

Abstract
Background and Aims
Bristol rock cress is among the few plant species in the British Isles considered to have a Mediterranean–montane element. Spatiotemporal patterns of colonization of the British Isles since the last interglacial and after the Last Glacial Maximum (LGM) from mainland Europe are underexplored and have not yet included such floristic elements. Here we shed light on the evolutionary history of a relic and outpost metapopulation of Bristol rock cress in the south-western UK.
Methods
Amplified fragment length polymorphisms (AFLPs) were used to identify distinct gene pools. Plastome assembly and respective phylogenetic analysis revealed the temporal context. Herbarium material was largely used to exemplify the value of collections to obtain a representative sampling covering the entire distribution range.
Key Results
The AFLPs recognized two distinct gene pools, with the Iberian Peninsula as the primary centre of genetic diversity and the origin of lineages expanding before and after the LGM towards mountain areas in France and Switzerland. No present-day lineages are older than 51 ky, which is in sharp contrast to the species stem group age of nearly 2 My, indicating severe extinction and bottlenecks throughout the Pleistocene. The British Isles were colonized after the LGM and feature high genetic diversity.
Conclusions
The short-lived perennial herb Arabis scabra, which is restricted to limestone, has expanded its distribution range after the LGM, following corridors within an open landscape, and may have reached the British Isles via the desiccated Celtic Sea at about 16 kya. This study may shed light on the origin of other rare and peculiar species co-occurring in limestone regions in the south-western British Isles.
Keywords: AFLPs, Arabis scabra, Brassicaceae, British Isles, evolutionary history, internal transcribed spacer 1 and 2, plastome sequences, Quaternary
INTRODUCTION
Climatic fluctuation during the Quaternary with its Pleistocene glaciation and deglaciation cycles drastically changed environmental conditions for plants (e.g. Willis and MacDonald, 2011; Allen et al., 2012), affecting sea levels (Richardson et al., 2014), opening and closing land bridges and corridors, and causing changes in landscape relief (Betzin et al., 2016; Santiso et al., 2016), sediments and soil types (e.g. Starnberger et al., 2013; Holtvoeth et al., 2017). As a result, species either had to migrate or adapt to these changing conditions, or both. Migration, survival and extinction gave rise to major southern glacial refugia in Europe, with the peninsulas of Iberia and Italy and the Balkans having been identified as the most important refugia for our present-day floras (Hewitt, 2000). Several studies have also recognized refugia north of these three regions including cryptic glacial refuge areas (e.g. Provan and Bennett, 2008; Tzedakis et al., 2013) and those characterized by prominent land features, e.g. ‘mountain islands’ (nunatak hypothesis, Brochmann et al., 2003) and coastal lines (Kadereit and Westberg, 2007). Moreover, the structure of refuge areas was often more complex [e.g. refugia within refugia in the Iberian Peninsula (Abellán and Svenning, 2014)] than previously expected and the assumption of having only southern refugia does not reflect adequately the complexity of the processes that shaped current patterns of biodiversity (Feliner, 2011).
In most phylogeographical studies, species distribution patterns and the occurrence of refuge areas are explained by the influence of the last three glaciation and deglaciation cycles, spanning almost 500 000 years (e.g. Hohmann and Koch, 2017), with the most prominent effect on present-day biota caused by the last glacial period (Weichselian in Central Europe or Würm glacial in the alpine region) and recolonization after the Last Glacial Maximum (LGM, 20 000–26 000 years ago). However, with mounting evidence of cryptic refugia and of potential periglacial survival there is an increasing demand for time-calibrated studies to test for alternative and/or additional recolonization scenarios.
Time-calibrated spatial scenarios can be developed in a comparative way by combining evidence from different datasets. This can be achieved, for example, by comparing dated macrofossil and pollen data with present-day species distribution data (Brewer et al., 2017), using past climate reconstructions in combination with ecological niche modelling (Svenning et al., 2011), or by extrapolation from known past spatio-temporal scenarios for ecologically similar species or ecologically linked species (Avise et al., 2016). A complementary method is to use genetic data to unravel spatial distribution patterns of genetic variation that results from demographic processes, and use the same dataset for temporal inferences (e.g. Durvasula et al., 2017; Koch et al., 2017a). However, resolving short periods of time such as glacial and postglacial scenarios by molecular marker systems requires a large number of loci, at best covering the entire nuclear genome randomly, using a technique such as amplified fragment length polymorphism (AFLP; Vos et al., 1995) which is an anonymous genome-wide DNA fingerprinting technique that can be used also in the context of unknown genomes. Furthermore, for estimating divergence times mutational models need to be available for the analysed data type, as is the case for DNA sequencing data. Therefore, in this study we use a combination of AFLP data for assessing gene-pool structure with sequencing of whole-plastid genomes by a genome-skimming approach (Straub et al., 2012) to estimate divergence times at high resolution.
This study focuses on A. scabra (Bristol rock cress), a short-lived perennial herb from the Brassicaceae family, which belongs to the so-called core-Arabis clade (Karl and Koch, 2013, 2014; Koch and Grosser, 2017). Despite detailed analysis of the systematics and evolutionary history of the genus Arabis, its taxonomic treatment still includes numerous paraphyletic groups (Koch et al., 2010; Karl and Koch, 2013), mainly because (1) there is hardly any morphological character or character combination exclusively identifying the various clades within Arabis (Koch et al., 2018), and (2) determination of related species within groups of taxa is often hindered, because characters are either missing (e.g. no fruits are available) or occur in rare combinations in the frequently formed hybrid individuals or populations (Koch and Grosser, 2017). This study relies almost exclusively on herbarium material in order to cover the entire distribution range of A. scabra and therefore the correct determination of voucher material is of utmost importance. The closest relatives of A. scabra are A. ciliata and A. serpyllifolia (Karl and Koch, 2014), but phylogenies derived from few nuclear and plastid DNA loci showed minor incongruencies, so relationships of A. scabra to other species and possible hybridization and introgression patterns are still unclear (Karl and Koch, 2014). However, cytogenetic analysis clearly showed that A. scabra is a diploid with 2n = 16 chromosomes (Pring, 1961; Koch et al., 1999), indicating that this taxon is not the result of a complex reticulate evolutionary history, as often found in other Arabis species assemblages.
Arabis scabra is a Mediterranean–montane element (Jalas and Suominen, 1994) growing in shallow soils, on scree and on rock edges within open woodland on carboniferous limestone. The Mediterranean distribution encompasses northern and north-eastern Iberian mountain ranges, including the Pyrenees. In France the species has a continuous distribution range throughout the south-eastern mountain ranges towards the most south-eastern edge of Switzerland (Jalas and Suominen, 1994). In north-western Europe A. scabra is known only from the Bristol area in the UK, and has been documented from the Avon Gorge since 1686 (Pring, 1961). Due to its particular disjunct distribution pattern, A. scabra is an interesting subject for studying more recent (postglacial) evolutionary histories. Arabis scabra belongs to the Lusitanian flora and floristically has been considered as an Erysimum duriaei element. This group of taxa encompasses ~140 European species, which are distributed primarily in the mountains of northern Spain, the Pyrenees, south-eastern France and the south-western edge of the Alps (Finnie et al., 2007). They are mostly non-woody, cold-adapted or cold-tolerant perennials and the vast majority are European endemics (86 %). Furthermore, they include three representatives with extreme cases of outlying populations found on the British Isles, namely A. scabra, Meconopsis cambrica and Saxifraga hirsuta. Past studies revealed that M. cambrica survived periglacially and was introduced to the British Isles before the LGM (Valtuena et al., 2012), while other species from the Lusitanian flora, such as Pinguicula grandiflora, Saxifraga spathularis, Daboecia cantabrica and Euphorbia hyberna, colonized the British Isles postglacially (Beatty and Provan, 2013, 2014; Beatty et al., 2015). For the other species sharing the present-day distribution pattern with A. scabra it is unclear whether there are parallel spatio-temporal patterns for colonization of the British Isles.
In this study we aim to define the genetic structure underlying the distribution of A. scabra and unravel its evolutionary history by applying a combination of AFLPs, sequencing of two loci from the plastid genome and the nuclear-encoded rDNA spanning ITS1 to 5.8 S rDNA-ITS2 for the entire dataset, and whole-plastome sequencing for a subset of 22 accessions, which were selected according to the results of AFLP analysis and geographical distribution. Furthermore, diagnostic SNPs based on the whole-plastome sequencing were analysed for the entire set of accessions included. Arabis scabra was sampled across its entire distribution range, mainly taking advantage of herbarium material to further highlight herbaria as primary biorepositories and sources of research material (Koch et al., 2017b).
Based on our combined datasets, we formulate a postglacial ‘out of Iberia’ hypothesis and show that populations from the British Isles originated from postglacial range expansion. Additionally, there is also evidence for multiple inter- and postglacial range expansion from the Iberian Peninsula as the primary centre of diversity towards the south-western Alps.
MATERIALS AND METHODS
Plant material, taxon sampling and experimental strategy
An initial screening of documented and sampled populations of Arabis scabra kept in various European herbaria was performed using the Global Biodiversity Information Portal (GBIF) (https://www.gbif.org/). In order to select accessions for molecular analysis we defined some criteria that would allow us to work only with high-quality and reliable material. (1) From all retained data points (1055 in total) we discarded all accessions with unknown or imprecise source information for our molecular analysis. (2) Remaining accessions were checked against the floristic literature to cover the entire distribution range of the species. (3) We further limited our sample collection to a geographically representative set focusing on accessions with well-preserved leaf material, thereby minimizing subsequent experimental failures due to low sample and DNA quality. All accessions fulfilling the three criteria defined above were subjected to DNA sequencing of markers from the nuclear genome (ITS; internal transcribed spacers 1 and 2 of ribosomal DNA) and plastid genome (trnL intron and trnLF intergenic spacer) for phylogenetic analyses in order to prove species identity and to identify potential events of hybridization and reticulate evolution. Only accessions that yielded high-quality PCR fragments and reliable DNA sequence information (86 individuals from 80 accessions) were used for AFLP analysis in order to characterize geographically structured gene pools. The final selection of accessions included nine individuals that were indicated and labelled on the vouchers as A. scabra but where some doubt remained about their identity. Nevertheless, these samples were included in the initial analysis.
A subset of 22 samples was selected based on the results derived from sequencing of plastidic and nrDNA markers as well as on AFLP data and geographical origin. These samples were subjected to a genome-skimming approach to assemble entire plastid genomes (Hohmann et al., 2015) for temporal high-resolution phylogenetic analysis. Finally, diagnostic SNPs were selected based on whole-plastome sequencing data, which were then sequenced for the complete set of A. scabra accessions. Accession details are provided in Supplementary Data Table S1. DNA sequence data from the entire genus Arabis have been published earlier (Karl and Koch, 2013, 2014; Koch and Grosser, 2017), so that no further outgroups were needed.
DNA extraction, amplification and sequencing of plastidic and nrDNA marker sequences
Total DNA was extracted using the Invisorb Spin Plant Mini Kit (Stratec Biomedical, Birkenfeld, Germany). PCR amplification of the phylogenetic and barcode markers used (nuclear ITS, plastid trnL intron and trnL-trnF intergenic spacer; hereafter named combined as the trnLF region) were performed in a volume of 25 μL, using 10 μm of each primer, a total of 2.0 mm MgCl2 and 0.5 U of Mango-Taq polymerase (Bioline, Luckenwalde, Germany). The primers used for ITS amplification were originally designed by White et al. (1990) with some modifications made (18F, 5′-GAA AGG AGA AGT GCT AAC AAG A-3′; 25R, 5′-GGG TAA TCC CGC CTG ACC TGG-3′). For some DNA fragments PCR quality was further increased to amplify ITS1 and ITS2 separately using internal primers ITS2a (5′-GCT GCG TTC TTC ATC GAT GC-3′) and ITS3 (5′-GCA TCG ATG AAG AAC GTA GC-3′), respectively. Amplification of the trnL intron and the trnL-trnF intergenic spacer was performed using primers c, d and e from Taberlet et al. (1991) (c, 5′-CGA AAT CGG TAG ACG CTA CG-3′; d, 5′-GGG GAT AGA GGG ACT TGA AC-3′; and e, 5′-GGT TCA AGT CCC TCT ATC ATC CC-3′) and a primer designed by Dobeš et al. (2004) (5′-GAT TTT CAG TCC TCT GCT CTA C-3′). Primers were extended by the M13 sequence for subsequent sequencing using M13 universal sequencing primers. The amplifications were run on a PTC 200 Peltier Thermal Cycler (MJ Research, Waltham, MA, USA) under the following conditions for ITS: 3 min of initial denaturation at 95 °C; 30 cycles of amplification for 30 s at 95 °C, 30 s at 44 °C and 1 min at 72 °C; and 5 min of final elongation at 72 °C. For the plastid loci, annealing temperature was increased to 50 °C. PCR success was checked with electrophoresis in a 1 % agarose gel in TAE buffer. PCR product clean-up was executed using the Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, USA). Custom Sanger sequencing was performed by GATC Biotech (Konstanz, Germany). The electropherograms were checked and trimmed using the program SeqMan DNASTAR Lasergene software package (DNASTAR, Madison, WI, USA).
Sequence alignment and phylogenetic analysis of plastidic and nrDNA marker sequences
The ITS DNA sequence data generated here were added to datasets published earlier and focusing on phylogenetic relationships among Eurasian Arabis hirsuta and related taxa (Karl and Koch, 2014). This dataset contained one accession of A. scabra, and also one representative of its closest relatives, Arabis ciliata and Arabis serpyllifolia. The final alignment had a length of 635 bp and contained 69 different sequences from 140 accessions and is available with the online material, which also includes GenBank accession codes for already published data (Supplementary Data Table S2). Plastid DNA sequences from the trnLF region were combined into one single concatenated alignment of 775 bp (alignment lengths of the trnL intron and trnL-trnF intergenic spacer were 327 and 448 bp, respectively). The NEXUS input file is provided in Supplementary Data Table S3 and comprises 70 different sequences from 141 accessions.
In our phylogenetic analyses based on ITS as well as trnLF data, Pseudoturritis turrita was used as an appropriate outgroup (Karl and Koch, 2013). We performed maximum likelihood (ML) as well as Bayesian inference (BI) analyses. The best-fitting nucleotide substitution model (SYM + I + G model for ITS and TPM1uf + G model for trnLF) was selected using MrModeltest 2.3 (Nylander, 2004), according to the Akaike information criterion (AIC). The ML analyses were performed in RAxML (Stamatakis, 2014) implemented in raxmlGUI (Silvestro and Michalak, 2012), with the search strategy set to rapid bootstrapping. Clade support was evaluated with a bootstrap of 1000 replicates. In the Bayesian analyses using MrBayes v. 3.2.6 (Ronquist et al., 2012), four simultaneous runs with four chains each were run for 50 million generations, sampling every 1000 trees. The first 25 % of these trees were discarded as burn-in when computing the consensus tree (50 % majority rule). For efficient swapping of the chains, the temperature of the heated chain was set to 0.005. Sufficient mixing of the chains was considered to be reached when the average standard deviation of split frequencies was below 0.01. Stationarity of the Markov chains was also confirmed in Tracer (http://tree.bio.ed.ac.uk/software/tracer/), and reliable effective sample size values (>200) were ensured.
Generation of AFLP data and genetic analysis
We successfully generated AFLP profiles for 86 individuals from 80 accessions of A. scabra and the various non-A. scabra (as an additional control group) following Vos et al. (1995) and Meudt and Clarke (2007). Genotyping error assessment was done as described by Bonin et al. (2004) with eight replicates included. Digestion of diluted genomic DNA (~30 ng) and ligation of dsDNA adaptors was performed simultaneously using endonucleases EcoRI and MseI, and T4 DNA Ligase (New England Biolabs, Frankfurt am Main, Germany). PCR reactions were carried out with AmpliTaq DNA Polymerase in AmpliTaq buffer II for PCR step 1 (pre-selective PCR) and with AmpliTaq Gold and AmpliTaq Gold Buffer (all from Life Technologies, Darmstadt, Germany) for PCR step 2 (selective PCR). Oligonucleotides and fluorescent-labelled oligonucleotides (with fluorophores FAM, HEX and ATTO550) were obtained from biomers.net (Ulm, Germany). A set of six combinations of selective primers was chosen for the study: (1) EcoRI + ACA (FAM)/MseI + CAT; (2) EcoRI + AAC (HEX)/MseI + CAT; (3) EcoRI + AGC (ATTO550)/MseI + CAT; (4) EcoRI + ACA (FAM)/MseI + CTC; (5) EcoRI + AAC (HEX)/MseI + CTC; and (6) EcoRI + AGC (ATTO550)/MseI + CTC. Thermocycling conditions for pre-selective PCR were as follows: 94 °C, 4 min; 12 × [94 °C, 30 s; 65–72 °C, 30 s (ramping of 0.7 °C per cycle); 72 °C, 1:30 min]; 20 × [94 °C, 30 s; 56 °C, 1 min, 72 °C, 1:30 min]; 72 °C, 1 min; final hold at 8 °C. Selective PCR was as follows: 72 °C, 2 min; 20 × [94 °C, 20 s; 56 °C, 30 s; 72 °C, 2 min]; hold at 72 °C for 60 min. Before fragment detection, amplicons were pooled, and an internal sizing standard (GS 500) was included to maintain consistency of band scoring between capillaries, since fragment migration occurs at different rates in each capillary. Raw data were corrected for optical interactions among the four detection channels (‘bleed-though’ or ‘pull-up’) by the service provider (GATC Biotech, Konstanz, Germany). Size calling and manual genotype calling were performed with GeneMarker 1.95 (SoftGenetics, State College, PA, USA) for fragments with a length of 65–550 bp. Scored data were exported as a binary data table for further analysis.
Because of the low levels of genetic polymorphism expected considering missing DNA sequence variation among the sequenced loci (ITS and trnL-trnLF) in A. scabra, the AFLP 0/1 matrix was critically inspected for putative genotyping errors: (1) general genotyping error, and (2) influence of quality of the herbarium material on banding profiles (number and length of fragments). For (1), the general genotyping error was estimated following the procedure described by Bonin et al. (2004) with eight replicates from A. scabra included. For a more general data inspection (2), principal component analysis (PCA) and eigendecomposition of the resulting association matrix were performed with the eigen() function in R (R Core Team, 2014) and total number of fragments and length of fragments were superimposed accordingly as described in Koch et al. (2017b) for quality control of the data. Finally, samples were omitted from the AFLP data set if they showed unusual fragment profiles as visualized by PCA (Supplementary Data Fig. S1).
Genetic assignment of accessions was inferred under the no admixture model implemented in STRUCTURE 2.3.4. (Pritchard et al., 2000) using the uncorrelated allele frequencies and the recessive alleles options (Falush et al., 2007). Burn-in period comprised 250 000 Markov chain Monte Carlo (MCMC) steps and data collection was carried out over another 250 000 steps. Ten replicate simulations were run for each value of K (the number of ancestral clusters assumed by STRUCTURE) ranging from 1 to 20. Individual ancestry was averaged across all replicate simulations for each value of K using CLUMPP Version 1.1.2 (Jakobsson and Rosenberg, 2007). A formal determination of the optimal value of K was carried out using Evanno’s mean ΔK (Evanno et al., 2005) and symmetrical similarity coefficients (Ehrich et al., 2007). A distance-based unrooted NeighborNet graph was generated with SplitsTree4 version 4.14.4 (Huson and Bryant, 2006) using Jaccard’s distance (Jaccard, 1901), which calculates the dissimilarity between asymmetrical binary variables, implemented in the vegan package from the software R Studio V0.0.98.501 (R Core Team, 2014). Estimation of molecular diversity indices (gene diversity, Tajima’s D, Watterson’s estimator θ S and θ π) were performed with Arlequin v. 3.5.2.2 (Excoffier and Lischer, 2010). Samples were also grouped according to major regions of the UK, Spain and France/Switzerland (Fig. 1). The significance of the D statistic was tested using the program Arlequin v. 3.5.2.2 (Excoffier and Lischer, 2010) by generating random samples under the hypothesis of selective neutrality and population equilibrium, using a coalescent simulation algorithm adapted from Hudson (1990).
Fig. 1.

(A) Distribution of sampled Arabis scabra accessions and their codes (combined sets of accessions are colour-coded and numbered) in Central Europe. The distribution of Arabis scabra according to Atlas florae Europaeae (Kurtto et al., 2013) is also indicated. (B) Bar plots of the respective assignment of AFLP genotypes to the two identified genetic clusters grouped by accession sets 1–10 (see also Fig. 4).
To further investigate the correlation of genetic dissimilarity (Jaccard distance) with geographical distance, we generated a Mantel correlogram (Legendre and Legendre, 2012) with distance classes of 50 km using the mantel.correlog function from the vegan package (Oksanen et al., 2017). The significance of Mantel’s r was tested by generating 1000 random permutations. Bonferroni’s correction was applied to P values for multiple testing.
Assembly of plastid genomes, phylogenetic network and tree analysis and estimation of divergence times
According to the results of AFLP analysis we selected a set of 22 accessions representing (1) the different gene pools and (2) the different regions [final coverage: UK, orange and blue nuclear gene pool; France, orange and blue gene pool; Spain, orange gene pool from the northern (ESPA-D) and the southern (ESPE-F) distribution area] to examine plastome DNA sequence variation at high temporal resolution (Fig. 1). We used the same DNA as for AFLP analysis. Libraries were sequenced at the Deep Sequencing Core Facility, University of Heidelberg, and were prepared from total genomic DNA with the Illumina TruSeq Kit (Illumina, San Diego, CA, USA) in paired-end mode with an insert size of 200–400 bp. Read length was 150 bp. Samples were barcoded for multiplexing and ~13 million reads were generated per sample. Sequencing was conducted on an Illumina HiSeq4000 machine. Assembly of plastid genomes generally followed the procedure described in Hohmann et al. (2015) and Novikova et al. (2016). Quality of the obtained sequencing data was assessed by using the program fastqc (Andrews, 2010) and sequences were trimmed for adapter contamination and quality using the program Trimmomatic (Bolger et al., 2014). Chloroplast sequences were obtained by a reference-based mapping approach. For this, reads were mapped on the plastid genome of Arabis alpina (GenBank accession number HF934132) using the program bwa (Li and Durbin, 2009). Duplicated sequences were removed using the SAMtools tool rmdup (Li et al., 2009). Mapping quality was improved using the gatk 3.8 tools RealignerTargetCreator and IndelRealigner (DePristo et al., 2011). In order to obtain only chloroplast sequences that are backed up by a sufficient sequencing depth, the gatk 3.8 tool CallableLoci (Van der Auvera et al., 2013) was used to indicate regions that have a sequencing depth >10 and a mapping quality >30. Based on this information callable regions were extracted. Finally, the new chloroplast sequences were generated using the gatk 3.8 tools UnifiedGenotyper for generating the genotypes and the FastaAlternateReferenceMaker for generating the new sequences in fasta format. Sequences were annotated in Geneious version 7.1.7 (Biomatters, Auckland, New Zealand) using Arabidopsis thaliana as reference (GenBank accession AP000423) and using the default alignment parameters, and then transferring the annotations. Sequences were then aligned using the program MAFFT version 7.017 (Katoh and Standley, 2013) and the resulting alignments were adjusted by hand using the program PhyDE (Müller et al., 2005). Entire plastomes of A. scabra were aligned to outgroup plastomes from Arabis stelleri (KY126841), Arabis hirsuta (NC009268), Arabis alpina (HF934132) and Draba nemorosa (NC009272). Arabis alpina and Draba nemorosa have both been shown to be in a sister group relationship to the so called core Arabis and were therefore chosen as an appropriate outgroup (Karl and Koch, 2013; Kiefer et al., 2014). Furthermore, Arabis hirsuta and Arabis stelleri were included in the analysis as additional taxa representing the core Arabis clade (Karl and Koch, 2013). In a first step the dataset of the ingroup (32 SNPs) was examined using the program TCS1.21 (Clement et al., 2000) for reconstructing a statistical parsimony network and identifying identical sequences.
In order to determine the best partition scheme as well as the most suitable molecular evolutionary model for each partition, the entire plastid genome alignment was analysed by PartitionFinder v1.1.1 (Lanfear et al., 2012), where the branchlengths parameter was set to linked. The program was run twice for each alignment, in one case exploring models covered by RAxML-ng (Kozlov et al., 2019) while in the other case suitable models for BEAST analysis (Drummond et al., 2012) were covered. The best partition scheme and associated models were then used as input information along with the annotated alignments for analysis by RAxML-ng (Kozlov et al., 2019) and BEAST (Drummond et al., 2012). In RAxML-ng 1000 bootstrap replicates were generated. BEAUTi was used for generating an input file for BEAST analysis. Models and partitions were set according to the result from the PartitionFinder analysis. The split between Arabis alpina and Draba nemorosa was calibrated according to the dates determined by Hohmann et al. (2015). Clocks and trees were treated as linked. The MCMC chain was run for 1 000 000 000 generations and every 100 000 generations a tree was sampled. In total eight runs were performed. Log files were examined using the program Tracer (Rambaut et al., 2018). The program logcombiner v. 1.7.5. (Drummond et al., 2012) was used to combine log and tree files. The first 25 % of the trees and log information was discarded as burn-in. Log files were then re-examined in Tracer to make sure that effective sample size values were all >200 and that the chosen burn-in was sufficient. In a last step, the program treeannotator v. 1.7.5. (Drummond et al., 2012) was used for plotting the estimated divergence times onto the topology of the best tree recovered. The tree was visualized using the program FigTree (Rambaut, 2018).
Generation and analysis of diagnostic markers defining A. scabra lineages
Network analysis recognized four groups of haplotypes among A. scabra accessions. Accordingly, four regions from the plastome alignment [Supplementary Data Table S4] were selected, which carry sequence variation, and this was used further to assign remaining accessions to these four haplotype groups. Primer combinations (after several rounds of optimization of primer sequence and PCR conditions) and PCR conditions to amplify and sequence these four regions were as follow: BLUE_for, 5′-TACCCAAAACAAACGCGCTA-3′, BLUE_rev, 5′-AACCTGCTAACGAACCGAAC-3′, annealing temperature 58 °C; GREEN_for, 5′-CCTCGTACGGCTCCA GAAAA-3′, GREEN_rev, 5′-GAGTACTTAAGGATTCTCTG TGT-3′, annealing temperature 55 °C; ORANGE_for, 5′-TCTTTCACATGTCTTCTGTCAA-3′, ORANGE_rev, 5′-TCAGTATCCAGGCTCCGTTT-3′, annealing temperature 55 °C; BRISTOL_for, 5′-GTATAGGTTCAAATCCTATTGGACGC-3′, BRISTOL_rev, 5′-TCAAGAACAACTGGAACGTTTTC-3′, annealing temperature 58 °C. All remaining A. scabra accessions were subjected to respective PCR reactions and PCR products were processed and sequenced as described above for ITS and trnLF regions. Subsequently accessions were assigned to lineages based on the sequenced markers.
RESULTS
Taxon identity and phylogenetic inferences based on plastid and nrDNA markers
Nine accessions originally determined as A. scabra did not result in DNA sequence data matching previously published sequences of A. scabra, which was checked with the phylogenetic placement tool in BrassiBase (Koch et al., 2012; Kiefer et al., 2014). All of them were determined to be other Arabis species. The nine non-A. scabra accessions belong to Arabis alpina (accession 32), Arabis collina (accession 007), Arabis juressi (accession 46), Arabis stellulata (accession 60), Arabis ciliata (accessions 25, 65, 74, 76) and Arabis serpyllifolia (accession 44). The number of ~10% of wrongly determined Arabis species in public herbaria was not unexpected, and this major error must be taken in account when utilizing data retrieved from databases such as GBIF or relying on original voucher information.
Analysis of all remaining accessions of A. scabra revealed that all individuals investigated were identical with respect to ITS (Fig. 2), trnL and trnLF (Fig. 3) sequences. Therefore, one representative sequence only was included in all phylogenetic reconstructions. According to our phylogenetic analysis A. scabra is most closely related to A. ciliata and A. serpyllifolia and the Iberian Arabis beirana and Arabis stenocarpa (trnLF data, Fig. 3). ITS data do recognize this monophyletic clade, but phylogenetic analysis also places A. vochinensis and North African Arabis doumetii and Arabis erubescens along an unresolved polytomy (Fig. 2). We therefore introduced the name ‘A. ciliata alliance’ for this group of taxa, which started to evolve ~2.4 mya during the onset of Pleistocene glaciation and deglaciation cycles (Koch et al., 2010). In particular, ITS data indicate a long and separate evolutionary history of A. scabra, and consequently any missing DNA sequence information from ITS, trnL and trnLF was unexpected and can be regarded as first evidence for severe range contractions and genetic bottlenecks during Pleistocene glaciation and deglaciation cycles. In the original dataset from Karl and Koch (2014) one A. scabra sequence from Spain was included carrying few sequence ambiguities, explaining the difference in phylogenetic reconstructions compared with all newly analysed accessions. New sequence information has been submitted to GenBank (Supplementary Data Table S1).
Fig. 2.

Bayesian analysis of the ITS DNA sequence data set. Bayesian posterior support is indicated along the branches. Divergence time estimates (mya) have been redrawn from various phylogenetic analyses (Karl et al., 2012; Karl and Koch, 2013, 2014). New DNA sequences are highlighted in red (Supplementary Data Tables S1 and S2). All Arabis scabra accessions analysed carried identical DNA sequences. *Conflicting phylogenetic signal among Arabis stellulata. #Different phylogenetic position of Arabis serpyllifolia compared with plastid data.
Fig. 3.

Bayesian analysis of the plastid trnLF DNA sequence data set. Bayesian posterior support is indicated along branches. New DNA sequences are highlighted in red (Supplementary Data Tables S1 and S3). All Arabis scabra accessions analysed carried identical trnLF DNA sequences.
AFLP gene pools and genetic analyses
Eighty-six individuals (including the nine non-A. scabra accessions) yielded a highly reproducible AFLP pattern with an error rate of 1.93 %. The 0/1 matrix resulting from the banding pattern consisted of 291 loci, of which 114 were segregating (Supplementary Data Table S5). A PCA approach was used to determine individuals showing unusual variation (Koch et al., 2017b). This analysis indicated five additional accessions from A. scabra (accessions 2.6, 2.7, 2.8, 70, 77; Supplementary Data Fig. S1), which could not be separated from technical artefacts due to low DNA quality. Consequently, these accessions were also omitted from the final AFLP analysis. The remaining 72 accessions of A. scabra (now excluding non-A. scabra and individuals with a low-quality banding pattern) were grouped into ten a priori and geographically defined sets (Fig. 1). Six of these sets were from Spain and comprised 42 accessions in total, two sets were from south-eastern France/Switzerland, comprising 16 accessions, and one set contained ten individuals from the UK. The remaining set (set 9) comprised four accessions geographically intermediate between the Spanish and south-western French sets. Distribution of the number of fragments and their frequencies indicated substantial differentiation within and among sets of accessions with respect to private and rare alleles, which occur in almost any accession and set of accessions (Table 1). Furthermore, comparison of the three major geographically defined sets of accessions placed the UK set at intermediate levels of genetic polymorphism intermediate between Spain and France/Switzerland.
Table 1.
Summary statistics of the AFLP data set. Sets of accessions are numbered 1–10 as shown in Fig. 1
| No. of fragments | |||||
|---|---|---|---|---|---|
| Accession set | N | Fixed | Polymorphic | No. of private fragments | No. of rare* fragments |
| UK (set 10) | 10 | 64 | 42 | 20 | 16 (16) |
| FRA | 20 | 49 | 45 | 10 | 9 (9) |
| Set 7 | 10 | 56 | 26 | 3 | 3 (3) |
| Set 8 | 6 | 60 | 25 | 4 | 5 (5) |
| Set 9 | 4 | 63 | 17 | 0 | 1 (1) |
| ESP | 42 | 51 | 71 | 35 | 33 (16) |
| Set 1 | 4 | 65 | 16 | 2 | 2 (2) |
| Set 2 | 9 | 64 | 23 | 3 | 1 (1) |
| Set 3 | 11 | 61 | 31 | 4 | 4 (4) |
| Set 4 | 7 | 60 | 22 | 5 | 5 (5) |
| Set 5 | 5 | 71 | 4 | 0 | 0 (0) |
| Set 6 | 6 | 68 | 27 | 12 | 4 (4) |
N, number of individuals.
*<10 %, number of unique fragments are given in brackets.
Estimation of population structure based on the AFLP data was done by STRUCTURE analysis. The optimal K was determined by applying two different methods. Calculation of the symmetrical similarity coefficient (SSC) resulted in optimal K = 2 (SSC > 0.99), while Evanno’s method of mean ΔK showed K = 3 to be the most suitable number of clusters. However, a pattern of two clusters did not conflict with the K = 3 result (Fig. 4, Supplementary Data Fig. S2).
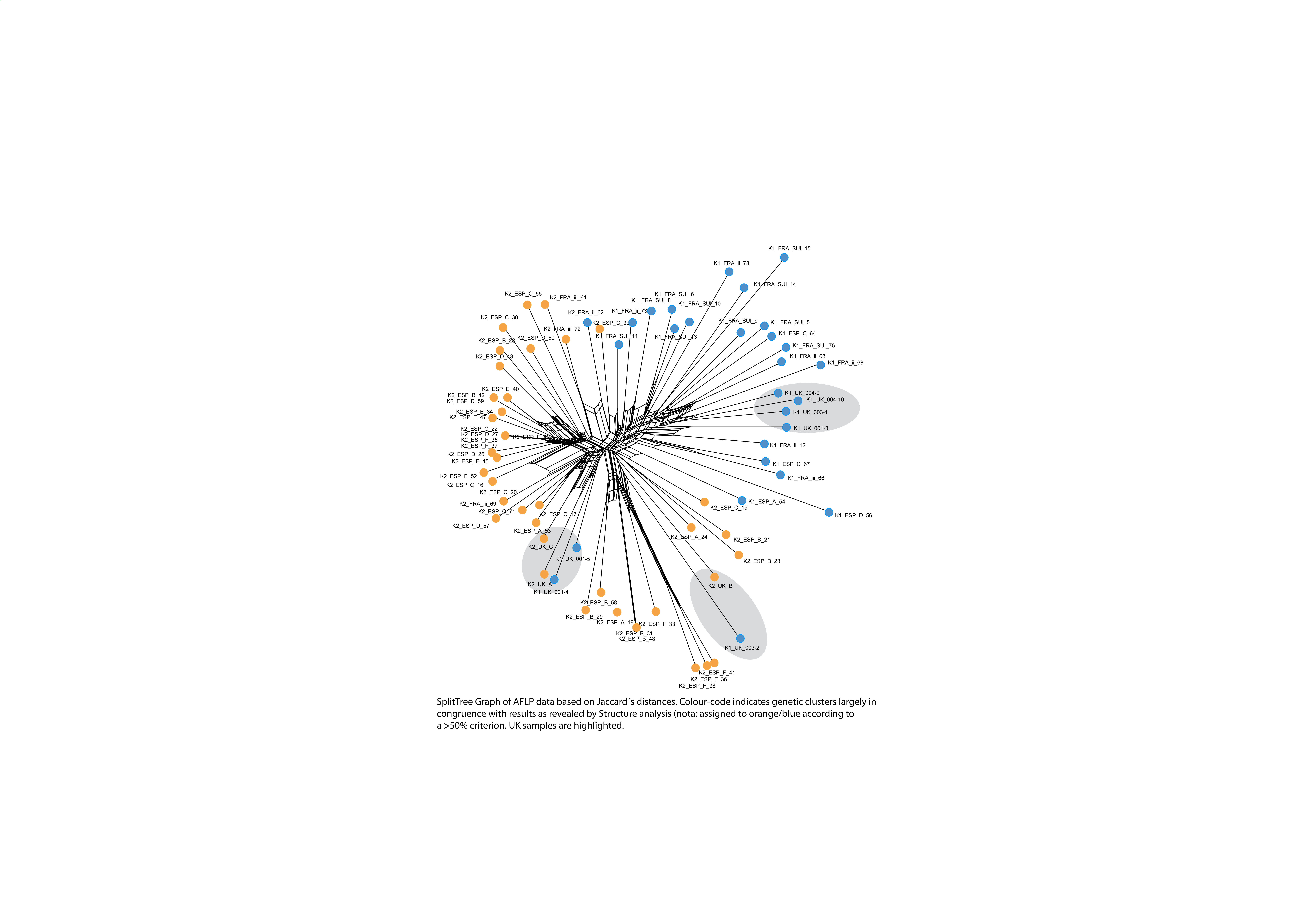
As the smallest best K is assumed to be the most suitable number of clusters, genetic structure is best described by two major gene pools, which essentially separate mountain regions of the Iberian Peninsula from the south-western French/Swiss distribution area. The geographically intermediate set 9 and accessions from the UK displayed an obvious mixture of these two gene pools. Results of the STRUCTURE analysis are confirmed by SplitsTree analysis considering that accessions with mixed gene pools have been assigned to the respective dominant genetic cluster (Supplementary Data Fig. S3).
Genetic diversity statistics (gene diversity, θ π, θ S, Tajima’s D) were calculated for accession sets 1–10 and for the main geographically defined regions (Spain and France/Switzerland; Table 2). Gene diversity in Spain (accession sets 1–6) and France/Switzerland (accession sets 7 and 8) is comparable to and correlates well with the two identified genetic clusters. The set of samples originating from the UK shows high genetic diversity, which is explained by a mixture of the two gene pools. Tajima’s D was calculated as a measure for indicating demographic expansion, contraction or introgression. We obtained various negative values of Tajima’s D indicating population size expansion; however, the values were only significant for the Spanish genetic cluster.
Table 2.
Genetic diversity statistics of the AFLP data set for individual sets of accessions (1–10) and sets combined into major geographical and genetic clusters. The three highest values are indicated in bold
| Distribution area (sample size) | S (polymorphic loci) | Gene diversity (s.d.) | θ π (s.d.) | θ S (s.d.) | Tajima’s D | |
|---|---|---|---|---|---|---|
| Accession set | ||||||
| 1 | ESP-A (4) | 16 | 0.078 (0.055) | 9.00 (6.28) | 8.73 (5.03) | 0.318 |
| 2 | ESP-B (9) | 23 | 0.083 (0.048) | 9.50 (4.81) | 8.46 (3.85) | 0.607 |
| 3 | ESP-C (11) | 31 | 0.067 (0.038) | 7.67 (3.87) | 10.58 (4.49) | −1.276 |
| 4 | ESP-D (7) | 22 | 0.062 (0.039) | 7.14 (3.81) | 8.97 (4.36) | −1.153 |
| 5 | ESP-E (5) | 4 | 0.014 (0.011) | 1.60 (1.12) | 1.92 (1.27) | −1.093 |
| 6 | ESP-F (6) | 27 | 0.121 (0.074) | 13.86 (7.27) | 11.82 (5.89) | 1.090 |
| 7 | FRA-A (10) | 26 | 0.065 (0.038) | 7.47 (3.81) | 9.19 (4.04) | −0.894 |
| 8 | FRA-B (6) | 25 | 0.092 (0.057) | 10.60 (5.63) | 10.94 (5.47) | −0.200 |
| 9 | FRA-C (4) | 17 | 0.076 (0.053) | 8.67 (5.08) | 9.57 (6.56) | −0.667 |
| 10 | UK (10) | 42 | 0.105 (0.059) | 12.04 (5.96) | 14.85 (6.32) | −0.928 |
| Major genetic clusters | ||||||
| 1–6 | ESP (42) | 71 | 0.077 (0.042) | 9.81 (4.58) | 16.50 (5.04) | −1.456* |
| 7–8 | FRA (16) | 38 | 0.086 (0.044) | 8.79 (5.16) | 11.45 (4.58) | −0.973 |
*P < 0.05.
Furthermore, a Mantel test was performed to correlate geographical with genetic distance. A significant positive correlation of genetic dissimilarity (Jaccard distance) with geographical distance was observed only at distances <300 km (total dataset, Fig. 5A), and Spanish populations contributed most to this significant signal (Fig. 5B, C). The distance of >300 km correlates well with the distance between the two geographically defined genetic clusters and might indicate substantial genetic isolation between clusters and only limited isolation by distance within genetic clusters. This result is in agreement with the weak genetic differentiation into two gene pools as revealed by the STRUCTURE analysis.
Fig. 5.

Mantel correlograms of genetic versus geographical distance showing the correlation coefficient r for three data sets: (A) all samples (ten regions); (B) Spanish accession sets (six regions); and (C) accession sets 7 and 8. Filled circles show significant positive or negative correlation at P < 0.05 (*) and P < 0.01 (**). Numbers indicate the total number of data pairs for a given distance class (50 km distance classes). Distribution of geographical distances is given as total counts and presented as bars.
Assembly and analysis of the plastid genomes
The entire plastid genome assembly of the 22 A. scabra accessions resulted in 22 different genomes (GenBank accession numbers in Supplementary Data Table S1). The average length of the plastid genome was 153 702 ± 13 bp. Within A. scabra 35 SNPs were detected, of which 29 were located in non-coding regions (Supplementary Data Table S4). The respective statistical parsimony network reconstructed by TCS1.21 recognized four major groups (Fig. 6). The result of the network analysis is fully consistent with the outcome of the RAxML analysis based on the entire plastome alignment and including the outgroup taxa (Supplementary Data Fig. S4). All accessions included in the AFLP analysis were screened by diagnostic SNP combinations differentiating the four groups of haplotypes identified by network analysis and phylogenetic tree reconstruction (Fig. 6B). The Iberian Peninsula harbours plastid types from groups II, III (Spain only) and IV. Towards Switzerland haplotypes from group II and IV are replaced by haplotype I. The accessions from near Bristol, UK, exclusively carry haplotype I plastid copies, indicating a geographical connection with either the South of France or the Iberian Peninsula. In order to determine split times of haplotype lineages a BEAST analysis was performed. The analysis resulted in an identical tree topology concerning the core Arabis clade (Supplementary Data Fig. S4). The two major A. scabra plastome haplotype groups diverged from each other ~51 ky ago. Both lineages split off in group I/II and III/IV 29 and 26 ky ago, respectively. Hence, it can be derived that a vicariance event happened ~26–29 ky ago, giving rise to haplotype III in Spain and haplotype I in Switzerland and neighbouring France close to the maximum glaciation in the Pyrenees, as a ‘glacial borderline’ separating the two areas (Fig. 6).
Fig. 6.

Results of TCS analysis (A) indicate two major clades (I/II and III/IV) based on plastome data. The root is indicated (*) and divergence time estimates are redrawn from BEAST analysis (Supplementary Data Fig. S4). (B) SNP screening for all Arabis scabra accessions and plastid haplotype assignment according to TCS analysis.
DISCUSSION
Phylogenetics and biogeographical information coincide
Arabis scabra and all the other Arabis species analysed herein (except A. alpina) belong to the ‘main Arabis clade’, which consists of ~65 species (Karl and Koch, 2013). The stem group age of this clade has been calculated to be 10–13 my, with a crown group age of ~7 my. The putative geographical origin of the crown group is uncertain and spans in particular the Caucasus, eastern Mediterranean and south-eastern European regions (Karl and Koch, 2013). With the onset of Pleistocene climatic fluctuations, evolutionary lineages from the main Arabis clade expanded into different regions, such as eastern Asia and North America, but also reached the western Mediterranean region and northern Africa, as exemplified herein by the A. ciliata alliance (Figs 2 and 3). As a consequence, there is substantial biogeographical–floristic coincidence with phylogenetic data considering the entire A. ciliata alliance: A. scabra and A. serpyllifolia are grouped within the ‘Erysimum duriaei’ floristic element group (Finnie et al., 2007). Further, A. ciliata shows close affinities with this distribution pattern (Finnie et al., 2007). Analysing distribution data from Atlas florae Europaeae, Finnie et al. (2007) found this ‘Erysimum duriaei’ floristic element as a group of 140 species primarily distributed in the mountains of northern Spain, the Pyrenees, south-eastern France and the south-western edge of the Alps. Among all European elements defined in that study, the ‘Erysimum duriaei’ element showed the highest level of endemism (86 % of the species) and the lowest percentage of trees and shrubs (together only 5 %). Furthermore, the vast majority of species are perennial (89 %). Two other species, endemic to Portugal, Arabis beirana and Arabis stenocarpa, fit with this pattern, and also Arabis vochinensis and Arabis stellulata have been assigned to the related ‘Salix serpyllifolia’ floristic elements, showing a wider occurrence in the mountains of central and southern Europe. However, key characteristics of the ‘Salix serpyllifolia’ floristic element, such as a high level of endemism (89 %), occurrence of trees (only 2 %) and dominance of perennial herbs (86 %), is very similar compared with the ‘Erysimum duriaei’ element. The North African species from Morocco, Arabis doumetii, Arabis erubescens and Arabis pubescens, were not classified by Finnie et al. (2007), but most likely they have close biogeographical affinities with the Iberian taxa. These deep phylogenetic footprints in present-day floristic biogeographical patterns have been described for some other evolutionary lineages of Brassicaceae. The largest genus of Brassicaceae, with more than 400 species (Draba), shows significant correlations of evolutionary lineages with world-wide floristic patterns dating back to the Pliocene (Jordon-Thaden and Koch, 2008), and also the genus Brassica (44 species) exhibits significant signatures (Arias et al., 2014) within the Mediterranean region, which also led to the recognition of a ‘Brassica cretica’ floristic element (Finnie et al., 2007). Other examples with different geographical–floristic affinities are from the genera Alyssum and Biscutella (Finnie et al., 2007), and the genus Cochlearia (Finnie et al., 2007; Koch, 2012). From all the different Arabis species (‘Erysimum duriaei’ and ‘Salix serpyllifolia’ floristic elements, A. ciliata alliance) it was only A. scabra that was able to colonize the British Isles. Only two other Arabis species are native to the British Isles, namely A. hirsuta and A. alpina, both belonging to different evolutionary lineages and floristic elements from Central Continental Europe and the alpine/arctic region (Karl et al., 2012; Karl and Koch, 2014; Koch and Grosser, 2017); and only A. alpina has a well-studied bio/phylogeographical history. Arabis alpina expanded its distribution range postglacially from refuge areas in Central Europe and rapidly colonized the entire arctic circle (Koch et al., 2006; Ehrich et al., 2007; Ansell et al., 2011; Karl et al., 2012), including the British Isles.
Phylogeographical evidence for late Pleistocene and postglacial range expansion
DNA sequence analyses of ITS and the initial screening from the plastid trnLF region revealed no sequence variation among all studied accessions of A. scabra. This clearly indicates a more recent Pleistocene evolutionary history of the gene pool, and the differentiation of the gene pool does not date back to the early Pleistocene close to the stem age of the species. A more detailed insight into the temporal dynamics of the evolution of the species is provided by whole-plastid genome sequences. Although we selected accessions from the entire distribution range of the taxon and AFLP gene pool, genetic variation among entire plastid genomes remained very low (35 SNPs in total), and accordingly the majority of SNPs are located in non-coding regions (29). If we assume a plastome-wide mutation rate of μ = 7.798 × 10−10 mutations/site/year (Koch et al. 2017b), an accumulation of 12 mutations per 100 000 years can be expected (or diverged genomes separated by 24 mutations) over the entire plastid genome. However, the mutation rate for non-coding regions is higher and has been calculated for Brassicaceae as μ = 2.004 × 10−10 and would result in ~3 times faster temporal scenarios (Koch et al. 2017b). The split between haplotypes from group I/II versus III/IV in A. scabra is characterized by a mean of 14 mutations (counting the Inverted Repeat only once), which provides a maximum of ~70 600 years ago; the application of the faster mutation rate provides an estimate of ~18 200 years ago (minimum–maximum, 18–71 kya). This temporal scenario is also supported by BEAST analysis using secondary calibrations and estimating a plastome stem group split 51 kya (Fig. 6).
Results of divergence time estimates fit with the expectation of a strong isolation effect during the last glaciation (20–100 kya) either between Spain and the south-western Alps (Pyrenees as barrier) or even among mountain ranges within the Iberian Peninsula. The low levels of genetic polymorphism within the two major plastid genome clades (Fig. 6) in combination with crown ages younger or close to the LGM (within groups II and IV 7000–10 000 years ago) can be best explained with a postglacial range expansion and migration scenario. However, what is the original refuge area? Either the Iberian Peninsula or the south-western Alps? AFLP data favour a Spanish refuge area, because the proportion of rare and unique alleles is higher there and is indicative of older and longer-term refuge areas compared with the French distribution range (Table 1). To fit all genetic data with alternative glacial–postglacial range contraction and expansion we have to consider two alternative scenarios (Fig. 7). The first scenario assumes that the last glaciation forced A. scabra into an Iberian and a French refuge region. Postglacially, A. scabra expanded its distribution range, gene pools might have even merged, and within an open landscape the species was able to reach also the British Isles. With further increasing temperatures the distribution range shrank and populations became isolated again (Fig. 7A, B, E). Alternatively, A. scabra survived the last glaciation maximum on the Iberian Peninsula. Postglacially, populations colonized the French and Swiss regions and genetic differentiation was structured along the colonizing gradient. This expansion also reached the British Isles and, similar to scenario 1, subsequent continued warming caused severe range contractions and populations were forced back into mountainous regions (Fig. 7C–E). AFLP data cannot readily distinguish between these two scenarios (Figs 1B and 7), whereas plastome data favour a multiple colonization scenario (inter- and postglacial) out of the Iberian Peninsula, and scenario 1 can therefore be considered the preferred explanation for the present-day distribution pattern and the phylogeographical history of A. scabra.
Fig. 7.

Hypothesized alternative scenarios of postglacial colonization of Arabis scabra in Central Europe and the settlement of British relic populations. Scenario 1 (A, B, E): (A) distribution of two main gene pools (blue and orange) at the LGM; (B) postglacial expansion and northward migration; (E) result of subsequent population retraction and isolation. Scenario 2 (C, D, E): (C) Distribution of one gene pool in Spain during the last glaciation; (D) postglacial expansion towards the south-western Alps and also northward migration. The limits of the British–Irish ice shield (hatched areas, after Sejrup et al., 2005) and Pyrenean glaciers (grey area, after Ehlers and Gibbard, 2004) at the LGM are indicated. The dashed lines indicate the LGM coastline (Taberlet et al., 1991). The European/alpine glaciers are not shown. The crosses represent our accessions.
Fig. 4.

Results of combined STRUCTURE runs of AFLP data for assumed number of genetic clusters ranging from K = 2 to K = 4, showing stable genetic assignment. LnP(D), mean posterior probability value per cluster K; SSC, mean symmetrical similarity coefficient.
Outpost or relict populations on the British Isles
Considering divergence time estimates from plastome data, we might have to assume severe regional structuring of genetic variation during the last glaciation on the Iberian Peninsula (e.g. separating subclades I and III geographically). The subsequent origin of further lineages and plastome types is dated close to the LGM and to dates more recent than the LGM (Fig. 6, clades II and IV). Furthermore, effective population sizes must have been small, because only little genetic variation has been accumulated and maintained in plastomes during the last glaciation, as is also true for AFLP data. Populations may have been always at risk of going extinct, and colonization may have happened by rare dispersal events on local and regional scales only, which is also seen in the Mantel correlograms (Fig. 5B). However, there must have been a migration corridor from Central Europe towards the British Isles. Present-day genetic variation in the Bristol region is high even compared with its putative source regions, indicating that either a past large effective population size and/or multiple colonization events generated high present-day genetic variation. Genetic variation is as high as in the putative mainland European source populations, which most likely underwent severe postglacial genetic bottlenecks. Arabis scabra is a short-lived biennial or rarely perennial plant. It grows on shallow soil, scree and rocky ledges, not competing with woodlands. This is true not only for the Avon Gorge near Bristol in the UK, but also for French Prealps and Spanish mountain regions such as the Pyrenees and Cantabrian mountains, with a strict preference for calcareous bedrock types and affiliation with vegetation types (basically lithophytes) such as Potentilletalia caulescentis Braun-Blanquet (Braun-Blanquet and Jenny, 1926). Plastome types from clade II in A. scabra may indicate a migration or dispersal corridor out of Spain and the Pyrenees. Respective affinities of the British Isles with the Lusitanian flora have been shown in a few phylogeographical studies (Valtuena et al., 2012; Beatty and Provan, 2013, 2014; Beatty et al., 2015; Martin et al., 2016), and most often colonization has been set in a postglacial temporal context (Pinguicula grandiflora, Saxifraga spathulata, Daboecia cantabrica, Euphorbia hyberna, Silene nutans) with the exception of Meconopsis cambrica as a glacial relict in the UK (Valtuena et al., 2012) or Galium fleurotii, for which periglacial refugia in unglaciated Great Britain have been assumed (Kolar et al., 2013). For several taxa multiple founder events in Great Britain have been postulated (Saxifraga spathularis, Daboecia cantabrica; likely in Meconopsis cambrica and Euphorbia hyberna). The same is true for Campanula rotundifolia (Sutherland and Galloway, 2018), which may have colonized south-western Great Britain postglacially via a corridor from the French Prealps and from south-western Germany towards Belgium. Arabis scabra plastome types from Bristol, UK, differ by one single mutation equalling ~7500 years of divergence. Of course, the number has neither a significance level nor any meaningful standard deviation (±7500 years), but does not violate a hypothesis that the species followed a colonization route from refugia in Central and south-western Europe, eventually crossing the dry Celtic Sea prior to the sea-level rise 16–15 kya (Clark et al., 2012). Interestingly, there are other species in the Bristol region (Cheddar Gorge and Mendip Hills), such as Cochlearia pyrenaica (Pyrenean scurvygrass) and Dianthus gratianopolitanus (Cheddar pink), showing a very similar spatiotemporal context (laboratory of M. A. Koch, unpubl. data). These species show a strict preference for calcareous bedrock types, as A. scabra does, which limits the present-day distribution in south-western Great Britain. The actual population size of A. scabra in the Bristol area varies from year to year, and counts of the whole population between 1977 and 1989 ranged from ~500 to 5400 individuals (A. scabra account in Online Atlas of the British and Irish Flora, Botanical Society of Britain and Ireland and the Biological Records Centre (BRC); https://www.brc.ac.uk/plantatlas/plant/arabis-scabra). Consequently, we have to consider A. scabra both, as an outpost of a former wider distribution area and a relict population that remarkably did not undergo a stronger bottleneck than populations in mainland Europe, possibly due to an almost annual life history, which may have helped the species to overcome short-term bottlenecks or fluctuations in population size or due to preservation of seeds in the soil seed bank, as presence in soil seed banks has also been shown for a related short-lived taxon (A. hirsuta; Thompson et al., 1997).
Conclusions
The short-lived perennial herb A. scabra, which is restricted to limestone throughout its entire distribution range, has expanded its distribution range after the LGM following corridors within an open landscape and may have reached the British Isles via the desiccated Celtic Sea at ~16 kya. This study may shed light on the origin of other rare and peculiar species co-occurring in limestone regions in the south-western British Isles, and indicate the importance of this region as a long-term stable refugium of plant biodiversity.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Table S1: detailed accession list and GenBank accession codes. Table S2: NEXUS file of Arabideae ITS sequence data. Table S3: NEXUS file of Arabideae trnLF plastid sequence data; Table S4: plastome SNP matrix. Table S5: NEXUS file of AFLP fragments. Figure S1: principal component analysis of AFLP data. Figure S2: simulation of population structure. Figure S3: split tree graph of AFLP data based on Jaccard’s distances. Figure S4: RAcML analysis bootstrap support and BEAST analysis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Peter Sack and Lisa Kretz for their help in the laboratory and herbarium curation and David Ibberson at the CellNetworks/Heidelberg Deep Sequencing Core facility. We also thank the curators of the herbaria E, G, VIT, VAL, MA, ARAN and MNHN for providing voucher material, and colleagues from Bristol Botanic Garden/Bristol University (Nicholas Wray and Antony Dodd) for providing fresh leaf material, dried specimens and seed material. Moreover, we are grateful to Florian Michling for discussions and support with AFLP analyses. The authors declare no conflict of interest. All sequence data have been deposited at GenBank.
LITERATURE CITED
- Abellán P, Svenning J-C. 2014. Refugia within refugia – patterns in endemism and genetic divergence are linked to Late Quaternary climate stability in the Iberian Peninsula. Biological Journal of the Linnean Society 113: 12–28. [Google Scholar]
- Allen GA, Marr KL, McCormick LJ, Hebda RJ. 2012. The impact of Pleistocene climate change on an ancient arctic-alpine plant: multiple lineages of disparate history in Oxyria digyna. Ecology and Evolution 2: 649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. [Google Scholar]
- Ansell SW, Stenøien HK, Grundmann M, et al. 2011. The importance of Anatolian mountains as the cradle of global diversity in Arabis alpina, a key arctic-alpine species. Annals of Botany 108: 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias T, Beilstein MA, Tang M, McKain MR, Pires JC. 2014. Diversification times among Brassica (Brassicaceae) crops suggest hybrid formation after 20 million years of divergence. American Journal of Botany 101: 86–91. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, et al. 2013. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics 43: 11.10.1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avise JC, Bowen BW, Ayala FJ. 2016. In the light of evolution X. Comparative phylogeography. Proceedings of the National Academy of Sciences of the USA 113: 7957–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GE, Povan J. 2013. Post-glacial dispersal, rather than in situ glacial survival, best explains the disjunct distribution of the Lusitanian plant species Daboecia cantabrica. Journal of Biogeography 40: 335–344. [Google Scholar]
- Beatty GE, Povan J. 2014. Phylogeographic analysis of two cold-tolerant plants with disjunct Lusitanian distributions does not support in situ survival during the last glaciation. Journal of Biogeography 41: 2185–2193. [Google Scholar]
- Beatty GE, Lennon JJ, O’Sullivan CJ, Provan J. 2015. The not-so-Irish spurge: Euphorbia hyberna (Euphorbiaceae) and the Littletonian plant ‘steeplechase’. Biological Journal of the Linnean Society 114: 249–259. [Google Scholar]
- Betzin A, Thiv M, Koch MA. 2016. Diversity hotspots of the laurel forest on Tenerife, Canary Islands: a phylogeographic study of Laurus and Ixanthus. Annals of Botany 118: 495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin A, Bellemain E, Bronken Eidesen P, Pompanon F, Brochmann C, Taberlet P. 2004. How to track and assess genotyping errors in population genetics studies. Molecular Ecology 13: 3261–3273. [DOI] [PubMed] [Google Scholar]
- Braun-Blanquet J, Jenny H. 1926. Vegetationsentwicklung und Bodenbildung in der alpinen Stufe der Zentralalpen. Denkschriften der Schweizerischen Naturforschenden Gesellschaft Zürich 63: 183–349. [Google Scholar]
- Brewer S, Giesecke T, Davis BAS, et al. 2017. Late-glacial and Holocene European pollen data. Journal of Maps 13: 921–928. [Google Scholar]
- Brochmann C, Gabrielsen TM, Nordal I, Landvik JY, Elven R. 2003. Glacial survival or tabula rasa? The history of North Atlantic biota revisited. Taxon 52: 417–450. [Google Scholar]
- Clark C, Hughes A, Greenwood S, Jordan C, Sejrup H. 2012. Pattern and timing of retreat of the last British-Irish ice sheet. Quaternary Science Reviews 44: 112–146. [Google Scholar]
- Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobeš C, Mitchell-Olds T, Koch M. 2004. Extensive chloroplast haplotype variation indicates Pleistocene hybridization and radiation of North American Arabis drummondii, A.× divaricarpa, and A. holboellii (Brassicaceae). Molecular Ecology 13: 349–370. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 29: 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durvasula A, Fulgione A Gütaker R, et al. 2017. African genomes illuminate the early history of Arabidopsis thaliana. Proceedings of the National Academy of Sciences of the USA 114: 5213–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers J, Gibbard P. 2004. Quaternary glaciations – extent and chronology. Part I: Europe. Developments in quaternary science: 2. Amsterdam: Elsevier. [Google Scholar]
- Ehrich D, Gaudeul M, Assefa A, et al. ; Intrabiodiv Consortium. 2007. Genetic consequences of Pleistocene range shifts: contrast between the Arctic, the Alps and the East African mountains. Molecular Ecology 16: 2542–2559. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HE. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. 2007. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Molecular Ecology Notes 7: 574–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliner GN. 2011. Southern European glacial refugia: a tale of tales. Taxon 60: 365–372. [Google Scholar]
- Finnie TJR, Preston CD, Hill MO, Uotila P, Crawley MJ. 2007. Floristic elements in European vascular plants: an analysis based on Atlas florae Europaeae. Journal of Biogeography 34: 1848–1872. [Google Scholar]
- Hewitt G. 2000. The genetic legacy of the quaternary ice ages. Nature 405: 907–913. [DOI] [PubMed] [Google Scholar]
- Holtvoeth J, Vogel H, Valsecchi V, et al. 2017. Linear and non-linear responses of vegetation and soils to glacial-interglacial climate change in a Mediterranean refuge. Scientific Reports 7: 8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann N, Koch MA. 2017. An Arabidopsis introgression zone studied at high spatio-temporal resolution: interglacial and multiple genetic contact exemplified using whole nuclear and plastid genomes. BMC Genomics 18: 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann N, Wolf EM, Lysak MA, Koch MA. 2015. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell 27: 2770–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 1990. Gene genealogies and the coalescent process. In: Futuyma DJ, Antonovics JD, eds. Oxford surveys in evolutionary biology. New York: Oxford University Press, 1–44. [Google Scholar]
- Huson DH, Bryant D. 2006. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23: 254–267. [DOI] [PubMed] [Google Scholar]
- Jaccard P. 1901. Distribution de la flore alpine dans le Bassin des Drouces et dans quelques regions voisines. Bulletin de la Société Vaudoise des Sciences Naturelles 37: 241–272. [Google Scholar]
- Jakobsson M, Rosenberg NA. 2007. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics (Oxford, England) 23: 1801–1806. [DOI] [PubMed] [Google Scholar]
- Jalas J, Suominen J. 1994. Atlas florae Europaeae. Distribution of vascular plants in Europe. 10: Cruciferae. Helsinki: Academic Bookstore. [Google Scholar]
- Jordon-Thaden I, Koch MA. 2008. Diversity patterns in the genus Draba: a first global perspective. Plant Ecology & Diversity 1: 255–263. [Google Scholar]
- Kadereit J, Westberg E. 2007. Determinants of phylogenetic structure: a comparative study of seven coastal flowering plant species across their European range. Watsonia 26: 229–238. [Google Scholar]
- Karl R, Koch MA. 2013. A world-wide perspective on crucifer speciation and evolution: phylogenetics, biogeography and trait evolution in tribe Arabideae. Annals of Botany 112: 983–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl R, Koch MA. 2014. Phylogenetic signatures of adaptation: the Arabis hirsuta species aggregate (Brassicaceae) revisited. Perspectives in Plant Ecology, Evolution and Systematics 16: 247–264. [Google Scholar]
- Karl R, Kiefer C, Ansell SW, Koch MA. 2012. Systematics and evolution of Arctic-Alpine Arabis alpina (Brassicaceae) and its closest relatives in the eastern Mediterranean. American Journal of Botany 99: 778–794. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer M, Schmickl R, German DA, et al. 2014. BrassiBase: introduction to a novel knowledge database on Brassicaceae evolution. Plant & Cell Physiology 55: e3. [DOI] [PubMed] [Google Scholar]
- Koch MA. 2012. Mid-Miocene divergence of Ionopsidium and Cochlearia and its impact on the systematics and biogeography of the tribe Cochlearieae (Brassicaceae). Taxon 61: 76–92. [Google Scholar]
- Koch M, Bishop J, Mitchell-Olds T. 1999. Molecular systematics and evolution of Arabidopsis and Arabis. Plant Biology 1: 529–537. [Google Scholar]
- Koch MA, Grosser J. 2017. East Asian Arabis species (Brassicaceae) exemplify past hybridization and subsequent emergence of three main evolutionary lineages in East Asia, America and the amphiberingian region. Botanical Journal of the Linnean Society 184: 224–237. [Google Scholar]
- Koch MA, Kiefer C, Ehrich D, Vogel J, Brochmann C, Mummenhoff K. 2006. Three times out of Asia Minor: the phylogeography of Arabis alpina L. (Brassicaceae). Molecular Ecology 15: 825–839. [DOI] [PubMed] [Google Scholar]
- Koch MA, Karl R, Kiefer C, Al-Shehbaz IA. 2010. Colonizing the American continent: systematics of the genus Arabis in North America (Brassicaceae). American Journal of Botany 97: 1040–1057. [DOI] [PubMed] [Google Scholar]
- Koch MA, Kiefer M, German D, et al. 2012. BrassiBase: tools and biological resources to study characters and traits in the Brassicaceae – version 1.1. Taxon 61: 1001–1009. [Google Scholar]
- Koch MA, Karl R, German DA. 2017a Underexplored biodiversity of Eastern Mediterranean biota: systematics and evolutionary history of the genus Aubrieta (Brassicaceae). Annals of Botany 119: 39–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Michling F, Walther A, Huang XC, Tewes L, Müller C. 2017b Early-Mid Pleistocene genetic differentiation and range expansions as exemplified by invasive Eurasian Bunias orientalis (Brassicaceae) indicates the Caucasus as key region. Scientific Reports 7: 16764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, German DA, Kiefer M, Franzke A. 2018. Database taxonomics as key to modern plant biology. Trends in Plant Science 23: 4–6. [DOI] [PubMed] [Google Scholar]
- Kolář F, Lučanová M, Vít P, et al. 2013. Diversity and endemism in deglaciated areas: ploidy, relative genome size and niche differentiation in the Galium pusillum complex (Rubiaceae) in Northern and Central Europe. Annals of Botany 111: 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35: btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtto A, Sennikov AN, Lampinen R. 2013. Atlas florae Europaeae. Helsinki: The Committee for Mapping the Flora of Europe & Societas Biologica Fennica Vanamo. [Google Scholar]
- Lanfear R, Calcott B, Ho SY, Guindon S. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29: 1695–1701. [DOI] [PubMed] [Google Scholar]
- Legendre P, Legendre L. 2012. Numerical ecology. Amsterdam: Elsevier. [Google Scholar]
- Meudt HM, Clarke AC. 2007. Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends in Plant Science 12: 106–117. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. ; 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H, Touzet P, Van Rossum F, Delalande D, Arnaud JF. 2016. Phylogeographic pattern of range expansion provides evidence for cryptic species lineages in Silene nutans in Western Europe. Heredity 116: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller J, Müller K, Quandt D, Neinhuis C. 2005. PhyDE – phylogenetic data editor, version 0.995. http://www.phyde.de. [Google Scholar]
- Novikova PY, Hohmann N, Nizhynska V, et al. 2016. Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nature Genetics 48: 1077–1082. [DOI] [PubMed] [Google Scholar]
- Nylander JAA. 2004. MrModeltest v2. Program distributed by the author. Evolutionary Biology Centre, Uppsala University. [Google Scholar]
- Oksanen J, Blanchet FG, Friendly M, et al. 2017. vegan: community ecology package. R package version 2.4–3. https://cran.r-project.org/web/packages/vegan. [Google Scholar]
- Pring ME. 1961. Biological flora of the British Isles: Arabis stricta Huds. Journal of Ecology 49: 431–437. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provan J, Bennett KD. 2008. Phylogeographic insights into cryptic glacial refugia. Trends in Ecology and Evolution 23: 564–571. [DOI] [PubMed] [Google Scholar]
- Rambaut A. 2018. FigTree v1.4.4. http://tree.bio.ed.ac.uk/software/figtree/. [Google Scholar]
- Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Systematic Biology syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team 2014. R: a language and environment for statistical computing. Vienna:R Foundation for Statistical Computing. [Google Scholar]
- Richardson JE, Bakar AM, Tosh J, et al. 2014. The influence of tectonics, sea-level changes and dispersal on migration and diversification of Isonandreae (Sapotaceae). Botanical Journal of the Linnean Society 174: 130–140. [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiso X, Lopez L, Retuerto R, Barreiro R. 2016. Phylogeography of a widespread species: pre-glacial vicariance, refugia, occasional blocking straits and long-distance migrations. AoB Plants 8: plw003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejrup HP, Hjelstuen BO, Torbjørn Dahlgren KI, et al. 2005. Pleistocene glacial history of the NW European continental margin. Marine and Petroleum Geology 22: 1111–1129. [Google Scholar]
- Silvestro D, Michalak I. 2012. raxmlGUI: a graphical front-end for RAxML. Organisms Diversity and Evolution 12: 335–337. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics (Oxford, England) 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starnberger R, Drescher-Schneider R, Reitner JM, Rodnight H, Reimer PJ, Spötl C. 2013. Late Pleistocene climate change and landscape dynamics in the Eastern Alps: the inner-alpine Unterangerberg record (Austria). Quaternary Science Reviews 68: 17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SC, Parks M, Weitemier K, Fishbein M, Cronn RC, Liston A. 2012. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. American Journal of Botany 99: 349–364. [DOI] [PubMed] [Google Scholar]
- Sutherland BL, Galloway LF. 2018. Effects of glaciation and whole genome duplication on the distribution of the Campanula rotundifolia polyploid complex. American Journal of Botany 105: 1–11. [DOI] [PubMed] [Google Scholar]
- Svenning JC, Fløjgaard SC, Marske KA, Nógues-Bravo D, Normand S. 2011. Applications of species distribution modelling to paleobiology. Quaternary Science Reviews 30: 2930–2947. [Google Scholar]
- Taberlet P, Gielly L, Pautou G, Bouvet J. 1991. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Molecular Biology 17: 1105–1109. [DOI] [PubMed] [Google Scholar]
- Thompson K, Bakker JP, Bekker RM. 1997. The soil seed banks of North West Europe: methodology, density and longevity. Cambridge: Cambridge University Press. [Google Scholar]
- Tzedakis PC, Emerson BC, Hewitt GM. 2013. Cryptic or mystic? Glacial tree refugia in northern Europe. Trends in Ecology and Evolution 28: 696–704. [DOI] [PubMed] [Google Scholar]
- Valtueña FJ, Preston CD, Kadereit JW. 2012. Phylogeography of a Tertiary relict plant, Meconopsis cambrica (Papaveraceae), implies the existence of northern refugia for a temperate herb. Molecular Ecology 21: 1423–1437. [DOI] [PubMed] [Google Scholar]
- Vos P, Hogers R, Bleeker M, et al. 1995. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Research 23: 4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TJ, Burns T, Lee S, Taylor J. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, eds. PCR protocols: a guide to methods and applications. San Diego: Academic Press, 315–322. [Google Scholar]
- Willis KJ, MacDonald GM. 2011. Long-term ecological records and their relevance to climate change predictions for a warmer world. Annual Review of Ecology, Evolution, and Systematics 42: 267–287. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.