

Abstract
Alkenes are an important class of compounds common among biologically active molecules and often used as intermediates in organic synthesis. Many alkenes exist in two stereoisomeric forms (E and Z), which have different structures and different properties. The selective formation of the two isomers is an important synthetic goal that has long inspired the development of new synthetic methods. However, the efficient synthesis of diastereopure, thermodynamically less stable, Z-alkenes is still challenging. Here, we demonstrate an efficient synthesis of diastereopure Z-alkenes (Z:E > 300:1) through a silver-catalyzed hydroalkylation of terminal alkynes, using alkylboranes as coupling partners. We also describe the exploration of the substrate scope, which reveals the broad functional group compatibility of the new method. Preliminary mechanistic studies suggest that a 1,2-metallate rearrangement of the silver borate intermediate is the key step responsible for the stereochemical outcome of the reaction.
Graphical Abstract

Alkenes serve as important intermediates in organic synthesis and are often found among pharmaceuticals and other biologically active compounds. The key feature of alkenes is the presence of a π bond, which hinders the rotation about the carbon-carbon σ-bond1 and confers structural rigidity to alkenes. The hindered rotation also leads to two distinct stereoisomeric forms of disubstituted alkenes, denoted as E and Z.2 An efficient synthesis of thermodynamically less stable Z-alkenes presents a complex set of challenges that continues to inspire the development of new synthetic methods.
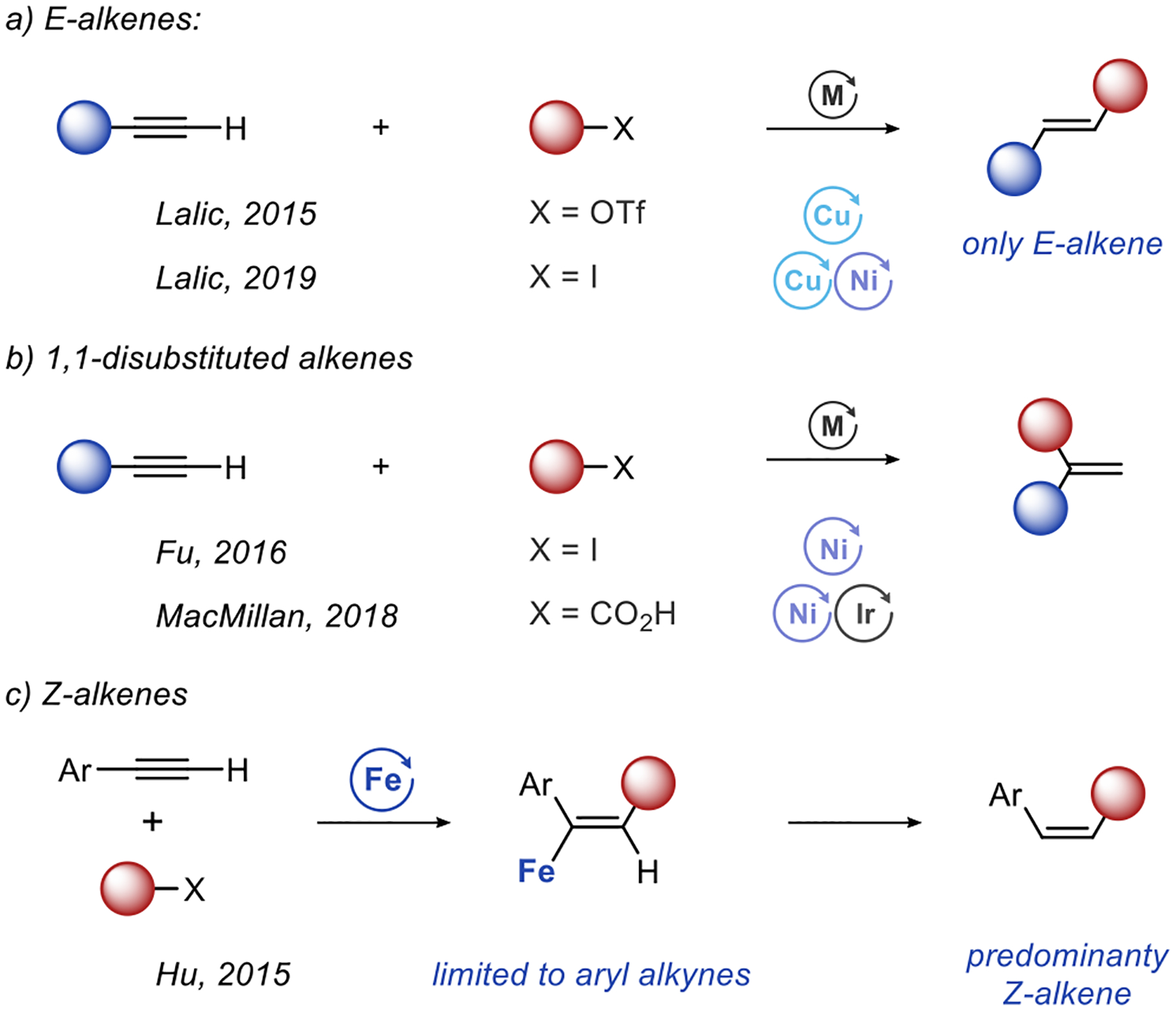
The efficient synthesis of Z-alkenes must accomplish three important goals. The most basic goal is to form a double bond. The synthesis should also exert complete control over the double bond geometry, so that only the Z isomer of the alkene is formed. The synthesis of diastereopure products is particularly important considering the difficulties often encountered in the separation of alkene isomers. Finally, a Z-alkene should be constructed from two smaller fragments through the formation of a new C-C σ−bond.3 Such a convergent approach to synthesis has long been recognized as key for the efficient build-up of molecular complexity.4 Ideally, all three goals of Z-alkene synthesis (π bond formation, formation of only one isomer, and C-C σ bond formation) should be accomplished in a single reaction. Unfortunately, this is difficult to achieve using standard catalytic methods for the synthesis of Z-alkenes,5 such as alkyne semireduction,6 cross-coupling reactions,7 alkene cross metathesis,8 or alkene isomerization.9
We were intrigued by the idea that hydroalkylation of alkynes is one reaction that may allow us to accomplish all three goals of Z-alkenes synthesis. Hydroalkylation has already proven effective in the synthesis of E-alkenes through reductive coupling of terminal alkynes with alkyl iodides10 or alkyl triflates (Scheme 1a).11 The same approach is also effective when applied to the synthesis of 1,1-disubstituted alkenes (Scheme 1b).12 Still, hydroalkylation reactions had limited success targeting Z-alkenes. Z-Selectivity has, so far, been achieved only in an iron-catalyzed radical hydroalkylation, reported by Hu et al. in 2015 (Scheme 1c). This method is limited to reactions of aryl acetylenes and yields mixtures of E- and Z-alkenes with varying selectivity.13
Scheme 1.
Hydroalkylation of Alkynes
Unfortunately, other mechanistic paradigms used to accomplish hydroalkylation favor the formation of E-alkenes. As a result, to achieve the synthesis of pure Z-alkenes we need a fundamentally new approach to hydroalkylation of alkynes.
Here, we describe a catalytic method for the synthesis of diastereopure Z-alkenes through the coupling of terminal alkynes and alkylboranes. The development of this method was inspired by a 1975 report from H. C. Brown (Scheme 2a).14 In the report, Brown showed that lithium acetylides readily add to alkylboranes to form borate complexes. Further reaction of the borate with a Brønsted acid leads to a 1,2-metallate shift15,16 and the formation of both isomers of alkenyl borane, with Z being the major isomer. The stereochemical outcome has been attributed to the nature of the 1,2-metalate shift, which is proposed to proceed through a carbocation intermediate.14c Subsequent protodeboration provides a mixture of Z- and E-alkenes.14c,17
Scheme 2.

New Approach to Hydroalkylation
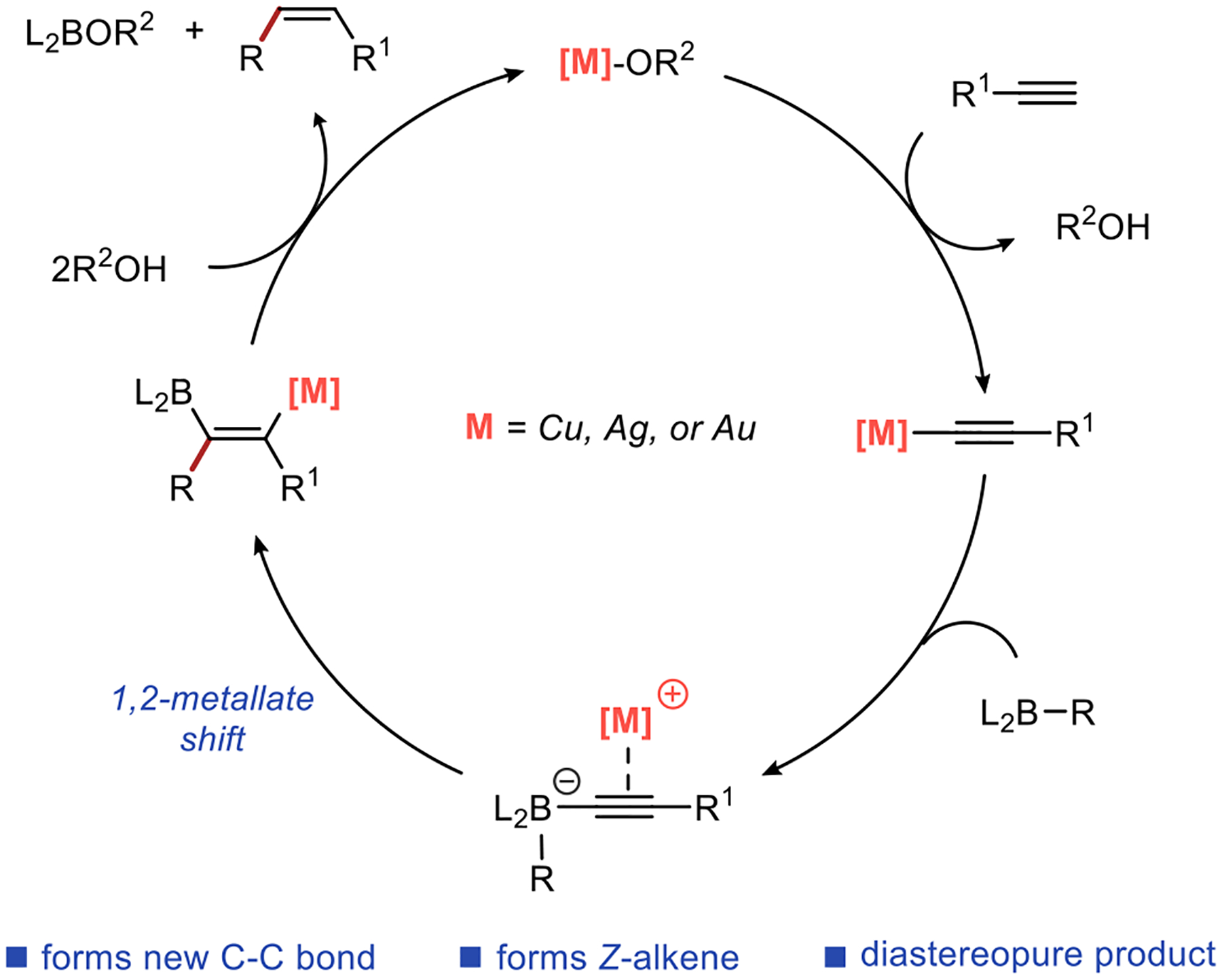
We reasoned that in the 1,2-metallate shift described by Brown, the Brønsted acid could be replaced by a cationic complex of a coinage metal (Scheme 2b). The 1,2-metallate shift promoted by π-acid coordination18,19 to the alkyne has been shown to require antiperiplanar orientation of the π-acid and the migrating group20 and would allow exclusive formation of the Z-alkene product. Furthermore, the coinage metal complex could be delivered in the form of a metal acetylide that would replace the lithium acetylide used in the reaction sequence described by Brown. Ultimately, these simple changes would allow us to develop a catalytic process that results in the exclusive formation of Z-alkenes.
Scheme 3 shows the postulated mechanism of such a catalytic reaction. In the presence of an alkoxide base, a coinage metal catalyst promotes acetylide formation, which is followed by addition to the alkyl borane. Upon addition, the metal counterion coordinates to the alkyne and promotes the 1,2-metallate shift through π-activation of the alkyne. Finally, alkene formation is accomplished by protodeboration and protodemetallation of the intermediate in the presence of an alcohol additive.
Scheme 3.
Proposed Catalytic Cycle
With the broad strategy outlined in Scheme 3 in mind, we explored the reaction of alkyne 1 with alkylborane 2 in the presence of a copper, silver, or gold catalyst (Table 1). Surprisingly, the silver catalyst performed the best, even though silver complexes are expected to be the least π-acidic.21 Crucially, only the Z isomer of alkene product 3 was formed in the reaction. Through further optimization of the reaction parameters, we found that a triazole carbene ligand and a nonpolar solvent provided the highest yield of the Z-alkene product. Under optimized conditions, complete conversion was achieved using a modest excess (1.3–1.5 equiv) of the alkylborane, which was prepared in situ from an alkene. Standard control experiments confirmed that the catalyst, base, and methanol were all necessary for the reaction (see SI).
Table 1.
Development of Hydroalkylation Reaction

|
Only Z-alkene detected in crude reaction mixtures by GC and 1H NMR analysis. The standard reaction conditions are bolded in red. aIsooctane was used instead of toluene. Ar = methyl-4-benzoate
The new transformation proved to be remarkably general, providing Z-alkenes as exclusive products with a wide range of substrates (Table 2). Functionalized alkyl alkynes could be successfully used in the reaction together with both electron-rich and electron-poor aryl alkynes. Alkylboranes prepared in situ from a variety of terminal alkenes, including 1,1-disubstituted alkenes, could be used as substrates. Furthermore, a wide range of functional groups was tolerated. Alkene products were formed in the presence of alcohols, aldehydes, ketones, esters, nitriles, alkenes, Boc-protected primary and secondary amines, anilines, alkyl bromides, alkyl chlorides, aryl iodides, aryl bromides, aryl boronic esters, epoxides, acetals, and alkyl and silyl ethers. Heterocycles such as pyridine, thiazole, benzimidazole, benzopyrazine, pyrimidine, pyridazine, thiophene, and furan were also compatible with the reaction conditions. The reaction could be performed on preparative scale, yielding one gram of alkene 3. Finally, in the synthesis of alkene 4, careful analysis of the crude reaction mixture by GC and GC/MS using authentic samples of Z and E isomers of the alkene revealed the presence of the very small amount of the E isomer (Z:E = 570:1). Similar GC analysis of crude reaction mixtures obtained in the synthesis of 3, 17, 35, 39, and 41, in all cases indicated Z:E ratio greater than 300:1.22
Table 2.
Reaction scope

|
Reactions performed on a 0.5 mmol scale. Reported are yields of isolated products after purification by silica gel chromatography. In all reactions, Z:E ratio of products estimated to be >300:1 (see text). Alkylboranes were prepared in situ by stirring corresponding alkene (1 equiv) and 9-borabicyclo[3.3.1]nonane dimer (0.45 equiv) in toluene (2M) at 60 °C. Reaction Conditions: TriAgCI (10 mol%), alkylborane (1.3 equiv), LiOt-Bu (1.5 equiv), MeOH (1.1 equiv), isooctane (5 mL), 45 °C, 16 h. a Conditions: IPrAgCI (10 mol%), alkyl borane (1.5 equiv), LiOf-Bu (2.0 equiv), MeOH (1.1 equiv), toluene (5 mL), 60 °C, 16 h. bR = propyl benzene, cYield determined by 1H NMR.
Several limitations in the scope of the hydroalkylation reaction were noted. While primary alkyl groups participate in the 1,2-metallate shift, secondary and tertiary alkyl, alkenyl, and aryl groups do not. Free amines, carboxylic acids, and carboxylates are not compatible with the reaction. We also noticed that the presence of an electron-withdrawing group two carbons away from the boron in alkyl boranes significantly lowers the yield of the reaction.
We have explored the application of the new method in the synthesis of biologically active compounds. Zucapsaicin is used in the treatment of osteoarthritis and neuropathic pain (Scheme 4a).23 The formal synthesis24 of this synthetic analogue of capsaicin was accomplished by the synthesis of 57 in 82% yield as a single isomer. A formal racemic synthesis of the natural product falcarindiol25 (Scheme 4b) was accomplished by preparing alkene 60 in 93% yield and 100% Z-selectivity. Falcarindiol has been shown to have a range of pharmacologically useful properties, including anti-cancer activity in breast cancer cells.26
Scheme 4.

Applications
Reactions were performed on a 0.5 mmol scale.aConditions: TriAgCI (10 mol%), Li-OtBu (1.5 equiv), MeOH (1.1 equiv), in Isooctane (5 mL), 45 °C, 16 h.
To probe the mechanism of the reaction, we explored the feasibility of the elementary steps in our postulated catalytic cycle (Scheme 5). In a stoichiometric reaction, we found that the silver catalyst, in the presence of an alkoxide, readily reacts with a terminal alkyne 61 to produce silver acetylide 62 (Scheme 5). The acetylide rapidly reacts with alkyl borane 2 to produce borate complex 63. The X-ray structure of this complex (Figure 1) indicates that the silver cation associated with the borate is coordinated to the alkyne and is therefore poised to promote the 1,2-metallate shift. However, the unfavorable orientation of the migrating alkyl group is consistent with the observed stability of this intermediate. In a control experiment, we confirmed that the silver borate complex is a viable catalyst for the hydroalkylation reaction (see SI).
Scheme 5.

Experiments Probing the Reaction Mechanism.
Figure 1.

X-ray structure of borate complex 63 with thermal ellipsoids at 50% probability. Disorder in one phenyl group and one isopropyl group has been omitted for clarity
These experimental observations provide direct support for some of the key aspects of the proposed reaction mechanism shown in Scheme 3 (M = Ag). Further studies are necessary to provide a more detailed understanding of the reaction mechanism, including the steps following the 1,2-metallate shift.
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. Forrest Michael for assistance in preparation of this manuscript. We also thank NIH for financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website
Experimental procedure and product characterization (pdf)
The authors declare no competing financial interests.
REFERENCES
- (1).Rabinovitch BS; Michel KW The Thermal Uni-molecular Cis-Trans Isomerization of Cis-Butene-21. J. Am. Chem. Soc 1959, 81, 5065. [Google Scholar]
- (2).Blackwood JE; Gladys CL; Loening KL; Petrarca AE; Rush JE Unambiguous Specification of Stereoisomerism About a Double Bond. J. Am. Chem. Soc 1968, 90, 509. [Google Scholar]
- (3).For selected examples of convergent synthesis of Z-alkenes, see:; a) Park BY; Nguyen KD; Chaulagain MR; Komanduri V; Krische MJ Alkynes as Allylmetal Equivalents in Redox-Triggered C–C Couplings to Primary Alcohols: (Z)-Homoallylic Alcohols Via Ruthenium-Catalyzed Propargyl C–H Oxidative Addition. J. Am. Chem. Soc 2014, 136, 11902; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kong JR; Krische MJ Catalytic Carbonyl Z-Dienylation Via Multicomponent Reductive Coupling of Acetylene to Aldehydes and A-Ketoesters Mediated by Hydrogen: Carbonyl Insertion into Cationic Rhodacyclopentadienes. J. Am. Chem. Soc 2006, 128, 16040; [DOI] [PubMed] [Google Scholar]; c) Thomas BN; Moon PJ; Yin S; Brown A; Lundgren RJ Z-Selective Iridium-Catalyzed Cross-Coupling of Allylic Carbonates and A-Diazo Esters. Chem. Sci 2018, 9, 238; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Armstrong MK; Goodstein MB; Lalic G Diastereodivergent Reductive Cross Coupling of Alkynes through Tandem Catalysis: Z- and E-Selective Hydroarylation of Terminal Alkynes. J. Am. Chem. Soc 2018, 140, 10233; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hodgson DM; Arif T Convergent Synthesis of Trisubstituted Z-Allylic Esters by Wittig−Schlosser Reaction. Org. Lett 2010, 12, 4204; [DOI] [PubMed] [Google Scholar]; f) Dong D-J; Li H-H; Tian S-K A Highly Tunable Stereoselective Olefination of Semistabilized Triphenylphosphonium Ylides with N-Sulfonyl Imines. J. Am. Chem. Soc 2010, 132, 5018; [DOI] [PubMed] [Google Scholar]; g) Reichard HA; Micalizio GC Metallacycle-Mediated Cross-Coupling with Substituted and Electronically Unactivated Alkenes. Chem. Sci 2011, 2, 573; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Das M; O’Shea DF Z-Stereoselective Aza-Peterson Olefinations with Bis(Trimethylsilane) Reagents and Sulfinyl Imines. Org. Lett 2016, 18, 336. [DOI] [PubMed] [Google Scholar]
- (4).a) Velluz L; Valls J; Nominé G Recent Advances in the Total Synthesis of Steroids. Angew. Chem., Int. Ed. Engl 1965, 4, 181; [Google Scholar]; b) Hendrickson JB Systematic Synthesis Design. 6. Yield Analysis and Convergency. J. Am. Chem. Soc 1977, 99, 5439. [Google Scholar]
- (5).Siau WY; Zhang Y; Zhao Y In Stereoselective Alkene Synthesis. Topics in Current Chemistry; Wang J, Ed.; Springer: Berlin, Heidelberg, 2012; Vol. 327. [DOI] [PubMed] [Google Scholar]
- (6).a) Oger C; Balas L; Durand T; Galano J-M Are Alkyne Reductions Chemo-, Regio-, and Stereoselective Enough to Provide Pure (Z)-Olefins in Polyfunctionalized Bioactive Molecules? Chem. Rev 2013, 113, 1313; [DOI] [PubMed] [Google Scholar]; b) Munslow I In Modern Reduction Methods; Andersson PG, Munslow I, Eds.; Wiley-VCH Verlag GmbH&Co. KgaA: Weiheim, Germany, 2008. [Google Scholar]
- (7).Meijere A; Bräse S; Oestreich M Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH Verlag GmbH & Co. KGaA: 2014, [Google Scholar]
- (8).For key references in Z-selective cross-metathesis, see:; a) Keitz BK; Endo K; Patel PR; Herbert MB; Grubbs RH Improved Ruthenium Catalysts for Z-Selective Olefin Metathesis. J. Am. Chem. Soc 2012, 134, 693; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meek SJ; O’Brien RV; Llaveria J; Schrock RR; Hoveyda AH Catalytic Z-Selective Olefin Cross-Metathesis for Natural Product Synthesis. Nature 2011, 471, 461; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xu C; Shen X; Hoveyda AH In Situ Methylene Capping: A General Strategy for Efficient Stereoretentive Catalytic Olefin Metathesis. The Concept, Methodological Implications, and Applications to Synthesis of Biologically Active Compounds. J. Am. Chem. Soc 2017, 139, 10919. [DOI] [PubMed] [Google Scholar]
- (9).a) Singh K; Staig SJ; Weaver JD Facile Synthesis of Z-Alkenes Via Uphill Catalysis. J. Am. Chem. Soc 2014, 136, 5275; [DOI] [PubMed] [Google Scholar]; b) Chen C; Dugan TR; Brennessel WW; Weix DJ; Holland PL Z-Selective Alkene Isomerization by High-Spin Cobalt(II) Complexes. J. Am. Chem. Soc 2014, 136, 945; [DOI] [PubMed] [Google Scholar]; c) Metternich JB; Gilmour R A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins. J. Am. Chem. Soc 2015, 137, 11254. [DOI] [PubMed] [Google Scholar]
- (10).Hazra A; Chen J; Lalic G Stereospecific Synthesis of E-Alkenes through Anti-Markovnikov Hydroalkylation of Terminal Alkynes. J. Am. Chem. Soc 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).a) Uehling MR; Suess AM; Lalic G Copper-Catalyzed Hydroalkylation of Terminal Alkynes. J. Am. Chem. Soc 2015, 137, 1424; [DOI] [PubMed] [Google Scholar]; b) Suess AM; Uehling MR; Kaminsky W; Lalic G Mechanism of Copper-Catalyzed Hydroalkylation of Alkynes: An Unexpected Role of Dinuclear Copper Complexes. J. Am. Chem. Soc 2015, 137, 7747. [DOI] [PubMed] [Google Scholar]
- (12).a) Lu X-Y; Liu J-H; Lu X; Zhang Z-Q; Gong T-J; Xiao B; Fu Y 1,1-Disubstituted Olefin Synthesis Via Ni-Catalyzed Markovnikov Hydroalkylation of Alkynes with Alkyl Halides. Chem. Commun 2016, 52, 5324; [DOI] [PubMed] [Google Scholar]; b) Till NA; Smith RT; MacMillan DWC Decarboxylative Hydroalkylation of Alkynes. J. Am. Chem. Soc 2018, 140, 5701; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang Z; Yin H; Fu GC Catalytic Enantioconvergent Coupling of Secondary and Tertiary Electrophiles with Olefins. Nature 2018, 563, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cheung CW; Zhurkin FE; Hu X Z-Selective Olefin Synthesis Via Iron-Catalyzed Reductive Coupling of Alkyl Halides with Terminal Arylalkynes. J. Am. Chem. Soc 2015, 137, 4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).a) Brown HC; Levy AB; Midland MM Reaction of Lithium Ethynyl- and Ethenyltrialkylborates with Acid. Valuable Route to the Markovnikov Alkenyl- and Alkylboranes. J. Am. Chem. Soc 1975, 97, 5017; [Google Scholar]; (b) Zweifel G; Arzoumanian H; Whitney CC A Convenient Stereoselective Synthesis of Substituted Alkenes Via Hydroboration-Iodination of Alkynes. J. Am. Chem. Soc 1967, 89, 3652; [Google Scholar]; (c) Miyaura N; Yoshinari T; Itoh M; Suzuki A Reaction of Lithium Alkynyltrialkylborates with Propionic Acid. General and Convenient Syntheses of Internal and Terminal Olefins Using Organoboranes. Tetrahedron Lett. 1974, 15, 2961. [Google Scholar]
- (15).a) Matteson DS; Mah RWH Neighboring Boron in Nucleophilic Displacement. J. Am. Chem. Soc 1963, 85, 2599; [Google Scholar]; b) Aggarwal Varinder K; Fang Guang Y; Ginesta X; Howells Dean M; Zaja M Toward an Understanding of the Factors Responsible for the 1,2-Migration of Alkyl Groups in Borate Complexes Pure and Applied Chemistry 2006, 78, 215. [Google Scholar]
- (16).For a review of catalyst-promoted 1,2-metallate shift, see:; Namirembe S; Morken JP Reactions of Organoboron Compounds Enabled by Catalyst-Promoted Metalate Shifts. Chem. Soc. Rev 2019, 48, 3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Binger P; Benedikt G; Rotermund GW; Köster R Borverbindungen, Xv. Alkalimetall-Alkyl-1-Alkinyl-Boranate. Justus Liebigs Ann. Chem 1968, 717, 21. [Google Scholar]
- (18).For selected examples of 1,2-metallate shift induced by π-activation of borate complexes, see:; a) Zhang L; Lovinger GJ; Edelstein EK; Szymaniak AA; Chierchia MP; Morken JP Catalytic Conjunctive Cross-Coupling Enabled by Metal-Induced Metallate Rearrangement. Science 2016, 351, 70; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Edelstein EK; Namirembe S; Morken JP Enantioselective Conjunctive Cross-Coupling of Bis(Alkenyl)Borates: A General Synthesis of Chiral Allylboron Reagents. J. Am. Chem. Soc 2017, 139, 5027; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lovinger GJ; Morken JP Ni-Catalyzed Enantioselective Conjunctive Coupling with C(sp3) Electrophiles: A Radical-Ionic Mechanistic Dichotomy. J. Am. Chem. Soc 2017, 139, 17293; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Chierchia M; Law C; Morken JP Nickel-Catalyzed Enantioselective Conjunctive Cross-Coupling of 9-BBN Borates. Angew. Chem., Int. Ed 2017, 56, 11870; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Myhill JA; Wilhelm-sen CA; Zhang L; Morken JP Diastereoselective and Enantioselective Conjunctive Cross-Coupling Enabled by Boron Ligand Design. J. Am. Chem. Soc 2018, 140, 15181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).For other related examples of transition metal-catalyzed 1,2-metallate shift, see:; a) Ishida N; Shimamoto Y; Murakami M Stereoselective Synthesis of (E)-(Trisubstituted Alkenyl)Borinic Esters: Stereochemistry Reversed by Ligand in the Palladium-Catalyzed Reaction of Alkynylborates with Aryl Halides. Org. Lett 2009, 11, 5434; [DOI] [PubMed] [Google Scholar]; b) Ishida N; Narumi M; Murakami M Synthesis of Amine−Borane Intramolecular Complexes through Palladium-Catalyzed Rearrangement of Ammonioalkynyltriarylborates. Org. Lett 2008, 10, 1279; [DOI] [PubMed] [Google Scholar]; c) Ishida N; Miura T; Murakami M Stereoselective Synthesis of Trisubstituted Alkenylboranes by Palladium-Catalysed Reaction of Alkynyltriarylborates with Aryl Halides. Chem. Commun 2007, 4381; [DOI] [PubMed] [Google Scholar]; d) Shimamoto Y; Sunaba H; Ishida N; Murakami M Regioselective Construction of Indene Skeletons by Palladium-Catalyzed Annulation of Alkynylborates with O-Iodophenyl Ketones. Eur. J. Org. Chem 2013, 2013, 1421. [Google Scholar]
- (20).Sebald A; Wrackmeyer B Novel Synthesis of Platinum(II) Alkenyl Compounds Via Organoboration of Platinum(II) Acetylides. J. Chem. Soc., Chem. Commun 1983, 309. [Google Scholar]
- (21).Shapiro ND; Toste FD Synthesis and Structural Characterization of Isolable Phosphine Coinage Metal π-Complexes. PNAS 2008, 105, 2779. [Google Scholar]
- (22). See SI for details.
- (23).Sałat K; Jakubowska A; Kulig K Zucapsaicin for the Treatment of Neuropathic Pain. Expert Opin. Investig. Drugs 2014, 23, 1433. [DOI] [PubMed] [Google Scholar]
- (24).Kaga H; Miura M; Orito K A Facile Procedure for Synthesis of Capsaicin. J. Org. Chem 1989, 54, 3477. [Google Scholar]
- (25).Mann TJ; Speed AWH; Schrock RR; Hoveyda AH Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis. Angew. Chem., Int. Ed 2013, 52, 8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lu T; Gu M; Zhao Y; Zheng X; Xing C Autophagy Contributes to Falcarindiol-Induced Cell Death in Breast Cancer Cells with Enhanced Endoplasmic Reticulum Stress. PLoS ONE 2017, 12, e0176348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.