

Abstract
The emergence of CRISPR-Cas9 gene-editing technologies and genome-wide CRISPR-Cas9 libraries enables efficient unbiased genetic screening that can accelerate the process of therapeutic discovery for genetic disorders. Here, we demonstrate the utility of a genome-wide CRISPR-Cas9 loss-of-function library to identify therapeutic targets for facioscapulohumeral muscular dystrophy (FSHD), a genetically complex type of muscular dystrophy for which there is currently no treatment. In FSHD, both genetic and epigenetic changes lead to misexpression of DUX4, the FSHD causal gene that encodes the highly cytotoxic DUX4 protein. We performed a genome-wide CRISPR-Cas9 screen to identify genes whose loss-of-function conferred survival when DUX4 was expressed in muscle cells. Genes emerging from our screen illuminated a pathogenic link to the cellular hypoxia response, which was revealed to be the main driver of DUX4-induced cell death. Application of hypoxia signaling inhibitors resulted in increased DUX4 protein turnover and subsequent reduction of the cellular hypoxia response and cell death. In addition, these compounds proved successful in reducing FSHD disease biomarkers in patient myogenic lines, as well as improving structural and functional properties in two zebrafish models of FSHD. Our genome-wide perturbation of pathways affecting DUX4 expression has provided insight into key drivers of DUX4-induced pathogenesis and has identified existing compounds with potential therapeutic benefit for FSHD. Our experimental approach presents an accelerated paradigm towards mechanistic understanding and therapeutic discovery of a complex genetic disease, which may be translatable to other diseases with well-established phenotypic selection assays.
One Sentence Summary:
Genome-wide CRISPR-Cas9 screens identify druggable pathways associated with facioscapulohumeral muscular dystrophy.
Introduction
Many genetic disorders have a well-defined clinical phenotype associated with specific mutations in a causative gene. However, in some forms of muscular dystrophy, there continues to be a large gap in knowledge between identification of causal genetic mutations and understanding the molecular pathways underlying the disease pathophysiology, which is vital for developing targeted therapies. The process of elucidating this link is challenging, particularly for rare diseases, and can take decades of basic science research for molecular pathways to be mapped and drug targets identified. In this study, we use a genetically complex type of muscular dystrophy, facioscapulohumeral muscular dystrophy (FSHD), to demonstrate how CRISPR-Cas9 screens, when applied on a genome-wide scale, can be a powerful tool for identifying targets for drug discovery. This approach, when coupled to a disease-relevant cell-based assay, rapidly identifies promising drug targets, thereby accelerating the process of therapeutic discovery for rare diseases.
FSHD is one of the most common autosomal dominant forms of muscular dystrophy, affecting both males and females at an incidence of one in eight thousand individuals (1). FSHD is a multifactorial genetic disorder linked to epigenetic dysregulation of D4Z4-repeats in the subtelomeric region of chromosome four (2–5). In healthy individuals, the D4Z4-repeat array ranges from 11–100 copies on both 4q chromosomes, whereas in individuals with FSHD1 (~95% of cases) the array is contracted to 1–10 copies on at least one 4q chromosome, resulting in the loss of repressive epigenetic regulation. For FSHD to manifest, this contraction must also occur in cis with the disease-permissive haplotype of 4qA(6). Individuals with FSHD2 (~5% of cases) are not associated with contractions of D4Z4 repeats but instead harbor loss-of-function mutations in either SMCHD1 (7) or DNMT3B (8), genes required for maintaining a repressive epigenetic state at D4Z4 arrays, and share the same genetic requirement for the disease-permissive haplotype (7). Together, these distinct pathogenic mechanisms underlying FSHD1 and FSHD2 lead to common epigenetic alterations that result in chromatin relaxation and aberrant expression of the DUX4 retrogene from the pathogenic locus. Although each D4Z4 repeat unit encodes a copy of DUX4, only the mRNA transcribed from the DUX4 gene embedded in the distal-most D4Z4 repeat unit is stably expressed due to the presence of a required polyadenylation signal conferred by the disease-permissive 4qA haplotype (6, 9–11). Thus, FSHD is both an epigenetic and genetic dominant gain-of function disease and therefore should be amenable to loss-of-function screens.
DUX4 is not normally expressed in adult somatic cells, whereas during early human development it activates the cleavage-stage zygotic transcription program (12, 13). Thus, DUX4 encodes a potent developmental transcription factor, and misexpression of DUX4 in FSHD skeletal muscle inappropriately activates this expression program and DUX4 target genes, which ultimately leads to pathology (14–18). Several studies have identified altered gene expression signatures triggered by misexpression of DUX4 via transcription expression profiling (16, 19–23), and these results have implicated multiple potential pathogenic pathways, including those associated with activation of stem cell and germline programs (20), repression of innate immunity(20), inhibition of myogenesis (24), oxidative stress, apoptosis, and inhibition of nonsense mediated RNA decay (25). Individually, these studies have yielded an abundance of data pertaining to DUX4-specific changes in different cell models; however, together there is minor congruence between different studies. Despite these experiments resulting in a comprehensive catalogue of both direct transcriptional targets of DUX4 and as genes dysregulated in the presence of DUX4, no actionable pathogenic gene targets for therapeutic intervention beyond DUX4 itself have emerged from these studies. However, DUX4-induced apoptosis is consistent across every system tested (16–18, 26) and thus is likely a key mediator of FSHD pathogenesis and a viable therapeutic target.
The emergence of CRISPR-Cas9 gene-editing and its utility on a genome-wide scale has been successful in elucidating cancer drug resistance genes (27–29) and pathogen invasion factors (30–33). A knockout CRISPR-Cas9 screen was also recently used to identify suppressors and enhancers of an amyotrophic lateral sclerosis (ALS) gene, C9ORF72, and the resulting dipeptide repeat protein toxicity that underlies neurodegeneration (34). Together, these studies have led to the discovery of novel genes and pathways in their respective biological context, accelerated by the unbiased nature of the genome-wide CRISPR-Cas9 screening approach. Therefore, we hypothesized that an unbiased CRISPR-Cas9 screen that systematically knocked out each human gene in the context of FSHD could be used to identify those genes involved in enabling DUX4-mediated cell death. The results of our screen point to perturbation of the hypoxia-signaling pathway as an effective therapy target to prevent DUX4-induced cell death and FSHD-associated pathologies in patient cells and zebrafish disease models. The success of our screen in its identification of therapeutic candidates for FSHD demonstrates that this approach can be applied to other diseases with a quantifiable cellular phenotype to accelerate the process of therapeutic discovery for rare disease.
Results
Genome-wide CRISPR-Cas9 loss-of-function screens identify DUX4-resistant gene knockouts
Aberrant DUX4 expression in cells from individuals with FSHD results in cell death (16), but its activation is sporadic and infrequent (1 in every 200–1000 cells) (13, 35, 36) and thereby does not provide a robust selection pressure for conducting genome-wide screens. Therefore, we used an immortalized myoblast line (MB135-DUX4i) with a doxycycline-inducible DUX4 transgene (18, 22) that ensures consistent DUX4 expression in all cells. Upon addition of doxycycline and subsequent DUX4 expression, most cells are dead within 48 hrs (Fig. 1A), thus providing a robust negative selection mechanism that could be leveraged to identify gene knockouts conferring resistance to DUX4-induced cell death in a CRISPR-Cas9 screen (Fig. 1B). We transduced the lentivirus-packaged GeCKO (Genome-scale CRISPR-Cas9 Knock Out) library into the MB135-DUX4i line at low multiplicity of infection (MOI) to target one virus entry per cell, achieving 300X library representation within the population. After seven days of puromycin selection and cell expansion for positively transduced cells, we harvested half of the population to provide a snapshot of the library representation prior to DUX4 selection (‘early time point’, ETP). To the remaining population, we subsequently activated DUX4 expression by adding doxycycline. After 48 hrs, dead cells were removed and the remaining DUX4-resistant cells were expanded. These rare surviving cells were hypothesized to be DUX4-resistant due to their CRISPR-Cas9-induced gene knockout.
Fig. 1. Genome-wide CRISPR-Cas9 knockout screen identifies genes linked to DUX4 resistance.

(A) Brightfield microscope image of MB135-DUX4i human myoblast line with doxycycline-inducible DUX4 expression used in screen. (Dox = doxycycline) (B) Flow diagram of CRISPR-Cas9 screening method. A CRISPR-Cas9 knockout library was packaged in lentivirus and transduced into cells at MOI of 0.3–0.4. Two cell populations were harvested for genomic DNA isolation and sequencing: control (Early Time Point, ETP) and post-DUX4-selection. Two independent screens were performed and analyzed. (C) Guide RNA expression amounts before and after DUX4 selection from one CRISPR-Cas9 knock-out screen. Guides from selected top genes are highlighted to show enrichment from multiple different guide RNA sequences. (D) P-value of genes derived from two independent CRISPR-Cas9 knockout screens as determined by the statistical package MaGECK (72). Inset represents gene hits with P < 0.01 in both experiments. (E) Representation of GeneMania functional associations between significant (P < 0.01) screen hits identified across two independent replicates. See fig. S1. (F) Schematic of HIF-mediated transcription. Under hypoxic conditions, HIF1A is stabilized and translocates into the nucleus to dimerize with ARNT, forming the HIF transcription factor. Together with co-activators CBP (CREBBP) and p300, HIFs induce transcription of hypoxia response genes, one of which is CDKN1A.
Sequencing libraries generated from the ETP and post-DUX4-selection populations were sequenced by the NextSeq Illumina platform to determine their sgRNAs and respective gene targets. The resulting sequencing reads were aligned to the sgRNA library sequences and counts of each sgRNA were determined for both ETP and post-DUX4-selection populations (Fig. 1C). Normalized read counts for each of the six distinct sgRNAs per gene were aggregated to perform statistical testing. The most significant gene hits (p<0.01) from two independent screens (library transduction, selection for DUX4 resistance and guide sequencing) (Fig. 1D) were investigated for functional associations using GeneMania (37) (fig. S1). The resulting groupings revealed potential pathways involved in regulating DUX4 cytotoxicity, and allowed for the prioritization of candidates for follow-up studies (Fig. 1E). From this analysis, we identified the most significant sub-network of genes emerging from our CRISPR-Cas9 screen as the hypoxia-signaling pathway (Fig. 1F). HIF1A and ARNT (HIF1B) are subunits of the HIF1 transcriptional complex that dimerize under hypoxic conditions (38). CBP (CREBBP) is a transcriptional co-activator of the HIF1 complex, and together they bind hypoxia response elements upstream of several genes essential for the cellular response to hypoxia (39). CDKN1A is one of many downstream target genes of the HIF1 complex and is reported to mediate hypoxia-related growth arrest (40). We chose to focus on our hypoxia signaling candidate genes in follow-up studies given that they are well characterized compared to other gene candidates from our screen, and can also be modulated by targeted chemical inhibitors.
DUX4 expression activates the cellular hypoxia signaling pathway responsible for cell death
We further investigated the role of the cellular hypoxia pathway and its link to DUX4 expression. In response to hypoxic stress, HIF1A protein is stabilized and enriches in the nucleus, which we observed via immunofluorescence in the presence of cobalt chloride, a chemical inducer of hypoxia (Fig. 2A). We also observed a similar pattern of protein stabilization and nuclear localization of HIF1A in response to DUX4 expression. Our high-magnification images show co-localization of HIF1A in nuclei with high DUX4 expression, but not in nuclei with low DUX4 expression, suggesting that a threshold of DUX4 expression triggers downstream activation of HIF1A, the master regulator of hypoxia signaling. Co-localization and enrichment of HIF1A was similarly observed in rare DUX4+ nuclei in myotubes from a patient with FSHD (Fig. 2B). We further assessed expression of hypoxia-responsive target genes (GLUT1 and PDK1) that signal metabolic changes during hypoxia and observed that their protein expression was also upregulated in the presence of DUX4 (fig. S2). Applying a cell-permeable hypoxia detection reagent that fluoresces in intracellular environments with low oxygen concentration to MB135-DUX4i cells showed that DUX4 expression is linked to intracellular hypoxic conditions compared to control conditions (Fig. 2C).
Fig. 2. Role of hypoxia response genes in DUX4-induced cell death.
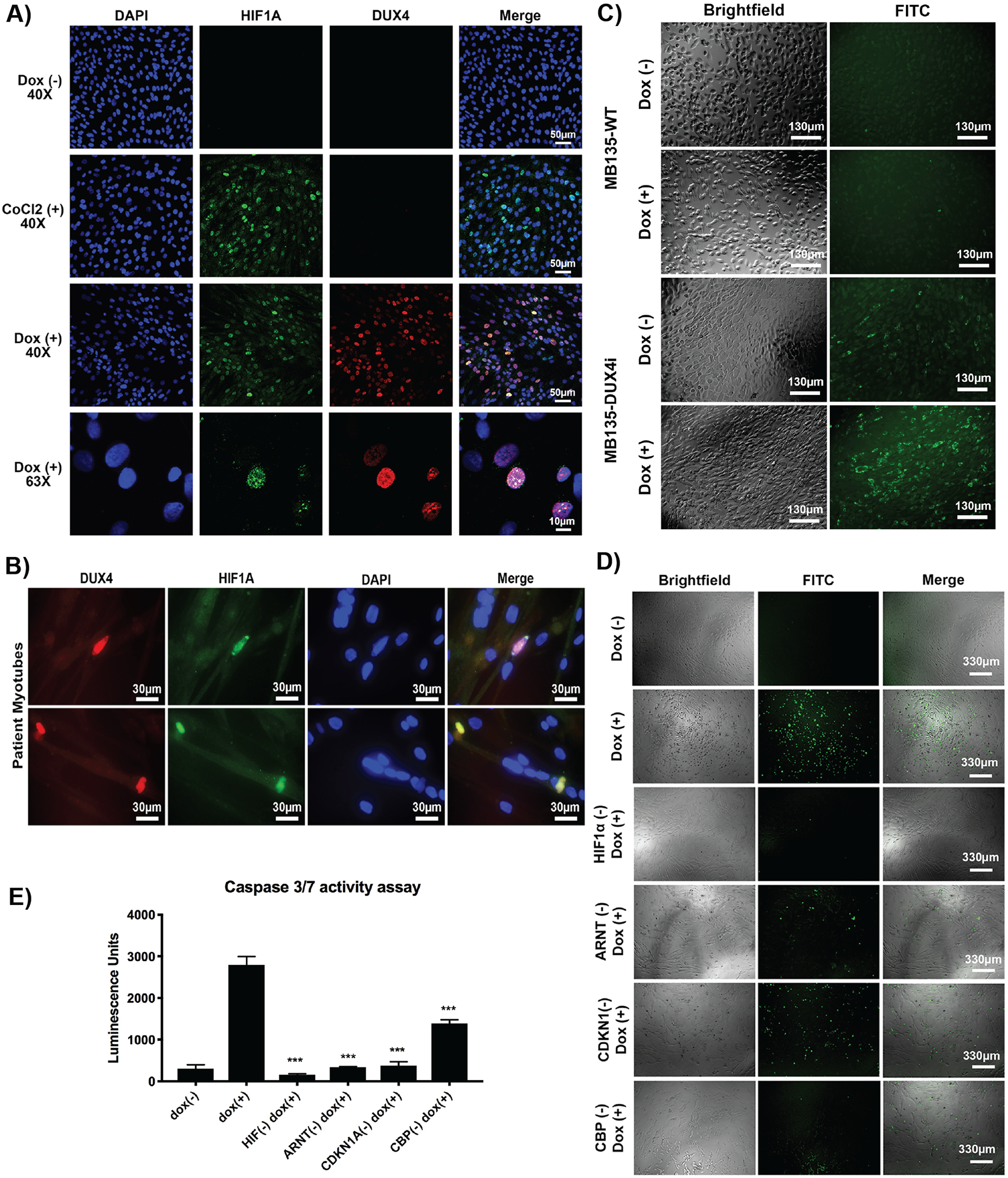
(A) Immunofluorescence of HIF1A in MB135-DUX4i cells under control (normoxia), CoCl2-treated (hypoxia), and doxycycline-treated (DUX4-activated) conditions after 48 hrs. (B) Immunofluorescence of FSHD patient myotubes (17ABIC) fixed after four days of differentiation and stained for DUX4 (red) and HIF1A (green). (C) Hypoxia reagent (green) applied to MB135-WT (top) and MB135-DUX4i (bottom) cells with and without doxycycline. (D) Individual gene knockouts were engineered into the MB135-DUX4i line. Cells were seeded and doxycycline added 6 hrs later to induce DUX4 expression for 48 hrs. CellEvent Caspase 3/7 FITC reagent was added to each well to visualize DUX4-induced cell death under a fluorescent microscope. (E) Cell death was quantified using a caspase 3/7 luminescence assay. Error bars represent standard error of the mean of six replicate wells. ***P < 0.01 using Mann-Whitney two-tailed test, compared to the original MB135-DUX4i line.
Our next investigation centered on validating the hypoxia-responsive genes emerging from our CRISPR-Cas9 screen and their link to DUX4-induced cell death. We selected sgRNA sequences that demonstrated enrichment in both screens and used these to generate single gene knockouts in the MB135-DUX4i line. Clonal populations were selected for expansion and Sanger sequenced to confirm integration of construct. Protein reduction was also confirmed via western blot (fig. S3). Using these single gene knockouts, we induced DUX4 expression and validated that they were indeed resistant to DUX4-induced cell death compared to control cells. Reduced cell death was observed qualitatively as minimal positive staining for caspase-3/7, a cell death marker, using a fluorescence detection reagent (Fig. 2D). Findings were confirmed quantitatively using a second caspase-3/7 luminescence assay (Fig. 2E). Our results demonstrate that knockout of key hypoxia signaling genes can desensitize cells to DUX4-induced toxicity and cell death. Of the many pathways activated in response to DUX4 expression, we propose that the hypoxia signaling pathway is the key driver that propagates the pathogenic signal leading to cell death.
Hypoxia signaling inhibitors attenuate DUX4-induced cell death
We next sought to mimic the DUX4 resistance property of our single gene knockout lines via a pharmacological approach, reasoning that effective compounds may serve as potential therapeutic candidates to treat FSHD. Studies have shown that the cellular hypoxia response, acting via HIF1 complex assembly and stabilization, can be disrupted using chemical inhibitors of the PI3K/Akt/mTOR (40–42) and Ras/MAPK (43, 44) signaling pathways. We tested several inhibitors’ ability to reduce DUX4-induced cell death using our MB135 iDUX4 line (Fig. 3A). Rapamycin (mTOR inhibitor) and wortmannin and LY294002 (PI3K inhibitors) are known to limit the synthesis of HIF1A protein (41). Herbimycin is used to inhibit receptor tyrosine kinases that activate PI3K/Akt/mTOR signal transduction, effectively blocking HIF1 complex stabilization (45). PD98059 and U0126 are MAPK kinase (MEK1 and MEK2) inhibitors that block HIF1A protein synthesis (46, 47). When cells were incubated in the presence of these HIF signaling inhibitors, we observed resistance to DUX4-induced cell death to varying degrees, mimicking our observation with our hypoxia gene knockout lines. The PI3K/Akt/mTOR inhibitors appeared more efficacious at a lower dose than the Ras/MAPK inhibitors and were thus selected for further analysis. Reduced cell death using these compounds was further validated using a secondary caspase 3/7 fluorescence assay (Fig 3B). We also tested the specificity of these compounds to inhibit DUX4-induced cell death in the presence of a pro-apoptotic agent, etoposide (a topoisomerase inhibitor), which causes accumulation of double-stranded DNA breaks, hence functioning via a different pathway than hypoxia-related apoptosis. Our results showed that these compounds were not effective in inhibiting etoposide-induced cell death, thus conferring specificity to inhibition of apoptotic signals derived from hypoxic-conditions underlying DUX4-induced cell death (fig. S4). Application of these inhibitors further resulted in reduced HIF1A nuclear expression, suggesting reduced overall hypoxic stress in DUX4-expressing cells (Fig 3C).
Fig. 3. Hypoxia signaling inhibitors prevent DUX4-induced cell death.
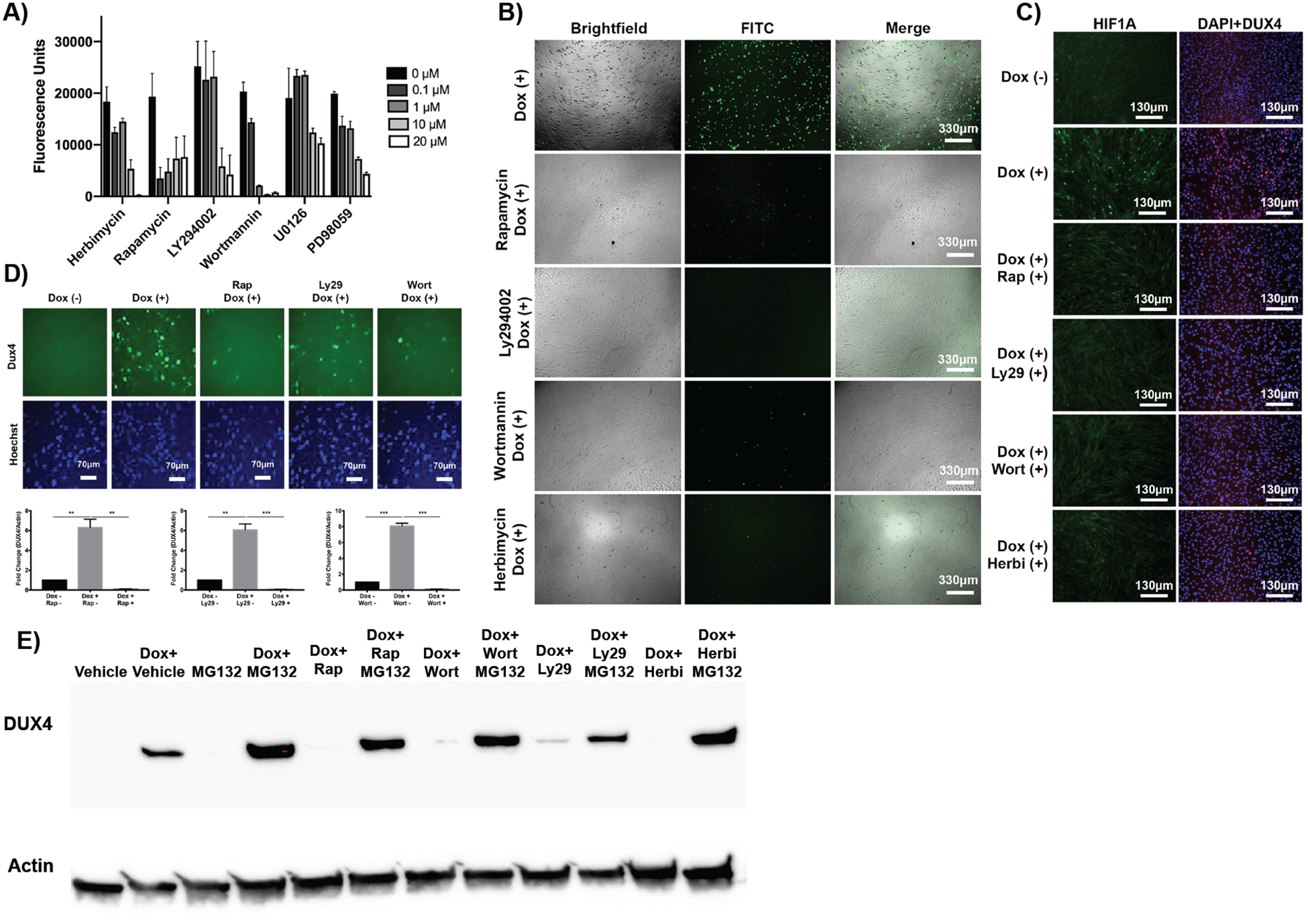
(A) Dose-response curves for known chemical inhibitors of hypoxia signaling. Cell death was quantified using a caspase 3/7 luminescence assay. (B) Fluorescence imaging analysis of DUX4-induced apoptosis in MB135-DUX4i cells incubated with PI3K/Akt/mTOR inhibitor compounds. Green cells are caspase-3/7 positive. (C) Fluorescence imaging analysis of hypoxia in MB135-DUX4i cells cultured with inhibitor compounds; HIF1A (green), DUX4 (red), DAPI (blue). (D) Immunofluorescence (P2B1 antibody, top) and quantification of western blot (E55 antibody, bottom) of DUX4 protein expression in the presence of signaling inhibitors in MB135-DUX4i cells. (E) Western blot for DUX4 protein with MG132 proteasome inhibition in the presence of inhibitors in MB135-DUX4i cells.
Unexpectedly, these inhibitors drastically reduced DUX4 protein in cells as measured by both immunofluorescence and western blot (Fig 3D). DUX4 reduction was most pronounced when inhibitors were added prior to activation of DUX4, but still exerted an effect up to 24 hrs post-DUX4 activation (fig. S5). Transcript of DUX4 was not altered in the presence of these compounds, indicating that they were not preventing the doxycycline inducibility of the DUX4 transgene itself (fig. S6). Further investigation into the reduced DUX4 protein expression in the presence of these compounds revealed an increase in DUX4 protein turnover. Application of a proteasome inhibitor (MG132) in the presence of the compounds restored DUX4 protein expression (Fig. 3E), thus indicating that these compounds are acting to increase DUX4 protein turnover to reduce the cellular hypoxia response and subsequent apoptosis.
Hypoxia signaling inhibitors reduce FSHD-related biomarkers in patient cells
Upon identifying pharmacological compounds that inhibit DUX4-induced cell death in the inducible DUX4 cell line, we next sought to investigate their potential therapeutic benefit in an in vitro model of FSHD using four different patient-derived myogenic cells. Given that DUX4 activation occurs in myotubes, we seeded patient myoblasts and switched to differentiation media to induce myoblast fusion over four days. On the fourth day, cells were treated with compounds for 12 hrs and then harvested for transcript quantification. DUX4 activation in patient cells is sporadic and infrequent, making it difficult to directly quantify DUX4 transcript and protein expression. Alternatively, as with previous published studies (48, 49), we assayed three well-established DUX4 target genes and FSHD disease biomarkers: ZSCAN4, TRIM43, and MBD3L5. The upregulation of these genes is closely linked to disease state and DUX4 expression in patients, and they are more readily detectable than DUX4 itself. Reduction in transcript expression of all three biomarker genes tested was observed across the four FSHD myogenic lines when treated with inhibitor compounds (Fig. 4, fig. S7). The observed reduction of these disease-related biomarkers to expression comparable to healthy control cells demonstrates the therapeutic potential of hypoxia-signaling inhibitors to ameliorate the molecular signatures associated with FSHD.
Fig. 4. Hypoxia signaling inhibitors reduce FSHD-related biomarkers in patient primary myotubes.
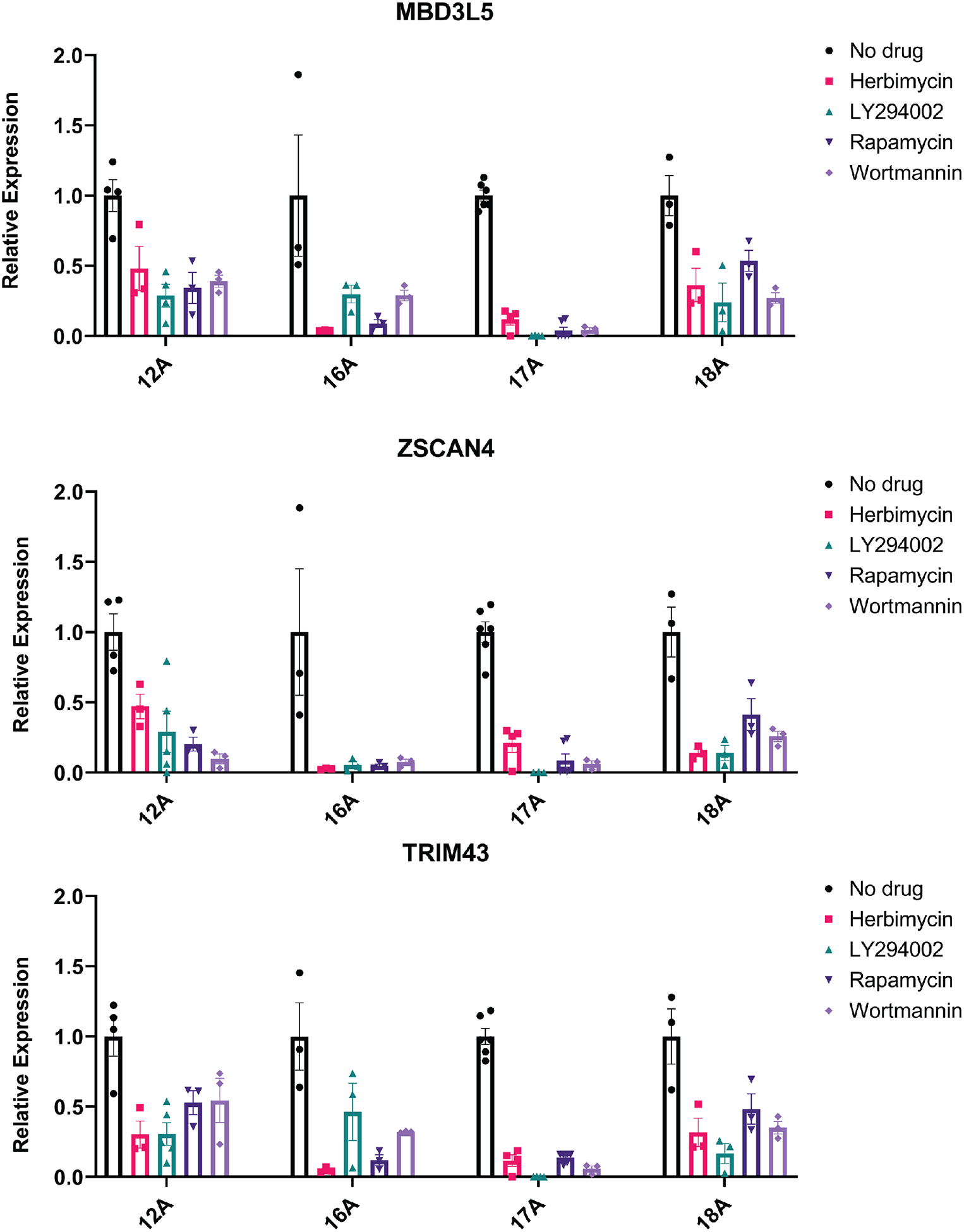
Transcript quantification of FSHD biomarkers MBD3L5, ZSCAN4, and TRIM43 from four patient donor muscle biopsies (12A, 16A, 17A, 18A from UMMS Wellstone cohort) with or without compound treatment for five days were normalized and summarized (original values of each line in fig. S7. Data are plotted as mean ± SEM. Statistical analysis: Two-way ANOVA and multiple comparisons test were performed and all treatment groups were significantly different from untreated control (P<0.0001).
Hypoxia signaling inhibitors improve phenotypes associated with DUX4 in zebrafish models
To assess the potential therapeutic benefit of hypoxia signaling inhibitor compounds in vivo, we next performed short-term drug treatments using two established DUX4 misexpression zebrafish models of FSHD (50, 51). In the first model, we induced misexpression of human DUX4 during zebrafish development by mRNA injection at the one-cell stage (50). This model results in abnormalities that recapitulate FSHD-related phenotypes including apoptosis and muscle degeneration. DUX4 mRNA injection at the one-cell stage results in asymmetric structural defects of the fin, facial, and trunk muscles with variable severity ranging from lethal to mild (Fig. 5A), as well as reduced physiological activity by day five post-fertilization. This model can be used to assess the efficacy of candidate compounds by treatment in water. Dosing of DUX4-injected eggs with rapamycin, wortmannin, and herbimycin, but not LY294002, during embryonic development reduced the ratios of affected vs unaffected fish in terms of their gross body phenotype after hatching (Fig. 5A). To assess whether these gross structural improvements translated into functional improvements, independent experiments were performed using the DanioVision platform. DUX4 injected embryos with or without inhibitor drugs in the water were individually placed in a wells of 48-well plates and fish swimming activity was recorded on day five post-fertilization. Rapamycin-, wortmannin-, and herbimycin-treated fish demonstrated a more active swim-activity heatmap compared to the untreated DUX4-injected cohort (Fig. 5B). Improvement in cumulative mobility was larger than expected given the smaller percentage improvement in gross structural defects (Fig. 5C). This suggests that the developmental structural defects in fish from the drug-treated groups were less severe in nature than those observed in the untreated DUX4-injected group, thus resulting in improved muscle function.
Fig. 5. Hypoxia signaling inhibitors ameliorate FSHD-associated phenotypes in two DUX4 zebrafish models.
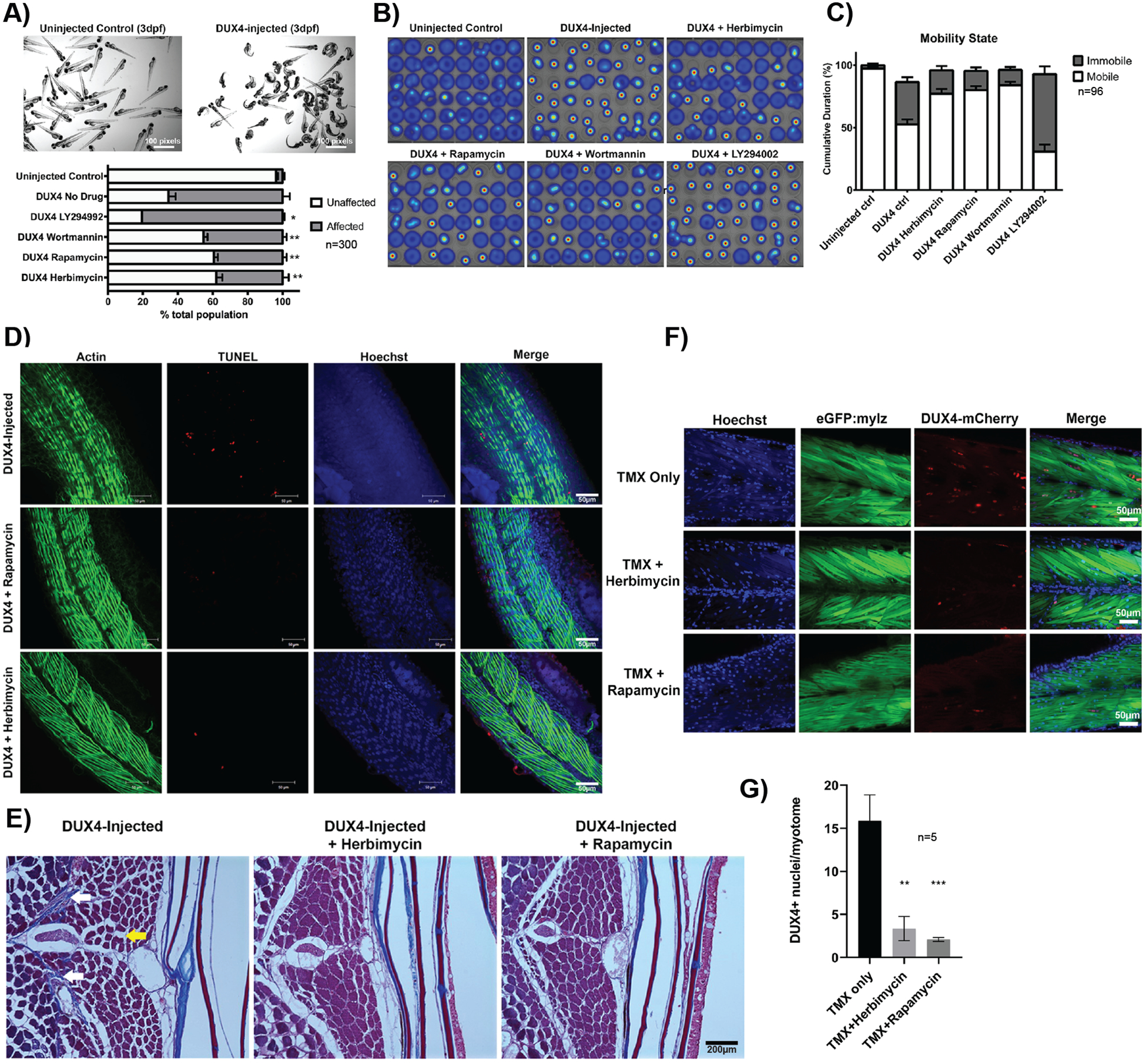
(A) Representative image of a pool of uninjected AB control and DUX4-injected zebrafish larvae at 3 days post-fertilization. Affected vs unaffected larvae counts with and without drug treatments are shown below (n=100, N=3). *P < 0.05, **P < 0.01 compared to DUX4-injected no-drug control (Mann-Whitney test across all replicates). (B) Representative heat maps of swim activity generated by DanioVision platform comparing uninjected, DUX4-injected control and DUX4-injected drug-treated fish over 15 mins. (C) Bar graph generated from swim activity data comparing ‘mobile’ vs ‘immobile’ periods for each fish over the 15 mins recording session in (B) (n=96). Data are plotted as mean ± SEM. Statistical analysis: Two-way ANOVA identified interaction between treatment and mobility with P<0.0001. Tukey’s multiple comparisons test revealed the uninjected and three drug treatment groups were all significantly different from no drug control (P<0.0001). (D) TUNEL staining (red) on DUX4-injected fish with and without drug treatment (rapamycin, herbimycin) post-drug treatment. Actin (phalloidin, green) and nuclei (hoechst, blue) are also stained. (E) Representative trichrome staining on aged DUX4-injected fish with and without inhibitors (herbimycin or rapamycin). White arrows indicate excess collagen and fibrosis, yellow arrow indicates fat infiltration. (F) Fluorescence images of DUX4-inducible fish treated with tamoxifen to activate DUX4 expression and fixed at day 4 post-fertilization. Muscle myosin light chain EGFP (green), Hoechst (blue), DUX4-mCherry (red). (G) Quantification of DUX4 positive nuclei per myotome in DUX4-induced fish with and without inhibitor treatment (n=5). Statistical analysis: One-way ANOVA and multiple comparisons test were performed, and drug treated groups were significantly different from no drug control (**P<0.01, *** P<0.001).
TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining on DUX4-injected fish revealed the presence of apoptotic fibers, which were reduced upon treatment with rapamycin and herbimycin (Fig. 5D). We subsequently aged a cohort of drug-treated and untreated DUX4-injected fish into adulthood and performed histological analysis at 8 months. Trichrome staining revealed a decrease in collagen, endomysial fibrosis, and fat infiltration in the fin muscle of herbimycin- and rapamycin-treated groups compared to the untreated DUX4-injected group (Fig. 5E). Our results indicate that drug treatment during developmental stages can prevent DUX4-induced muscle pathology in adulthood. To assess the direct effect of herbimycin and rapamycin on DUX4 expression, we used our inducible DUX4 transgenic model (51) in which the addition of tamoxifen resulted in muscle-specific expression of a DUX4-mCherry fusion protein that could be visualized (Fig. 5F). Quantification of DUX4 positive nuclei (Fig. 5G) showed decreased expression of DUX4 in drug-treated fish compared to untreated, mimicking our observation of reduced DUX4 in response to inhibitor treatment in our in vitro studies.
Discussion
Here, we used FSHD as a model disease to demonstrate how genome-wide CRISPR-Cas9 screens can be used to identify gene targets and pathways to accelerate drug discovery and testing for rare diseases. FSHD is a genetically complex type of muscular dystrophy with a shortage of therapies in development compared to other forms of muscular dystrophies (52–54). This is in part due to the lack of understanding of the link between DUX4 misexpression and disease pathology, and thus a shortage of ‘druggable’ pathways emerging from basic science research in the field. In contrast to previous transcriptome-based studies that report dysregulation of many genes and pathways in response to DUX4 expression (16, 19–22, 55), our genome-wide CRISPR-Cas9 knockout screen identified specific genes and pathways that enabled resistance to DUX4-induced cell death when modulated, thus providing a strategy to identify actionable drug targets for further characterization.
The identification of several hypoxia response genes, in particular both subunits of the HIF1 complex (HIF1A and HIF1B/ARNT), from our CRISPR-Cas9 screen led to the discovery that the cellular hypoxia response pathway plays a key pathogenic role in mediating DUX4-induced cell death. Perturbation of this signaling pathway in DUX4-expressing cells by pharmacological compounds circumvented the hypoxic response and cell death, suggesting that DUX4-associated pathogenesis in individuals with FSHD may be targeted by similar approaches.
Although the hypoxia signaling pathway has not been a targeted focus area in FSHD research, HIF1A has been cited in two previous studies. The first study is a meta-analysis performed across multiple FSHD sample datasets, which reported HIF1A as one of the most rewired genes with increased activity in FSHD (56). The second is a microarray study in which HIF-signaling genes were observed to be overrepresented among FSHD dysregulated genes (57). Our discovery opens therapeutic prospects for individuals with FSHD using existing drugs that modulate this pathway, many of which have been characterized in the context of cancer therapy (41). Although we specifically focused on inhibitors of the PI3K/Akt/mTOR pathway (58), other HIF signaling inhibitors including cardiac glycosides, topoisomerase inhibitors, and MAPK pathway inhibitors, may also exert a similar therapeutic benefit (42, 59, 60). A recent publication reported that inhibition of p38 MAPK is effective at suppressing DUX4 expression at the transcript level (61). p38 is also known to be a key regulator of the cellular hypoxic response (62–64), and thus inhibition of MAPK may also exert beneficial effects on the downstream pathogenicity associated with hypoxia signaling.
For rapid clinical translation, testing should prioritize FDA-approved HIF signaling inhibitors (42), with specific focus on those that are suitable for lifetime dosing in patients, such as rapamycin, and demonstrate efficacy in FSHD mouse models. These candidate inhibitors may be effective as a prophylactic to promote DUX4 protein turnover and/or minimize pathogenic signals that may lead to cell or myofiber death in patients throughout the course of their lives. Our finding that FSHD biomarker gene expression can be reduced in patient myotubes with the addition of inhibitor compounds post-differentiation, when myonuclei have been primed for DUX4 activation, indicates that the inhibitors have the potential to exert a therapeutic benefit even when applied after initiation of DUX4-triggered pathogenic signals.
Further fine mapping of the molecular signals linking hypoxia signaling and cell death in the context of DUX4 expression will be pursued in future studies and is anticipated to provide more in-depth mechanistic insight into FSHD pathogenicity and additional therapeutic targets. Specifically, the role of p21 as a downstream mediator of hypoxia-signaling (p53-independent) in DUX4-expressing cells warrants further investigation, given that several studies have reported upregulation of p21 in FSHD cell and animal models (65–67). In addition, the identification of CBP as a candidate gene emerging from our knockout screen supports recent findings that p300/CBP compound inhibitors prevent DUX4-induced cytotoxicity (68). Previous literature identifying DUX4 interaction with co-activators p300/CBP to mediate chromatin opening and transcriptional activation of target genes may explain the link between DUX4 expression and activation of the hypoxia signaling cascade that ultimately results in cell death (69). Furthermore, validation of other unexplored hits from our CRISPR-Cas9 screen will likely reveal more druggable pathways to test in patient cells and in vivo models of FSHD. Several of our other top CRISPR-Cas9 screen hits (DOT1L, SIN3B, AMOTL2) are found highly expressed in the testis, correlating with reported germ line expression of DUX4 in the testis – known as an active area of developmental apoptosis (13, 18). Other gene clusters emerging from our screen with potential functional correlation include two groups of transcriptional activation genes: MED24, MED25, and MED16, and TADA1, TADA2B, and TAF6L.
Although we have successfully identified a potential therapeutic avenue for FSHD, there are limitations to the screening approach. First, the screen was based on cells surviving a toxic dose of DUX4 expression and DUX4-induced cell death may not be the sole factor contributing to disease pathology in patients with FSHD. This suggests that preventing DUX4-induced cell death may not be entirely effective in halting FSHD pathology in patients, and that additional studies may be required to further elucidate the complete underlying pathogenic mechanism prior to clinical translation. The second limitation is inherent in the method of DUX4 induction used in our CRISPR-Cas9 screen, which involves the use of doxycycline-inducible transcriptional activation (Tet-On system). In our pilot CRISPR-Cas9 screens in which baculovirus was used to induce widespread DUX4 expression, we observed enrichment of known virus uptake genes (B3GAT3, SLC35B2, B4GALT7)(70). These genes were interpreted to be false positive hits given that their knockout would presumably interfere with baculovirus entry into cells, thus circumventing DUX4 expression to yield a false positive ‘resistance’ outcome. Likewise, it is possible that some of our CRISPR-Cas9 screen hits derived using the dox-inducible DUX4 cell line may perturb the activation of the Tet-On system, thus inhibiting DUX4 transcription to yield a false positive hit. Our control experiments have demonstrated that perturbation of the hypoxia signaling pathway does not inhibit the dox-inducibility of the DUX4 transgene. As we have demonstrated, assessment of DUX4 transcript expression for candidate compounds during screen validation steps is important to avoid false positive hits.
In conclusion, our knockout CRISPR-Cas9 screen produced a valuable resource for mining potential therapeutic targets for FSHD. Further mapping of functional associations between hits may yield additional ‘druggable’ pathways. Although we have identified several therapeutic targets from our screen, our current dataset is limited to DUX4 resistance conferred by inactivation of genes. Future screening approaches using CRISPR-Cas9 technology can be used to systematically discover DUX4 resistance acquired through other types of genetic perturbations to reveal additional therapeutic targets. Our successful demonstration of using genome-wide CRISPR-Cas9 screen as a therapeutics discovery platform for FSHD may serve as a model for finding potential druggable targets for other rare genetic diseases with a high-throughput assayable cellular phenotype.
Materials and Methods
Study design
The objective of this study was to identify key drivers of DUX4-induced cell death that can yield insight into FSHD pathogenesis and serve as therapeutic targets. We performed a genome-wide CRISPR screen identifying genes whose knockout conferred survival in the presence of DUX4 misexpression. To select gene candidates for follow-up studies, we performed pathway analysis and identified several candidates involved in the regulation of the cellular hypoxic response pathway. We subsequently used a combination of genetic and pharmacologic tools to validate these candidates. Single gene knockout cell lines and pharmacological compounds targeting the hypoxia response pathway were used in the presence of DUX4 to confirm resistance. To test compounds for potential efficacy on three FSHD disease biomarkers, we used four myogenic lines derived from patients. To test the efficacy of these compounds in an in vivo model, we selected two different DUX4 expression models in zebrafish: mRNA injection and an inducible transgenic model. Doses of each compound were selected at the maximal non-toxic concentration in wild type fish embryos. Sample size was calculated to achieve 80% power with a type I error rate of 5% and an anticipated difference of ~20% in percentage of larvae pathology. The duration of treatment was determined before starting each experiment. Fish embryos were randomly assigned to control or treatment groups. Assays were performed non-blinded. All data points available were included in the final analysis.
GeCKOv2 library preparation and lentivirus packaging
GeCKO human libraries A & B were obtained from Addgene and amplified according to provided protocol (71). Briefly, library components A & B were electroporated into NEB DH5a cells and plated across multiple plates for overnight growth. Once >50X colonies per construct for each library component were established, colonies were collected across all plates and resuspended in LB for maxi prep. For lentivirus packaging of plasmid library, HEK293T cells were grown to 60% confluence in nine T225 flasks. Cells in each flask were transfected with 20 μg library A, 20 μg library B, 20 μg pVSV-g (envelope plasmid), 30 μg pspax2 (packaging plasmid) using Fugene transfection reagent. After 72 hrs, supernatant was collected and filtered using a 0.45μm vacuum filter and incubated with Lenti-X concentrator (Clontech) overnight. The next day, the supernatant was centrifuged at 1800 g for 1 hr at 4°C, and the resulting pellet was resuspended in DMEM and stored in −80°C. Virus preps were titered to determine a MOI range of 0.3–0.4 prior to screen.
Genome-wide knock-out screen for DUX4 resistance
Genome-wide knock-out screen was conducted using an immortalized human myoblast line with a doxycycline-inducible DUX4 transgene (MB135-DUX4i blasticidin). For each screen, 5 × 106 cells were plated across eleven T150 plates – ten plates were transduced with GeCKOv2 lentivirus and one was used as the untransduced control. The next day, media with puromycin (2 μg/ml) were added to all plates. After all cells had died in the control plate, remaining live cells were split between sixteen T150 plates – eight were harvested and frozen down as Early Time Point (ETP) sample as soon as they reached 4 × 107 cells (300X library representation), while the rest were maintained for an additional week (passaged to ensure no less than 300X library threshold). Next, doxycycline (2 μg/ml) was added to activate DUX4 expression. Remaining DUX4 ‘resistant’ cells were expanded for two weeks and frozen down upon reaching 4 × 107 cells. Genomic DNA extraction was performed on both ETP and DUX4 populations using protocols described in Chen et al. (28). PCR reactions were performed to amplify sgRNA insert and attach sequencing barcodes, also according to protocols in Chen et al. (28).
Next Generation sequencing and analysis
Experimental replicate samples from two independent screens consisting of two ETP samples and two corresponding DUX4 resistant samples were multiplexed and sent to Broad Institute for sequencing using the Illumina Nextseq-75 platform to produce 75 bp single end reads on one lane of a flowcell. PhiX was added at 35% due to low sample complexity. The sequencing data was demultiplexed using custom scripts and the 20 bp sgRNA sequences were aligned to the GeCKOv2 library sequences using bowtie. A custom script was used to count the aligned reads to each of the sgRNA and MAGeCK was then used to identify genes in DUX4 that were enriched relative to the ETP. The sequencing data generated from the screen and raw sgRNA counts have been deposited to GEO series GSE133332. This includes the output of MAGeCK and R code to generate Fig. 1, C and D: https://github.com/leklab/fshd_ko_screen. The 18 genes that were significant (p<0.01) in both experiments (Fig. 1D) were entered into GeneMania web interface (http://genemania.org) selecting only pathway analysis. The HIF-signaling pathway was highlighted as the most significant gene set (FDR = 0.000392) in the network topology diagram (Fig. 1E).
Generation of single CRISPR-Cas9 knockouts
Guide sequences (table S1) used to generate single gene knock-out lines were cloned into the lenti-CRISPR_v2 plasmid backbone (Addgene #52961) and packaged into lentivirus similar to GeCKO library preparation. To create individual knock-out lines, MB135-DUX4i cells were transduced with lentivirus for CRISPR-Cas9s targeting each gene. The next day, cells were put on puromycin selection (2 μg/ml) for one week. To generate clonal lines, cells were diluted to a concentration of 5 cells in 10 ml and distributed evenly across a 96-well plate. After 3 weeks, clonal populations were selected and harvested for western blot quantification of protein knock-out.
Western blot analysis
Cells were harvested in RIPA (Thermofisher #89900) buffer supplemented with Protease Inhibitor cocktail (Thermofisher #32963) and PhosStop phosphatase inhibitor (Thermofisher # 4906845001). Protein assay was performed using DC assay kit (Bio-Rad #5000112). Samples were run on a 4–20% TGX gel (Bio-Rad #4561096) and transferred using the Bio-Rad Transblot Turbo. Membranes were blocked in 5% milk in TBS-tween-20 before addition of primary antibodies overnight at 4°C on a rocker. Primary antibody list and concentrations are detailed in table S2. After TBS-tween-20 washes, secondary HRP antibody (1:2000) was added, and blots were incubated for 1 hr at room temperature on a rocker. After additional TBS-tween-20 washes, membranes were incubated in ECL blotting substrate (Bio-Rad #1705062) for 5 mins, and imaged using Bio-Rad Chemidoc. Protein quantification and analysis was performed using Bio-Rad’s Image Lab software and GraphPad.
Apoptosis measurements
Apoptosis assays were conducted on cells seeded in clear 96-well plates (10,000 cells per well). For testing of signaling inhibitor drugs, cells were resuspended in respective drugs prior to seeding: Rapamycin (1 μM, Cell Signaling #9904S), Herbimycin (10 μM, Santa Cruz #3516), Wortmannin (1 μM, Cell Signaling #9951S), LY294002 (1 μM, Cell Signaling #9901S). To induce DUX4 expression, doxycycline (2 μg/ml) was added 6 hrs later. For caspase 3/7 fluorescence visualization, a single drop of the CellEvent caspase 3/7 reagent (Thermofisher #R37111) was added to each well 48 hrs after addition of doxycycline and incubated in a tissue culture incubator for 15 mins. The Echo Revolve microscope was used to visualize and image fluorescence in the FITC channel for caspase 3/7 apoptotic cells. For caspase 3/7 quantification assays, the Caspase-Glo 3/7 (Promega #G8090) luminescence reagent was added to cells 48 hrs after addition of doxycycline. The Biotek plate reader was used to take measurements on the luminescence channel every 30 mins for 2 hrs. Peak luminescence values were used for analysis.
Immunofluorescence
5×104 cells per chamber were plated on 4-chambered slides. The following day, media was replaced with media containing the indicated test compounds. Four hours later, 2 μg/mL doxycycline was added to the relevant samples. 24 hrs after the addition of DOX, samples were washed with PBS and fixed with 4% paraformaldehyde in PBS for 10 mins at room temperature with agitation. Slides were then washed 3 × 3 mins with PBS and were permeabilized for 15 mins with 0.1% Triton X-100 in PBS. Slides were blocked for at least one hour in 2% blocking buffer (2% horse serum, 2% goat serum, 2% BSA, 0.2% Triton X-100 in PBS) and were incubated overnight with primary antibodies in 2% blocking buffer at 4˚C with agitation. Slides were washed 3 × 3 mins with PBS, incubated with 1:300 dilution of Alexa Fluor secondary antibody for 2 hrs at RT with agitation, and stained with 1 μg/ml Hoechst for 10 min and mounted using fluoro-gel, or mounted using VECTASHIELD with DAPI. Imaging was performed using a Leica DMR fluorescence microscope or the Echo Revolve. Confocal microscopy was performed on selected samples using the Leica SP5 at the Yale CCMI Advanced Light Microscopy core.
Hypoxia detection experiments
MB135-DUX4i cells were plated at 90% confluence in a 96-well plate and doxycycline (2 μg/ml) added after cells had attached. Image-iT Green hypoxia reagent (Thermofisher #I14834) was added 24 hrs later (5 μM final concentration) and cells were incubated at 37°C for 1 hr before imaging. Green fluorescence was visualized and captured using the Echo fluorescent microscope.
Cell culture
MB135-DUX4i myoblast line was kindly provided by Prof. S. Tapscott (University of Washington). Cells were cultured in F10 media (Thermofisher #11550–043) supplemented with 20% FBS (Atlanta Biologicals), antibiotics (Thermofisher #15240062), 1μM dexamethasone (Sigma #D4902), 10 ng/ml Fibroblast Growth Factor (Promega #G5071). DUX4 expression was induced by addition of 2 μg/ml doxycycline hyclate (Sigma #D9891). Primary myogenic lines (17U, 17A, 18A, 12A, 16A) were kindly provided by the Wellstone Center for FSHD research (University of Massachusetts, Worcester) and were cultured in skeletal muscle growth media (Promocell #C-23060) supplemented with 20% FBS (Atlanta Biologicals #S11150) and antibiotics (Thermofisher #15240062). To induce myotube formation in primary lines, cells were switched to skeletal muscle differentiation media (Promocell #C-23061).
DUX4 expression analysis
Drug compounds were added to MB135-DUX4i media (5 μM wort, 10 μM Ly29, 10 nM Rapa, 0.1% DMSO vehicle). After four hours, doxycycline was added to the appropriate samples. Samples were harvested 24 hrs later for RNA extraction and 0.3 μg of total RNA was used for cDNA synthesis. 10 ng of cDNA was used as template in PCR reactions using DUX4 and GAPDH primers listed in Table S3.
FSHD biomarker analysis
Cells from four different individuals with FSHD (17A, 18A, 12A, 16A) and control (17U) lines from the UMMS Wellstone Center biobank were used for biomarker analysis. Cells were grown to confluence in growth media before switching to differentiation media for four days. On day four, inhibitor compounds were added over night and cells were harvested for RNA extraction the next day. TRIzol total RNA extraction (Thermofisher #15596018) and SuperScript III (Thermofisher #18080085) cDNA synthesis were employed according to manufacturer’s protocols. Biomarker qPCR reactions were performed using Taqman Master Mix (Thermofisher #4304437) and run on a StepOnePlus Real-Time PCR System (Thermofisher). Results were analyzed using StepOne Software, and visualized graphically using Microsoft Excel and Graphpad Prism 8. Specific Taqman assays for ZSCAN4 (Hs00537549_m1, Thermofisher), MBD3L5 (Hs04190573_mH, Thermofisher) and TRIM43 (Hs00299174_m1, Thermofisher) were used. Expression was normalized to PPIA (Hs99999904_m1, Thermofisher), which was found to not change in response to drug treatment. Reactions were set up in duplicate and repeated. Biomarker expression of each compound treatment group was normalized to the untreated cells from the same donor before being summarized in final figures since baseline quantities of biomarker expression vary from patient to patient.
Zebrafish drug studies using DUX4-injection model
Zebrafish (Danio Rerio) wild-type AB strain was maintained in the Boston Children’s Hospital Zebrafish facility under approved protocols. Zebrafish embryos were raised at 28.5°C and cared for as previously described (50, 51). Briefly, for DUX4-injection model, human DUX4 mRNA was synthesized by in vitro transcription. AB fish embryos at the one-cell stage were injected with 0.1 pg of DUX4 mRNA and 0.1% phenol red. DUX4 injected eggs were mixed and randomly assigned to drug or no-drug groups. Drug-treated groups received inhibitor compound in their fish water at the following concentrations: rapamycin at 100 nM, LY294002 at 10 μM, wortmannin at 50 nM, and herbimycin at 10 nM, starting at 6 hrs post-injection and maintained for 5 days. Dead eggs were cleared each day and recorded. On day 5, fish were phenotyped by examination under a light microscope. Each drug was tested in three replicates of 100 eggs and across three independent repeats. To assess swim activity, DUX4 injected eggs were harvested and randomly assigned to treatment groups. At day 1, individual eggs were selected at random to fill a 48-well plate. The DanioVision platform was used to record swimming activity for 15 mins in the dark on day 5. Distance and velocity data were collected and analyzed according to the manual. Heatmaps and mobility state data were produced by the DanioVision software. Adult DUX4-injected fish with or without drug treatment were harvested at 8 months (n=3 each group), fixed in Bouin’s solution, and processed for paraffin embedding and sectioning. Histological analysis with H & E and Masson Trichrome stainings were performed on slides as previously described (51). TUNEL staining was performed on zebrafish embryos at 24 hrs post-drug treatment according to the Click-iT Plus (Thermofisher #C10617) TUNEL assay user guide.
Zebrafish drug studies using inducible transgenic model
Inducible DUX4 transgenic strain was used to visualize and quantify DUX4 protein expression in response to herbimycin and rapamycin treatment. This strain was generated as previously described (51). Briefly, tamoxifen addition resulted in CreERt2-mediated loxP excision of the mylz-EGFP signal and subsequent activation of the DUX4-mCherry fusion transgene, thus turning on quantifiable DUX4 expression in the fish muscle. All eggs were switched to water with 20 μM of tamoxifen at 6 hrs post fertilization and randomly assigned to drug or no-drug groups. In the drug-treated groups, at 12 hrs post fertilization rapamycin was added to the tamoxifen water at a final concentration of 100 nM and herbimycin at 10 nM. Tamoxifen was removed from water after 24 hrs while drugs were maintained until day 4. On day 4, fish were sorted to isolate those that contain both GFP-positive heart (Cre transgene) and GFP-positive body (DUX4-inducible transgene), and then euthanized and fixed in PFA. Nuclei were visualized with Hoechst dye (1:5000). Muscle (mylz-EGFP, green) and DUX4 (DUX4-mCherry, red) labeling in the transgenic line did not require any additional antibodies for visualization. To visualize DUX4 expression, images of fluorescence slides were captured on a Zeiss LSM 700 laser scanning confocal microscope at BCH Cellular Imaging core.
Statistical analysis
Statistical analysis was performed in GraphPad Prism 8. Specifically, Mann-Whitney U (two-tailed test) was used to determine significance for caspase assays (p<0.01); for the primary cell biomarker analysis, two way ordinary ANOVA (with treatment and cell line as two factors) and Holm-Sidak’s multiple comparison to the untreated control group was performed; and for the zebrafish drug studies analysis, one way Welch and Brown-Forsythe ANOVA test and Dunnett’s multiple comparison to the no drug control group was performed. P values from the analysis are included in the figure legends.
Supplementary Material
Fig. S1. GeneMania functional associations between top screen hits
Fig. S2. Changes in GLUT1 and PDK1 expression in the presence of DUX4
Fig. S3. Western blot protein quantification of single gene knock-outs
Fig. S4. Effect of inhibitors on etoposide-induced cell death
Fig. S5. Western blot time-course of rapamycin dosing
Fig. S6. DUX4 transcript expression with inhibitor treatment
Fig. S7. FSHD biomarkers expression in four patient lines with inhibitor treatment
Table S1. Oligo sequences for single gene knock-out lines
Table S2. Antibody concentrations
Table S3. Primer sequences used for RT-PCR
Acknowledgments:
We thank Prof. S. Tapscott for kindly sharing the MB135-DUX4i blast line. FSHD patient lines were shared by Prof. C. Emerson’s lab and the Wellstone Center for FSHD.
Funding:
AL is funded by the MDA Development Grant (MDA514330) and was previously supported by the FSH Society (FSHS-82015-04). YZ is funded by American Heart Association postdoctoral fellowship (AHA18POST34070039). JC is funded through The Chris Carrino Foundation for FSHD and an MDA Development Grant (MDA631018). NES is supported by NIH/NHGRI (R00HG008171, DP2HG010099), NIH/NCI (R01CA218668), DARPA (D18AP00053), the Sidney Kimmel Foundation, the Melanoma Research Alliance, and the Brain and Behavior Foundation. LMK is funded by an MDA research grant (MDA381140). AL, LMK, KRW, ODK are funded through the NIH P50 grant (2P50HD060848-12). PLJ is funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (R01AR062587). The cellular imaging core at BCH is supported by IDDRC NIH grant (5U54HD090255-04).
Footnotes
Competing interests:
LMK and AL filed a patent application (WO/2019/051290) based on data presented here. NES consults for Vertex, Pfizer and Agenus. LMK consults for Sarepta and Dyne Therapeutics. PLJ is on the Scientific Advisory Board for Fulcrum Therapeutics.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. The sequencing data generated from the screen and raw sgRNA counts have been deposited to GEO series GSE133332.
References
- 1.Deenen JCW, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJGM, Bakker E, Weinreich SS, Verbeek ALM, van Engelen BGM, Population-based incidence and prevalence of facioscapulohumeral dystrophy., Neurology 83, 1056–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, Gruter A, Hofker MH, Moerer P, Williamson R, van Ommen GB, Padberg GW, Frants RR, Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy, Nat. Genet 2, 26–30 (1992). [DOI] [PubMed] [Google Scholar]
- 3.van Deutekom JCT, Bakker E, Lemmers R, van der Wielen MJR, Bik E, Hofker MH, Padberg GW, Frants RR, Evidence for subtelomeric exchange of 3. 3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1, Hum. Mol. Genet 5, 1997–2003 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Winokur ST, Bengtsson U, Feddersen J, Mathews KD, Weiffenbach B, Bailey H, Markovich RP, Murray JC, Wasmuth JJ, Altherr MR, Schutte BC, The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: implications for a role of chromatin structure in the pathogenesis of the disease, 2, 225–234 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Himeda CL, Jones PL, The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy, Annu. Rev. Genomics Hum. Genet, 1–27 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Lemmers RJLF, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM, A unifying genetic model for facioscapulohumeral muscular dystrophy., Science 329, 1650–3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemmers RJLF, Tawil R, Petek LM, Balog J, Block GJ, Santen GWE, Amell AM, van der Vliet PJ, Almomani R, Straasheijm KR, Krom YD, Klooster R, Sun Y, den Dunnen JT, Helmer Q, Donlin-Smith CM, Padberg GW, van Engelen BGM, de Greef JC, Aartsma-Rus AM, Frants RR, de Visser M, Desnuelle C, Sacconi S, Filippova GN, Bakker B, Bamshad MJ, Tapscott SJ, Miller DG, van der Maarel SM, Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2., Nat. Genet 44, 1370–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Boogaard ML, Lemmers RJLF, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, van der Vliet PJ, Straasheijm KR, van den Akker RFP, Kriek M, Laurense-Bik MEY, Raz V, van ostaijen-ten MM, Hansson KBM, van der Kooi EL, Kiuru-Enari S, Udd B, van Tol MJD, Nishino I, Tawil R, Tapscott SJ, van Engelen BGM, van der Maarel SM, Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy, Am. J. Hum. Genet, 1020–1029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabriels J, Beckers MC, Ding H, De Vriese A, Plaisance S, Van Der Maarel SM, Padberg GW, Frants RR, Hewitt JE, Collen D, Belayew A, Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element, Gene 236, 25–32 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, Sauvage S, Mattéotti C, van Acker AM, Leo O, Figlewicz D, Barro M, Laoudj-Chenivesse D, Belayew A, Coppée F, Chen Y-W, DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1., Proc. Natl. Acad. Sci. U. S. A 104, 18157–62 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hewitt JE, Lyle R, Clark LN, Valleley EM, Wright TJ, Wijmenga C, Van Deutekom JCT, Francis F, Sharpe PT, Hofker M, Frants RR, Williamson R, Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy, 3, 1287–1295 (1994). [DOI] [PubMed] [Google Scholar]
- 12.Hendrickson PG, Doráis JA, Grow EJ, Whiddon JL, Lim J, Wike CL, Weaver BD, Pflueger C, Emery BR, Wilcox AL, Nix DA, Peterson CM, Tapscott SJ, Carrell DT, Cairns BR, Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL / HERVL retrotransposons, Nat. Publ. Gr 49, 925–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG, Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene., PLoS Genet. 6, e1001181 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, Simsek T, Garner HR, Tassin A, Perlingeiro RR, Kyba M, An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies, EMBO J. 27, 2766–2779 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallace LM, Liu J, Domire JS, Garwick-Coppens SE, Guckes SM, Mendell JR, Flanigan KM, Harper SQ, RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy., Mol. Ther 20, 1417–23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rickard AM, Petek LM, Miller DG, Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways., Hum. Mol. Genet, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Mattéotti C, Arias C, Corona ED, Nuñez NG, Leo O, Wattiez R, Figlewicz D, Laoudj-Chenivesse D, Belayew A, Coppée F, Rosa AL, The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein, Neuromuscul. Disord 17, 611–623 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Shadle SC, Zhong JW, Campbell AE, Conerly ML, Jagannathan S, Wong CJ, Morello TD, van der Maarel SM, Tapscott SJ, DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy, PLoS Genet. 13, 1–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao Z, Snider L, Balog J, Lemmers RJLF, Van Der Maarel SM, Tawil R, Tapscott SJ, DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle., Hum. Mol. Genet 23, 5342–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, van der Maarel SM, Ruzzo WL, Gentleman RC, Tawil R, Tapscott SJ, DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy., Dev. Cell 22, 38–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Den Heuvel A, Mahfouz A, Kloet SL, Balog J, Van Engelen BGM, Tawil R, Tapscott SJ, Van Der Maarel SM, Single-cell RNA sequencing in facioscapulohumeral muscular dystrophy disease etiology and development, Hum. Mol. Genet 28, 1064–1075 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jagannathan S, Shadle SC, Resnick R, Snider L, Tawil RN, Van Der Maarel M, Bradley RK, Tapscott SJ, Model systems of DUX4 expression recapitulate the transcriptional profile of FSHD cells, Hum. Mol. Genet 25, 4419–4431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young JM, Whiddon JL, Yao Z, Kasinathan B, Snider L, Geng LN, Balog J, Tawil R, van der Maarel SM, Tapscott SJ, DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis., PLoS Genet. 9, e1003947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knopp P, Krom YD, Banerji CRS, Panamarova M, Moyle LA, Den Hamer B, DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis, J. Cell Sci, 3816–3831 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Q, Snider L, Jagannathan S, Tawil R, van der Maarel SM, Tapscott SJ, Bradley RK, A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy., Elife 4, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, Guttridge D, Yang J, Harper SQ, DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo., Ann. Neurol 69, 540–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shalem O, Sanjana NE, Hartenian E, Shi X, a Scott D, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, Genome-scale CRISPR-Cas9 knockout screening in human cells., Science 343, 84–7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, Lee H, Zhang F, Sharp PA, Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis, Cell 160, 1246–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X, Robitaille M, Brown KR, Jaksani S, Overmeer R, Boj SF, Adams J, Pan J, Clevers H, Sidhu S, Moffat J, Angers S, Genome-wide CRISPR screens reveal a Wnt – FZD5 signaling circuit as a druggable vulnerability of RNF43 -mutant pancreatic tumors, 23 (2017), doi: 10.1038/nm.4219. [DOI] [PubMed] [Google Scholar]
- 30.Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe-molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia-beltran W, Piechocka-trocha A, Krogan NJ, Marson A, Sabatini DM, Lander ES, A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors, Nat. Genet, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sidik SM, Huet D, Ganesan SM, Carruthers VB, Niles JC, Lourido S, Sidik SM, Huet D, Ganesan SM, Huynh M, Wang T, Nasamu AS, Article A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes Article A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes, Cell 166, 1423–1430.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han J, Perez JT, Chen C, Andrade J, Manicassamy B, Han J, Perez JT, Chen C, Li Y, Benitez A, Kandasamy M, Lee Y, Genome-wide CRISPR / Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication Resource Genome-wide CRISPR / Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication, CellReports 23, 596–607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim HS, Lee K, Bae S, Park J, Lee C, Kim E, Kim M, Kim S, Kim C, Kim J, CRISPR/Cas, CRISPR screen, Whole Genome Sequencing, RNA virus, Poliovirus, Enterovirus D68, host - pathogen interaction, sialic acid, 1–16 (2017). [Google Scholar]
- 34.Kramer NJ, Haney MS, Morgens DW, Jovičić A, Couthouis J, Li A, Ousey J, Ma R, Bieri G, Tsui CK, Shi Y, Hertz NT, Tessier-lavigne M, Ichida JK, Bassik MC, Gitler AD, CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity, Nat. Genet 50 (2018), doi: 10.1038/s41588-018-0070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones TI, Chen JCJ, Rahimov F, Homma S, Arashiro P, Lou Beermann M, King OD, Miller JB, Kunkel LM, Emerson CP, Wagner KR, Jones PL, Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis., Hum. Mol. Genet 21, 4419–30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tassin A, Laoudj-Chenivesse D, Vanderplanck C, Barro M, Charron S, Ansseau E, Chen Y-W, Mercier J, Coppée F, Belayew A, DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy?, J. Cell. Mol. Med 17, 76–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warde-farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, Maitland A, Mostafavi S, Montojo J, Shao Q, Wright G, Bader GD, Morris Q, The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function, 38, 214–220 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majmundar AJ, Wong WJ, Simon MC, Review Hypoxia-Inducible Factors and the Response to Hypoxic Stress, Mol. Cell 40, 294–309 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dengler VL, Galbraith MD, Espinosa JM, Dengler VL, Galbraith MD, Espinosa JM, Transcriptional regulation by hypoxia inducible factors, Crit. Rev. Biochem. Mol. Biol 9238 (2014), doi: 10.3109/10409238.2013.838205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris AL, Radcliffe J, HYPOXIA — A KEY REGULATORY FACTOR IN TUMOUR GROWTH, 2, 38–47 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Masoud GN, Li W, HIF-1 α pathway: role, regulation and intervention for cancer therapy, Acta Pharm. Sin. B 5, 378–389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu C, Huang R, Khuc T, Shou D, Bullock J, Griffin S, Zou C, Little A, Astley H, Xia M, Identification of approved and investigational drugs that inhibit hypoxia-inducible factor-1 signaling, Oncotarget 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wigerup C, Påhlman S, Bexell D, Pharmacology & Therapeutics Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer, Pharmacol. Ther 164, 152–169 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J, MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300, J. Biol. Chem 278, 14013–14019 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fisher RNÃ,BJ, Iii AAF, Regulation of hypoxia inducible factor-1 by nitric oxide in contrast to hypoxia in microvascular endothelium, FEBS Lett. 549, 99–104 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Fukuda R, Hirota K, Fan F, Do Jung Y, Ellis LM, Semenza GL, Insulin-like Growth Factor 1 Induces Hypoxia-inducible Factor 1-mediated Vascular Endothelial Growth Factor Expression, Which is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells *, 277, 38205–38211 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Liu L, Ning X, Han S, Zhang H, Sun L, Shi Y, Sun S, Guo C, Yin F, Qiao T, Wu K, Fan D, Hypoxia induced HIF-1 accumulation and VEGF expression in gastric epithelial mucosa cells: Involvement of ERK1/2 and PI3K/Akt, Mol. Biol 42, 403–412 (2008). [PubMed] [Google Scholar]
- 48.Zhang Y, King OD, Rahimov F, Jones TI, Ward CW, Kerr JP, Liu N, Emerson CP, Kunkel LM, a Partridge T, Wagner KR, Human skeletal muscle xenograft as a new preclinical model for muscle disorders., Hum. Mol. Genet 23, 3180–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen JC, King OD, Zhang Y, Clayton NP, Spencer C, Wentworth BM, Emerson CP, Wagner KR, Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics., Mol. Ther, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitsuhashi H, Mitsuhashi S, Lynn-Jones T, Kawahara G, Kunkel LM, Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy., Hum. Mol. Genet 22, 568–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pakula A, Lek A, Widrick J, Mitsuhashi H, Gwilt KMB, Gupta VA, Rahimov F, Criscione J, Zhang Y, Gibbs D, Murphy Q, Manglik A, Mead L, Kunkel L, Transgenic zebraf ish model of DUX4 misexpression reveals a developmental role in FSHD pathogenesis, Hum. Mol. Genet 28, 320–331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aartsma-Rus A, Straub V, Hemmings R, Haas M, Schlosser-Weber G, Stoyanova-Beninska V, Mercuri E, Muntoni F, Sepodes B, Vroom E, Balabanov P, Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues, Nucleic Acid Ther. 27, 251–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crone M, Mah JK, Current and Emerging Therapies for Duchenne Muscular Dystrophy, Curr. Treat. Options Neurol 20, 31 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Duan D, Systemic AAV Micro-dystrophin Gene Therapy for Duchenne Muscular Dystrophy, Mol. Ther 26, 2337–2356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rahimov F, King OD, Leung DG, Bibat GM, Emerson CP, Kunkel LM, Wagner KR, Transcriptional profiling in facioscapulohumeral muscular dystrophy to identify candidate biomarkers., Proc. Natl. Acad. Sci. U. S. A 109, 16234–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banerji CRS, Knopp P, Moyle LA, Severini S, Orrell RW, Teschendorff AE, Zammit PS, Zammit PS, β-catenin is central to DUX4 -driven network rewiring in facioscapulohumeral muscular dystrophy, (2014), doi: 10.1098/rsif.2014.0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsumagari K, Chang S-C, Lacey M, Baribault C, V Chittur S, Sowden J, Tawil R, Crawford GE, Ehrlich M, Gene expression during normal and FSHD myogenesis., BMC Med. Genomics 4, 67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.F. A. and Jiang B-H, Oxygen-independent Regulation of HIF-1: Novel Involvement of PI3K/AKT/mTOR Pathway in CancerCurr. Cancer Drug Targets 13, 245–251 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Lim J, Lee E, You H, Lee JW, Park J, Chun Y, Ras-dependent induction of HIF-1 a 785 via the Raf / MEK / ERK pathway: a novel mechanism of Ras-mediated tumor promotion, Oncogene 785, 9427–9431 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Xia M, Bi K, Huang R, Cho M, Sakamuru S, Miller SC, Li H, Sun Y, Printen J, Austin CP, Inglese J, Identification of small molecule compounds that inhibit the HIF-1 signaling pathway, Mol. Cancer 13, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhong JW, Tawil R, Tapscott SJ, Sverdrup FM, Clinically advanced p38 inhibitors suppress DUX4 expression in cellular and animal models of facioscapulohumeral muscular dystrophy, J. Pharmacol. Exp. Ther (2019), doi: 10.1124/jpet.119.259663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu L, Pathak PS, Fukumura D, Hypoxia-Induced Activation of p38 Mitogen-Activated Protein Kinase and Phosphatidylinositol 3′-Kinase Signaling Pathways Contributes to Expression of Interleukin 8 in Human Ovarian Carcinoma Cells, Clin. Cancer Res 10, 701–707 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-johnson D, Selective Activation of p38α and p38γ by Hypoxia, J. Biol. Chem 274, 23570–23576 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Park EC, Rongo C, The p38 MAP kinase pathway modulates the hypoxia response and glutamate receptor trafficking in aging neurons, Elife 5, 1–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winokur ST, Chen Y-W, Masny PS, Martin JH, Ehmsen JT, Tapscott SJ, van der Maarel SM, Hayashi Y, Flanigan KM, Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation., Hum. Mol. Genet 12, 2895–907 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Davidovic L, Durand N, Khalfallah O, Tabet R, Barbry P, Bardoni B, Mari B, Sacconi S, A Novel Role for the RNA – Binding Protein FXR1P in Myoblasts Cell-Cycle Progression by Modulating p21 / Cdkn1a / Cip1 / Waf1 mRNA Stability, 9 (2013), doi: 10.1371/journal.pgen.1003367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bosnakovski D, Gearhart MD, Toso EA, Recht OO, Cucak A, Jain AK, Barton MC, Kyba M, p53-independent DUX4 pathology in cell and animal models of facioscapulohumeral muscular dystrophy, 1211–1216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bosnakovski D, Silva MT, Sunny ST, Ener ET, Toso EA, Yuan C, Cui Z, Walters MA, Jadhav A, Kyba M, HUMAN GENETICS A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death, 300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi SH, Gearhart MD, Cui Z, Bosnakovski D, Kim M, Schennum N, Kyba M, DUX4 recruits p300 / CBP through its C-terminus and induces global H3K27 acetylation changes, 44, 5161–5173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosmarin DM, Carette JE, Olive AJ, Starnbach MN, Brummelkamp TR, Attachment of Chlamydia trachomatis L2 to host cells requires sulfation, 1–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanjana NE, Shalem O, Zhang F, Improved vectors and genome-wide libraries for CRISPR screening, Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, Liu XS, MAGeCK enables robust identification of essential genes from genome-scale CRISPR / Cas9 knockout screens, 1–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. GeneMania functional associations between top screen hits
Fig. S2. Changes in GLUT1 and PDK1 expression in the presence of DUX4
Fig. S3. Western blot protein quantification of single gene knock-outs
Fig. S4. Effect of inhibitors on etoposide-induced cell death
Fig. S5. Western blot time-course of rapamycin dosing
Fig. S6. DUX4 transcript expression with inhibitor treatment
Fig. S7. FSHD biomarkers expression in four patient lines with inhibitor treatment
Table S1. Oligo sequences for single gene knock-out lines
Table S2. Antibody concentrations
Table S3. Primer sequences used for RT-PCR