

Abstract
Circulating tumor cells (CTCs) exist in a microenvironment quite different from the solid tumor tissue microenvironment. They are detached from matrix and exposed to the immune system and hemodynamic forces leading to the conclusion that life as a CTC is “nasty, brutish, and short.” While there is much evidence to support this assertion, the mechanisms underlying this are much less clear. In this chapter we will specifically focus on biomechanical influences on CTCs in the circulation and examine in detail the question of whether CTCs are mechanically fragile, a commonly held idea that is lacking in direct evidence. We will review multiple lines of evidence indicating, perhaps counterintuitively, that viable cancer cells are mechanically robust in the face of exposures to physiologic shear stresses that would be encountered by CTCs during their passage through the circulation. Finally, we present emerging evidence that malignant epithelial cells, as opposed to their benign counterparts, possess specific mechanisms that enable them to endure these mechanical stresses.
Keywords: Circulating tumor cell, Metastasis, Hemodynamic forces, Fluid shear stress
11.1. Introduction
Metastasis is responsible for the lethal consequences of most solid tumor types and remains, in many respects, a poorly understood biological process. To colonize organ sites remote from the primary tumor, cancer cells must travel through the bloodstream, a microenvironment wholly unlike the solid tissues from which they were derived. Within this microenvironment, these migrant cancer cells are referred to as circulating tumor cells or CTCs. Although CTCs may have first been described as early as 1869 [1], they remain enigmatic in many ways, in part, due to difficulties in studying these rare cells. Among the open and actively investigated questions related to CTCs are: (1) How long do they spend in the circulation and navigate it? (2) How does the genotypic/phenotypic diversity of CTCs relate to that of the primary tumor or metastases? (3) What is the capacity of individual CTCs for metastatic colonization? and (4) How does the microenvironment of the circulation influence the biology of CTCs? Regarding the latter question, this chapter will focus on how hemodynamic forces interact with the particular biology of CTCs in a manner that is relevant to metastasis.
Metastasis has long been regarded as an inefficient process when considered from the perspective of CTCs [2]. For example, Zeidman observed in 1950 that only tens of lung tumors resulted from >105 cancer cells injected intravenously, and Fidler found that intravenously injected B16 melanoma cells rapidly die, with only about 1% cells surviving to 24 h [3, 4]. Shedding rates of 3–4 × 106 cancer cells/day/g of tumor tissue have been measured in experimental tumor models [5]. Moreover, peritoneovenous shunts to relieve ascites in cancer patients release billions to trillions of cancer cells into the circulation without resulting in observable metastases in the lungs and other organs in some patients [6]. This leads to the conclusion that the overwhelming majority of CTCs do not produce clinically observable metastases, but why? There are several, non-mutually exclusive explanations for this. Foremost among them is that only a subset of CTCs are capable of growing into a metastasis due to both cell-intrinsic and microenvironmental mechanisms [7, 8]. First articulated as the “seed and soil hypothesis” by Paget, this is now a widely held and idea, although many questions remain [9, 10]. Additionally, CTCs may rapidly succumb to destructive mechanisms including (1) anoikis or programmed cell death due to their detachment from extracellular matrix, though cancer cells often have some intrinsic resistance to this [11], (2) exposure to immune system-mediated destruction when separated from the immune-privileged microenvironment of the primary tumor [8], and (3) mechanical destruction due to hemodynamic forces including deformation in the microvasculature [12]. Comparatively speaking, far less is known about these destructive mechanisms. In this chapter, we will focus on the latter issue, specifically the question of whether CTCs are mechanically fragile and findings that support the concept that CTCs, as compared to benign counterparts, may possess biologic mechanisms that provide resistance to destructive mechanical forces. We will first begin with a consideration of the mechanical challenges CTCs are confronted with in the circulation.
11.2. Biomechanics of the Circulation: Strain and Stress
The circulation is a remarkable, highly evolved system for the efficient transport of blood cells, gasses, nutrients, and hormones that provides tissues with nourishment and can aid in the organisms’ defense, homeostasis, and growth, among other critical roles. It is estimated that a red blood cell in a human makes an entire circuit in about 1 min, during which it passes twice through the heart and microcirculation—pulmonary and periphery. Here we will consider three main aspects of the circulation that impinge on the mechanical stability of CTCs: (1) fluid shear stress imparted by blood flow, (2) deformation in the microcirculation, and (3) forces generated by adhesive interactions between circulating cells and the vascular wall.
11.2.1. Hemodynamic Shear Stress: Going with the Flow
The circulation is typically thought of as two units, the cardiovascular system, which delivers blood to the tissues, and the lymphatic system, which distributes lymph. The cardiovascular system consists of plasma, red blood cells (RBC), leukocytes, and platelets that are cycled throughout the tissues in the pulmonary and systemic circulation. The lymph is recycled blood plasma that has filtered through interstitial fluid, before eventually draining into lymphatic ducts and finally the subclavian veins.
Within these systems, flow is driven by either gravitational or pressure gradient forces. In a given blood vessel, the rate of change of pressure along the length of the vessel in a specific direction gives rise to the pressure gradient, forcing flow through the vessel. However, viscosity, a measure of a fluids resistance to deformation, opposes the pressure gradient, creating shear stresses between neighboring layers of fluid moving at different velocities. The magnitude of fluid shear stress (FSS) a given fluid layer experiences is dependent on the viscosity of the fluid and the shear rate.
τ is the wall fluid shear stress (WSS), μ is the viscosity, and the shear rate is δu/δy. Stress is force acting on a surface divided by the area of the surface. The shear rate is the derivative of fluid velocity perpendicular to a boundary. Under the assumptions that flow in a straight, horizontal tube with a Newtonian fluid is steady (not accelerating) and laminar, one can solve for the Hagen-Poiseuille equation using the Navier-Stokes equations in cylindrical coordinates. Newtonian fluids behave with a viscosity μ that is independent of shear stress. In fluids with a solid boundary, a no-slip condition can be applied that states that the fluid will have a velocity of zero next to the boundary.
Δp is the change in pressure across the tube, L is the length of the vessel, Q is the flow rate, and d is the diameter. This results in a parabolic velocity profile across a vessel for Newtonian fluids in laminar flow. Thus, FSS varies linearly with respect to the radial position of a fluid layer, with the maximum FSS seen at the wall. The viscosity of blood, however, varies with shear rate of ~100 s−1 or less before behaving like a Newtonian fluid [1]. In the microcirculation, Poiseuille flow is not valid, as blood behaves in a non-Newtonian manner and the interaction of RBCs alters the dynamics of the flow. Laminar flow, the norm within much of the circulation, occurs when there is no disruption between layers of fluid, resulting in parallel layers of fluid flow. Fluid flow can transiently become turbulent and chaotic. The Reynolds number characterizes this flow pattern of a fluid and can be used to predict the transition between laminar and turbulent flow.
ρ is the density, μ is the average velocity of the fluid, and L is the length of the vessel. Re greater than 2000 suggests transition to turbulent flow. Re is less than one within the microcirculation where viscous forces dominate. Turbulence within the circulation is rare, but it is present momentarily in the ascending aorta, aortoiliac bifurcation, or during the opening and closing of heart valves, among other locations [13]. When turbulence occurs, it increases the frequency of cell damage. At levels of WSS between 2000 and 4000 dyn/cm2, turbulence resulted in significantly greater hemolysis than laminar shear flows of similar magnitudes [14]. However, the brevity of turbulent flow likely limits the extent of cellular damage. Flow destabilization occurs in very brief ~150 ms time intervals and is immediately followed by low velocity and Re flows.
The location of the primary tumor often dictates the initial route of metastasis through the circulation and thus the stresses a CTC will encounter. Two paths of metastasis within the circulation are lymphatic and hematogenous spread. Average WSS in the lymphatics is 0.64 dyn/cm2 with peaks between 4 and 12 dyn/cm2 [15, 16]. Lymphatic capillaries are typically 15–75 μm with the lower order vessels becoming increasingly larger with the thoracic duct being ~5 mm. On the other hand, during hematogenous dissemination, CTCs will pass through the cardiovascular system and be exposed to a pulsatile and more mechanically stressful environment. In humans, although a commonly cited value for overall mean arterial WSS is ~15 dyn/cm2, considerable variation exists both regionally and locally within the arterial tree with a linear, inverse relationship between average WSS and arterial lumen diameter [17]. A range of 10–70 dyn/cm2 has been reported for normal arteries, whereas venous WSS is much lower in the range of 1–6 dyn/cm2 [18, 19]. The WSS encountered in the microcirculation (arterioles, capillaries, venules) is estimated between 3 and 140 dyn/cm2 in a low Re environment [20]. CTCs must also pass through the heart, which represents a very dynamic FSS environment with wide local variations that have been challenging to precisely measure experimentally. Various experimental and computational efforts indicate maximum WSS near the tips of the ventricular surface of valve leaflets to be in the range of 79–100 dyn/cm2 [21]. Turbulent, high Re flows may exist briefly around heart valves and into the ascending aorta [13]. However, it is difficult to quantify FSS levels on blood cells under these circumstances, and a wide range of estimates are reported up to 520 dyn/cm2 [22]. Pathological conditions can modify the degree of FSS present in the circulation. Computational simulations indicate that aortic coarctation and coronary stenosis can produce local WSS values in the range of 1000–3000 dyn/cm2 [23, 24]. Moreover, mechanical heart valves are known to produce FSS in the range of 1500–4500 dyn/cm2 [25]. Exercise can increase, and conversely anesthesia can reduce mean arterial WSS [17]. A summary of the FSS encountered in different areas of the circulation is shown in the y-axis of Fig. 11.1a.
Fig. 11.1.
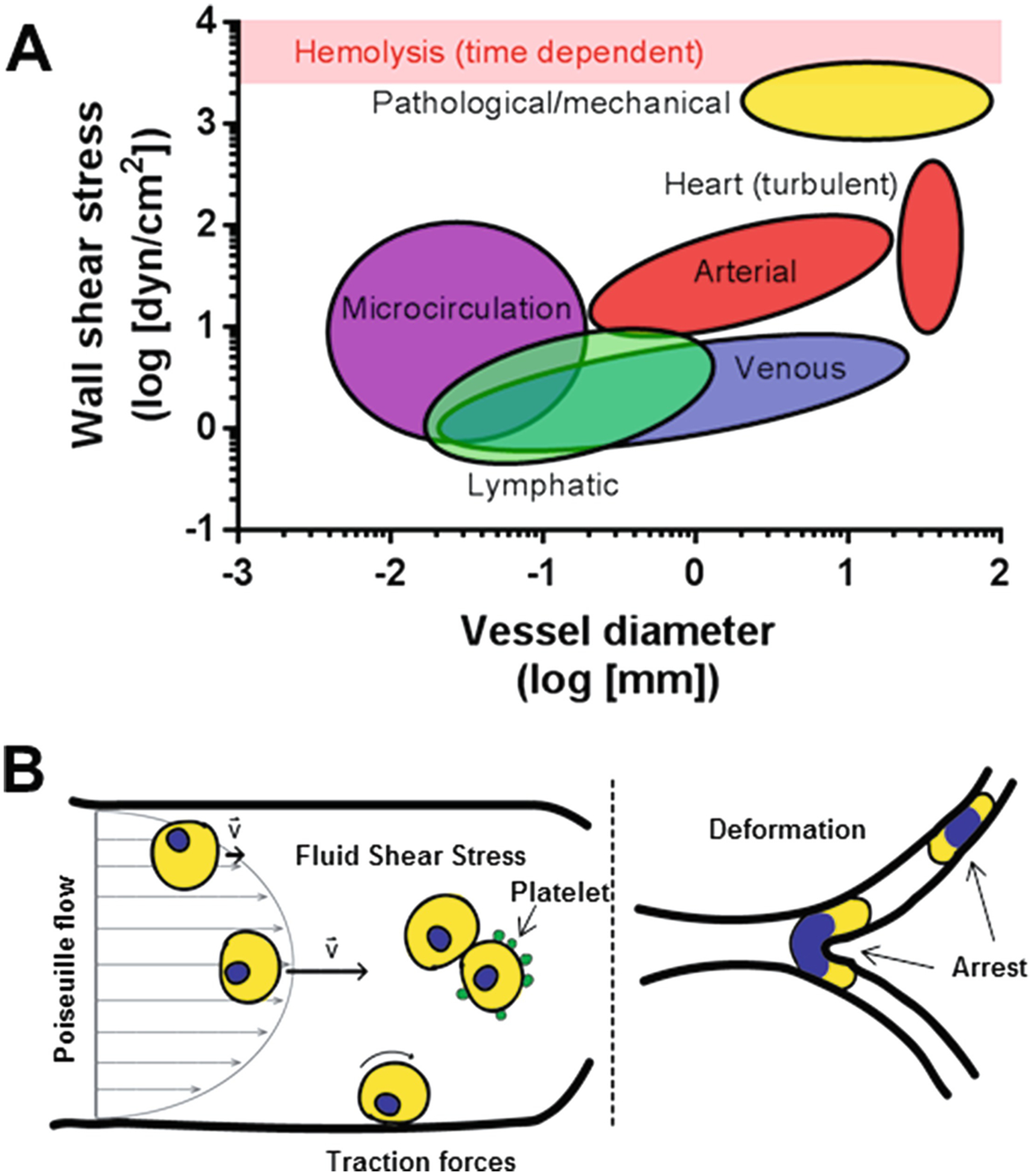
(a) Range of wall shear stress (WSS) related to vessel diameter in physiologic and pathophysiologic conditions based on [13–25]. Ovals represent approximate values in different parts of the vasculature. The level of WSS associated with hemolysis is indicated by the red bar, though it is important to note that this is dependent on the time of exposure. (b) Representation of different hemodynamic forces encountered by CTCs. Fluid shear stress (FSS) can be calculated under conditions of Poiseuille flow where the maximum is at the vessel wall and the minimum is at the vessel axis. CTCs may associate with each other or with other blood components such as platelets which could influence the level of FSS encountered. Traction forces, generated by adherence of CTCs to the vessel wall under flow, may be encountered by CTCs under certain circumstances. Once in the microcirculation the arrest and deformation of the relatively large CTCs may also be destructive
It is also important to consider how FSS varies among species since experimental models for CTCs involve a variety of animal species. Generally speaking for mammals, for a given arterial vessel, WSS is inversely proportional to animal size [17]. For example, in the common carotid artery, average mean WSS is 11.6 dyn/cm2 for human, 15.8 dyn/cm2 for dogs, 23.3 dyn/cm2 for rabbits, 46.6 dyn/cm2 for rats, and 64.8 dyn/cm2 for mice. Zebra fish embryos are a potentially useful model for the study of CTCs [26, 27]. Much of the research focus in this organism has been on the role of hemodynamic forces in cardiac development [28]. As the animal develops, peak WSS in the AV canal measured by digital particle image velocimetry ranged from 2.5 dyn/cm2 at 37 hpf to 76 dyn/cm2 at 4.5 dpf [29].
11.2.2. Cell Deformation in the Microvasculature: Size Matters
The diameters of vessels within the circulatory system vary across several orders of magnitude from <5 μm to >30 mm. A summary of the size of vessels encountered in different areas of the circulation is shown in x-axis of Fig. 11.1a. Normal red blood cells have diameters between 6 and 8 μm. In blood vessels smaller than the diameter of a RBC, the cell is able to rapidly deform to squeeze through. All leukocytes are too large to pass undeformed through much of the microcirculation. Basophils, neutrophils, and eosinophils are 14–16 μm, 12–14 μm, and 12–17 μm in diameter, respectively. Lymphocytes are 10–14 μm in diameter. The largest of the white blood cells, monocytes/macrophages, have diameters of 15–25 μm. Platelets can pass through the microvasculature unhindered as their diameters are 2–4 μm. In comparison, cultured cancer cells are generally 15–20 μm in diameter. CTC cell size has not been extensively documented. One study evaluating CTC size in a small sample of patients found the mean diameter to be slightly larger than leukocytes ranging between 10 and 15 μm, although the size distribution was overlapping [30]. Another study focusing on measurements of nuclear size of prostate cancer CTCs found that they could be clustered into three distributions with means at 6.82 μm, 10.63 μm, and 21.63 μm [31]. Interestingly, visceral metastasis was more common in those patients with CTCs in the smallest size distribution.
Due to the size constraint of the microcirculation, blood cells must be able to deform in order to pass through the small diameter vessels. The velocity of leukocytes within the microcirculation is significantly less compared to red blood cells, which can traverse capillary beds in seconds [32]. RBCs, which can rapidly alter their shape, navigate capillaries ~300 times faster than leukocytes [33]. Nonetheless, leukocytes can negotiate capillaries, although they are frequently briefly arrested in the microvasculature [34]. While in these small vessels, both RBCs and leukocytes experience stress on their membranes and cytoskeleton as they are deformed. Membrane unfolding of leukocytes allows a large area expansion necessary for the shape changes associated with deformation in vessels smaller than the diameter of the cell [35]. The area expansion modulus for neutrophils was 3.9 × 10−5 N/m [36]. On the other hand, red blood cells had a modulus of 0.3–0.6 N/m and a nearly incompressible membrane [35, 37]. RBCs instead have a tremendous ability to change the geometry of the cell while maintaining a near constant membrane surface area. The bending modulus has been reported as 10−19 N/m, indicating a remarkable ability to rapidly deform. RBC cytoplasmic viscosity has been measured as 0.006 Pa*s [37]. On the other hand, neutrophil cytoplasmic viscosity was 135–200 Pa*s [38]. With respect to the cytoskeletal makeup of red blood cells, it is clear that the spectrin is important in strengthening the membrane and providing it with durability and flexibility, though the dynamics of the spectrin cytoskeleton remains largely unknown [39]. RBCs are also extraordinarily resistant to high levels of fluid shear stress, with hemolysis detectable at 1500 dyn/cm2, a supraphysiologic magnitude, after 2 min exposures [40]. In addition, hemolysis has been reported at brief, millisecond pulses of 4500–5600 dyn/cm2 [41, 42]. Thus, hemolysis is dependent on both exposure time and magnitude of shear [43]. Compared to blood cells, the deformability of CTCs is less clear. However, an initial foray into this question has revealed some interesting findings [44]. In mouse breast cancer models, CTCs were less deformable than blood cells as measured by a suspended microchannel resonator, but CTCs were not different from the cell line used to initiate the tumors. Interestingly, however, based on a limited set of observations, CTC’s from prostate cancer patients exhibited similar deformability to blood cells. Further analysis of the deformability of CTCs is warranted as this issue is likely to be a critically important determinant for how CTCs negotiate the microvasculature, as well as the relevance of models employing cancer cell lines as discussed below.
Due to these differences in physical properties, the flow patterns of the respective cells differ. RBCs exhibit two types of motion, tumbling and tank-treading, at low and high shear rates, respectively [45, 46]. Tank-treading, where the cell is in a fixed orientation and the membrane rotates around the cell body, allows the transfer of shear forces into the cell interior. The whole cell then partakes in the flow, resulting in reduced apparent blood viscosity and lower flow resistance [35]. In small diameter vessels, the apparent viscosity is also reduced, called the Fåhræus-Lindqvist effect, through the orientation of erythrocytes in the center of the vessel and formation of rouleaux, leaving the plasma along the wall of the vessel. This effect also causes margination of leukocytes, allowing interaction with the endothelium in small diameter vessels [47]. Shunting of leukocytes has been witnessed with cells preferentially being shuttled into vessels with the highest flow rate at bifurcations, possibly bypassing smaller diameter capillaries where cell arrest would occur [35]. Computational analysis also predicts margination of CTCs [48]. Despite the different mechanisms employed, blood cells are able to freely circulate continuously, which as described below, is not obviously the case for CTCs.
11.2.3. Leukocytes and Traction Forces
In response to inflammatory stimuli, leukocytes engage in molecularly choreographed adhesive interactions with vessel walls where traction forces against the vessel wall and fluid shear also generating torque from the passing blood flow come to bear on these cells. Generally, this process involves selectin-mediated rolling in the lower-shear environment of postcapillary venules, followed by integrin-mediated tight adhesion to the endothelium and ultimately extravasation. A detailed discussion of the biomechanical implications of this behavior is beyond the scope of this chapter, but the reader is referred to a recent review [49]. There is evidence that leukocyte-like behaviors may be evident in CTCs under some circumstances which is discussed in more detail below. Decades of research on blood cell biology and hemodynamics have revealed considerable information about the mechanobiology of the circulating microenvironment in which CTCs reside. A summary of the primary forces to which CTCs are exposed is shown in Fig. 11.1b.
11.3. Circulating Tumor Cells
CTC’s are of inherent biological and clinical interest since they represent a definable intermediate in the metastatic cascade [7]. The CTC microenvironment presents a fundamentally different lifestyle for cancer cells accustomed to a solid tumor microenvironment—detached from a solid matrix, diluted into a dynamic river of blood cells, and distributed within some 105 km of the vasculature. Moreover, the transition to becoming a CTC is likely to be abrupt, and while the time spent in free circulation in larger vessels and in the heart is likely relatively short, CTCs may dwell within the microvasculature for longer periods of time. CTCs are rare (~1–10 CTC/109 blood cells) which is a central challenge to studying them, particularly in vivo, contributing to many open questions [9]. Most of the focus to date on CTCs has been on developing methods to isolate them and to characterize them genetically [7, 50]. Indeed, it has only recently been experimentally demonstrated that isolated human CTC preparations include cells capable of initiating metastasis, at least in immunocompromised mice [51, 52]. This effort is driven by the promise that CTCs could provide a “liquid biopsy” that can be collected in a minimally invasive manner and better represent tumoral heterogeneity than solid tissue biopsies [53]. Numerous technical and biological challenges remain before this promise is broadly realized in the clinic [54]. To understand the biomechanical influences on CTCs, it is important to first discuss what is currently known about the natural history of CTCs.
11.3.1. Becoming a CTC
Intravasation is recognized as necessary step for distant metastasis and generating CTCs. Intravasation can occur early and throughout tumor progression, including even non-transformed epithelial cells [55–57]. There is currently evidence to support that intravasation can result from both an active process of cancer cell invasion into blood vessels as well as a passive process of cancer cell shedding into the circulation driven by mechanical stress on the tumor and/or disorganization within the tumor microenvironment [58].
Cancer cells are thought to intravasate within the poorly formed and functioning tumor (primary and metastatic) vasculature. This can happen at both invasive margins of tumors and within the core of the tumor [59]. However, evidence indicating that CTCs are detectable from preneoplastic lesions indicates that a mature tumor-associated vasculature per se is not prerequisite [56, 57]. In fact, epithelial cells can be found in the circulation of patients with benign inflammatory conditions in which blood vessels may have enhanced permeability [60]. CTCs can also originate from micrometastases, and not just clinically observable tumors [61], suggesting the possibility that they might traverse microvascular beds in a stepwise fashion. Direct observations, via intravital microscopy, provide evidence for an active intravasation process, facilitated by perivascular macrophages [62]. Epithelial-tomesenchymal transition (EMT) a cancer cell phenotype that can promote invasive characteristics has also been implicated in intravasation [63]. Numerous accounts of CTCs bearing markers of EMT have been reported, for example [64, 65]. However, it is important to note that EMT may not be required for metastasis in all contexts [66–68].
CTCs might also be generated without requiring active cancer cell migration [58]. Early studies showed that a high rate of cancer cell shedding is observed in perfused experimental tumors [5, 69]. Liotta et al. showed that mechanical trauma to the tumor could increase the number and size of cell clumps released into the venous effluent of experimental tumors [69]. Some studies have shown that surgical interventions can increase CTC numbers perioperatively, presumably due to manipulation of the tumor [70]. This data suggests the possibility that routine mechanical forces, combined with the disorganized vasculature of tumors, could also contribute to the genesis of CTCs.
CTCs are now routinely observed to be as both single cells and less frequently as clusters with more than 50 cells, also known as circulating tumor microemboli (CTM). CTM have been observed for some time and in addition to cancer cells, can include platelets, leukocytes, and perhaps other cells [8, 69, 71–73]. However, it is not yet entirely clear how (active migration of cell groups or passive release?) or where (in what size vessels?) CTM intravasate directly from tumors (primary or metastatic), or alternatively, whether they are the result of proliferation of CTCs elsewhere in the vasculature. CTM have garnered considerable attention as it has been demonstrated in experimental models that CTM have a greater capacity for metastasis than individual CTCs [69, 71, 74]. One hypothesis is that these multicellular clusters are protected from the destructive forces mentioned above, including anoikis, immune—and mechanical—insults. In an impressive set of experiments, Aceto et al. showed that CTM in a mouse model of breast cancer are of predominantly oligoclonal origin from a mixture of cells in the primary tumor, indicating that they are not derived from the proliferation of single CTC [74].
11.3.2. Life (and Death) in the Circulation
Once CTCs have entered the circulation, most available evidence indicates that they have only a brief stay there, though measuring CTC half-life in the circulation has been quite challenging. Knowing CTC half-life is important, as it comes to bear on understanding the temporal nature of CTC clearance mechanism(s). Using a flow cytometry-based method to quantify CTCs, Meng et al. estimated the half-life of CTCs following surgical excision of primary breast tumors to be on the order of 1–2 h [61]. Using different methods to enumerate CTCs, another study corroborated a rapid decline of CTCs following primary prostate cancer resection [75]. Aceto et al. used in vivo flow cytometry to estimate the half-life of CTCs and CTMs from a bolus intravenous injection in immunocompromised mice and found the half-life of ~30 min and ~10 min, respectively [74]. Numerous caveats exist with these measurements, and they do not alone reveal whether CTCs are continuously circulating or whether the time spent in free circulation is punctuated with periods of relative immobility arrested in the microvasculature. As mentioned above, and detailed below, one fundamental and rapid (seconds) mechanism acting on the clearance of CTCs is size restriction in the microvasculature, which at least temporarily removes these cells from free circulation. Moreover, the time frames of other clearance mechanisms (e.g., anoikis and immune destruction) are not well defined.
It is important to note that not all CTCs are viable. Numerous reports indicate a significant number of dead and dying CTCs in cancer patients e.g., [76–78]. However, it is not entirely clear from these studies the extent to which dead/dying CTCs result from destructive mechanisms in the host or are an artifact of the methods used to isolate and characterize CTCs. Some cell death may result from the CTC isolation methods including the reagents used, duration of isolation, and perhaps exposure to FSS, though the magnitude of exposure in most CTC isolation protocols is significantly less than might be encountered in the circulation. Moreover, passive intravasation mechanisms discussed above may already shed dead and dying cells directly into the circulation [79]. In short, we do not yet have an adequate understanding of many details involved with CTC clearance to develop an adequate model of CTC half-life.
Once cancer cells enter the circulation and become CTCs, it is very likely that they do not persist in a freely circulating state for very long. Escape from the immediate tumor vasculature may be influenced by spatial and temporal heterogeneity in tumor blood flow patterns [80, 81]. This suggests the possibility that CTCs or CTM may dwell for some period of time within the tumor microcirculation before exiting to the systemic circulation. Consistent with this interpretation, Liotta et al. found that passage of clumps of cancer cells into the venous effluent of experimental tumors could be restricted by the size of vessels present [69]. After CTCs enter transport vessels, they would be expected to move at blood velocity, which increases from 0.3 cm/s in the capillaries to 40 cm/s in the aorta. Since total transit time of a red blood cell is on the order of 1 min, for most CTCs, this is likely to be just seconds, depending on the distance between where they are generated and the first microvascular bed encountered.
When entering the systemic circulation, CTCs are subject to existing blood flow patterns which can be used to predict the exposure of various organs to CTCs. Indeed, metastasis often presents in the first microvascular bed encountered based on these patterns [82]. For example, colon cancer frequently metastasizes to the liver, where the splanchnic circulation delivers CTCs, and the lumbar spine is a frequent site of prostate cancer metastasis, to which CTCs are delivered by Batson’s plexus, a valveless venous system draining the pelvic floor [83]. Ewing took examples such as this as an alternative Paget’s “seed and soil” hypothesis [84]. However, although this might explain a proclivity for some organs to be involved in metastasis, it cannot alone explain all patterns of metastasis as it is clear that it can occur in sites other than the first microvascular bed encountered [82, 85, 86].
11.3.3. Vascular Arrest and Transit
Consideration of both first principles and available experimental data indicates that size restriction in the microvasculature is the most likely initial fate befalling CTCs, likely occurring within seconds of release into the circulation. As mentioned above, cancer cells from solid tumors are large (15–20 μm in diameter) relative to capillaries (3–8 μm in diameter), and thus the microcirculation represents a CTC filter. Fidler’s early work with radiolabeled B16 melanoma cells showed that the majority (>50%) of intravenously injected cells are present in the lungs 1 min postinjection with a much smaller amount (~2%) detectable in the liver and only ~1% remaining in the blood sample taken [4]. The number of cells detectable in the lungs was relatively stable for 1 h postinjection and then began to decrease such that by 24 h only ~1% remained with concomitant evidence of cell death. Similarly, radiolabeled colon cancer cells are efficiently trapped in the lung or liver of rats 30 min after injection and after intravenous or portal vein injection, respectively [87]. Other studies using quantitative PCR to measure tumor cells have shown that over 85% of injected cells are present in the lung 5 min postinjection with about 20% of those cells persisting to 24 h [88]. In contrast to these studies, Chambers and colleagues, using an accounting method involving co-injection 9 μm fluorescent microspheres, which lodge in the microvasculature as a reference, found a much higher percentage of B16-F10 cells (98%) which are present in the lung at 1 h post-intravenous injection with 83% persisting as solitary cells by 24 h [89]. Similar results had been obtained previously by this group using a portal vein injection to seed cells into the liver [90]. While these studies may differ quantitatively in regard to the fate of cancer cells at 24 h and beyond (e.g., whether they die or are displaced), there is clearly good agreement that, at a short interval, a majority of cells are arrested in the lung. An important caveat to all of the studies mentioned above is that they involve bolus injections of dissociated, cultured cancer cells into mice. These may not adequately model what happens to CTCs released from tumors. Moreover, outside of the lung and liver, we do not have quantitative data supporting the degree to which CTCs are arrested in the microcirculation of other organs and tissues.
Although the evidence above supports the idea that most cancer cells are arrested, at least transiently, in the first microvascular bed encountered, some clearly do escape. As mentioned above, metastasis is not simply restricted to the first microvascular bed encountered based on blood flow patterns, and the fact that CTCs are detectable in the arm vein of cancer patients is further evidence of this. Cancer cells are deformable and motile, and these properties could contribute to their ability to negotiate the microcirculation barrier. However, there is considerable uncertainty about the frequency of escape from the first encounter with the microvasculature and the extent to which it is a passive stochastic vs. an active biological process. Some studies have attempted to address this question.
Early microcinematic studies suggested that cancer cells can traverse the mesenteric capillary bed, consistent with other experimental evidence, with considerable distortion, though cancer cells could not be unambiguously identified in these studies [91]. A more recent and particularly heroic study from Keinast et al. has shed light on the early minutes to months in the life of individual cancer cells introduced into the mouse brain [92]. Initial cancer cell arrest and distortion occurred in small cancer cell-sized diameter vessels and particularly at vascular branch points. Only ~10% of cells remained fixed at the initial site of arrest for >60 s. Other cells were observed to relocate from an initial site of arrest and lodge in another for up to 48 h. In multiple lung and melanoma cell models, between 37.6% and 63.8% of individual intravascular arrested cells were either displaced or died during the week-long observation window, though the authors could not distinguish between these two fates. What was clear in this study, however, is that continuously growing macrometastases only arose from cells that productively extravasated. In contrast, studies in perfused lung model have indicated that cancer cells can proliferate in vessel lumens [93], raising the possibility that CTMs could arise from this source. We will not discuss extravasation, clearly an active biological process, further in this chapter, but the reader is referred to a recent review and other chapters in this volume for more details [94].
Little is known about how CTCs move following a period of arrest in the microvasculature. The work cited above from Keinast et al. indicates that in the immediate minutes following arrival in the microvasculature, CTCs are not tightly adherent to the endothelium and by distorting can be pushed forward by hemodynamic pressure [92]. The question of when and where adhesive interactions begin between CTCs and the vascular wall is important and still elusive. CTC adhesion to the endothelium can be influenced by inflammation (for review [8]). In fact, in IL-1α-treated mice, cancer cells were observed to arrest in vessels much larger than the cancer cell diameter [95]. Similar behavior has been observed in isolated perfused rodent lungs [93]. It is important to point out that in many of the intravital microscopy studies to date, leukocyte-like rolling behavior on the endothelium has not been observed in cancer cells arriving in microvascular beds [92, 96–98]. However, a couple of studies have reported this behavior in inflamed vasculature [99, 100]. Barthel et al. found that PC-3 prostate cancer cells engineered to express E-selectin ligands were observed rolling in TNF-α-inflamed postcapillary venules of the mouse cremaster muscle [99]. Thus, the conditions under which CTCs may develop leukocyte-like behaviors in vivo is underexplored. If leukocyte-like behavior of CTCs is restricted to inflamed postcapillary venules where leukocyte trafficking predominates, an important question is how many CTCs can negotiate the initial size restriction in capillaries to access this microenvironment. These studies highlight the need to carefully consider the models employed and circumstances under which short-term adhesive interactions between CTCs and the endothelium are studied.
Whether and how CTCs can continue movement within the lumen of the microvessel through active means are also poorly understood. Entenberg et al. reported that size-arrested cells in the lung microvasculature initially exhibit a highly protrusive phenotype, reminiscent of their behavior in the primary tumor, which diminishes over 24 h, but this is not associated with luminal translocation of the cells [101]. However, in another study, Yamauchi et al., using video microscopy of a skin flap preparation, showed that some HT-1080 fibrosarcoma cells within 2 h of arrival in the microvascular bed were observed to translocate at a velocity of 13.2 μm/h, in a manner that depended on capillary diameter [102]. Below 8 μm no migration was observed. It is unknown whether movement such as this requires traction forces or involves intracellular contractility and/or amoeboid migration. Interestingly, Hung et al. have shown that the mechanosensor Piezo1 can trigger myosin II-based contractility in cancer cells in confined spaces [103]. Thus, the extent of intraluminal migration of individual CTCs in this confined space may depend on local features of the microvascular bed as well as the migratory behavior of CTCs [104].
The microvasculature poses an even more daunting obstacle to CTMs due to their larger size. Studies evaluating the metastatic potential of CTMs have focused on metastasis of intravenously injected CTMs to the lung, which does not require transcapillary passage [69, 71, 74]. It is not firmly established whether CTMs can pass this first microvascular bed encountered as an intact cell assemblage. However, suggesting that this is possible, Au et al. showed that CTMs could traverse 5–10 μm microfluidic capillary tubes under physiologic pressure in whole blood and remain intact [26]. Similar behavior was observed for cancer cell clusters in zebra fish embryos. It will be interesting to determine if this behavior explains how CTMs are able to be detected in venous blood draws. Alternatively, these large cell assemblages may traverse small (50–100 μm) physiologic arteriovenous shunts in the lungs or extremities [105–107]. In summary, it remains largely unclear whether transit across the microvasculature is simply a stochastic process, i.e., some cells entering the microvascular bed accomplish through chance, or if biological processes can heavily influence this. Toward the latter idea, a “circulator” phenotype has been postulated where CTCs avoid arrest in a highly efficient manner [85]. However, given the available data, a picture emerges that most CTCs will arrest, at least temporarily, when they encounter the microvasculature. Thus, brief periods of free movement within the circulation are punctuated with longer stays in capillary lumens in which some cells will extravasate, some will die (as discussed below), and some will transit to the next capillary bed. This process is summarized in Fig. 11.2.
Fig. 11.2.
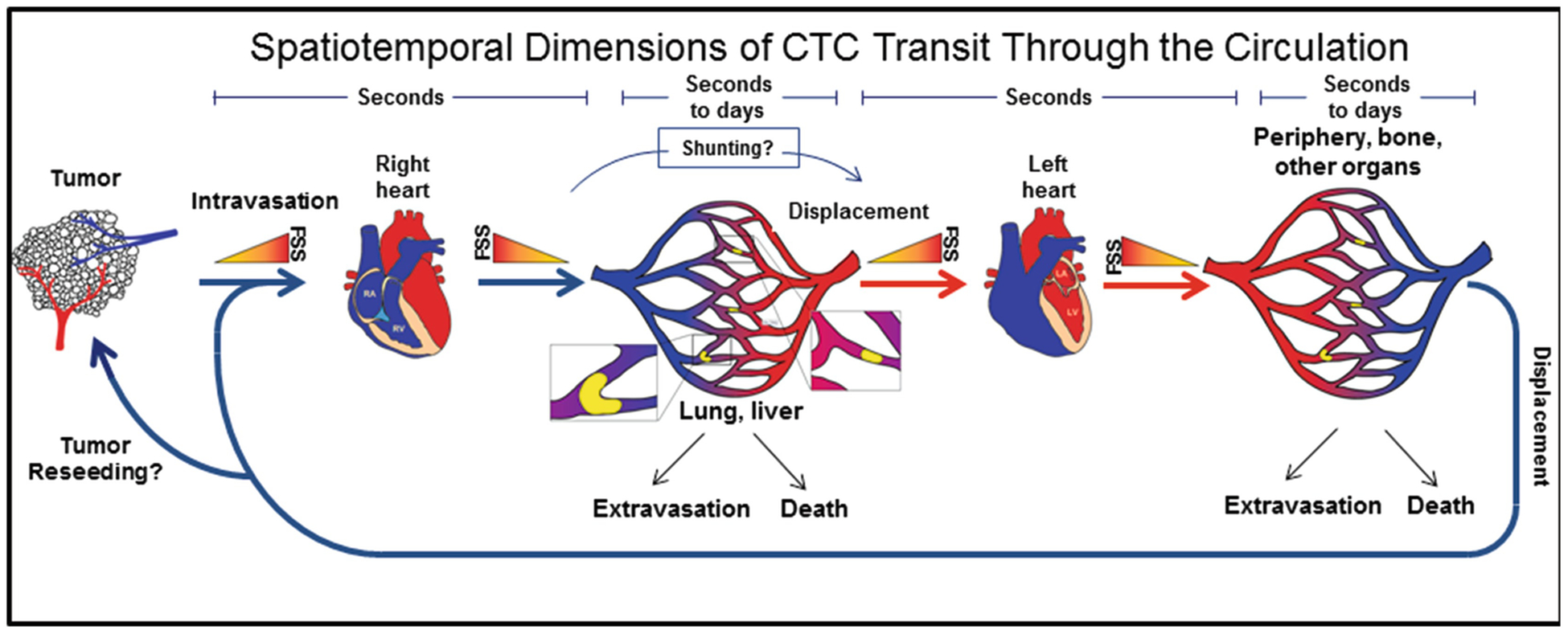
Dynamics of CTCs passage through the circulation. CTCs released from the primary tumor travel through the venous circulation, through increasing levels of FSS, to the right heart and then through descending levels of FSS to the lungs (or directly to the liver for some tumors) in a matter of seconds. Upon reaching the microcirculation, most intact cells will lodge for some period of time ranging from seconds to days, before undergoing cell death, extravasation or displacement. It is unclear at present to what extent CTCs might avoid lodgement in the microcirculation via arteriovenous shunts. Displaced cells would travel to the left heart, where they could encounter the maximum level of FSS around valve leaflets, and then out to the periphery where they may lodge in another microvascular bed and repeat the cycle, perhaps even reseeding existing primary or metastatic tumors
11.4. Are CTC’s Mechanically Fragile?
In many reviews on metastasis are versions of the following phrase: “tumor cells in the circulation must overcome the damage incurred by hemodynamic shear forces” [108]. While this makes intuitive sense, cancer cells from solid organs are relatively large and do not obviously have the same membrane-cytoskeletal and biophysical properties of blood cells that protect them from hemodynamic shear; there is precious little evidence to support this idea. Likewise, in much of the work directed at developing methods to isolate CTCs, it is often mentioned that CTCs are “fragile.” Though in this case, fragile could have different meanings—mechanically fragile (i.e., CTCs are easily physically destroyed) or biologically fragile (i.e., CTCs do not survive long when exposed to isolation techniques, perhaps undergoing apoptosis). In the preceding section, we discussed experiments that indicate that at least in the moments that it takes to traverse the tail vein, pass through the right heart and become entrapped in the lung microvasculature, most cancer cells remain intact, providing evidence that under these circumstances, CTCs are not mechanically fragile. In this section, we will look more closely at various biomechanical influences on CTCs and whether or not they are mechanically fragile. We will also present evidence that cancer cells, unlike their benign counterparts, are surprisingly resistant to brief pulses of high-level fluid shear stress (FSS). This is an active biological mechanism common to transformed cells and may explain why, counterintuitively, CTCs are mechanically stable when exposed to hemodynamic shear. Essentially, we propose to address the question, “Why aren’t CTCs mechanically fragile?”
11.4.1. Death by Deformation in the Microvasculature?
It has been repeatedly observed that cancer cells are distorted when they enter the microcirculation as the size differential suggests that they must. Often they can be observed tortuously bent around vessel bifurcations. This led Weiss to hypothesize that lethal deformation of cancer cells within the microvasculature could be a significant contributor to metastatic inefficiency [12]. In this view, rapid cancer cell deformation in the microvasculature causes cells to stretch, expanding plasma membrane surface area until some critical point, where it irreversibly ruptures. Since cancer cells arrested in the microcirculation typically occlude blood flow, the force driving this deformation is presumably blood pressure, not shear induced by passing blood flow or adhesive interactions with the endothelium. However, plasma may continue to flow around the occluding cells [109, 110]. Evidence supporting this hypothesis is drawn from in vitro studies on the passage of hypotonically swelled cancer cells through microporous filters [111]. This was also investigated in vivo by observing acridine orange-stained fibrosarcoma cells observed in a mouse cremaster muscle preparation [112]. A loss of membrane integrity was observed by uptake of ethidium bromide, pre-loaded into the mice. These authors found that ~80% of the deformed, arrested cells exhibited a loss of membrane integrity in less than 30 min. In contrast, Morris et al. found that 97% of calcein AM-labeled B16-F10 melanoma and D2A1 murine mammary carcinoma cells excluded ethidium bromide for up to 2 h in the liver and in an extensively deformed state in muscle [109]. The discrepancy was attributed to toxicity of the acridine orange dye [113]. Moreover, Morris et al. present a theoretical framework for why deformation-driven membrane rupture is unlikely [113]. Thus, the preponderance of evidence indicates that it is unlikely that deformation of CTCs in the microvasculature leads, within minutes, to the destruction of cancer cells.
It is somewhat less clear, however, whether deformation of the cell and its organelles might contribute to lethality on longer time scales. As mentioned above, there is uncertainty about the kinetics of cell clearance from the lung microvasculature and the extent to which this involves cell displacement or death. Compression can lead to a transient induction of autophagy within 30 min [114], which might be an adaptive response to help promote the survival of cancer cells deformed within the microcirculation. Several investigators using intravital video microscopy have observed that parts of cells arrested in the microcirculation break free in a process termed “clasmatosis,” which occurs within minutes of arrest in the microcirculation [109, 110]. The membrane-bound fragments are small, 3–5 μm in diameter, and do not appear to result from apoptotic membrane blebbing, and their production is enhanced in the presence of vascular flow [110]. Interestingly, Headly et al. have shown that local myeloid cells efficiently ingest these particles and suggest that this may play a role in the immune response to metastatic cancer cells which would occur on a longer time scale [110]. There is also evidence that some cells arrested in the microvasculature undergo apoptotic cell death within the first 48 h of arrest [115, 116]. Cells arrested in the lumen may be deprived of matrix engagement leading to anoikis or other survival factors present in the solid tumor microenvironment. In addition, the deformation caused by size constriction in the microvasculature may ultimately lead to mechanical rupture of the nucleus [117]. Within the cell, the nucleus is a large and relatively rigid structure which is rate-limiting for migration through pores. The Lammerding and Piel groups showed that migration of cancer cells through short channels up to 5 μm diameter caused rupture of the nuclear envelope leading to DNA damage and, less frequently, cell death [118, 119]. The damage was repairable through a mechanism that is also involved in postmitotic membrane resealing. Whether this behavior is relevant to deformed cancer cells in the microvasculature has not been investigated, these findings suggest another possible cause of cell death—or mutagenesis—in CTCs arrested in the microcirculation. Finally, given that some CTCs are likely to die during arrest in the microcirculation, it is interesting to speculate about the extent to which they contribute to circulating tumor DNA.
11.4.2. Cancer Cells and Fluid Shear Stress?
Cancer cells are already exposed to fluid shear before entering the circulation in the form of interstitial fluid flows, producing cell surface fluid shear on the order of (0.1–10 dyn/cm2). This is biologically important on a number of fronts [120]. Much of this work concerns the effects that this relatively low, continuous fluid shear has on cell signaling and consequent behavior of cancer cells, e.g., [121, 122]. However, since this chapter is primarily concerned with the biomechanics of the CTC microenvironment, we will not pursue this topic further here.
When cancer cells enter the bloodstream, they are exposed to an environment of greatly varying levels of fluid shear stress (FSS) as detailed above (Fig. 11.1a). The ramifications of this on CTC biology are only beginning to be understood in part because current in vitro models do not adequately replicate the spatiotemporal dynamics of the circulation and how CTCs experience it (Fig. 11.2). Additionally, most of the in vitro experiments do not include blood, and it remains difficult to study the particular influence of FSS on CTCs in vivo. From the first moments that cancer cells enter the circulation, they are exposed to FSS as they emerge into the vessel lumen. Video microscopy showed that initial cell protrusions into blood vessels were fragmented, with non-metastatic cells exhibiting more of this behavior than a metastatic variant, suggesting that this was driven partly by vascular flow [123]. Once in the venous circulation, CTCs would experience an escalating level of FSS as flow velocity increases to 15 cm/s in the vena cava before entering the right heart (Fig. 11.2). The actual FSS a CTC would encounter during laminar flow in the transport vessels would depend on the CTCs radial position in the vessel, its size, and flow velocity. However, as CTCs are large relative to most blood cells, there is an expectation that they would be driven toward the higher shear environment of the vessel wall through margination [48]. Importantly, this phase of the journey would last only seconds, unless adhesive interactions with the endothelium prevail somewhere to slow the movement of the cancer cell. Below, we will consider data from various model systems on the biological effects of FSS CTCs and comment on their inherent limitations.
11.4.2.1. Parallel Plate Flow Chambers and Microchannels
Parallel plate flow chambers and versions in microfluidic formats have been used extensively to evaluate the effects of fluid shear stress on mammalian cells, in particular endothelial cells which are constantly exposed to FSS in the vascular wall. An advantage of this model is that laminar flow can be delivered in a controlled manner at known levels to cells adherent to one side of the chamber. Obviously, if cancer cells are the adherent party, this is not necessarily an accurate model to study the effects of FSS on CTCs in free circulation. Adherent cells might utilize different mechanisms to sense and respond to FSS. Nevertheless, some insights have been gained from these studies. Several studies have indicated that exposure of cancer cells to relatively low microvascular and venous FSS (WSS up to 5.6 dyn/cm2) for periods of up to 1 h can activate cell adhesion, motility, and invasive mechanisms that might promote cell adhesion to endothelial cells and extravasation [124–127]. A speculative interpretation of these data is that as cancer cells intravasate and are exposed to hemodynamic shear, while still being attached to the vessel wall, exposure to FSS might prime these cells for further adhesive and invasive behavior relevant to extravasation. It is worth noting that none of these studies indicate that exposure to FSS at this level is associated with damage to cancer cells. However, it has been reported that somewhat higher levels of FSS (WSS = 12 dyn/cm2) exposure for 24–48 h results in a G2/M cell cycle arrest [128]. However, it is not clear to us how this sort of exposure would relate to CTCs in vivo.
Parallel plate flow chambers and microchannels have also been used to investigate adhesive interactions between free-flowing cancer cells and other immobilized cell types including endothelial cells [129–132], PMNs [133], and platelets [134] or ligands [135–137]. One such study showed that platelets facilitate melanoma cell attachment to a collagen I-coated surface at 2 dyn/cm2 in a manner that depends on β3 integrin [137]. Although possible, CTCs are unlikely to encounter sites of vascular injury while in free circulation. This behavior could also serve to stabilize adhesive interactions in arrested cells or during extravasation. Importantly, studies in this model have also demonstrated the potential for cancer cells to exhibit leukocyte-like rolling behavior on endothelial cells under microvascular-venous levels of FSS (WSS up to ~5 dyn/cm2). In particular, these studies illustrate roles for selectin-mediated adhesion in this process [130–132, 134, 136]. Indeed, CTCs from prostate cancer patients exhibit E-selectin-dependent rolling behavior in microtubes and IL-1β-stimulated endothelial cells under these flow conditions [138]. Cancer cell aggregates, representing CTMs, can also exhibit rolling behavior on E-selectin-coated surfaces [48]. However, how CTMs experience and respond to FSS is largely unexplored experimentally. These studies, as well as computational modeling of CTC behavior, have been well reviewed recently and will not be detailed further here [139–141].
11.4.2.2. Cone and Plate Viscometer
Cone and plate viscometers have an experimental advantage in the capability to provide a uniform fluid shear environment to cancer cell suspensions. This is closer to what one envisions for CTCs that are freely circulating. In the earliest study of this kind, Brooks examined the effects of FSS on B16 melanoma cells [142]. At a shear rate of 2250 s−1 (~29 dyn/cm2), only 20–50% viability was lost at 1 h, the first time point taken, and it took 5.5 h of continuous exposure to reduce viability to zero. Given the discussion above, it is exceedingly unlikely that most CTCs remain in continuous circulation for this period of time and that their exposure to this level of shear would only occur very briefly. Thus, rather than establishing the fragility of cancer cells, to the contrary, this study indicates that they are quite robust when confronted with the FSS anticipated physiologically. More recently, Egan et al. have used this experimental model to investigate the effects of platelets in protecting ovarian cancer cells [143]. At both venous (200 s−1; 1.5 dyn/cm2) and arterial (1500 s−1; 12 dyn/cm2) shear rates administered over 10 min, cell death (loss of membrane integrity) reaching a maximum of 30% by 10 min was detected by LDH release; however, this did not scale, as one might expect, with increasing shear rate. Addition of platelets at various ratios decreased LDH release at the higher, but not lower shear rate. The reasons for the relatively rapid loss of viability at lower shear rates in this study compared to the aforementioned study and others cited below applying relatively high levels of shear, where a substantial loss of viability is not observed, are unclear. These results await further confirmation in additional cell lines and means to assess cell viability.
This experimental model has also been used to gain other insights relevant to the CTC microenvironment. Exposure of esophageal cancer cells to a venous shear rate of 200 s−1 for 10–15 min induced ROCK-dependent membrane blebbing and primed invasive behavior of these cells [126]. The Konstantopoulos group has shown that low shear-induced collisions facilitate platelet and PMN binding to colon carcinoma cells on a time scale (30–300 s) relevant to consider for freely circulating CTCs [144, 145]. These studies indicate that heterotypic CTC-blood cell aggregates could form rapidly within the circulation and facilitate lodgement in the microvasculature and extravasation and promote the survival of CTCs. Another unexpected aspect of FSS has been reported by the King group using this model. They found that exposure to microvascular levels of FSS (2.0 dyn/cm2) for 60–120 min potentiated TRAIL-induced apoptosis, suggesting a particular vulnerability for CTCs [146].
11.4.2.3. Continuous Flow Circuits
Continuous flow circuits also allow for the biology of cancer cells to be probed when the cells are in suspension. Here, a cell suspension is continuously flowed through tubing by means of a peristaltic pump, although this model, too, fails to capture the entire dynamics of the circulatory system. Also, in this type of model, cells are exposed to a range of FSS levels depending on their local conditions, as they would be in circulation, not uniform levels as with the models above. Although Brooks mentioned using this type of model in 1984 [142], it has not been extensively utilized until recently. Fan et al. developed a circuit that included a 20 μm wide microfluidic constriction [147]. This system could generate a maximum WSS at the constriction of 60.5 dyn/cm2. Brief periods of circulation (2 min), even at the maximum level, did not affect HCT116 colon cancer cell viability. Whereas a significant loss of viability was observed at all levels of FSS evaluated at 20 h compared to cells that were not subjected to flow. Extended FSS exposure also resulted in lingering effects on cell proliferation in those cells that survived.
Using a continuous flow circuit that incorporated a 500 μm wide observation channel, Fu et al. observed, using a FRET-based apoptosis reporter, that exposure of up to 30 dyn/cm2 resulted in elevated apoptosis rates in non-metastatic MCF7 cells as compared to metastatic MDA-MB-231 breast cancer cells [27]. Peak apoptosis rates were observed at around 16 h of continuous circulation and were significantly higher than static cells in nonadhesive cultures. Elevated apoptosis was correlated with a loss of cell viability, and this could be blocked by caspase inhibition. Moreover, apoptotic cancer cells were observed freely circulating in zebra fish embryos 24 h postinjection. These results extend previous findings demonstrating an apoptotic mode of cell death and are in agreement with others showing that extended time in continuous circulation is necessary to induce this. Furthermore, they showed that exposure to FSS triggers apoptotic cell death via elevated mitochondrial ROS production. This group went on to show that exposure to FSS for 6 h in this system primed the migratory and invasive characteristics of breast cancer cells in a manner that depended on ROS production [148]. Using a somewhat different system, this same group showed that higher levels of FSS (60 dyn/cm2) which are achievable in the femoral artery during exercise resulted in necrotic cell death, as evidenced by propidium iodide uptake and loss of viability in the MTT assay, over a time course of 2–18 h [149]. Taken together, these studies provide convincing evidence that physiologic levels of FSS applied continuously for hours can induce ROS-driven apoptotic cell death and at higher levels necrotic cell death. However, the model, and hence interpretations, is limited by the fact that, as argued above, available evidence indicates that CTCs do not freely circulate (at constant levels of FSS) for hours. Rather, size restriction in the microvasculature is likely to limit this for most CTCs to seconds of free circulation through larger vessels and the heart.
11.4.2.4. Syringe and Needle
We developed a simple model involving pump-controlled flow of a cell suspension through a syringe and small diameter needle to specifically interrogate the effects of brief but high level FSS [150]. As mentioned above, the physiological range of FSS spans four orders of magnitude, with the highest levels of FSS represented near the walls of arterioles and around heart valves. These environs might only be encountered briefly by CTCs while in the circulation. However, this model also afforded the opportunity to push the limits of FSS that might be encountered by CTCs to really test the hypothesis that cancer cells are mechanically fragile under these extreme circumstances. In this model, a flow rate of 250 μL/s through a 1.27 cm long, 150 μm diameter needle applies a brief (~1 ms) “pulse” of FSS ranging between 750 and 6300 dyn/cm2. This includes the levels of force necessary to rupture red cell membranes on a millisecond time scale (4500–5600 dyn/cm2) [41, 42]. A limitation of this model is that the actual FSS applied to individual cancer cells varies depending on their radial position within the flow profile. Unlike the other models described above, this is a high Reynolds number environment (Re = 1980) near the border of transition between laminar and turbulent flow. The surprising findings from these studies were that cancer cell lines from many tissue origins exhibited remarkable resistance to high, but brief, repeated pulses of FSS, as compared to non-transformed counterparts which did exhibit mechanical fragility—loss of plasma membrane integrity, and fragmentation and loss of viability measured by a variety of assays [150]. Extensive controls were performed to establish that loss of cell viability is a direct result of exposure to FSS and not other variables. By way of comparison, both freshly isolated leukocytes and red blood cells were considerably more resistant to damage/death than cancer cells in this model. Moreover, these studies demonstrated that resistance to FSS is a property of cellular transformation driven by multiple oncogenic pathways and that exposure to a single pulse of high-level FSS at this level induced resistance to subsequent, repeated pulses, implying a physiological response to increase resistance to FSS. What we know about the mechanisms involved in resistance to this type of FSS is described in more detail below.
Other groups have also independently reported similar results using this model. Mitchell et al. essentially corroborated our findings showing that malignant breast cancer cell lines, as compared to the non-transformed MCF-10A cell line are more resistant to FSS. They went on to show that depletion of nuclear lamins A and C from breast cancer cells led to increased sensitivity to FSS, in the form of increased apoptosis, but not necrosis, measured 2 h after exposure to FSS. These results that the loss of structural integrity of the nucleus and/or lamin-dependent gene expression play a role in the survival of cells confronted with this mechanical insult and is resonant with finding cited above that the mechanical challenge of navigating narrow pores also show lamin A-/C-dependent effects on cell survival [117]. In a more recent study, Vennin et al. used the syringe and needle model to show that pre-treatment of mouse KPC pancreatic cancer cells with Fasudil, a ROCK inhibitor, sensitizes cells to FSS-reducing subsequent attachment to cell-derived matrix and proliferation and increasing apoptosis and propidium iodide uptake [152]. These results are in accord with our previous finding that another ROCK inhibitor, Y-27632, sensitized cells to FSS-induced loss of viability [150].
In a variation of the syringe and needle model, Triantafillu et al. attached a 46 cm section of 125 μm diameter tubing to a syringe and drove cancer cell suspensions through this conduit with WSS values from 20 to 60 dyn/cm2 [153]. In this configuration, cells are exposed to FSS for an average of between ~1.6 and 4.6 s. The temporal dimension and magnitude of FSS in this model are reflective of a short trip through the arterial circulation, albeit at a continuous level of FSS. At 20 dyn/cm2 immortalized but non-transformed mammary epithelial cells exhibited an ~50% decrease in cell viability, whereas two of three malignant breast cancer cell lines, MDA-MB-231 and MCF7, did not show a significant loss of viability in comparison to static cells in suspension, both by trypan blue and clonogenic assays. Taken together, studies in this type of model indicate that malignant cells exhibit resistance to FSS as compared to benign epithelial cells. Moreover, both the duration and magnitude of exposure are important determinants that can distinguish the differential behavior of benign and malignant epithelial cells when confronted with FSS.
11.4.3. Resistance to Fluid Shear Stress Is a Conserved Biophysical Property of Cancer Cells
This chapter has summarized multiple lines of evidence that cancer cells are not, contrary to popularly held opinion, inherently mechanically fragile and widely subject to rapid (second to minutes) destruction by physiologic hemodynamic shear forces. This conclusion is supported by quantitative assessments of experimental CTCs in various metastasis models, intravital imaging efforts, and numerous in vitro studies that have examined a wide range of FSS exposures (magnitude and time) in a wide variety of cancer cell lines. So is this true for actual CTCs? This question has not yet been addressed directly, though as mentioned above, CTCs are repeatedly regarded as “fragile” by the investigators who routinely work with them. It is important to point out that most if not all of the work supporting the mechanical robustness of cancer cells involves established cancer cell lines, which when introduced into the circulation, a high proportion of the cells are viable. It is possible that adaptation to cell culture with repeated in vitro passaging somehow selects for cells that are more mechanically robust than CTCs or cancer cells as they exist in patient’s tumors.
The possibility is that all of the aforementioned data is the result of a cell culture artifact notwithstanding; one way to reconcile these differing perspectives (Are CTCs mechanically robust or not?) is if dead and/or dying CTCs are mechanically fragile. Indeed, there is support for this view. As discussed above, there is abundant evidence that many CTCs are not viable and/or apoptotic. Apoptosis results numerous changes is cellular physiology. Among these, it has been observed that during staurosporine-induced apoptosis, there is a rapid (30–60 min) decrease in cell stiffness (Young’s modulus) with concomitant changes in cytoarchitecture including actin depolymerization and disruption of nuclear lamins [154]. Both of these features have been shown to increase sensitivity of cancer cells to FSS in the needle and syringe model [150, 151]. Alternatively, as our studies and others indicate, benign epithelial cells that may be included within the CTC population are mechanically fragile [150, 151, 153]. Hence, an apparent mechanical fragility of CTCs could be a direct result of biological fragility—their propensity to already be dead or dying when they enter the circulation or die rapidly (within hours) when disconnected from the tumor microenvironment—or their lack of intrinsic FSS resistance mechanisms as in the case of benign epithelial cells.
Thus a question becomes, “Why aren’t viable cancer cells, and by extension viable CTCs, mechanically fragile?” We are beginning to tackle this question. What is emerging is that resistance to FSS is a basic biophysical property of the transformed cell phenotype, perhaps common in all cancer cells [150]. We have identified two features of transformed cancer cells that may act in concert or independently to promote FSS resistance: (1) the ability to rapidly repair damage to the plasma membrane and (2) the ability to rapidly modulate membrane-cytoskeletal features in response to FSS exposure, so as to prevent further damage. Below, we will summarize our findings and what is known about the mechanisms involved.
An immediate, and potentially catastrophic, effect of exposure to excessive FSS is a breach in the plasma membrane. The ability to maintain the integrity of the plasma membrane is of fundamental importance to a cell. Even a small breach in this barrier can rapidly result in death due to disruption of necessary ion gradients, oxidation, and loss of vital intracellular substrates. Many bacterial toxins act to create pores in the plasma membrane. Thus it is not surprising that cellular mechanisms exist to rapidly repair the plasma membrane, for a recent review of these mechanisms see [155]. We detected evidence of membrane repair in cancer cells exposed to brief pulses of FSS in the needle and syringe model cells that took up membrane-impermeant propidium iodide but otherwise remained viable [150]. Moreover, we showed that extracellular Ca++, but not other divalent cations, was critical for maintaining cell viability in re sponse to FSS in this context. In fact, Ca++-dependent resistance to FSS was observed at much lower levels of FSS which might more commonly be encountered by CTCs in the needle and syringe model. Extracellular Ca++ entering through membrane wounds is known to trigger membrane resealing events [156, 157]. These can act either by forming a patch of various types to repair larger holes or by reducing membrane tension to facilitate resealing [155]. The extent to which differences in membrane repair efficiency can explain the differential sensitivity of benign and malignant epithelial cells is not yet known, and the mechanisms involved in repairing membrane damage in this context are under active investigation.
Another surprising finding of our initial study was that cancer cells exposed to a single, initial pulse of FSS exhibited increased resistance to multiple repeated pulses of FSS-up to ten evaluated [85]. This was evidenced by reduced propidium iodide uptake in viable cells after the initial pulse. Moreover, cancer cells exhibited a biphasic loss of cell viability in response to a train of FSS pulses, with a more precipitous drop of viability initially, followed by a slower phase. This behavior was not simply due to selection of a more FSS-resistant population of cells. This finding suggested that cancer cells might rapidly (the interval between pulses was ~90 s) modulate their membrane-cytoskeletal properties in response to an initial pulse of FSS, becoming more resistant to damage in subsequent pulses. Consistent with this interpretation, we found that pre-treatment with non-cytotoxic exposures of the actin-depolymerizing drug cytochalasin D, or the ROCK inhibitor Y-27632, sensitized cancer cells to FSS-induced cell death [126]. Moreover, in a follow-up study, Chivukula et al. using a micropipette aspiration technique applied to suspended cells following exposure to FSS, showed that prostate cancer cells, but not a benign counterpart, demonstrate a 77% increase in Young’s modulus after exposure to high-level FSS in the needle and syringe model [158]. This adaptive response in the biophysical properties of cancer cells in response to FSS is likely to be related to their ability to avoid damage from subsequent pulses of FSS. Interestingly though, cell stiffness per se is not a simple determinant of resistance to FSS. In this study, we found, as others have shown in adherent cells using other methods [159], that cancer cells under static conditions are less stiff than benign epithelial cells (~20 vs 50 Pa, respectively). Cancer cells stiffen in response to FSS (~35 Pa), but they do not reach the level of benign cells. Despite this, cancer cells survive exposure to high FSS while benign cells do not. It is not clear how cancer cells sense exposure to brief pulses of high level FSS. One possibility is Ca++ enters through plasma membrane wounds. However, we noted that after ten pulses, the total number of viable cells exceeds the number of viable cells that have taken-up PI, suggesting the possibility that a mechanosensory channel is involved. The mechanistic basis for these biophysical changes in cancer cells in response to FSS is also under investigation. A summary of current findings related to the mechanism(s) of FSS is shown in Fig. 11.3.
Fig. 11.3.
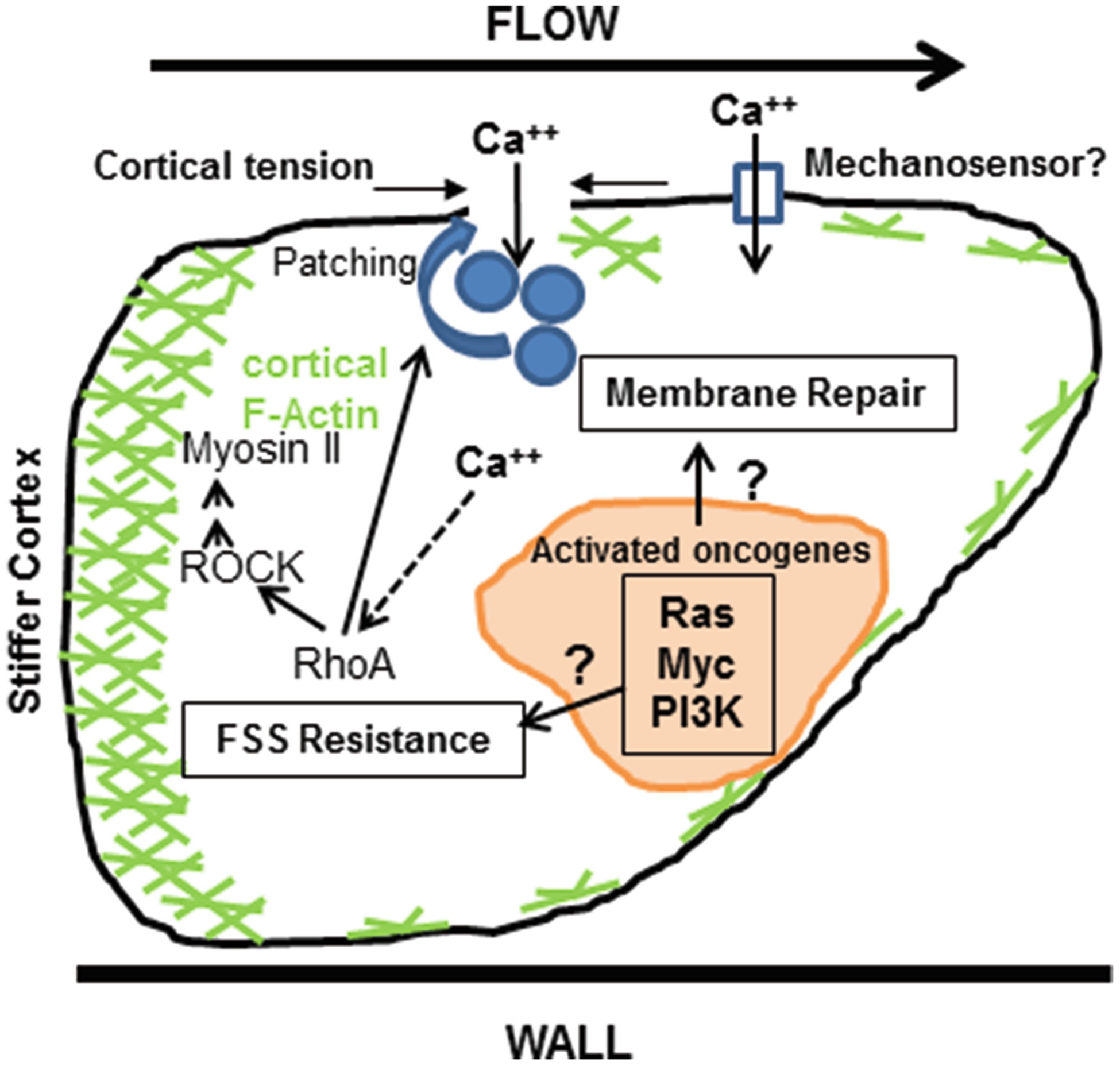
Mechanisms of FSS resistance may involve enhanced membrane repair, or increased resistance to FSS-induced membrane damage. There is evidence supporting both mechanisms to date. Exposure to FSS may cause damage to the plasma membrane which can be rapidly repaired either via calcium triggered membrane patching or by increased cortical tension and membrane biophysics. Extracellular calcium entry through transient membrane wounds, or an as yet unidentified mechanosensor, may trigger actin remodeling and activity of the RhoA-myosin II axis resulting in increased cellular contractility and less membrane damage, and/or increase membrane repair. Given the widespread nature of FSS resistance in transformed cells an important question is how well know oncogenic signaling pathways might influence these processes
11.5. Conclusions
CTCs have captured the attention of cancer biologists for decades, and recent technical advances have made the study of these elusive cells, although still challenging, tractable for some. Given that these cells afford a window into metastasis, they may hold keys to understanding this process in more detail as well as providing a venue for the “liquid biopsy” that could have clinical utility. CTCs are a natural area for interdisciplinary research among biologists and engineers, both for the technical aspects of CTC isolation and to understand the different forces at play on these cells while they are in the circulation. The main emphasis of this chapter has been to explore to what extent these forces are capable of mechanically damaging or destroying CTCs while they are in the circulation. Metastasis is clearly an inefficient process, and most available evidence indicates that CTCs have a relatively short half-life while in the circulation, but the extent to which this is due to mechanical fragility of CTCs has not been thoroughly elucidated.
In this chapter, we reviewed a variety of evidence that viable cancer cells, and by extension viable CTCs, are not appreciably destroyed by the magnitude and duration of exposure to fluid shear stresses that would be encountered on their journey through the circulation. We have suggested that apparent mechanical fragility may be a secondary effect of a loss of cell viability due to other causes. Dead and dying cells that may enter the circulation passively or succumb to immune attack or deprivation of matrix attachment or other factors present in the solid tumor microenvironment after they enter the circulation may be much more mechanically fragile than viable CTCs. To the contrary, we are beginning to elucidate mechanisms in cancer cells that confer resistance to fluid shear stress, not present in benign epithelial cells. These mechanisms appear to be the product of cellular transformation, tied to common oncogenic pathways and thus may be common to many cancers, but the detailed mechanistic connections are not yet clear. Thus, although intuition leads one to conclude that CTCs may not be built to withstand hemodynamic stresses like blood cells, available evidence suggests otherwise. It is important to stress that most of this evidence is derived from the study of cultured cancer cell lines, both in in vivo and in vitro models, and whether they extend to CTCs in cancer patients remains to be determined. However, if this is indeed the case, one could envision that therapeutic strategies to interfere with cancer cell-intrinsic resistance to fluid shear stress could, in effect, create a more formidable barrier to hematogenous metastasis.
Here we have also discussed a number of still very poorly understood aspects of CTC biology as it relates to the microenvironment of the circulation. Several of these questions are: (1) Can we more precisely define the fates of individual CTCs as they traverse the vasculature? That is, what is the probability of displacement from the first microvascular bed encountered vs. extravasation or cell death? (2) How, indeed, do relatively large cancer cells negotiate a capillary bed and avoid size-based entrapment? Can some CTCs freely circulate like blood cells? There are already some hints that both the size and deformability of CTCs may be different from cancer cell lines commonly employed in animal models. The size restriction problem is compounded in the case of CTMs and CTCs complexed with platelets or leukocytes. Do these manage to transit the microcirculation as well, or are they primed for arrest and extravasation? (3) When and under what circumstances do adhesive interactions between CTCs and the vascular endothelium and other cell types occur in vivo, and what roles do they play? (4) How can these basic insights into the behavior of CTCs in the circulation be used to advance the management of cancer patients, including how the answers to the questions above might vary by cancer type and state of disease progression? These broad questions await answers which may require further technology developments to enable the study of actual CTCs in cancer patients as well as refinements in the computational, in vitro, and animal models, including long-duration intravital microscopy, used for experimental study of CTCs. This old field is likely to be a fruitful area of collaboration for biologists and engineers for many years to come.
Contributor Information
Benjamin L. Krog, Department of Molecular Physiology and Biophysics, Carver College of Medicine, University of Iowa, Iowa City, IA, USA
Michael D. Henry, Department of Molecular Physiology and Biophysics, Carver College of Medicine, University of Iowa, Iowa City, IA, USA Department of Pathology and Urology, Carver College of Medicine, University of Iowa, Iowa City, IA, USA; Holden Comprehensive Cancer Center, Carver College of Medicine, University of Iowa, Iowa City, IA, USA.
References
- 1.Ashworth TR (1869) A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J 14:146–147 [Google Scholar]
- 2.Weiss L (1990) Metastatic inefficiency. Adv Cancer Res 54:159–211 [DOI] [PubMed] [Google Scholar]
- 3.Zeidman I, Mc CM, Coman DR (1950) Factors affecting the number of tumor metastases; experiments with a transplantable mouse tumor. Cancer Res 10(6):357–359 [PubMed] [Google Scholar]
- 4.Fidler IJ (1970) Metastasis: quantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2’-deoxyuridine. J Natl Cancer Inst 45(4):773–782 [PubMed] [Google Scholar]
- 5.Butler TP, Gullino PM (1975) Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res 35(3):512–516 [PubMed] [Google Scholar]
- 6.Tarin D et al. (1984) Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Res 44(8):3584–3592 [PubMed] [Google Scholar]
- 7.Pantel K, Speicher MR (2016) The biology of circulating tumor cells. Oncogene 35(10):1216–1224 [DOI] [PubMed] [Google Scholar]
- 8.Labelle M, Hynes RO (2012) The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov 2(12):1091–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paget S (1989) The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev 8(2):98–101 [PubMed] [Google Scholar]
- 10.Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3(6):453–458 [DOI] [PubMed] [Google Scholar]
- 11.Buchheit CL, Weigel KJ, Schafer ZT (2014) Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer 14(9):632–641 [DOI] [PubMed] [Google Scholar]
- 12.Weiss L (1991) Deformation-driven, lethal damage to cancer cells. Its contribution to metastatic inefficiency. Cell Biophys 18(2):73–79 [DOI] [PubMed] [Google Scholar]
- 13.Stein PD, Sabbah HN (1976) Turbulent blood flow in the ascending aorta of humans with normal and diseased aortic valves. Circ Res 39(1):58–65 [DOI] [PubMed] [Google Scholar]
- 14.Kameneva MV et al. (2004) Effects of turbulent stresses upon mechanical hemolysis: experimental and computational analysis. ASAIO J 50(5):418–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margaris KN, Black RA (2012) Modelling the lymphatic system: challenges and opportunities. J R Soc Interface 9(69):601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon JB et al. (2006) Lymph flow, shear stress, and lymphocyte velocity in rat mesenteric prenodal lymphatics. Microcirculation 13(7):597–610 [DOI] [PubMed] [Google Scholar]
- 17.Cheng C et al. (2007) Large variations in absolute wall shear stress levels within one species and between species. Atherosclerosis 195(2):225–235 [DOI] [PubMed] [Google Scholar]
- 18.Malek AM, Alper SL, Izumo S (1999) Hemodynamic shear stress and its role in atherosclerosis. JAMA 282(21):2035–2042 [DOI] [PubMed] [Google Scholar]
- 19.Brass LF, Diamond SL (2016) Transport physics and biorheology in the setting of hemostasis and thrombosis. J Thromb Haemost 14(5):906–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Popel AS, Johnson PC (2005) Microcirculation and Hemorheology. Annu Rev Fluid Mech 37:43–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao K, Bukac M, Sucosky P (2016) Three-dimensional macro-scale assessment of regional and temporal wall shear stress characteristics on aortic valve leaflets. Comput Methods Biomech Biomed Engin 19(6):603–613 [DOI] [PubMed] [Google Scholar]
- 22.Antiga L, Steinman DA (2009) Rethinking turbulence in blood. Biorheology 46(2):77–81 [DOI] [PubMed] [Google Scholar]
- 23.Strony J et al. (1993) Analysis of shear stress and hemodynamic factors in a model of coronary artery stenosis and thrombosis. Am J Phys 265(5 Pt 2):H1787–H1796 [DOI] [PubMed] [Google Scholar]
- 24.Keshavarz-Motamed Z, Garcia J, Kadem L (2013) Fluid dynamics of coarctation of the aorta and effect of bicuspid aortic valve. PLoS One 8(8):e72394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dasi LP et al. (2009) Fluid mechanics of artificial heart valves. Clin Exp Pharmacol Physiol 36(2):225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Au SH et al. (2016) Clusters of circulating tumor cells traverse capillary-sized vessels. Proc Natl Acad Sci U S A 113(18):4947–4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu A et al. (2016) High expression of MnSOD promotes survival of circulating breast cancer cells and increases their resistance to doxorubicin. Oncotarget 7(31):50239–50257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yalcin HC et al. (2017) Heart function and hemodynamic analysis for zebrafish embryos. Dev Dyn 246(11):868–880 [DOI] [PubMed] [Google Scholar]
- 29.Hove JR et al. (2003) Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421(6919):172–177 [DOI] [PubMed] [Google Scholar]
- 30.Ozkumur E et al. (2013) Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Sci Transl Med 5(179):179ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen JF et al. (2015) Subclassification of prostate cancer circulating tumor cells by nuclear size reveals very small nuclear circulating tumor cells in patients with visceral metastases. Cancer 121(18):3240–3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hogg JC et al. (1994) Erythrocyte and polymorphonuclear cell transit time and concentration in human pulmonary capillaries. J Appl Physiol (1985) 77(4):1795–1800 [DOI] [PubMed] [Google Scholar]
- 33.Chien S (1987) Red cell deformability and its relevance to blood flow. Annu Rev Physiol 49:177–192 [DOI] [PubMed] [Google Scholar]
- 34.Lien DC et al. (1990) Neutrophil kinetics in the pulmonary microcirculation. Effects of pressure and flow in the dependent lung. Am Rev Respir Dis 141(4 Pt 1):953–959 [DOI] [PubMed] [Google Scholar]
- 35.Schmid-Schonbein GW (1993) The damaging potential of leukocyte activation in the microcirculation. Angiology 44(1):45–56 [DOI] [PubMed] [Google Scholar]
- 36.Hochmuth RM (2000) Micropipette aspiration of living cells. J Biomech 33(1):15–22 [DOI] [PubMed] [Google Scholar]
- 37.Wan J, Forsyth AM, Stone HA (2011) Red blood cell dynamics: from cell deformation to ATP release. Integr Biol (Camb) 3(10): 972–981 [DOI] [PubMed] [Google Scholar]
- 38.Hochmuth RM (1993) Measuring the mechanical properties of individual human blood cells. J Biomech Eng 115(4B):515–519 [DOI] [PubMed] [Google Scholar]
- 39.Lux SEt (2016) Anatomy of the red cell membrane skeleton: unanswered questions. Blood 127(2):187–199 [DOI] [PubMed] [Google Scholar]
- 40.Leverett LB et al. (1972) Red blood cell damage by shear stress. Biophys J 12(3):257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams AR, Hughes DE, Nyborg WL (1970) Hemolysis near a transversely oscillating wire. Science 169(3948):871–873 [DOI] [PubMed] [Google Scholar]
- 42.Rooney JA (1970) Hemolysis near an ultrasonically pulsating gas bubble. Science 169(3948):869–871 [DOI] [PubMed] [Google Scholar]
- 43.Sutera SP, Mehrjardi MH (1975) Deformation and fragmentation of human red blood cells in turbulent shear flow. Biophys J 15(1):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw Bagnall J et al. (2015) Deformability of tumor cells versus blood cells. Sci Rep 5:18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abkarian M, Faivre M, Viallat A (2007) Swinging of red blood cells under shear flow. Phys Rev Lett 98(18):188302. [DOI] [PubMed] [Google Scholar]
- 46.Fischer TM (2004) Shape memory of human red blood cells. Biophys J 86(5):3304–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldsmith HL, Spain S (1984) Margination of leukocytes in blood flow through small tubes. Microvasc Res 27(2):204–222 [DOI] [PubMed] [Google Scholar]
- 48.King MR et al. (2015) A physical sciences network characterization of circulating tumor cell aggregate transport. Am J Physiol Cell Physiol 308(10):C792–C802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sundd P et al. (2011) Biomechanics of leukocyte rolling. Biorheology 48(1):1–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu M et al. (2011) Circulating tumor cells: approaches to isolation and characterization. J Cell Biol 192(3):373–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baccelli I et al. (2013) Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol 31(6):539–544 [DOI] [PubMed] [Google Scholar]
- 52.Hodgkinson CL et al. (2014) Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med 20(8):897–903 [DOI] [PubMed] [Google Scholar]
- 53.Alix-Panabieres C, Pantel K (2016) Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov 6(5):479–491 [DOI] [PubMed] [Google Scholar]
- 54.Bardelli A, Pantel K (2017) Liquid biopsies, what we do not know (yet). Cancer Cell 31(2):172–179 [DOI] [PubMed] [Google Scholar]
- 55.Husemann Y et al. (2008) Systemic spread is an early step in breast cancer. Cancer Cell 13(1):58–68 [DOI] [PubMed] [Google Scholar]
- 56.Rhim AD et al. (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148(1–2):349–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Podsypanina K et al. (2008) Seeding and propagation of untransformed mouse mammary cells in the lung. Science 321(5897):1841–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bockhorn M, Jain RK, Munn LL (2007) Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? Lancet Oncol 8(5):444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deryugina EI, Kiosses WB (2017) Intratumoral cancer cell intravasation can occur independent of invasion into the adjacent stroma. Cell Rep 19(3):601–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pantel K et al. (2012) Circulating epithelial cells in patients with benign colon diseases. Clin Chem 58(5):936–940 [DOI] [PubMed] [Google Scholar]
- 61.Meng S et al. (2004) Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res 10(24):8152–8162 [DOI] [PubMed] [Google Scholar]
- 62.Wyckoff JB et al. (2007) Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 67(6):2649–2656 [DOI] [PubMed] [Google Scholar]
- 63.Chiang SP, Cabrera RM, Segall JE (2016) Tumor cell intravasation. Am J Physiol Cell Physiol 311(1):C1–C14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Armstrong AJ et al. (2011) Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res 9(8):997–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu M et al. (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339(6119):580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tarin D, Thompson EW, Newgreen DF (2005) The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res 65(14):5996–6000 discussion 6000–1 [DOI] [PubMed] [Google Scholar]
- 67.Fischer KR et al. (2015) Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527(7579):472–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng X et al. (2015) Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527(7579):525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liotta LA, Saidel MG, Kleinerman J (1976) The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Res 36(3):889–894 [PubMed] [Google Scholar]
- 70.Kaifi JT et al. (2016) Perioperative circulating tumor cell detection: current perspectives. Cancer Biol Ther 17(8):859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fidler IJ (1973) The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. Eur J Cancer 9(3):223–227 [DOI] [PubMed] [Google Scholar]
- 72.Molnar B et al. (2001) Circulating tumor cell clusters in the peripheral blood of colorectal cancer patients. Clin Cancer Res 7(12):4080–4085 [PubMed] [Google Scholar]
- 73.Stott SL et al. (2010) Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci U S A 107(43):18392–18397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aceto N et al. (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158(5):1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stott SL et al. (2010) Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med 2(25):25ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Larson CJ et al. (2004) Apoptosis of circulating tumor cells in prostate cancer patients. Cytometry A 62(1):46–53 [DOI] [PubMed] [Google Scholar]
- 77.Mehes G et al. (2001) Circulating breast cancer cells are frequently apoptotic. Am J Pathol 159(1):17–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kallergi G et al. (2013) Apoptotic circulating tumor cells in early and metastatic breast cancer patients. Mol Cancer Ther 12(9):1886–1895 [DOI] [PubMed] [Google Scholar]
- 79.Swartz MA et al. (1999) Cells shed from tumours show reduced clonogenicity, resistance to apoptosis, and in vivo tumorigenicity. Br J Cancer 81(5):756–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kamoun WS et al. (2010) Simultaneous measurement of RBC velocity, flux, hematocrit and shear rate in vascular networks. Nat Methods 7(8):655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuan F et al. (1994) Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res 54(17):4564–4568 [PubMed] [Google Scholar]
- 82.Weiss L (1992) Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin Exp Metastasis 10(3):191–199 [DOI] [PubMed] [Google Scholar]
- 83.Batson OV (1967) The vertebral system of veins as a means for cancer dissemination. Prog Clin Cancer 3:1–18 [PubMed] [Google Scholar]
- 84.Ewing J (1928) Neoplastic diseases. A treatise on tumors. Am J Med Sci 176(2):278 [Google Scholar]
- 85.Scott J, Kuhn P, Anderson AR (2012) Unifying metastasis – integrating intravasation, circulation and end-organ colonization. Nat Rev Cancer 12(7):445–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scott JG et al. (2014) A filter-flow perspective of haematogenous metastasis offers a non-genetic paradigm for personalised cancer therapy. Eur J Cancer 50(17):3068–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mizuno N et al. (1998) Mechanism of initial distribution of blood-borne colon carcinoma cells in the liver. J Hepatol 28(5):878–885 [DOI] [PubMed] [Google Scholar]
- 88.Qian B et al. (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 4(8):e6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cameron MD et al. (2000) Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60(9):2541–2546 [PubMed] [Google Scholar]
- 90.Luzzi KJ et al. (1998) Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153(3):865–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeidman I (1961) The fate of circulating tumors cells. I. Passage of cells through capillaries. Cancer Res 21:38–39 [PubMed] [Google Scholar]
- 92.Kienast Y et al. (2010) Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16(1):116–122 [DOI] [PubMed] [Google Scholar]
- 93.Al-Mehdi AB et al. (2000) Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med 6(1):100–102 [DOI] [PubMed] [Google Scholar]
- 94.Reymond N, d’Agua BB, Ridley AJ (2013) Crossing the endothelial barrier during metastasis. Nat Rev Cancer 13(12):858–870 [DOI] [PubMed] [Google Scholar]
- 95.Scherbarth S, Orr FW (1997) Intravital video microscopic evidence for regulation of metastasis by the hepatic microvasculature: effects of interleukin-1alpha on metastasis and the location of B16F1 melanoma cell arrest. Cancer Res 57(18):4105–4110 [PubMed] [Google Scholar]
- 96.Ito S et al. (2001) Real-time observation of micrometastasis formation in the living mouse liver using a green fluorescent protein gene-tagged rat tongue carcinoma cell line. Int J Cancer 93(2):212–217 [DOI] [PubMed] [Google Scholar]
- 97.Thorlacius H et al. (1997) Tumor cell arrest in the microcirculation: lack of evidence for a leukocyte-like rolling adhesive interaction with vascular endothelium in vivo. Clin Immunol Immunopathol 83(1):68–76 [DOI] [PubMed] [Google Scholar]
- 98.Enns A et al. (2004) Integrins can directly mediate metastatic tumor cell adhesion within the liver sinusoids. J Gastrointest Surg 8(8):1049–1059 discussion 1060 [DOI] [PubMed] [Google Scholar]
- 99.Barthel SR et al. (2009) Alpha 1,3 fucosyltransferases are master regulators of prostate cancer cell trafficking. Proc Natl Acad Sci U S A 106(46):19491–19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heidemann F et al. (2014) Selectins mediate small cell lung cancer systemic metastasis. PLoS One 9(4):e92327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Entenberg D et al. (2015) In vivo subcellular resolution optical imaging in the lung reveals early metastatic proliferation and motility. Intravital 4(3). 10.1080/21659087.2015.1086613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yamauchi K et al. (2005) Real-time in vivo dual-color imaging of intracapillary cancer cell and nucleus deformation and migration. Cancer Res 65(10):4246–4252 [DOI] [PubMed] [Google Scholar]
- 103.Hung WC et al. (2016) Confinement sensing and signal optimization via Piezo1/PKA and myosin II pathways. Cell Rep 15(7):1430–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Paul CD, Mistriotis P, Konstantopoulos K (2017) Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer 17(2):131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tobin CE (1966) Arteriovenous shunts in the peripheral pulmonary circulation in the human lung. Thorax 21(3):197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rhodes BA et al. (1972) Arteriovenous shunt measurements in extremities. J Nucl Med 13(6):357–362 [PubMed] [Google Scholar]
- 107.Eldridge MW et al. (2004) Exercise-induced intra-pulmonary arteriovenous shunting in healthy humans. J Appl Physiol (1985) 97(3):797–805 [DOI] [PubMed] [Google Scholar]
- 108.Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147(2):275–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Morris VL et al. (1993) Early interactions of cancer cells with the microvasculature in mouse liver and muscle during hematogenous metastasis: videomicroscopic analysis. Clin Exp Metastasis 11(5):377–390 [DOI] [PubMed] [Google Scholar]
- 110.Headley MB et al. (2016) Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531(7595):513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weiss L, Harlos JP, Elkin G (1989) Mechanism of mechanical trauma to Ehrlich ascites tumor cells in vitro and its relationship to rapid intravascular death during metastasis. Int J Cancer 44(1):143–148 [DOI] [PubMed] [Google Scholar]
- 112.Weiss L et al. (1992) Lethal deformation of cancer cells in the microcirculation: a potential rate regulator of hematogenous metastasis. Int J Cancer 50(1):103–107 [DOI] [PubMed] [Google Scholar]
- 113.Weiss L (1993) Deformation-driven destruction of cancer cells in the microvasculature. Clin Exp Metastasis 11(5):430–436 [DOI] [PubMed] [Google Scholar]
- 114.King JS, Veltman DM, Insall RH (2011) The induction of autophagy by mechanical stress. Autophagy 7(12):1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Varghese HJ et al. (2002) Activated ras regulates the proliferation/apoptosis balance and early survival of developing micrometastases. Cancer Res 62(3):887–891 [PubMed] [Google Scholar]
- 116.Kim JW et al. (2004) Rapid apoptosis in the pulmonary vasculature distinguishes non-metastatic from metastatic melanoma cells. Cancer Lett 213(2):203–212 [DOI] [PubMed] [Google Scholar]
- 117.Isermann P, Lammerding J (2017) Consequences of a tight squeeze: nuclear envelope rupture and repair. Nucleus 8:1–7. 10.1080/19491034.2017.1292191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Denais CM et al. (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352(6283):353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Raab M et al. (2016) ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352(6283):359–362 [DOI] [PubMed] [Google Scholar]
- 120.Swartz MA, Lund AW (2012) Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer 12(3):210–219 [DOI] [PubMed] [Google Scholar]
- 121.Shieh AC et al. (2011) Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res 71(3):790–800 [DOI] [PubMed] [Google Scholar]
- 122.Lee HJ et al. (2017) Fluid shear stress activates YAP1 to promote cancer cell motility. Nat Commun 8:14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wyckoff JB et al. (2000) A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res 60(9):2504–2511 [PubMed] [Google Scholar]
- 124.Korb T et al. (2004) Integrity of actin fibers and microtubules influences metastatic tumor cell adhesion. Exp Cell Res 299(1):236–247 [DOI] [PubMed] [Google Scholar]
- 125.von Sengbusch A et al. (2005) Focal adhesion kinase regulates metastatic adhesion of carcinoma cells within liver sinusoids. Am J Pathol 166(2): 585–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lawler K et al. (2006) Mobility and invasiveness of metastatic esophageal cancer are potentiated by shear stress in a ROCK- and Ras-dependent manner. Am J Physiol Cell Physiol 291(4):C668–C677 [DOI] [PubMed] [Google Scholar]
- 127.Yang H et al. (2016) Mechanosensitive caveolin-1 activation-induced PI3K/Akt/mTOR signaling pathway promotes breast cancer motility, invadopodia formation and metastasis in vivo. Oncotarget 7(13):16227–16247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chang SF et al. (2008) Tumor cell cycle arrest induced by shear stress: roles of integrins and Smad. Proc Natl Acad Sci U S A 105(10):3927–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Giavazzi R et al. (1993) Rolling and adhesion of human tumor cells on vascular endothelium under physiological flow conditions. J Clin Invest 92(6):3038–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burdick MM et al. (2003) Colon carcinoma cell glycolipids, integrins, and other glycoproteins mediate adhesion to HUVECs under flow. Am J Physiol Cell Physiol 284(4):C977–C987 [DOI] [PubMed] [Google Scholar]
- 131.Dimitroff CJ et al. (2004) Rolling of human bone-metastatic prostate tumor cells on human bone marrow endothelium under shear flow is mediated by E-selectin. Cancer Res 64(15):5261–5269 [DOI] [PubMed] [Google Scholar]
- 132.Barthel SR et al. (2008) Analysis of glycosyltransferase expression in metastatic prostate cancer cells capable of rolling activity on microvascular endothelial (E)-selectin. Glycobiology 18(10):806–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liang S, Slattery MJ, Dong C (2005) Shear stress and shear rate differentially affect the multi-step process of leukocyte-facilitated melanoma adhesion. Exp Cell Res 310(2):282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.McCarty OJ et al. (2000) Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood 96(5):1789–1797 [PubMed] [Google Scholar]
- 135.Haier J, Nicolson GL (2000) Tumor cell adhesion of human colon carcinoma cells with different metastatic properties to extracellular matrix under dynamic conditions of laminar flow. J Cancer Res Clin Oncol 126(12):699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li J et al. (2013) Human fucosyltransferase 6 enables prostate cancer metastasis to bone. Br J Cancer 109(12):3014–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Felding-Habermann B et al. (1996) Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J Biol Chem 271(10): 5892–5900 [DOI] [PubMed] [Google Scholar]
- 138.Gakhar G et al. (2013) Circulating tumor cells from prostate cancer patients interact with E-selectin under physiologic blood flow. PLoS One 8(12):e85143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wirtz D, Konstantopoulos K, Searson PC (2011) The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat Rev Cancer 11(7):512–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mitchell MJ, King MR (2013) Computational and experimental models of cancer cell response to fluid shear stress. Front Oncol 3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cheung LS et al. (2011) Biophysics of selectinligand interactions in inflammation and cancer. Phys Biol 8(1):015013. [DOI] [PubMed] [Google Scholar]
- 142.Brooks DE (1984) The biorheology of tumor cells. Biorheology 21(1–2):85–91 [DOI] [PubMed] [Google Scholar]
- 143.Egan K, Cooke N, Kenny D (2014) Living in shear: platelets protect cancer cells from shear induced damage. Clin Exp Metastasis 31(6):697–704 [DOI] [PubMed] [Google Scholar]
- 144.McCarty OJ et al. (2002) Fluid shear regulates the kinetics and molecular mechanisms of activation-dependent platelet binding to colon carcinoma cells. Biophys J 83(2):836–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jadhav S, Konstantopoulos K (2002) Fluid shear-and time-dependent modulation of molecular interactions between PMNs and colon carcinomas. Am J Physiol Cell Physiol 283(4):C1133–C1143 [DOI] [PubMed] [Google Scholar]
- 146.Mitchell MJ, King MR (2013) Fluid shear stress sensitizes cancer cells to receptor-mediated apoptosis via Trimeric death receptors. New J Phys 15:015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fan R et al. (2016) Circulatory shear flow alters the viability and proliferation of circulating colon cancer cells. Sci Rep 6:27073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ma S et al. (2017) Hemodynamic shear stress stimulates migration and extravasation of tumor cells by elevating cellular oxidative level. Cancer Lett 388:239–248 [DOI] [PubMed] [Google Scholar]
- 149.Regmi S, Fu A, Luo KQ (2017) High shear stresses under exercise condition destroy circulating tumor cells in a microfluidic system. Sci Rep 7:39975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Barnes JM, Nauseef JT, Henry MD (2012) Resistance to fluid shear stress is a conserved biophysical property of malignant cells. PLoS One 7(12):e50973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mitchell MJ et al. (2015) Lamin A/C deficiency reduces circulating tumor cell resistance to fluid shear stress. Am J Physiol Cell Physiol 309(11):C736–C746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vennin C et al. (2017) Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med 9(384). 10.1126/scitranslmed.aai8504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Triantafillu UL et al. (2017) Fluid shear stress induces cancer stem cell-like phenotype in MCF7 breast cancer cell line without inducing epithelial to mesenchymal transition. Int J Oncol 50(3):993–1001 [DOI] [PubMed] [Google Scholar]
- 154.Pelling AE et al. (2009) Mechanical dynamics of single cells during early apoptosis. Cell Motil Cytoskeleton 66(7):409–422 [DOI] [PubMed] [Google Scholar]
- 155.Jimenez AJ, Perez F (2017) Plasma membrane repair: the adaptable cell life-insurance. Curr Opin Cell Biol 47:99–107 [DOI] [PubMed] [Google Scholar]
- 156.Steinhardt RA, Bi G, Alderton JM (1994) Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263(5145):390–393 [DOI] [PubMed] [Google Scholar]
- 157.Miyake K, McNeil PL (1995) Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J Cell Biol 131(6 Pt 2):1737–1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chivukula VK et al. (2015) Alterations in cancer cell mechanical properties after fluid shear stress exposure: a micropipette aspiration study. Cell Health Cytoskelet 7:25–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Suresh S (2007) Biomechanics and biophysics of cancer cells. Acta Biomater 3(4):413–438 [DOI] [PMC free article] [PubMed] [Google Scholar]