

Abstract
The lung surfactant proteins are recognized as critical not only for their role in lowering lung surface tension but also in innate host defense. Reports have shown that some asthmatic patients have decreased levels of one member of this protein family in particular, Surfactant Protein-A (SP-A). Our studies set out to determine the contribution of SP-A to the response of a key effector cytokine in asthma, Interleukin (IL)-13. Our studies employ both animal models sufficient and deficient in SP-A challenged with IL-13 and primary epithelial cells from participants with asthma that are exogenously treated with SP-A in the context of IL-13 challenge. The inflammatory response and mucin production were assessed in both model systems. As compared to WT mice, we show that the activity of IL-13 is dramatically augmented in SP-A−/− mice, which have significantly increased neutrophil and eosinophil recruitment, mucin production and asthma-associated cytokines in the bronchoalveolar lavage fluid. In parallel, we show asthma-associated factors are attenuated in human cells from asthma subjects when exogenous SP-A is added during IL-13 challenge. While many of these phenotypes have previously been associated with STAT6 signaling, SP-A inhibited IL-13-induced STAT3 phosphorylation in mice and in human epithelial cells while having little effect on STAT6 phosphorylation. In addition, when either STAT3 or IL-6 were inhibited in mice, the phenotypes observed in SP-A−/− mice were significantly attenuated. These studies suggest a novel mechanism for SP-A in asthma as a modulator of IL-13-induced inflammation via mediating downstream IL-6/STAT3 signaling.
Introduction
Asthma is a chronic disease characterized by persistent symptoms, airflow limitation and frequent exacerbations and affects 5-10% of the US population (1, 2). Asthma remains one of the most common respiratory disease in both children and adults and is triggered by infection, environmental allergens or other stimuli (3, 4). Asthma is responsible for significant health care costs and morbidity. While understanding of the disease has increased, asthma still remains poorly understood and difficult to manage due to the heterogeneity of the disease.
Interleukin (IL)-13 is a type 2 cytokine that is crucial to the generation of many features of the asthma phenotype including goblet cell hyperplasia, increased airway hyperactivity and tissue remodeling (5, 6). IL-13 has also been shown to disrupt the barrier integrity of bronchial epithelial cells, which may contribute to the airway inflammation in allergic asthma (7, 8). Additionally, IL-13 increases susceptibility of human airway epithelium to rhinovirus infection (9) and suppresses double stranded RNA-induced IFN-λ production in a mouse model of asthma (10), resulting in poor control of viral infection and subsequent exacerbations. Previous studies have determined that IL-13-dependent signaling pathways are altered in asthma (11–18), which result in enhanced inflammatory responses. The mechanisms driving this enhanced response are several including a defective host innate immune response in asthma.
A key player in pulmonary innate immunity is surfactant protein A (SP-A), a member of the collectin family of proteins. SP-A is synthesized and secreted by alveolar type II cells, airway club cells and submucosal cells (19, 20) and serves as a first line of defense against inhaled pathogens. Encoded by SP-A1 and SP-A2 genes in humans, the mature SP-A protein is organized into an octadecamer structure that is derived from a combination of products from both genes. Both in vitro and in vivo studies show that SP-A functions as an opsonin and binds to the lipids and proteins on pathogens such as Mycoplasma pneumoniae, a pathogen associated with asthma exacerbations, and functions to attenuate M. pneumoniae pathogenicity (21, 22) and abrogates lung inflammation (23). We have shown that the anti-inflammatory function of SP-A is defective in asthma in the setting of M. pneumoniae infection (24) and that those patients with asthma who are obese demonstrate significantly lower levels of SP-A as compared to those patients with asthma who are not obese (25).
Using SP-A deficient mice and human airway epithelial cells from asthmatic participants, we show that lack of SP-A results in a dramatically heightened response to IL-13. Our studies suggest a novel mechanism for SP-A protection against non-infectious lung injury in the context of increased amounts of IL-13, a type 2 cytokine up-regulated in many asthmatic patients. In both mouse models and human cells from asthmatic participants, SP-A functions as a protective factor which we believe involves inhibition of STAT3 signaling that may be dependent upon IL-6 production.
Materials and Methods
Study Approval.
All murine experiments were carried out according to IACUC approved protocols at Duke University or the University of Arizona and comply with ARRIVE guidelines and are in accordance with National Institutes of Health guides for the care and use of Laboratory animals. The human subjects participation was on Institutional Review Board approved protocols at either Duke University or University of Arizona and were carried out in accordance with the Code of Ethics of the World Medical Association.
Mouse models.
SP-A−/− mice on C57BL/6 background that were backcrossed for 14 generations as previously described (26) were bred in-house and wild-type (WT) C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and bred in house to control for microbiome diversity. No inherent abnormalities in lung function or inflammation have been described for SP-A deficient mice in an unchallenged state and mice breed and age with no discernable differences from WT mice. Age-matched approximately 6-8 week old male mice were used for experiments. Mice were anesthetized under inhaled isoflourane and given 3.9 μg of recombinant IL-13 in 50 μl of sterile saline via oropharyngeal delivery as previously described (27). For studies to assess early indicators of inflammation, mice received 1 dose of IL-13 and were assessed ~24 hrs later. For experiments to assess later indicators of inflammation, mice received IL-13 for 3 consecutive days and were assessed on day 4 (24 hrs after the last IL-13 dose). Lyophilized IL-13 was purchased from Peprotech and resuspended immediately prior to use in sterile saline without a carrier protein. At the desired time point, mice were euthanized by a lethal dose of Urethane and the lungs lavaged with PBS (0.1 mM EDTA) and lung tissue was obtained for further analysis. Differential cells counts were analyzed from the lavage fluid after H&E staining. Viability was assessed by Trypan blue exclusion. A subset of mice received the STAT3 inhibitor S31-210 (Sigma; 5 mg/kg) or IL-6 inhibitor LMT-28 (Sigma; 1 mg/kg), based on doses used in previous methods, via ip injection 2 hours prior to receiving a one time oropharyngeal dose of IL-13 (28).
Mouse tracheal cell isolation.
Tracheal epithelial cells were isolated and grown according to previously published methods (29). Briefly, after mice were sacrificed, tracheas were removed by dissection and immediately placed in Ham’s F-12 medium on ice. A longitudinal incision was made exposing the mucosal lining and excess connective tissue, muscle, vasculature, and nerves were removed. The tracheas were gently digested in Ham’s F-12 medium containing 0.1% protease solution (Sigma) for 45 min at 37°C, after which digestion was inhibited with the addition of FBS. The tracheas were transferred to a dish containing medium, and the mucosal linings were scraped with a pipette tip. Cells were collected, transferred to a 15-ml conical tube, and centrifuged (900 rpm, 5 min, 4°C). The cell pellet was resuspended in 5 ml Versene (Life Technologies) for 15 min at 37°C, after which it was washed. Finally, the cell pellet was resuspended in 6 ml 10% FBS–Ham’s F-12 medium and ready for plating into transwells.
Culture media and supplements.
DMEM–Ham’s F-12 medium was used for both the harvest and culture of MTECs. The culture medium was supplemented with 250 ng/ml amphotericin B solution (HyClone), 20 ng/ml cholera toxin (List Biological), 104 μg/ml bovine pituitary extract (Lonza), 5 μg/ml insulin (Sigma), 5 μg/ml human apo-transferrin (Sigma), 0.1 μM dexamethasone (Sigma), 5 ng/ml mouse epidermal growth factor (Sigma), 0.01 μM retinol (Sigma), 20 U/ml nystatin (Sigma), and 50 μg/ml gentamicin. FBS (Atlanta Biologicals) was used to prepare 10 and 5% serum-rich media used at different steps in the ALI culture protocol.
In vitro culture of MTECs.
Costar Transwell (12 mm, 0.4 μm pores) 12-well plates were used to culture the MTECs according to methods previously described (29). Transwell membranes were coated with rat tail collagen (300 μg/ml in 0.02 N glacial acetic acid) and conditioned with Ham’s F-12 medium. Cells were plated evenly between the 12 wells in 500 μl 10% FBS culture media. The plate was incubated at 37°C in an air–5% CO2 atmosphere for 72 h, without changing the medium. After the initial seeding period of 72 hr, the medium on both the apical and basal sides of the membrane was replaced every other day. When the cells reached 80% confluence (6–8 days), the culture medium was changed to 5% FBS and replaced daily, only on the basolateral side, establishing an ALI. When the cells reached full confluence, the medium was changed to serum-free Ham’s F-12 culture medium and replaced daily. The cells were maintained at ALI for a minimum of 14 days.
RT-PCR.
Mouse tissues and human bronchial epithelial cells were collected into 1 ml of TRI Reagent® (Sigma). RNA was isolated using the standard TRI reagent/chloroform extraction method. DNA was synthesized from 1 µg of total RNA using Bio-Rad™ cDNA Synthesis kit. Real-time polymerase chain reaction (RT-PCR) was performed using Bioline 2x SensiFAST SYBR no-ROX mix. The samples were analyzed for expression levels of mouse MUC5AC using forward and reverse primers specific to the gene (forward 5’ GAG GGC CCA GTG AGC ATC TCC 3’, reverse 5’ TGG GAC AGC AGC AGT ATT CAG T 3’). The relative levels of expression obtained were normalized to the mammalian housekeeper gene Cyclophilin using primers specific to the gene (forward 5’ AGC ACT GGA GAG AAA GGA TTT GG 3’, reverse 5’ TCT TCT TGC TGG TCT TGC CAT T 3’). For human epithelial cells, MUC5AC and eotaxin were assessed using TaqMan™ probes (Applied Biosystems), and each condition is compared to its unexposed negative control after standardization to the housekeeper gene, GAPDH (Applied Biosystems).
Histological analysis.
Mice were euthanized and the left lung lobes were dissected and immersed in 10% buffered formalin for fixation. After 3 days, the lung lobes were transferred from formalin to 70% ethanol, then routinely processed and paraffin embedded for PAS staining by AML Laboratories (St. Augustine, FL). Stained sections were scored blinded according to standard methods by two independent investigators (23).
ELISA.
Mouse BAL samples were examined for IL-6, KC and eotaxin (R&D Systems) according to standard methods. Human IL-8 (R&D Systems) secreted into the apical compartment of transwells was examined according to standard methods. A subset of IL-13 treated BAL samples from SP-A−/− mice were further analyzed in the presence or absence of exogenously added SP-A (10 μg/ml) to in order to determine if the presence of SP-A altered IL-6 levels as detected by ELISA.
Research Participants.
Participants were recruited from the population in Durham, North Carolina and Tucson, Arizona and the surrounding areas. Informed consent was obtained from each participant (18-65 years of age). Asthmatic participants met GINA criteria for mild and moderate asthma including the presence of reversibility of airflow obstruction or airways responsiveness with a provocative concentration of methacholine resulting in a 20% fall in FEV1 (PC20 FEV1) of ≤ 8 mg/ml or < 16 mg/ml if they were taking inhaled corticosteroids (30). The presence of atopy was determined using skin testing and peripheral eosinophils and were measured in a subset of participants. Healthy participants had no evidence of airflow obstruction, and no history of pulmonary disease. Exclusion criteria included an exacerbation of asthma within four weeks of study requiring antibiotics and/or corticosteroids, greater than 10-pack year history of tobacco use or any cigarette use in the last year and any other significant medical conditions.
Research bronchoscopy and cell culture.
Participants underwent bronchoscopy with endobronchial-protected brushing and bronchoalveolar lavage (BAL), as previously described (31). The brushing of the proximal airways to obtain bronchial epithelial cells was performed under direct visualization using a separate protected cytologic brush for each pass, for a total of eight passes. BAL was performed via instillation of warm sterile saline in 60-ml aliquots, with return via gentle hand suction, for a total of 300 ml. Participants were discharged when their FEV1 achieved 90% of their pre-bronchoscopy, post-albuterol value.
Human airway epithelial cell air-liquid interface (ALI) experiments.
Freshly isolated airway bronchial epithelial cells from endobronchial brushing were cultured with BEGM (Lonza, Walkersville, MD) as previously described (31). Culture media contained the following supplements: Bovine pituitary extract, Insulin, Gentamycin-amphotericin, Retinoic Acid, Transferrin, Triiodothyronine, Epinephrine, and human Epidermal Growth Factor. After reaching confluence, cells were trypsinized and seeded onto collagen-coated polyester Transwell insert membranes of 12-mm diameter, at a concentration of 4 × 104/well. Then the cells were cultured at air–liquid interface for 2 weeks to allow for differentiation.
Typical expansion of airway epithelial cells from a single patient yielded ~8 × 10^6 cells. Within each participant sample of cells, as many experiments as possible were performed, including IL-13-induced IL-8 production, MUC5AC expression, and STAT3/6 phosphorylation. Conditions were performed in triplicate with appropriate unexposed controls and western blots repeated at least three times using cells for a given participant. Every effort was made to perform these experiments in cells from every participant, and generally cells from 5-10 participants were used for each experiment.
SP-A preparation.
SP-A was purified from the BAL fluid of patients with alveolar proteinosis that were seen at Duke University Medical Center and were under IRB approval. Surfactant was isolated from BAL samples by butanol extraction and octylglucoside solubilization using previously described methods (32). Extracted SP-A was resuspended in 5 mM Tris pH 7.4 and passed over a polymyxin B-agarose column to reduce endotoxin contamination. SP-A used in assays had final endotoxin concentrations of <0.01 pg/mg SP-A as determined by the Limulus amoebocyte lysate assay (QCL-1000, BioWhittaker (Lonza)). Human SP-A extracted by our group is available and is currently shipped upon request to labs both nationally and internationally for use in the research setting.
MUC5AC gene expression and IL-8 production by human airway epithelial cells.
IL-8 in apical supernatant was determined by ELISA 48 hrs after IL-13 challenge (50 ng/ml on the basolateral surface). Muc5AC gene expression was determined by RT-PCR relative to GAPDH after chronic IL-13 challenge for 5 days (10 ng/ml on the basolateral surface every 48 hours x 2). In our studies, a longer exposure to IL-13 is required for optimal Muc5AC upregulation in human epithelial cells at ALI. SP-A (20 μg/ml) or vehicle was added 30 min prior to IL-13 challenge. After standardization to the housekeeping gene, data are displayed as fold relative to the non-IL-13 challenged control for each respective participant set, with the standard error of the mean. IL-8 and was measured in apical supernatants by ELISA and Muc5AC was determined by RT-PCR of cell lysates.
Western analysis of STAT activation.
Human bronchial epithelial cells (BECs) obtained from asthmatic and normal participants were grown at an ALI for 2 weeks prior to IL-13 challenge (10 ng/ml) in the presence or absence of oligomeric SP-A (20 μg/ml). Total cell lysates were analyzed for STAT3 or STAT6 phosphorylation determined by Western blot and compared to total STAT3 or STAT6 30 min after IL-13 challenge. Western blots are representative of n=3 asthma and 3 normal participants. For collection, Radioimmunoprecipitation assay (RIPA) buffer (Millipore) with protease inhibitors (Roche) was added to each transwell for cell lysis and protein extraction. Each sample was sonicated and insoluble material was removed by centrifugation. The protein concentration of each lysate was determined by BCA and an equal amount of lysate was loaded onto electrophoretic gels for each sample to be analyzed by Western. Antibodies for phospho-STAT3, STAT3, phospho-STAT6, STAT6 and β-actin were all used according to manufacturer’s (Cell Signaling) recommendations and analyzed in the order listed respectively for each blot.
Human bronchial epithelial cell IL-6 receptor expression.
Human bronchial epithelial cells from asthmatic and normal participants were grown at ALI for 2 weeks after which lystates and apical supernatants from untreated samples were collected. Lysates and supernatants were assayed for baseline expression levels of gp130 and soluble IL-6 receptor (sIL-6r) using western blotting and ELISA, respectively. Western blotting was performed using a gp130 antibody (Cell Signaling) and the previously listed protocol. Soluble IL-6 receptor levels in apical supernatants were determined using an ELISA Quantikine kit (R&D Systems).
SP-A binding assay.
A 96-well half-area plate was coated with 2 ug/ml of IL-6 (Peprotech) and allowed to incubate overnight at 4° C. The plate was then washed twice and blocked for 2 hours at 37° C. Increasing concentrations of SP-A protein was then added to the plate and incubated for 2 hours at 37° C. The plate was then washed five times and then incubated with HRP-conjugated anti-SP-A antibody (Abcam) for 1 hour at 37° Celsius. Plate was then washed five times after which TMB substrate (R&D Systems) was added for 10 minutes and then neutralized using 2.0 N sulfuric acid. Absorbance was read at 490 nM (BioTek).
Statistical analysis.
For murine experiments, analyses were performed using Graph Pad Prism and Student’s T-Test or One-way ANOVA, as necessary. F-test was used to compare variances between data analyzed and Welch’s correction applied if variances were significantly different (p<0.05) between group. Airway epithelial cell data were compared using Stident’s T-Test or One Way ANOVA, or Wilcoxon if data were not nornally distributed. Statistical significance was defined as a p-value ≤ 0.05.
Results
SP-A protects against IL-13 challenge in mice.
In order to evaluate the importance of SP-A during IL-13 challenge, we examined the response to the cytokine in a mouse model using mice harboring either WT or null alleles (SP-A−/−) for the surfactant protein. Mice were challenged with IL-13 on three consecutive days using established methods (Fig. 1A) (27). Twenty-four hours after the last challenge, mice were euthanized and indices of inflammation were examined: inflammatory cells in BAL, mucin production, IL-6, KC and eotaxin.
Figure 1, SP-A deficient mice are more susceptible to IL-13 challenge than WT mice.
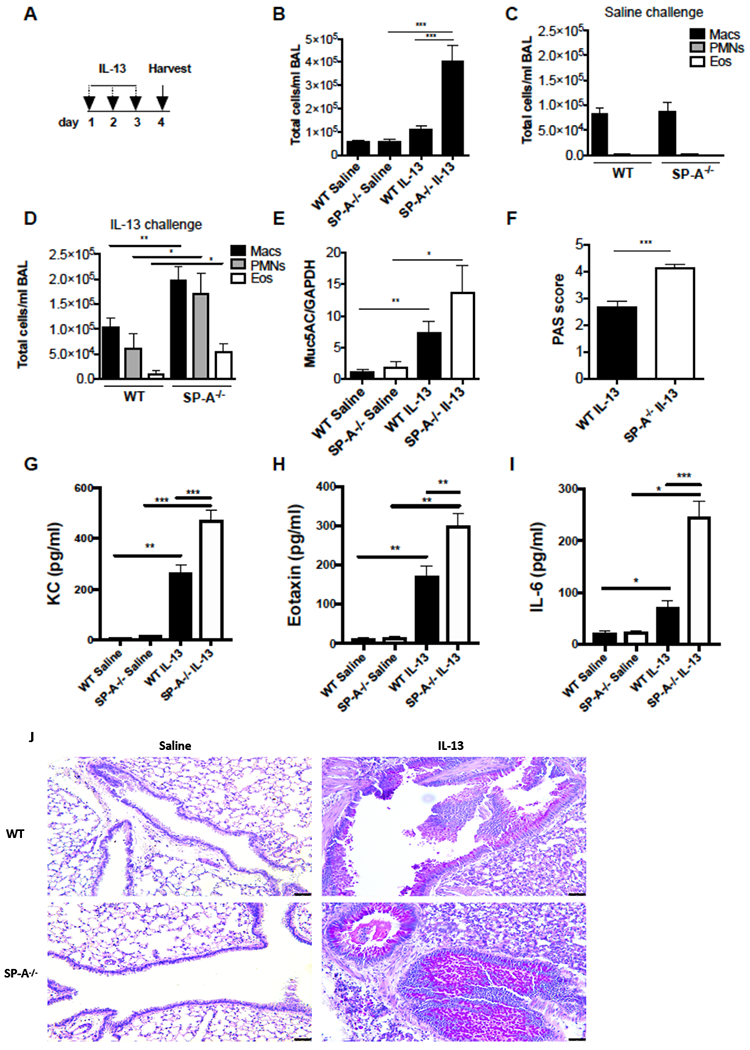
A) WT and SP-A mice were challenged with IL-13 for 3 consecutive days via oropharyngeal instillation. B) Total cells present in the BAL were examined on day 4 in saline challenged versus IL-13 challenged mice. Differential cell populations were determined by cell staining for macrophages, neutrophils and eosinophils in BAL of C) saline challenged or D) IL-13 challenged mice. E) Muc5AC gene expression was quantified by RT-PCR of lung tissue and represented as fold of the saline treated control. F) PAS scores from IL-13 challenged mice were quantified by lung histological staining. G) KC, H) Eotaxin and I) IL-6 in BALF were quantified by ELISA. J) Representative micrographs of PAS stained lungs in WT and SP-A−/− mice after IL-13 challenge. B-I data were obtained from n=8,8 saline treated and n=20,20 IL-13 treated mice across three separate experiments, *p<0.05, **p<0.01, ***p<0.001.
Total leukocytes in the BAL of WT mice increased only modestly with IL-13 challenge, but the total cells in lavages of SP-A−/− mice were significantly elevated compared to SP-A−/− saline controls and WT IL-13 challenged mice (Fig. 1B). Lavages from saline challenged mice consisted almost exclusively macrophages and were not different between the WT and SP-A−/− control mice (Fig. 1C). Lavages from IL-13 challenged mice had increased neutrophils and eosinophils, both of which were significantly elevated in SP-A−/− mice as compared to WT mice (Fig. 1D). There were very few to no lymphocytes detected in any of the BALs (not shown).
IL-13 challenge resulted in up-regulation of Muc5AC RNA expression in lung tissue from both WT and SP-A−/− mice compared to saline controls (Fig. 1E). Mucin production was significantly more pronounced in IL-13 challenged SP-A−/− mice as compared to WT challenged mice (Fig. 1F,J). Blinded scoring of PAS stained histological lung sections were between 0 and 1 for all of the control saline challenged mice and there was no difference between WT and SP-A−/− at baseline (data not shown).
Cytokines associated with neutrophil and eosinophil recruitment, KC and eotaxin (CCL11), respectively were significantly higher in IL-13 challenged SP-A−/− mice as compared to WT mice (Fig. 1G,H). IL-6, a cytokine known to mobilize neutrophils from the bone marrow and lead to downstream activation of JAK-STAT signaling, was also upregulated in BAL from IL-13 challenged SP-A−/− as compared to WT challenged mice (Fig. 1I).
Inhibition of STAT3 signaling during IL-13 challenge attenuates phenotypes in SP-A−/− mice.
Since the majority of the phenotypes we observed in the SP-A−/− mice have been previously associated with JAK-STAT signaling (33, 34), we choose to examine the activation of the STAT3 and STAT6 pathways in the IL-13 challenged lungs. We did not detect STAT3 or STAT6 phosphorylation in the vehicle treated animals. However, upon IL-13 challenge we saw an increase in both STAT3 and STAT6 phosphorylation in the lung lysates of WT and SP-A−/− mice. Interestingly, while STAT6 phosphorylation was similar between IL-13 challenged WT and SP-A−/− mice, STAT3 phosphorylation was significantly higher in SP-A−/− mice as compared to WT mice in the challenge model (Fig. 2A).
Figure 2, Inhibition of Stat3 signaling attenuates IL-13 driven inflammation in SP-A−/− mice.
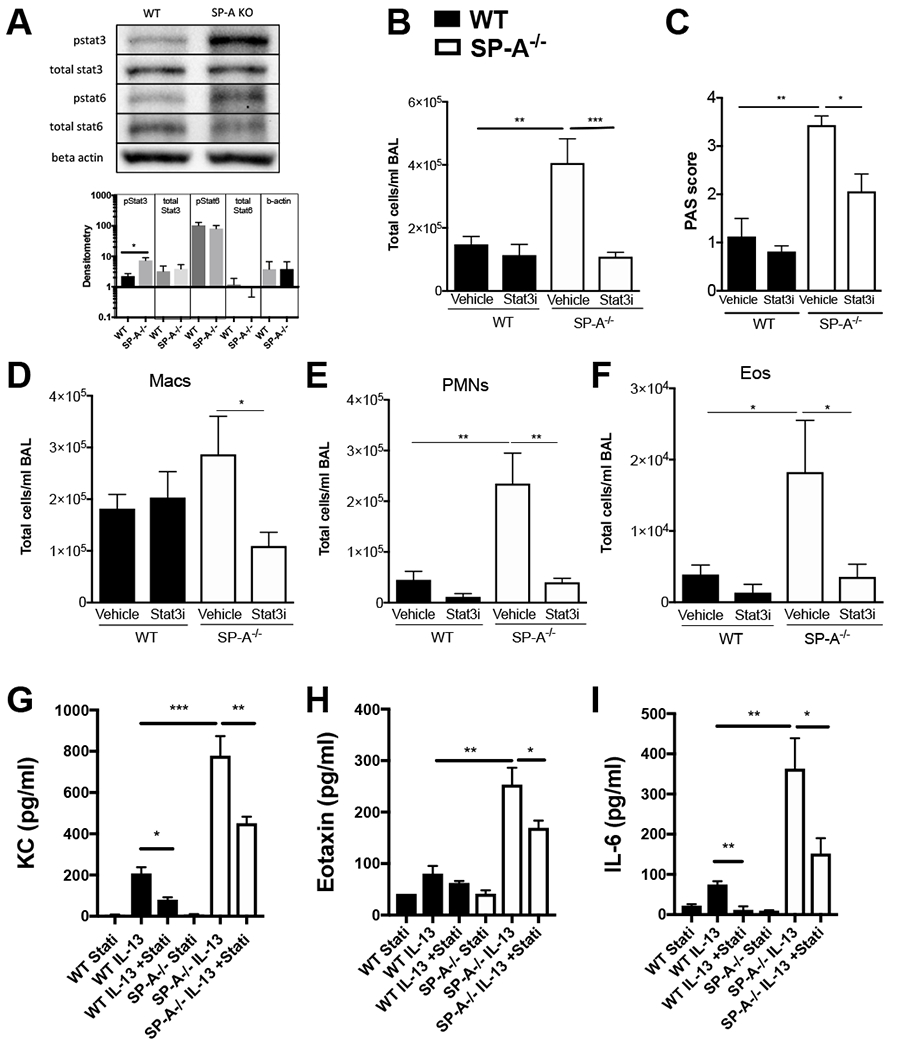
A) WT, SP-A−/− mice were exposed to IL-13 for 3 days and Stat3 and Stat6 phosphorylation was assessed from whole lung lystates by Western blot. n=8,8 lysates per group; representative bands shown from two independent blots. No Stat3 or Stat6 phosphorylation was detected in saline control treated mice. B-I) WT (black bars) or SP-A−/− (white bars) mice received vehicle or Stat3 inhibitor (5 mg/kg body weight) via ip injection 2 hrs prior to a one time airway challenge with IL-13. Twenty-four hours later, B) total cells in BAL were counted and C) mucin production was assessed from PAS stained histological sections. By differential staining, BAL cells consisted of D) macrophages (Macs), E) neutrophils (PMNs), and F) eosinophils (Eos). G) KC, H) Eotaxin and I) IL-6 were determined in BAL of SP-A−/− mice treated with and without Stat3 inhibitor prior to IL-13 by ELISA. n=8-12/per group, *p<0.05, **p<0.01, ***p<0.001.
In order to determine the role of STAT3 signaling in the enhanced inflammatory response to IL-13 in SP-A−/− mice, we examined the effect of STAT3 inhibition during IL-13 challenge. Since these signaling events were expected to occur early in the inflammatory process, these studies with the inhibitor were conducted at an earlier timepoint of 24 hrs after a one time IL-13 challenge. The inhibitor was given one time, 2 hrs prior to IL-13 challenge. While the STAT3 inhibitor had little effect on the WT IL-13 challenged mice, inhibition of STAT3 in IL-13 challenged SP-A−/− mice resulted in significant reduction of total cells in BAL as compared to the vehicle treated controls (Fig. 2B). Macrophages (Fig. 2D), neutrophils (Fig. 2E) and eosinophils (Fig. 2F) were significantly reduced in the STAT3 inhibitor treated IL-13 challenged SP-A−/− mice as compared to vehicle treated IL-13 challenged controls. The reduction in cellular inflammation in the STAT3 inhibitor group of SP-A−/− mice was at similar levels detected in the WT challenged mice.
Mucin production was also significantly reduced in SP-A−/− mice that were treated with the STAT3 inhibitor prior to IL-13 challenge as compared to vehicle treated IL-13 challenged controls (Fig. 2C). There was no observable effect in regards to cell recruitment in BAL or mucin production of vehicle or STAT3 inhibitor in unchallenged mice in either the WT or SP-A deficient mice (data not shown). While levels of KC and IL-6 were significantly elevated in SP-A−/− as compared to WT mice, (Fig. 2G,I), both cytokines were significantly reduced in both the WT and SP-A deficient mice that received the STAT3 inhibitor as compared to their respective vehicle controls. In contrast, eotaxin was only significantly reduced in SP-A−/− mice given the STAT3 inhibitor (Fig. 2H).
Inhibition of IL-6 during IL-13 challenge attenuates phenotypes in SP-A−/− mice.
IL-6 may directly induce mucin and is also known to be an important initiator of STAT3 signaling (35). Since our findings demonstrate that SP-A−/− mice have significantly increased IL-6 levels after IL-13 challenge, we next sought to determine if the mechanism of action for SP-A in limiting IL-13-induced STAT3 signaling was through regulation of IL-6 production. As shown in Fig. 1I and 2I SP-A−/− mice had significantly higher levels of IL-6 in BAL post IL-13 challenge as compared to WT mice. We next chose to inhibit IL-6 signaling during IL-13 challenge to see if we could atteunate the phenotypes observed in SP-A−/− mice. Mice were given an IL-6 antagonist (LMT-28) that targets the IL-6 receptor β subunit glycoprotein 130, 2 hours prior to challenge with IL-13 and assessed for inflammation 24 hours post-challenge as previously described (28). Again, since these signaling events were expected to occur early in the inflammatory process, these studies with the inhibitor were conducted at an earlier timepoint of 24 hrs after a one time IL-13 challenge. Macrophages in the BAL were not significantly different when IL-6 was inhibited (Fig. 3A,B); however, neutrophils (Fig. 3C) and eosinophils (Fig. 3D) were significantly lower in SP-A−/− mice that received the IL-6 inhibitor prior to IL-13 challenge compared to those receiving vehicle control. There were no significant changes in eosinophils or neutrophils in WT IL-13 challenged mice given the IL-6 inhibitor. Similarly, Muc5AC (Fig. 3E) and eotaxin (Fig. 3G) were significantly decreased in only the SP-A−/− mice receiving the IL-6 inhibitor as compared to those receiving the vehicle control. While KC significantly increased in the absence of SP-A during IL-13 challenge as compared to SP-A sufficient mice, levels of KC were not altered by the IL-6 inhibitor in either genotypes of mice (Fig. 3F).
Figure 3, Inhibition of IL-6 signaling attenuates IL-13 driven inflammation in SP-A−/− mice.
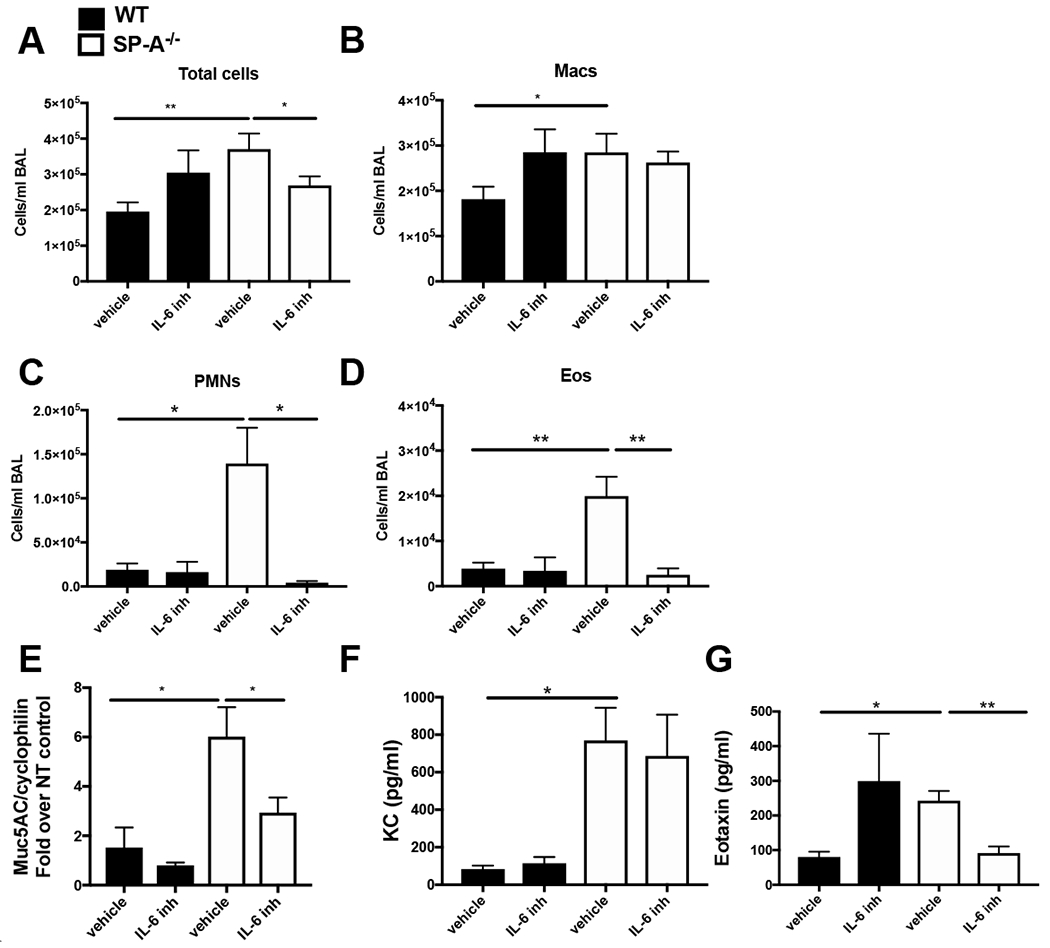
A-F) WT (black bars) and SP-A−/− (white bars) mice were injected with either an IL-6 inhibitor (or vehicle control) 2 hrs prior to a one time airway challenge with IL-13. Twenty-four hours post challenge A) total cells in the BAL which consisted of B) macrophages (Macs), C) neutrophils (PMNs), D) eosinophils (Eos) were examined. E) Muc5AC gene expression was examined by RT-PCR in lung tissue. F) KC, and G) Eotaxin levels were examined by ELISA. n=9,9/per group, *p<0.05, **p<0.01, ***p<0.001.
SP-A attenuates IL-13-induced inflammation in bronchial epithelial cells from asthmatic participants.
We next sought to determine if our findings in mice were translatable to humans. In order to do this, freshly isolated bronchial epithelial cells from well-phenotyped participants with and without mild-moderate asthma (Table 1) were obtained by bronchoscopy and cultured at an air liquid interface (ALI) for 14 days to induce differentiation, and then exposed to IL-13. IL-13-challenged cells cultured from non-asthmatic participants had similar levels of IL-8 production as compared to their non-stimulated controls (fold change of non-asthmatic control, ~1.1). However, cells cultured from asthmatic participants demonstrated a significant increase in IL-8 when stimulated with IL-13 as compared to their asthmatic respective control (Fig. 4A). Both non-asthmatic and asthmatic cells responded to IL-13 challenge by up-regulating Muc5AC RNA relative to non-stimulated controls (Fig. 4C). Cells from asthmatic participants had a modestly greater response as compared to those from non-asthmatic participants, but the values did not reach statistical significance. The addition of exogenous SP-A prior to stimulation with IL-13 resulted in significantly lower expression of IL-8 (Fig. 4B) and Muc5AC RNA (Fig. 4D) by bronchial epithelial cells from asthmatic participants.
Table 1.
Demographics for patients that underwent bronchoscopy for analysis of bronchial epithelial cells.
| PARTICIPANT DEMOGRAPHICS | ASTHMA | NORMAL |
|---|---|---|
| Males | 5 | 3 |
| Females | 9 | 6 |
| White | 5 | 5 |
| Black | 8 | 4 |
| Hispanic | 0 | 0 |
| Asian | 1 | 0 |
| Age | 27 ± 2 | 32 ± 3 |
| FEV1 (%) | 91 ± 2.7 | 101 ± 4.3 |
| FVC (%) | 101 ± 2.2 | 100 ± 4.0 |
| FEV1/FVC (%) | 76 ± 1.6 | 84 ± 1.4 |
| PC20 (mg/ml) | 1.4 ± 0.3 | N/A |
| *Age of onset | ||
| > 12 years old | 2 | N/A |
| < 12 years old | 11 | N/A |
| ACQ | 1.4 ± 0.1 | N/A |
| Exacerbations per year | 1.0 ± 0.3 | N/A |
| eNO | 42 ± 15 | 13 ± 2 |
| Atopy (% positive) | 79 | 44 |
| Peripheral Eosinophil (%) | 3.4 ± 0.9 | 1.1 ± 0.2 |
| Peripheral Neutrophils (%) | 56 ± 2.0 | 60.6 ± 3.9 |
| MEDICATIONS | ||
| Short acting beta agonist | 13 | N/A |
| Leukotriene modifier | 1 | N/A |
| Inhaled corticosteroid (ICS) | N/A | N/A |
| Antihistamine | 5 | N/A |
| Combination therapy (ICS/LABA) | 1 | N/A |
| Short acting anticholinergic | N/A | N/A |
| Mean ± SE |
Age not available for 1 patient.
Figure 4, SP-A attenuates IL-13 induced inflammation in cultured bronchial epithelial cells from asthmatic participants.
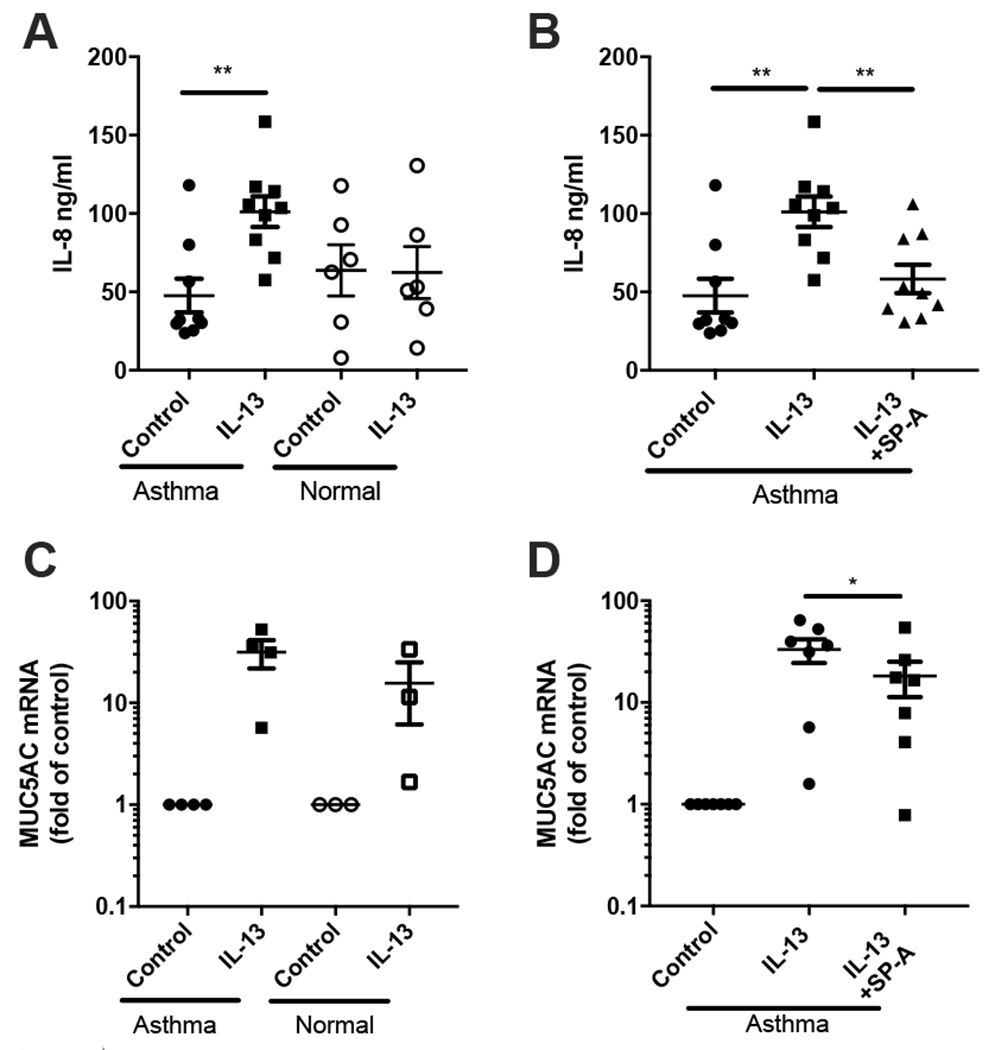
BECs from asthmatic and normal participants obtained by bronchoscopy were cultured at ALI for 2 weeks after which they were stimulated with IL-13 in the presence or absence of oligomeric full-length SP-A isolated from APP patients (20 μg/ml), which was added 30 min prior to each IL-13 challenge. A) IL-8 in AS was determined by ELISA 48 hrs after IL-13 challenge (10 ng/ml) from cells of asthmatic and normal participants. B) IL-8 in AS from IL-13 stimulated asthma cells, in the presence or absence of SP-A. IL-8 data are displayed as fold relative to the non-IL-13 challenged control for each respective patient set, with the standard error of the mean. C) Muc5AC gene expression was determined by RT-PCR relative to GAPDH after chronic IL-13 challenge for 5 days (10 ng/ml) from BEC of asthmatic and normal participants. D) Muc5AC gene expression from IL-13 challenged BEC from asthmatic participants in the presence or absence of SP-A. Experiments were performed in triplicate for each set of patient cells based upon availability of adequate cell number for experiments. Each dot represents the average value for one patient, *p<0.05,**p<0.01.
SP-A attenuates STAT3 phosphorylation in human bronchial epithelial cells from asthmatic participants.
Previous work supports the concept that bronchial epithelial STAT3 phosphorylation is required for allergic inflammation in the house dust mite model of asthma (36, 37). We therefore examined the effect of SP-A on IL-13-induced STAT signaling in our human primary bronchial epithelial cells from asthmatic participants that were grown at an ALI. In agreement with previous findings examining IL-13 signaling in different cell types (38–40), IL-13 challenge in human bronchial epithelial cells also increased STAT3 phosphorylation quickly, after only ~30 min (Fig. 5A). The addition of exogenous SP-A prior to IL-13 challenge resulted in decreased STAT3 phosphorylation as compared to the no SP-A control condition (Fig. 5A). Interestingly, phosphorylation of STAT6 was not different in asthma and non-asthma cells and was not attenuated by exogenous SP-A in asthma cells.
Figure 5, SP-A inhibits IL-13-induced STAT3 phosphorylation in asthma.
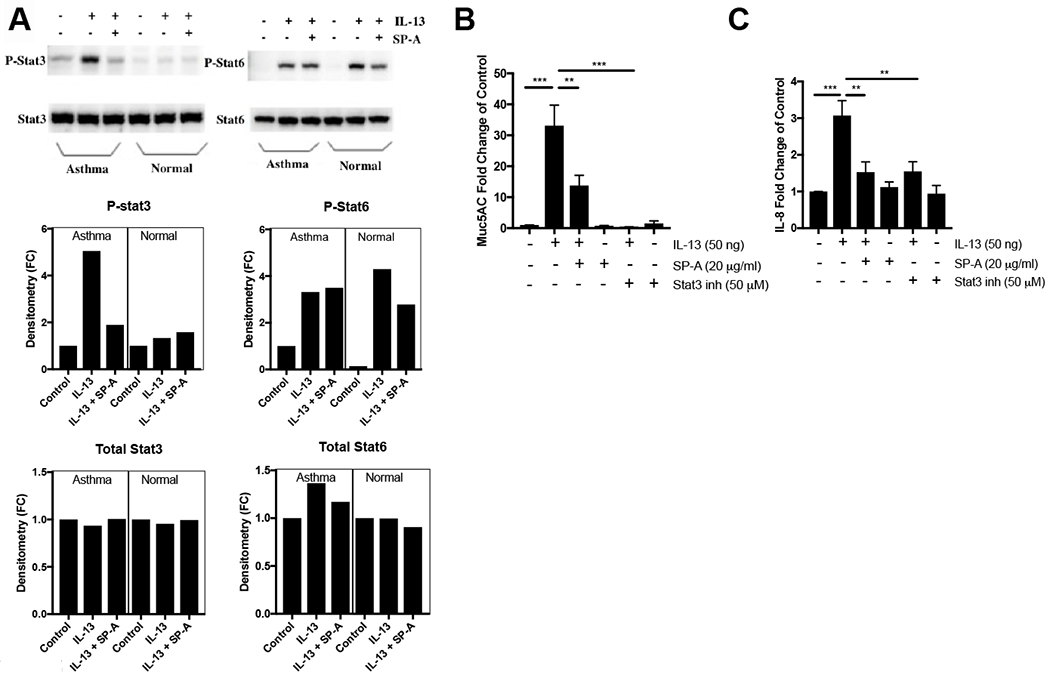
Human bronchial epithelial cells (BECs) obtained from asthmatic and normal participants were grown at an ALI for 2 weeks prior to IL-13 challenge (10 ng/ml) in the presence or absence of control oligomeric SP-A (20 μg/ml). A) Total cell lysates were analyzed for STAT3 or STAT6 phosphorylation determined by Western blot and compared to total STAT3 or STAT6 30 min after IL-13 challenge. Western blots are representative of n=3 asthma and 3 normal participants on two independent blots. B) Muc5AC gene expression was determined after stimulation with IL-13 using a 5 day protocol (10 ng/ml at 48 hours x 2, with trypsinization at 5 days) and C) IL-8 secretion were examined 48 hours from BEC that were challenged with IL-13 in the presence or absence of either SP-A or a STAT3 inhibitor added 30 minutes prior to IL-13.
Additional studies in human bronchial epithelial cells were conducted in which either SP-A or a STAT3 inhibitor (S31-210) were added to the ALI cells prior to IL-13 challenge. There was no upregulation of either IL-8 or Muc5AC in response to IL-13 when STAT3 was inhibited (Fig. 5B,C). In these studies, SP-A inhibited IL-8 and Muc5AC production similar to levels observed when a STAT3 inhibitor was present.
IL-6 receptor expression does not differ between asthma and normal bronchial epithelial cells.
While it is appreciated that epithelial cells from asthmatics produce IL-6, it has been noted that these cells may loose the ability to produce IL-6 after culturing and differentiation (41). Therefore, we next examined expression of the receptors for IL-6 to determine if they were differentially expressed in cells from asthma as compared to normals. For these studies, naïve human BECs that had been differentiated at an ALI for 2 week were used. Lysates were assayed for baseline expression levels of gp130 and apical supernatants were assayed for sIL-6r by ELISA. While the levels of both GP130 and sIL-6r were quite variable between patient samples, we did not see any difference in their expression levels between asthma and normal participants (Table 2).
Table 2. IL-6 receptor expression in normal and asthma.
GP130 was probed by western blot and relative densitometry reported relative to GAPDH. sIL-6r was assessed by ELISA.
| IL-6 receptors | Normal | Asthma |
|---|---|---|
| GP130 (arbitrary units) | 3.5 ± 2 | 4.1 ± 2.8 |
| sIL-6r (pg/ml) | 240 ± 54 | 239 ± 28 |
Mouse tracheal epithelial cells lacking SP-A have enhanced IL-6 production compared to cells from WT mice.
Our studies thus far suggest that the heightened response to IL-13 observed in SP-A−/− mice may be due to increased IL-6 production and IL-6 signaling through STAT3. We next sought to determine if IL-6 was differentially produced from IL-13 stimulated epithelial cells from WT and SP-A−/− mice that were grown at an ALI. Interestingly, under baseline conditions (no challenge) there was a trend for more IL-6 production in the apical supernatants in the cells grown from SP-A−/− mice, although this did not reach statistical significance (p=0.06). Upon stimulation with IL-13 for 48 hrs, IL-6 production in the apical compartment from SP-A−/− MTECS was significantly greater as compared to WT stimulated cells (Fig. 6). There were no changes or differences in IL-6 levels in the basolateral compartment under any of the conditions and genotypes tested.
Figure 6, Increased IL-6 production from IL-13 challenged MTECs.
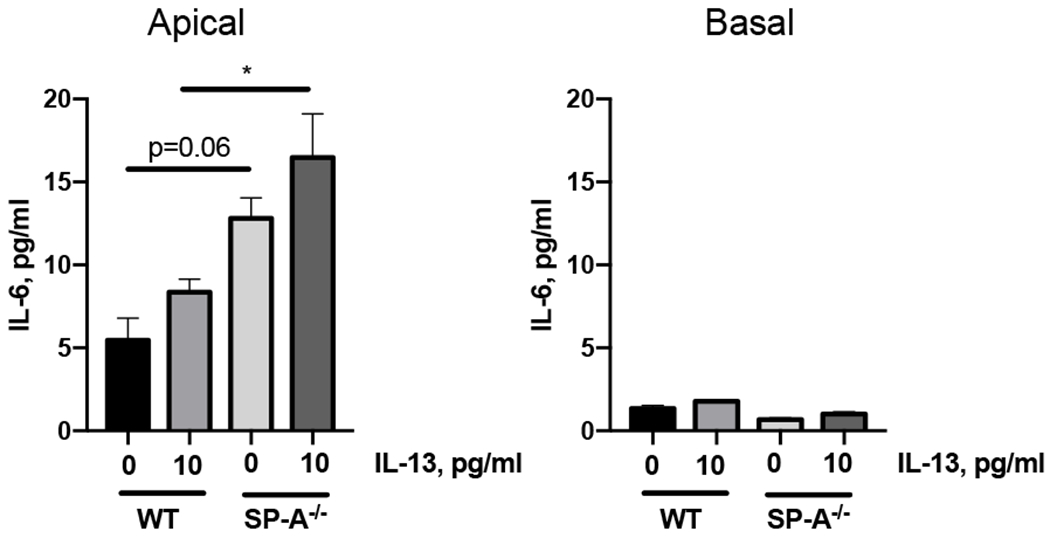
Mouse tracheal epithelial cells were harvested from WT and SP-A−/− mice and grown at an ALI for 2 weeks prior to IL-13 challenge (10 ng/ml, basolaterally). Apical and basolateral supernatants were harvested for analysis after 48 hrs of challenge. IL-6 levels were determined by ELISA. *p<0.05, n=3 for each condition.
SP-A binds to IL-6 in a dose-dependent manner.
In order to determine if the phenotypes we observed could in part be explained by SP-A binding to IL-6, we set out to determine if SP-A bound to IL-6 using a plate bound assay. Using purified human SP-A within the normal physiological range of what is measured at the air-liquid interface, we found that when added to immobilized plate-bound human IL-6, SP-A bound in a dose-dependent manner (Fig. 7).
Figure 7, SP-A binds to IL-6 in a dose-dependent manner.
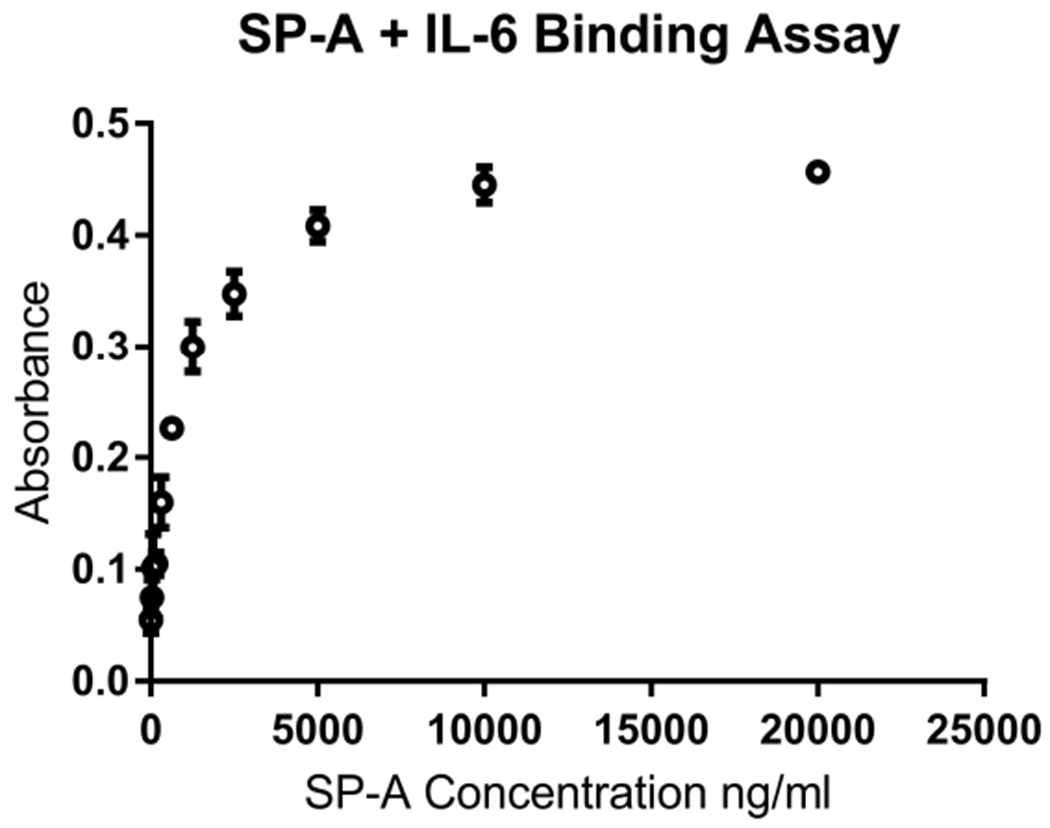
A 96-well plate was coated with IL-6 and increasing concentrations of SP-A protein were added. An HRP-conjugated anti-SP-A antibody and TMB substrate were used to detect SP-A binding at an absorbance of 490 nM.
Since SP-A bound to IL-6, we wanted to exclude the possibility that SP-A binding to IL-6 would interfere with IL-6 detection by ELISA. In order to test this, we took BAL fluid from SP-A deficient mice that had been challenged with IL-13 and added a physiologic dose of exogenous SP-A (in the range of binding as determined in the new figure 7). The IL-6 levels were similar between the SP-A-treated and non-treated with an average change of 1.28 pg/ml (SE±3.45 pg/ml) for 8 samples tested (data not shown).
Discussion
Here we provide novel findings from our investigation regarding the role of SP-A in mediating the inflammatory responses to IL-13 using both mouse models and primary human bronchial epithelial cells. Compared to SP-A sufficient mice, SP-A−/− mice had dramatic responses to IL-13 challenge, which were attenuated when either STAT3 or IL-6 signaling pathways were inhibited prior to challenge. Taken together, our findings suggest that SP-A plays a protective role in the context of increased IL-13 by limiting an overzealous inflammatory response that is driven by IL-6 and STAT3 signaling in the absence of SP-A.
While SP-A is well-recognized as an important mediator of innate immunity in the lung (reviewed in (42)), less is known about the role of SP-A in chronic inflammatory lung diseases, such as asthma. Early work examined levels of SP-A in asthmatic versus normal fluid obtained from BAL (43–45). Overall, the studies of SP-A levels in human BAL provided contradictory results, with reports of higher and lower levels of SP-A levels in asthma compared to controls. This discrepancy could in part be attributed to the variability within clinical asthma which is now more accurately described by the specific asthma phenotypes which are driven by specific mechanisms or endotypes (46). In this study, we focus upon asthma with type 2 inflammation with atopy and early onset disease, but we have also shown that SP-A is reduced in a specific subset of asthmatic patients who are obese (25).
Regardless of SP-A levels, the role of SP-A in asthma has been the topic of investigation by our group and others using SP-A gene deficient mice challenged in models of allergic airways disease (23, 47). SP-A−/− mice have increased hallmarks of asthma including BAL eosinophilia, mucin production, IL-4, IL-5 and IgE as compared to WT mice (47). On a mechanistic level, SP-A has been shown to bind to eosinophils (Ca2+ dependent and receptor-mediated) and prevent degranulation (23) while promoting their apoptosis (48). In this investigation, we determined that SP-A modulates IL-13-driven signaling in both mice and human airway bronchial epithelial cells via reduced phosphorylation and activation of STAT3, a critical mediator in allergic disease (36), while having little to no effect on STAT6 signaling.
In asthma, STAT activation occurs with IL-13 and IL-13 receptor (IL-4Rα and IL-13Rα1) interactions lead to Janus kinase-mediated tyrosine phosphorylation of the IL-4Rα chain, and subsequent activation of the transcription factors STAT1, 3, and 6 (49, 50). We expected that SP-A treatment would inhibit IL-13 signaling by simply blocking the interaction of the IL-13 ligand with its cognate receptor. Unexpectedly, we observed that not only was IL-13 signaling in the presence of SP-A intact, but that there was differential regulation of the IL-13-induced STAT-3 phosphorylation states specifically in asthmatic bronchial epithelial cells and in mouse models. IL-13 stimulation led to a significantly augmented STAT3 phosphorylation upon IL-13 stimulation and the presence of SP-A led to inhibition of that signaling. Thus, unexpectedly, SP-A did not merely block access of the ligand to its receptor. On the contrary, the presence of SP-A allowed IL-13 signaling, but fundamentally altered the balance of the STAT3 and STAT6 pathways, which we hypothesize, restores homeostasis.
One possible interpretation of the mechanism by which SP-A inhibits IL-13-mediated effects via STAT3 and not STAT6 is that an IL-13 induced cytokine that (re)-utilize STAT3 signaling, such as IL-6, is increased and has exaggerated activity in the absence of SP-A. Indeed we did see increased IL-6 in the SP-A−/− mice after IL-13 challenge and neutralization of IL-6 during the IL-13 challenge in SP-A−/− attenuated the phenotypes back to those observed in WT mice. Thus, the inhibiting effects of SP-A may not completely affect the response directly to IL-13, but more to cytokines down-stream of IL-13, such as IL-6. In support of this hypothesis, we discovered that SP-A binds to IL-6 in a dose-dependent manner which could thereby be the main mechanism by which SP-A limits the response in the context of high IL-13 levels.
Another alternative for the protective mechanisms of SP-A could be in part due to its ability to negatively regulate CD4+T cells during IL-13-induced inflammation and enhance IL-4Rα-mediated macrophage activation (51). Recent publications show that M2-activated macrophages are involved in suppression of the proinflammatory cytokine response and resolution of tissue damage, and IL-13 strongly induces chemokine expression (i.e., CCL2, CCL17, CCL11, and CCL24) in WT mice (52). This potential explanation, while outside the scope of these studies, warrants further consideration in future studies.
A limitation of our study is the small sample size of human asthmatic epithelial cells grown at an ALI in which to test our hypothesis. Due to the challenging nature of these cells, not all experiments could be validated in every patient cell sample that was examined. Another limitation is that while we saw increased IL-6 production in our IL-13 challenged mouse models, we failed to detect differences in IL-6 in the human BECs after IL-13 challenge. This could be attributed to differences in response to IL-13 from a “whole organism” perspective that contains a functioning immune system versus what we observe locally in primary isolated and cultured epithelial cells, which have been shown to lose the ability to produce IL-6 (41).
Previous findings by ourselves and others highlight the importance of SP-A in asthma (24, 25, 47, 53). Our findings in mice and in human cells from asthma participants add to this knowledge by demonstrating that in the absence of SP-A, the response to a critical asthma cytokine, IL-13, is significantly heightened. Clinically, this could contribute to reduced lung function and exacerbations in individuals with low levels or dysfunctional SP-A. More studies are needed to determine if such an association exists. By gaining a better understanding of how specifically SP-A is able to downregulate STAT3 and not STAT6 phosphorylation indirectly through mediation of IL-6, we may be able to achieve better treatment strategies for those asthma sufferers with the type 2 asthma endotype.
Key Points:
SP-A is a collectin and plays a key role in innate immunity in the lung.
SP-A modulates inflammation in airway epithelial cells from patients with asthma.
SP-A modulates IL-13-induced inflammation through downstream IL-6/STAT3 signaling.
Acknowledgments
Funding: HL125602 (Ledford), AI125357 (Kraft, Voelker, Chu) and Arizona Biomedical Research Commission grant ADHS16-162519 (Kraft, Ledford).
Footnotes
Disclosures: Drs. Ledford and Kraft are co-founders of RaeSedo, LLC, a company to develop novel peptidomimetic based therapeutics derived from an active area of SP-A. No peptidomimetic studies are reported in this manuscript but results with full-length SP-A described herein provided rationale for the establishment of RaeSedo, LLC.
Some of the experiments reported herein were conducted at Duke University prior to Drs. Ledford and Kraft relocating to the Unversity of Arizona and as such were under IRB or IACUC approvals from Duke University at the time of study.
REFERENCES
- 1.Dougherty RH, and Fahy JV. 2009. Acute exacerbations of asthma: epidemiology, biology and the exacerbation-prone phenotype. Clin Exp Allergy 39: 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peat JK, Woolcock AJ, and Cullen K. 1987. Rate of decline of lung function in subjects with asthma. Eur J Respir Dis 70: 171–179. [PubMed] [Google Scholar]
- 3.Bousquet J, Bousquet PJ, Godard P, and Daures JP. 2005. The public health implications of asthma. Bull World Health Organ 83: 548–554. [PMC free article] [PubMed] [Google Scholar]
- 4.Mannino DM, Homa DM, Akinbami LJ, Moorman JE, Gwynn C, and Redd SC. 2002. Surveillance for asthma--United States, 1980–1999. MMWR Surveill Summ 51: 1–13. [PubMed] [Google Scholar]
- 5.Djukanovic R, Wilson JW, Britten KM, Wilson SJ, Walls AF, Roche WR, Howarth PH, and Holgate ST. 1992. Effect of an inhaled corticosteroid on airway inflammation and symptoms in asthma. The American review of respiratory disease 145: 669–674. [DOI] [PubMed] [Google Scholar]
- 6.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, and Donaldson DD. 1998. Interleukin-13: central mediator of allergic asthma. Science 282: 2258–2261. [DOI] [PubMed] [Google Scholar]
- 7.Saatian B, Rezaee F, Desando S, Emo J, Chapman T, Knowlden S, and Georas SN. 2013. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 1: e24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramirez-Icaza G, Mohammed KA, Nasreen N, Van Horn RD, Hardwick JA, Sanders KL, Tian J, Ramirez-Icaza C, Johnson MT, and Antony VB. 2004. Th2 cytokines IL-4 and IL-13 downregulate paxillin expression in bronchial airway epithelial cells. J Clin Immunol 24: 426–434. [DOI] [PubMed] [Google Scholar]
- 9.Lachowicz-Scroggins ME, Boushey HA, Finkbeiner WE, and Widdicombe JH. 2010. Interleukin-13-induced mucous metaplasia increases susceptibility of human airway epithelium to rhinovirus infection. Am J Respir Cell Mol Biol 43: 652–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moriwaki A, Matsumoto K, Matsunaga Y, Fukuyama S, Matsumoto T, Kan-o K, Noda N, Asai Y, Nakanishi Y, and Inoue H. 2011. IL-13 suppresses double-stranded RNA-induced IFN-lambda production in lung cells. Biochemical and biophysical research communications 404: 922–927. [DOI] [PubMed] [Google Scholar]
- 11.Prieto J, Lensmar C, Roquet A, van der Ploeg I, Gigliotti D, Eklund A, and Grunewald J. 2000. Increased interleukin-13 mRNA expression in bronchoalveolar lavage cells of atopic patients with mild asthma after repeated low-dose allergen provocations. Respiratory medicine 94: 806–814. [DOI] [PubMed] [Google Scholar]
- 12.Howard TD, Koppelman GH, Xu J, Zheng SL, Postma DS, Meyers DA, and Bleecker ER. 2002. Gene-gene interaction in asthma: IL4RA and IL13 in a Dutch population with asthma. American journal of human genetics 70: 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chibana K, Trudeau JB, Mustovich AT, Hu H, Zhao J, Balzar S, Chu HW, and Wenzel SE. 2008. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology 38: 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingram JL, Huggins MJ, Church TD, Li Y, Francisco DC, Degan S, Firszt R, Beaver DM, Lugogo NL, Wang Y, Sunday ME, Noble PW, and Kraft M. 2011. Airway fibroblasts in asthma manifest an invasive phenotype. Am J Respir Crit Care Med 183: 1625–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsukura S, Stellato C, Georas SN, Casolaro V, Plitt JR, Miura K, Kurosawa S, Schindler U, and Schleimer RP. 2001. Interleukin-13 upregulates eotaxin expression in airway epithelial cells by a STAT6-dependent mechanism. Am J Respir Cell Mol Biol 24: 755–761. [DOI] [PubMed] [Google Scholar]
- 16.Richter A, Puddicombe SM, Lordan JL, Bucchieri F, Wilson SJ, Djukanovic R, Dent G, Holgate ST, and Davies DE. 2001. The contribution of interleukin (IL)-4 and IL-13 to the epithelial-mesenchymal trophic unit in asthma. Am J Respir Cell Mol Biol 25: 385–391. [DOI] [PubMed] [Google Scholar]
- 17.Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, and Izuhara K. 2006. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. The Journal of allergy and clinical immunology 118: 98–104. [DOI] [PubMed] [Google Scholar]
- 18.Van der Pouw Kraan TC, Van der Zee JS, Boeije LC, De Groot ER, Stapel SO, and Aarden LA. 1998. The role of IL-13 in IgE synthesis by allergic asthma patients. Clinical and experimental immunology 111: 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auten RL, Watkins RH, Shapiro DL, and Horowitz S. 1990. Surfactant apoprotein A (SP-A) is synthesized in airway cells. American journal of respiratory cell and molecular biology 3: 491–496. [DOI] [PubMed] [Google Scholar]
- 20.Goss KL, Kumar AR, and Snyder JM. 1998. SP-A2 gene expression in human fetal lung airways. American journal of respiratory cell and molecular biology 19: 613–621. [DOI] [PubMed] [Google Scholar]
- 21.Piboonpocanun S, Chiba H, Mitsuzawa H, Martin W, Murphy RC, Harbeck RJ, and Voelker DR. 2005. Surfactant protein A binds Mycoplasma pneumoniae with high affinity and attenuates its growth by recognition of disaturated phosphatidylglycerols. J Biol Chem 280: 9–17. [DOI] [PubMed] [Google Scholar]
- 22.Kannan TR, Provenzano D, Wright JR, and Baseman JB. 2005. Identification and characterization of human surfactant protein A binding protein of Mycoplasma pneumoniae. Infect Immun 73: 2828–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ledford JG, Mukherjee S, Kislan MM, Nugent JL, Hollingsworth JW, and Wright JR. 2012. Surfactant protein-A suppresses eosinophil-mediated killing of Mycoplasma pneumoniae in allergic lungs. PLoS One 7: e32436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Voelker DR, Lugogo NL, Wang G, Floros J, Ingram JL, Chu HW, Church TD, Kandasamy P, Fertel D, Wright JR, and Kraft M. 2011. Surfactant protein A is defective in abrogating inflammation in asthma. Am J Physiol Lung Cell Mol Physiol 301: L598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lugogo N, Francisco D, Addison KJ, Manne A, Pederson W, Ingram JL, Green CL, Suratt BT, Lee JJ, Sunday ME, Kraft M, and Ledford JG. 2018. Obese asthmatic patients have decreased surfactant protein A levels: Mechanisms and implications. J Allergy Clin Immunol 141: 918–926 e913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, Wert SE, Stripp BR, Morris RE, Glasser SW, Bachurski CJ, Iwamoto HS, and Whitsett JA. 1996. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci U S A 93: 9594–9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollingsworth JW, Theriot BS, Li Z, Lawson BL, Sunday M, Schwartz DA, and Walker JK. 2010. Both hematopoietic-derived and non-hematopoietic-derived {beta}-arrestin-2 regulates murine allergic airway disease. Am J Respir Cell Mol Biol 43: 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong SS, Choi JH, Lee SY, Park YH, Park KY, Lee JY, Kim J, Gajulapati V, Goo JI, Singh S, Lee K, Kim YK, Im SH, Ahn SH, Rose-John S, Heo TH, and Choi Y. 2015. A Novel Small-Molecule Inhibitor Targeting the IL-6 Receptor beta Subunit, Glycoprotein 130. J Immunol 195: 237–245. [DOI] [PubMed] [Google Scholar]
- 29.Ledford JG, Voelker DR, Addison KJ, Wang Y, Nikam VS, Degan S, Kandasamy P, Tanyaratsrisakul S, Fischer BM, Kraft M, and Hollingsworth JW. 2015. Genetic variation in SP-A2 leads to differential binding to Mycoplasma pneumoniae membranes and regulation of host responses. J Immunol 194: 6123–6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.https://ginasthma.org. accessed Jan 2020 GINA website.
- 31.Kraft M, Adler KB, Ingram JL, Crews AL, Atkinson TP, Cairns CB, Krause DC, and Chu HW. 2008. Mycoplasma pneumoniae induces airway epithelial cell expression of MUC5AC in asthma. Eur Respir J 31: 43–46. [DOI] [PubMed] [Google Scholar]
- 32.McIntosh JC, Mervin-Blake S, Conner E, and Wright JR. 1996. Surfactant protein A protects growing cells and reduces TNF-alpha activity from LPS-stimulated macrophages. Am J Physiol 271: L310–319. [DOI] [PubMed] [Google Scholar]
- 33.Pernis AB, and Rothman PB. 2002. JAK-STAT signaling in asthma. J Clin Invest 109: 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vale K 2016. Targeting the JAK-STAT pathway in the treatment of ‘Th2-high’ severe asthma. Future Med Chem 8: 405–419. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, and Stark GR. 2013. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A 110: 16975–16980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simeone-Penney MC, Severgnini M, Tu P, Homer RJ, Mariani TJ, Cohn L, and Simon AR. 2007. Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J Immunol 178: 6191–6199. [DOI] [PubMed] [Google Scholar]
- 37.Gavino AC, Nahmod K, Bharadwaj U, Makedonas G, and Tweardy DJ. 2016. STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 71: 1684–1692. [DOI] [PubMed] [Google Scholar]
- 38.Xu B, Bhattacharjee A, Roy B, Xu HM, Anthony D, Frank DA, Feldman GM, and Cathcart MK. 2003. Interleukin-13 induction of 15-lipoxygenase gene expression requires p38 mitogen-activated protein kinase-mediated serine 727 phosphorylation of Stat1 and Stat3. Mol Cell Biol 23: 3918–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russell MA, Cooper AC, Dhayal S, and Morgan NG. 2013. Differential effects of interleukin-13 and interleukin-6 on Jak/STAT signaling and cell viability in pancreatic beta-cells. Islets 5: 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, and Cathcart MK. 2013. IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med 54: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marini M, Vittori E, Hollemborg J, and Mattoli S. 1992. Expression of the potent inflammatory cytokines, granulocyte-macrophage-colony-stimulating factor and interleukin-6 and interleukin-8, in bronchial epithelial cells of patients with asthma. J Allergy Clin Immunol 89: 1001–1009. [DOI] [PubMed] [Google Scholar]
- 42.Wright JR 2005. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol 5: 58–68. [DOI] [PubMed] [Google Scholar]
- 43.van de Graaf EA, Jansen HM, Lutter R, Alberts C, Kobesen J, de Vries IJ, and Out TA. 1992. Surfactant protein A in bronchoalveolar lavage fluid. J Lab Clin Med 120: 252–263. [PubMed] [Google Scholar]
- 44.Cheng G, Ueda T, Numao T, Kuroki Y, Nakajima H, Fukushima Y, Motojima S, and Fukuda T. 2000. Increased levels of surfactant protein A and D in bronchoalveolar lavage fluids in patients with bronchial asthma. Eur Respir J 16: 831–835. [DOI] [PubMed] [Google Scholar]
- 45.Erpenbeck VJ, Schmidt R, Gunther A, Krug N, and Hohlfeld JM. 2006. Surfactant protein levels in bronchoalveolar lavage after segmental allergen challenge in patients with asthma. Allergy 61: 598–604. [DOI] [PubMed] [Google Scholar]
- 46.Wenzel S 2012. Severe asthma: from characteristics to phenotypes to endotypes. Clin Exp Allergy 42: 650–658. [DOI] [PubMed] [Google Scholar]
- 47.Pastva AM, Mukherjee S, Giamberardino C, Hsia B, Lo B, Sempowski GD, and Wright JR. 2011. Lung effector memory and activated CD4+ T cells display enhanced proliferation in surfactant protein A-deficient mice during allergen-mediated inflammation. J Immunol 186: 2842–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dy ABC, Arif MZ, Addison KJ, Que LG, Boitano S, Kraft M, and Ledford JG. 2019. Genetic Variation in Surfactant Protein-A2 Delays Resolution of Eosinophilia in Asthma. J Immunol 203: 1122–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Litonjua AA, Tantisira KG, Lake S, Lazarus R, Richter BG, Gabriel S, Silverman ES, and Weiss ST. 2005. Polymorphisms in signal transducer and activator of transcription 3 and lung function in asthma. Respir Res 6: 52: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fulkerson PC, Zimmermann N, Hassman LM, Finkelman FD, and Rothenberg ME. 2004. Pulmonary chemokine expression is coordinately regulated by STAT1, STAT6, and IFN-gamma. J Immunol 173: 7565–7574. [DOI] [PubMed] [Google Scholar]
- 51.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, Tucci A, Hughes LD, Kong Y, Weinstein JS, Licona-Limon P, Schmid ET, Pelorosso F, Gagliani N, Craft JE, Flavell RA, Ghosh S, and Rothlin CV. 2017. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356: 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munitz A, Brandt EB, Mingler M, Finkelman FD, and Rothenberg ME. 2008. Distinct roles for IL-13 and IL-4 via IL-13 receptor alpha1 and the type II IL-4 receptor in asthma pathogenesis. Proc Natl Acad Sci U S A 105: 7240–7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dy ABC, Tanyaratsrisakul S, Voelker DR, and Ledford JG. 2018. The Emerging Roles of Surfactant Protein-A in Asthma. J Clin Cell Immunol 9: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]