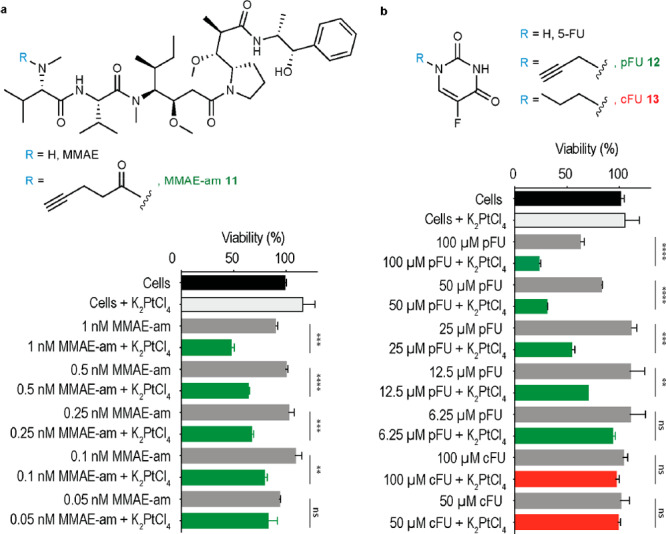
Platinum-mediated decaging in cells. HeLa cells were incubated with different concentrations of MMAE-am 11a or pFU 12b for 3 days with or without K2PtCl4 (20 μM, twice a day). Compound 13, a nondecaging alkyl-FU derivative, was used as a negative control. Toxicity was determined by AlamarBlue assay. Error bars represent ± s.d. (n = 3). Each experiment was repeated three times. The statistical significance of the differences between groups was evaluated with the unpaired t test. Statistical results: ns > 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001.
It is important to note that for both prodrugs the addition of K2PtCl4 did not restored their toxicity to the level observed for unmodified MMAE and 5-FU drugs (Figures S39 and S40). Although a 2-fold increase in toxicity for the prodrug activation may look modest, it is important to mention that this is considered relevant given the slow reaction rates possible at the low concentration of K2PtCl4 complex tolerated by cells. Indeed, this low reagent concentration was necessary to ensure the platinum complex remained nontoxic. On top of this, in vitro studies with probes 7 and 9 revealed that the presence of nucleophiles (e.g., glutathione) ends in lower conversions into the corresponding decaged products. It should be noted, however, that even in the presence of high concentrations of glutathione (e.g., 1.5 mM) the reaction still proceeds with moderate rates (t1/2 = 197 min for 7 + K2PtCl4; t1/2 = 246 min for 9 + K2PtCl4; Table S5). Regarding CisPt, we found that the reaction is more susceptible to the presence of nucleophiles (e.g., t1/2 = 921 min for 7 + CisPt in the presence of 0.5 mM of glutathione; Table S6). This deactivation of the metals in the presence of nucleophiles is in line with the modest decaging yields observed in the cell studies. This is an issue that could be further improved, for example, by using platinum-based nanoparticles known to have reduced toxicity and higher payload concentrations or by using platinum complexes stabilized with different organic ligands in a way to optimize the metal reactivity.62 Our data, however, demonstrate that decaging reactions with platinum complexes are possible in cell culture and could achieve release of sufficient amounts of the active drug in cells to induce cell death.
Platinum Decaging of ADC
Next, we decided to extend the tertiary amide caging group for chemically controlled drug-release from an ADC. The caging group of MMAE-am 11 was adapted for this purpose because MMAE is a common payload in ADC design.60 Ideally a CisPt-cleavable ADC would be stable to cleavage by endogenous extra- or intracellular conditions. For this reason, we decided to use a carbonyl acrylic bioconjugation handle63,64 coupled to MMAE for antibody modification (Figure 4a; SI for synthesis).
Figure 4.
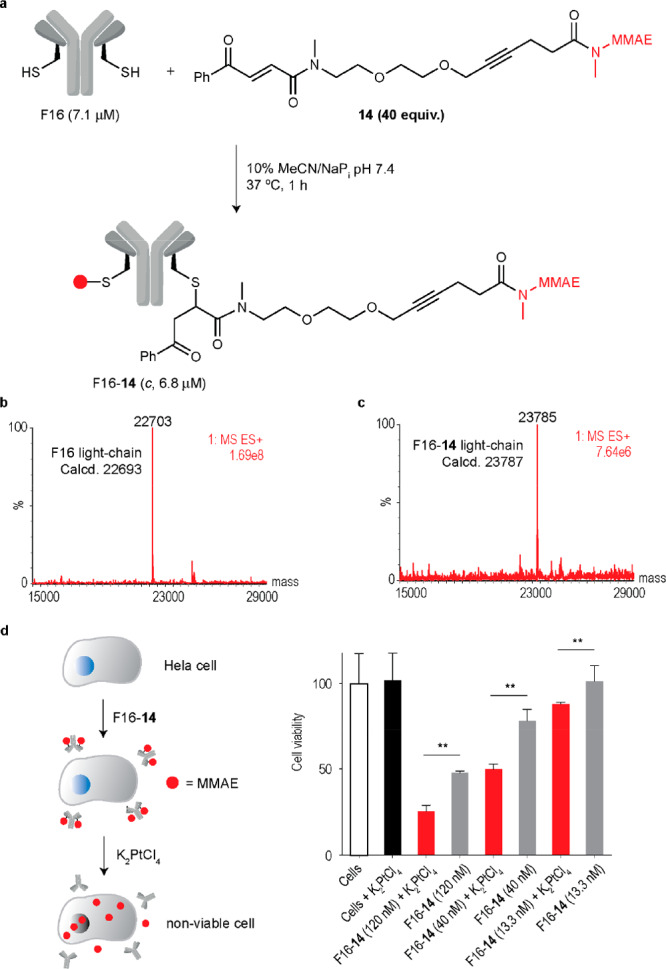
Platinum-mediated drug decaging from a noninternalizing ADC. a. Cysteine-selective and irreversible modification of the noninternalizing antibody F16 (anti tenascin-C) in IgG format with MMAE conjugating linker 14. IgG(F16) contains a single reactive cysteine at the C-terminal extremity of the light chain ideal for cysteine-specific modification. Briefly, a solution of F16 (7.1 μM) in sodium phosphate buffer (NaPi) pH 7.4 was treated with 14 (40 equiv) in MeCN to a final concentration of 10% v/v. The reaction was heated to 37 °C for 1 h, and reaction progress was monitored by LC–MS. The ADC was purified by dialysis into fresh NaPi buffer pH 7.4 with a 10 kDa MWCO overnight. b. Deconvoluted ESI–MS mass spectrum of the light-chain of F16. c. Deconvoluted ESI–MS mass spectrum of the light-chain of F16-14 that shows an exact drug-to-light-chain ratio of 1. d. Schematic of the platinum-mediated decaging of MMAE from a noninternalizing ADC. e. Cell viability of HeLa cells after treatment with F16-14 and subsequent decaging efficiency upon treatment with 20 μM K2PtCl4, twice daily. Cell viability was measured at day 3 by using AlamarBlue reagent. The statistical significance of the differences between groups was evaluated by using the unpaired t test. A p value <0.05 (**) was considered statistically significant. Error bars represent ± s.d. (n = 3). Experiments were performed three times.
To test the susceptibility of the conjugating linker to platinum decaging, compound 14 and K2PtCl4 (10 equiv) were incubated in DMF/water (1:1) at 37 °C for 18 h and analyzed by LC–MS (Figure S41). Release of MMAE was observed with complete consumption of 14 along with two potential intermediates (Figure S41). We then went on and selected the noninternalizing F16 antibody for modification, which is specific to the alternatively spliced A1 domain of tenascin-C, found overexpressed in most solid tumors.65 A noninternalizing ADC ensures that as little ADC as possible will be metabolized by the cells and that the maximum possible drug release is due to extracellular decaging with platinum complexes. Site-selective conjugation is expected to occur at the engineered cysteine residues in each light-chain of F16 enabling the construction of a chemically defined ADC. Furthermore, the newly formed C–S bond between the linker and the antibody is stable and does not undergo thiol-exchange reactions as in the case of frequently used maleimides.63,64 Complete conversion to a homogeneous ADC was achieved after reaction of F16 for 1 h at 37 °C with the carbonyl acrylic MMAE drug linker 14 in sodium phosphate buffer pH 7.4 as assessed by LC–MS (Figure 4b,c). Importantly, the heavy chain remained unmodified as expected considering the absence of reactive cysteines in the structure (Figures S42 and S43). Next, we performed the decaging in cells to release MMAE from the ADC (Figure 4d). With a cancer cell line (HeLa cells) as a model, we found F16-14 to be more toxic to cells at submicromolar concentrations in the presence of nontoxic amounts of the platinum complex K2PtCl4 (Figure 4d). This tertiary amide decaging reaction should stimulate platinum-mediated MMAE delivery from antibodies in the context of targeted cancer therapeutics. Furthermore, a small model protein (ubiquitin-K63C) engineered with a single cysteine residue66 was modified with linker 14 for ease of analysis by LC–MS. When attempting decaging in vitro with CisPt, loss of MMAE followed by further degradation of the linker could be observed by LC–MS, which provides further evidence for the efficient release of the secondary amine drug from the protected tertiary amide protected conjugate (Figures S44–S47).
Cisplatin-Mediated Prodrug Decaging in Vivo
To test the in vivo efficacy of pFU and its combinatorial effect with CisPt, we used the zebrafish larvae xenograft model.67 This model is a fast in vivo platform with resolution to analyze crucial hallmarks of cancer, such as metastatic and angiogenic potentials but it is also highly sensitive to discriminate differential anticancer therapy responses with single-cell resolution.68−71 We first attempted to visualize the CisPt reaction by decaging fluorogenic probe 9 in larval zebrafish (Figure 5). This probe shows an increase in fluorescence of 22-fold upon removal of the propargyl group (Figure S48). For in vivo imaging, a set of zebrafish larvae were incubated with probe 9 for 24 h, washed for 1 h in embryonic medium and then further incubated with dimethyl sulfoxide (DMSO) or C isPt for 24 h (Figure 5a). Probe 9 and CisPt were used at the highest nontoxic concentration to the zebrafish embryos (9, 1 μM; CisPt, 34 μM; Figure S49). As shown in Figure 5b, the control group displays nearly no background fluorescence, but the CisPt-treated group showed an increased fluorescence (Figure 5c). This implies that probe 9 and CisPt are tissue-permeable and capable of reacting in vivo.
Figure 5.
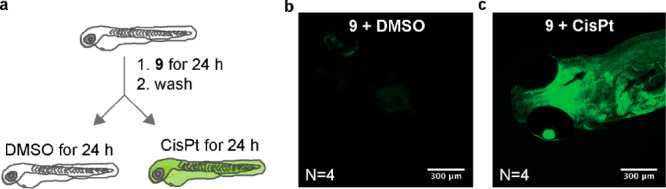
CisPt Decages the Fluorogenic Probe 9 in vivo. Zebrafish larvae were exposed to 9 diluted in embryonic medium for 24 h, followed by a 1 h wash in embryonic medium. Larvae were randomly distributed into two conditions: DMSO or CisPt for 24 h (a). Confocal image of zebrafish larvae exposed to 9 + DMSO (b) and 9 + CisPt (c).
Before measuring efficacy of CisPt depropargylation, we assessed the maximum tolerated concentration for each compound: pFU 12, cFU 13, CisPt, pFU + CisPt, and cFU + CisPt in nontumor zebrafish larvae (Figure S49). Next, colorectal cancer (CRC) HCT116 zebrafish xenografts were generated as previously described.67 Briefly, 24 h post injection (hpi), xenografts were randomly distributed into different treatments: DMSO (control), pFU (1.65 mM), cFU (1.65 mM), CisPt (0.034 mM), pFU + CisPt (1.65 mM + 0.034 mM), and cFU + CisPt (1.65 mM + 0.034 mM). Xenografts were analyzed at 4, 6, and 7 days post injection (dpi), i.e., 3, 5, and 6 days post treatment (dpt), respectively (Figure 6). At 3dpt (4dpi) (Figure 6 I), in the single treatments with pFU or CisPt, we could not observe any significant reduction of mitotic index (Figure 6 I, m), induction of apoptosis (activated caspase 3, Figure 6 I, n) or reduction of tumor size (Figure 6 I, o). In contrast, the combinatorial treatment—pFU + CisPt—induced a significant antitumoral synergistic effect manifested by a ∼2 fold increase in apoptosis (Figure 6 I, n; DMSO versus pFU + CisPt **P = 0.0033; pFU versus pFU + CisPt ***P = 0.0006) accompanied by 25% reduction of tumor size (Figure 6 I, o; DMSO versus pFU + CisPt *P = 0.0279; Figure 6 I, a versus d). However, if the duration of the treatment is increased 2 (Figure 6 II) or 3 (Figure 6 III) additional days, then we could detect some toxicity in single treatment (Figure 6 II, p–r; Figure 6 III, s–u). Nevertheless, the combination of pFU with CisPt induced a clear pronounced antitumor synergistic effect. At 5 dpt (6 dpi), the combinatorial treatment led to a reduction of proliferation (Figure 6 II, p; DMSO versus pFU + CisPt ****P < 0.0001; pFU versus pFU + CisPt *P = 0.0104), a ∼4 fold increase in cell death by apoptosis (Figure 6 II, q; DMSO versus pFU + CisPt ****P < 0.0001; pFU versus pFU + CisPt ****P < 0.0001) and a ∼38% reduction of tumor size (Figure 6 II, r; DMSO versus pFU + CisPt ****P < 0.0001; Figure 6 II, e versus h). Finally, the 6 days of treatment (7 dpi) culminates in a ∼45% tumor shrinkage (Figure 6 III, u; DMSO versus pFU + CisPt **P = 0.0010; Figure 6 III, i versus l).
Figure 6.

CisPt-mediated prodrug decaging in zebrafish xenografts. HCT116 human CRC cells were fluorescently labeled with lipophilic CM-DiI (shown in red) and injected into the perivitelline space (PVS) 2 days post fertilization (dpf) Tg(Fli1:eGFP) zebrafish larvae. Zebrafish xenografts were randomly distributed into treatment groups, daily treated with DMSO, CisPt, pFU, and pFU+CisPt and analyzed at 4, 6, or 7dpi for proliferation, apoptosis and tumor size. At 4 dpi, 6 dpi, and 7dpi, zebrafish xenografts were imaged by stereoscope (a–l) and by confocal microscopy (a′–l′ DAPI plus DiI, a′′–l′′ maximum projection of activated caspase 3). Proliferation (mitotic figures: m; p, *P = 0.0104, ***P = 0.0004, ****P < 0.0001; s, **P = 0.0023, *** P = 0.0002), apoptosis (activated caspase 3: n, **P = 0.0033, ***P = 0.0006; q, *P = 0.0126, ****P < 0.0001; t, **P = 0.0068) and tumor size (n° of tumor cells: o, *P = 0.0279; r, ****P < 0.0001; u, *P = 0.0411, **P = 0.0010) were analyzed and quantified. Graphs represent fold induction (normalized values to controls) of Avg ± SEM. The number of xenografts analyzed is indicated in the representative images and each dot represents one zebrafish xenograft. Statistical analysis was performed using an unpaired test. Statistical results: ns > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001. All images are anterior to the left, posterior to right, dorsal up, and ventral down. Scale bar 50 μm.
Importantly, by comparing the combined treatment of the nondecaging compound cFU with CisPt to the prodrug pFU with CisPt, it is clear that pFU was able to induce a more significant cytostatic (block proliferation) and cytotoxic effect (apoptosis and reduction of tumor size) than the control cFU at both 5 dpf (6 dpi) and 6 dpt (7 dpi; Figures S50 and S51). Also, the combined effect of pFU + CisPt was more pronounced than the combination of 5-FU + CisPt, regarding proliferation (DMSO versus pFU + CisPt ****P < 0.0001; DMSO versus FU + CisPt *P = 0.0104; Figure S50i) and tumor size (DMSO versus pFU + CisPt ****P < 0.0001; DMSO versus FU + CisPt *P = 0.0273; Figure S50k). This might be related with the increased permeability of pFU (versus FU), which results in a more efficient intracellular delivery of FU after Pt decaging. In conclusion, our results show the efficient activation of the anticancer pFU in the presence of nontherapeutic amounts of the anticancer drug CisPt in an in vivo setting.
Conclusions
In summary, we present a new decaging reaction of alkynes with platinum complexes for the release of secondary amines from otherwise stable tertiary amides, both in mammalian cell culture and in living organisms. This reaction was shown to proceed by platinum-mediated intramolecular cyclization mechanism. Our data suggest that water, a necessary solvent in chemical biology applications, is working as a metal-activating agent. Molecular electronic structure calculations further corroborated the mechanism of the reaction which was also supported by LC–MS characterization of the intermediates. The reaction can proceed catalytically under certain conditions and was later extended to N-propargyl groups with comparable efficacies to that of palladium-mediated depropargylation. The caging group was adapted for the synthesis of a noninternalizing ADC, which results in drug release upon treatment with platinum complexes in cancer cells. The reaction was also adapted and demonstrated to function in a colorectal cancer zebrafish xenograft model with nontoxic amounts of CisPt to activate a prodrug of anticancer agent 5-FU, which led to a significant tumor reduction in vivo.
The work disclosed here represents a significant addition to the toolbox of decaging strategies for chemical biology applications. Indeed, the platinum-mediated cleavable reaction can be accomplished in aqueous systems having high concentrations of salts with high yields and reaction rates, similar to those observed for the standard palladium decaging metal. The reaction is, however, susceptible to the presence of nucleophiles resulting in slower rates (∼6–15 times slower). We further demonstrate the compatibility of the reaction in cellular environments. Although the reaction is suitable for drug activation on cells inducing cytotoxicity, the presence of a range of biomolecules/nucleophiles significantly reduces the overall yield. These results are suggestive of instability of the Pt complexes, probably by formation of bioinorganic complexes. Although the active aqua Pt species have a limited lifetime in cell media, they persist long enough to be partially effective.
Our work was conceived on the hypothesis that platinum complexes could be used for prodrug activation on tumors during CisPt chemotherapy. The instability of the platinum complexes in physiological/biological conditions preclude the application envisioned. Further studies are needed to obtain Pt complexes compatible for such in vivo applications, but these results set the stage for future developments on platinum-mediated decaging reactions.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement Nos 675007 and 702574. We also thank the EPSRC (doctoral studentship to B.S.), FCT Portugal (FCT Investigator to G.J.L.B., IF/00624/2015; FCT Stimulus to B.L.O., CEECIND/02335/2017; LISBOA-01-0145-FEDER-022170 and FCT-PTDC/MEC-ONC/31627/2017 to R.F.), CNPq (PhD scholarship to F.S.S.S., 140485/2017-1; research grant to G.F.C., 311963/2017-0), the Champalimaud Foundation and Congento (R.F.), H2020 (EU_FT-ICR_MS, GA 731077, C.C.) and CAPES (Print call 88887.310560-00 to J.B.D. and G.J.L.B.) for funding. We also thank Prof. Dario Neri’s laboratory for the generous gift of F16 antibody, Dr Ester Jiménez-Moreno for providing ubiquitin, the Champalimaud Fish and Rodents Facility for their excellent animal care, the Portuguese Mass Spectrometry Network (LISBOA-01-0145- FEDER-022125), SeTIC/UFSC, special staff members (Bruno Amattos, Guilherme Arthur Geronimo and Stephanie Andreon) for supercomputing resources, infrastructure and assistance, and Dr Vikki Cantrill for her help with the editing of this manuscript. G.J.L.B. is a Royal Society University Research Fellow (URF\R\180019) and the recipient of a European Research Council Starting Grant (TagIt, GA 676832).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c01622.
Author Present Address
○ João Conde: NOVA Medical School, Faculdade de Ciências Médicas, Universidade Nova de Lisboa, 1169-056 Lisboa, Portugal
Author Contributions
∇ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144 (5), 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Chudasama V.; Maruani A.; Caddick S. Recent advances in the construction of antibody-drug conjugates. Nat. Chem. 2016, 8 (2), 114–119. 10.1038/nchem.2415. [DOI] [PubMed] [Google Scholar]
- Caculitan N. G.; dela Cruz Chuh J.; Ma Y.; Zhang D.; Kozak K. R.; Liu Y.; Pillow T. H.; Sadowsky J.; Cheung T. K.; Phung Q.; Haley B.; Lee B.-C.; Akita R. W.; Sliwkowski M. X.; Polson A. G. Cathepsin B Is Dispensable for Cellular Processing of Cathepsin B-Cleavable Antibody–Drug Conjugates. Cancer Res. 2017, 77 (24), 7027–7037. 10.1158/0008-5472.CAN-17-2391. [DOI] [PubMed] [Google Scholar]
- Dorywalska M.; Dushin R.; Moine L.; Farias S. E.; Zhou D.; Navaratnam T.; Lui V.; Hasa-Moreno A.; Casas M. G.; Tran T.-T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15 (5), 958–970. 10.1158/1535-7163.MCT-15-1004. [DOI] [PubMed] [Google Scholar]
- Rodrigues T.; Bernardes G. J. L. Development of Antibody-Directed Therapies: Quo Vadis?. Angew. Chem., Int. Ed. 2018, 57 (8), 2032–2034. 10.1002/anie.201712185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargh J. D.; Isidro-Llobet A.; Parker J. S.; Spring D. R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48 (16), 4361–4374. 10.1039/C8CS00676H. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen P. R. Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol. 2016, 12 (3), 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- Rossin R.; van Duijnhoven S. M.; Ten Hoeve W.; Janssen H. M.; Kleijn L. H.; Hoeben F. J.; Versteegen R. M.; Robillard M. S. Triggered Drug Release from an Antibody-Drug Conjugate Using Fast ″Click-to-Release″ Chemistry in Mice. Bioconjugate Chem. 2016, 27 (7), 1697–1706. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Versteegen R. M.; Wu J.; Khasanov A.; Wessels H. J.; Steenbergen E. J.; Ten Hoeve W.; Janssen H. M.; van Onzen A.; Hudson P. J.; et al. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9 (1), 1484. 10.1038/s41467-018-03880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46 (16), 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Mondal M.; Liao R.; Xiao L.; Eno T.; Guo J. Highly Multiplexed Single-Cell In Situ Protein Analysis with Cleavable Fluorescent Antibodies. Angew. Chem., Int. Ed. 2017, 56 (10), 2636–2639. 10.1002/anie.201611641. [DOI] [PubMed] [Google Scholar]
- Nani R. R.; Gorka A. P.; Nagaya T.; Kobayashi H.; Schnermann M. J. Near-IR Light-Mediated Cleavage of Antibody-Drug Conjugates Using Cyanine Photocages. Angew. Chem., Int. Ed. 2015, 54 (46), 13635–13638. 10.1002/anie.201507391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X.; Pan Z.; Yu B.; De La Cruz L. K.; Zheng Y.; Ke B.; Wang B. Click and release: bioorthogonal approaches to ″on-demand″ activation of prodrugs. Chem. Soc. Rev. 2019, 48 (4), 1077–1094. 10.1039/C8CS00395E. [DOI] [PubMed] [Google Scholar]
- Davies S.; Stenton B. J.; Bernardes G. J. L. Bioorthogonal Decaging Reactions for Targeted Drug Activation. Chimia 2018, 72 (11), 771–776. 10.2533/chimia.2018.771. [DOI] [PubMed] [Google Scholar]
- Zhang D. A.-O.; Dragovich P. S.; Yu S. F.; Ma Y.; Pillow T. H.; Sadowsky J. D.; Su D.; Wang W.; Polson A.; Khojasteh S. C.; Hop C. Exposure-Efficacy Analysis of Antibody-Drug Conjugates Delivering an Excessive Level of Payload to Tissues. Drug Metab. Dispos. 2019, 47 (10), 1146–1155. 10.1124/dmd.119.087023. [DOI] [PubMed] [Google Scholar]
- Durbin K. R.; Phipps C.; Liao X.. Mechanistic Modeling of Antibody-Drug Conjugate Internalization at the Cellular Level Reveals Inefficient Processing Steps. Mol. Cancer Ther. 2018, 17 ( (6), ), 1341–1351 10.1158/1535-7163.MCT-17-0672. [DOI] [PubMed] [Google Scholar]
- Yang M.; Li J.; Chen P. R. Transition metal-mediated bioorthogonal protein chemistry in living cells. Chem. Soc. Rev. 2014, 43 (18), 6511–6526. 10.1039/C4CS00117F. [DOI] [PubMed] [Google Scholar]
- Soldevila-Barreda J. J.; Metzler-Nolte N. Intracellular Catalysis with Selected Metal Complexes and Metallic Nanoparticles: Advances toward the Development of Catalytic Metallodrugs. Chem. Rev. 2019, 119 (2), 829–869. 10.1021/acs.chemrev.8b00493. [DOI] [PubMed] [Google Scholar]
- Völker T.; Meggers E. Transition-metal-mediated uncaging in living human cells—an emerging alternative to photolabile protecting groups. Curr. Opin. Chem. Biol. 2015, 25, 48–54. 10.1016/j.cbpa.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Miller M. A.; Askevold B.; Mikula H.; Kohler R. H.; Pirovich D.; Weissleder R. Nano-palladium is a cellular catalyst for in vivo chemistry. Nat. Commun. 2017, 8 (1), 15906. 10.1038/ncomms15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusop R. M.; Unciti-Broceta A.; Johansson E. M.; Sanchez-Martin R. M.; Bradley M. Palladium-mediated intracellular chemistry. Nat. Chem. 2011, 3 (3), 239–243. 10.1038/nchem.981. [DOI] [PubMed] [Google Scholar]
- Martinez-Calvo M.; Couceiro J. R.; Destito P.; Rodriguez J.; Mosquera J.; Mascarenas J. L. Intracellular Deprotection Reactions Mediated by Palladium Complexes Equipped with Designed Phosphine Ligands. ACS Catal. 2018, 8 (7), 6055–6061. 10.1021/acscatal.8b01606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray T. L.; Salji M.; Brombin A.; Perez-Lopez A. M.; Rubio-Ruiz B.; Galbraith L. C. A.; Patton E. E.; Leung H. Y.; Unciti-Broceta A. Bright insights into palladium-triggered local chemotherapy. Chem. Sci. 2018, 9 (37), 7354–7361. 10.1039/C8SC02291G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson J. C.; Macleod K. G.; Rybski W.; Fraser C.; Torres-Sanchez C.; Patton E. E.; Bradley M.; Carragher N. O.; Unciti-Broceta A. Extracellular palladium-catalysed dealkylation of 5-fluoro-1-propargyl-uracil as a bioorthogonally activated prodrug approach. Nat. Commun. 2014, 5, 3277. 10.1038/ncomms4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. T.; Carragher N. O.; Unciti-Broceta A. Palladium-mediated dealkylation of N-propargyl-floxuridine as a bioorthogonal oxygen-independent prodrug strategy. Sci. Rep. 2015, 5 (1), 9329. 10.1038/srep09329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenton B. J.; Oliveira B. L.; Matos M. J.; Sinatra L.; Bernardes G. J. L. A thioether-directed palladium-cleavable linker for targeted bioorthogonal drug decaging. Chem. Sci. 2018, 9 (17), 4185–4189. 10.1039/C8SC00256H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streu C.; Meggers E. Ruthenium-induced allylcarbamate cleavage in living cells. Angew. Chem., Int. Ed. 2006, 45 (34), 5645–5648. 10.1002/anie.200601752. [DOI] [PubMed] [Google Scholar]
- Volker T.; Dempwolff F.; Graumann P. L.; Meggers E. Progress towards bioorthogonal catalysis with organometallic compounds. Angew. Chem., Int. Ed. 2014, 53 (39), 10536–10540. 10.1002/anie.201404547. [DOI] [PubMed] [Google Scholar]
- Tomas-Gamasa M.; Martinez-Calvo M.; Couceiro J. R.; Mascarenas J. L. Transition metal catalysis in the mitochondria of living cells. Nat. Commun. 2016, 7 (1), 12538. 10.1038/ncomms12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Lopez A. M.; Rubio-Ruiz B.; Sebastian V.; Hamilton L.; Adam C.; Bray T. L.; Irusta S.; Brennan P. M.; Lloyd-Jones G. C.; Sieger D.; Santamaria J.; Unciti-Broceta A. Gold-Triggered Uncaging Chemistry in Living Systems. Angew. Chem., Int. Ed. 2017, 56 (41), 12548–12552. 10.1002/anie.201705609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubokura K.; Vong K. K.; Pradipta A. R.; Ogura A.; Urano S.; Tahara T.; Nozaki S.; Onoe H.; Nakao Y.; Sibgatullina R.; Kurbangalieva A.; Watanabe Y.; Tanaka K. In Vivo Gold Complex Catalysis within Live Mice. Angew. Chem., Int. Ed. 2017, 56 (13), 3579–3584. 10.1002/anie.201610273. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu Y.; Fan X.; Wang J.; Ngai W. S. C.; Zhang H.; Li J.; Zhang G.; Lin J.; Chen P. R. Copper-Triggered Bioorthogonal Cleavage Reactions for Reversible Protein and Cell Surface Modifications. J. Am. Chem. Soc. 2019, 141 (43), 17133–17141. 10.1021/jacs.9b05833. [DOI] [PubMed] [Google Scholar]
- Labinger J. A. Platinum-Catalyzed C–H Functionalization. Chem. Rev. 2017, 117 (13), 8483–8496. 10.1021/acs.chemrev.6b00583. [DOI] [PubMed] [Google Scholar]
- Basu U.; Banik B.; Wen R.; Pathak R. K.; Dhar S. The Platin-X series: activation, targeting, and delivery. Dalton Trans. 2016, 45 (33), 12992–13004. 10.1039/C6DT01738J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone T. C.; Suntharalingam K.; Lippard S. J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116 (5), 3436–3486. 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. I.; Christodoulou J.; Parkinson J. A.; Barnham K. J.; Tucker A.; Woodrow J.; Sadler P. J. Cisplatin binding sites on human albumin. J. Biol. Chem. 1998, 273 (24), 14721–14730. 10.1074/jbc.273.24.14721. [DOI] [PubMed] [Google Scholar]
- Florea A.-M.; Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3 (1), 1351–1371. 10.3390/cancers3011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny R. G.; Marmion C. J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens?. Chem. Rev. 2019, 119 (2), 1058–1137. 10.1021/acs.chemrev.8b00271. [DOI] [PubMed] [Google Scholar]
- Dasari S.; Tchounwou P. B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D. J.; Molepo J. M.; Green R. M.; Montpetit V. A.; Hugenholtz H.; Lamothe A.; Mikhael N. Z.; Redmond M. D.; Gadia M.; Goel R. Factors affecting platinum concentrations in human surgical tumour specimens after cisplatin. Br. J. Cancer 1995, 71 (3), 598–604. 10.1038/bjc.1995.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hennik M. B.; van der Vijgh W. J.; Klein I.; Elferink F.; Vermorken J. B.; Winograd B.; Pinedo H. M. Comparative pharmacokinetics of cisplatin and three analogues in mice and humans. Cancer Res. 1987, 47 (23), 6297–6301. [PubMed] [Google Scholar]
- Jacobs S.; McCully C. L.; Murphy R. F.; Bacher J.; Balis F. M.; Fox E. Extracellular fluid concentrations of cisplatin, carboplatin, and oxaliplatin in brain, muscle, and blood measured using microdialysis in nonhuman primates. Cancer Chemother. Pharmacol. 2010, 65 (5), 817–824. 10.1007/s00280-009-1085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A. V.; Hambley T. W. Platinum Drug Distribution in Cancer Cells and Tumors. Chem. Rev. 2009, 109 (10), 4911–4920. 10.1021/cr9001066. [DOI] [PubMed] [Google Scholar]
- Holding J. D.; Lindup W. E.; Bowdler D. A.; Siodlak M. Z.; Stell P. M. Disposition and tumour concentrations of platinum in hypoalbuminaemic patients after treatment with cisplatin for cancer of the head and neck. Br. J. Clin. Pharmacol. 1991, 32 (2), 173–179. 10.1111/j.1365-2125.1991.tb03878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol J. L.; Cupissol D.; Gestin-Boyer C.; Bres J.; Serrou B.; Michel F. B. Tumor-tissue and plasma concentrations of platinum during chemotherapy of non-small-cell lung cancer patients. Cancer Chemother. Pharmacol. 1990, 27 (1), 72–75. 10.1007/BF00689280. [DOI] [PubMed] [Google Scholar]
- Del Monte U. Does the cell number 10(9) still really fit one gram of tumor tissue?. Cell Cycle 2009, 8 (3), 505–506. 10.4161/cc.8.3.7608. [DOI] [PubMed] [Google Scholar]
- Abu Sohel S. M.; Liu R.-S. Carbocyclisation of alkynes with external nucleophiles catalysed by gold, platinum and other electrophilic metals. Chem. Soc. Rev. 2009, 38 (8), 2269–2281. 10.1039/b807499m. [DOI] [PubMed] [Google Scholar]
- Tomás-Mendivil E.; Toullec P. Y.; Díez J.; Conejero S.; Michelet V.; Cadierno V. Cycloisomerization versus Hydration Reactions in Aqueous Media: A Au(III)-NHC Catalyst That Makes the Difference. Org. Lett. 2012, 14 (10), 2520–2523. 10.1021/ol300811e. [DOI] [PubMed] [Google Scholar]
- Belger K.; Krause N. Smaller, faster, better: modular synthesis of unsymmetrical ammonium salt-tagged NHC–gold(i) complexes and their application as recyclable catalysts in water. Org. Biomol. Chem. 2015, 13 (31), 8556–8560. 10.1039/C5OB01286D. [DOI] [PubMed] [Google Scholar]
- Alemán J.; del Solar V.; Navarro-Ranninger C. Anticancer platinum complexes as non-innocent compounds for catalysis in aqueous media. Chem. Commun. 2010, 46 (3), 454–456. 10.1039/B917758B. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Brindisi M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58 (7), 2895–2940. 10.1021/jm501371s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J.; Xu M.; Parvez S.; Peterson R. T.; Franzini R. M. Bioorthogonal Removal of 3-Isocyanopropyl Groups Enables the Controlled Release of Fluorophores and Drugs in Vivo. J. Am. Chem. Soc. 2018, 140 (27), 8410–8414. 10.1021/jacs.8b05093. [DOI] [PubMed] [Google Scholar]
- Morihiro K.; Ankenbruck N.; Lukasak B.; Deiters A. Small Molecule Release and Activation through DNA Computing. J. Am. Chem. Soc. 2017, 139 (39), 13909–13915. 10.1021/jacs.7b07831. [DOI] [PubMed] [Google Scholar]
- Corinti D.; Coletti C.; Re N.; Piccirillo S.; Giampà M.; Crestoni M. E.; Fornarini S. Hydrolysis of cis- and transplatin: structure and reactivity of the aqua complexes in a solvent free environment. RSC Adv. 2017, 7 (26), 15877–15884. 10.1039/C7RA01182B. [DOI] [Google Scholar]
- Berners-Price S. J.; Appleton T. G.. The Chemistry of Cisplatin in Aqueous Solution. In Platinum-Based Drugs in Cancer Therapy; Kelland L. R., Farrell N. P., Eds.; Humana Press: Totowa, NJ, 2000; pp 3–35. [Google Scholar]
- Dolezel P.; Kuban V. Mass spectrometric study of platinum complexes based on cisplatin. Chem. Zvesti 2002, 56 (4), 236–240. [Google Scholar]
- Vidal C.; Tomás-Gamasa M.; Destito P.; López F.; Mascareñas J. L. Concurrent and orthogonal gold(I) and ruthenium(II) catalysis inside living cells. Nat. Commun. 2018, 9 (1), 1913. 10.1038/s41467-018-04314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho S. E.; Schneider F. S. S.; de Oliveira D. C.; Tripodi G. L.; Eberlin M. N.; Caramori G. F.; de Souza B.; Domingos J. B. Mechanism of Palladium(II)-Mediated Uncaging Reactions of Propargylic Substrates. ACS Catal. 2019, 9 (5), 3792–3799. 10.1021/acscatal.9b00210. [DOI] [Google Scholar]
- Senter P. D.; Sievers E. L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30 (7), 631–637. 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- Kelland L. R.; Abel G.; McKeage M. J.; Jones M.; Goddard P. M.; Valenti M.; Murrer B. A.; Harrap K. R. Preclinical Antitumor Evaluation of Bis-acetato-ammine-dichloro-cyclohexylamine Platinum(IV): an Orally Active Platinum Drug. Cancer Res. 1993, 53 (11), 2581–2586. [PubMed] [Google Scholar]
- Cheng Q.; Liu Y. Multifunctional platinum-based nanoparticles for biomedical applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9 (2), e1410 10.1002/wnan.1410. [DOI] [PubMed] [Google Scholar]
- Bernardim B.; Cal P. M. S. D.; Matos M. J.; Oliveira B. L.; Martínez-Sáez N.; Albuquerque I. S.; Perkins E.; Corzana F.; Burtoloso A. C. B.; Jiménez-Osés G.; et al. Stoichiometric and irreversible cysteine-selective protein modification using carbonylacrylic reagents. Nat. Commun. 2016, 7 (1), 13128. 10.1038/ncomms13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardim B.; Matos M. J.; Ferhati X.; Compañón I.; Guerreiro A.; Akkapeddi P.; Burtoloso A. C. B.; Jiménez-Osés G.; Corzana F.; Bernardes G. J. L. Efficient and irreversible antibody–cysteine bioconjugation using carbonylacrylic reagents. Nat. Protoc. 2019, 14 (1), 86–99. 10.1038/s41596-018-0083-9. [DOI] [PubMed] [Google Scholar]
- Gébleux R.; Stringhini M.; Casanova R.; Soltermann A.; Neri D. Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int. J. Cancer 2017, 140 (7), 1670–1679. 10.1002/ijc.30569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.; Sun S.; Jiménez-Moreno E.; Neves A. A.; Bernardes G. J. L. Site-selective installation of an electrophilic handle on proteins for bioconjugation. Bioorg. Med. Chem. 2018, 26 (11), 3060–3064. 10.1016/j.bmc.2018.02.028. [DOI] [PubMed] [Google Scholar]
- Fior R.; Póvoa V.; Mendes R. V.; Carvalho T.; Gomes A.; Figueiredo N.; Ferreira M. G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (39), E8234 10.1073/pnas.1618389114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio M.; Ablain J.; Chuan Y.; Langenau D. M. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer 2020, 20 (9), 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio M.; Zon L. I. Fishing for answers in precision cancer medicine. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (39), 10306. 10.1073/pnas.1713769114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.; Brunson D. C.; Tang Q.; Do D.; Iftimia N. A.; Moore J. C.; Hayes M. N.; Welker A. M.; Garcia E. G.; Dubash T. D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177 (7), 1903–1914. 10.1016/j.cell.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa B.; Ferreira S.; Póvoa V.; Cardoso M. J.; Vieira S.; Stroom J.; Fidalgo P.; Rio-Tinto R.; Figueiredo N.; Parés O.; Greco C.; Ferreira M. G.; Fior R. Developments in zebrafish avatars as radiotherapy sensitivity reporters — towards personalized medicine. EBioMedicine 2020, 51, 102578. 10.1016/j.ebiom.2019.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.