

Abstract
Despite existing therapy, patients with heart failure (HF) suffer substantial morbidity and mortality, highlighting the urgent need to identify novel pathophysiological mechanisms as well as therapies. Traditional models for pharmacologic intervention have targeted neurohormonal axes and hemodynamic disturbances in HF. However, several studies have now highlighted the potential for ketone metabolic modulation as a promising treatment paradigm. During the pathophysiologic progression of HF, the failing heart reduces fatty acid and glucose oxidation, with associated increases in ketone metabolism. Recent studies indicate that enhanced myocardial ketone use is adaptive in HF, and limited data demonstrate beneficial effects of exogenous ketone therapy in studies of animal models and humans with HF. This review will summarize current evidence supporting a salutary role for ketones in HF including: 1) normal myocardial ketone utilization, 2) alterations in ketone metabolism in the failing heart, 3) effects of therapeutic ketosis in animals and humans with HF, and 4) the potential significance of ketosis associated with sodium-glucose co-transporter 2 (SGLT2) inhibitors. Although a number of important questions remain regarding the use of therapeutic ketosis and mechanism of action in HF, current evidence suggests potential benefit, particularly in HF with reduced ejection fraction, with theoretical rationale for its use in HF with preserved ejection fraction. While early in its study and development, therapeutic ketosis across the spectrum of HF holds significant promise.
Keywords: heart failure, ketone, SGLT2, metabolism, fatty acid oxidation, glucose oxidation
INTRODUCTION
Heart failure (HF) affects 6.2 million people in the United States.1 Paradigm shifts in treatment approaches may help reduce the residual morbidity common to HF with reduced ejection fraction (HFrEF) and preserved ejection fraction (HFpEF). At its most basic level, HF can be viewed as a state of metabolic insufficiency, along with the well-known functional impairment. Though original descriptions of HF focused on hemodynamic maladaptations, and treatment has generally targeted the renin-angiotensin-aldosterone and sympathetic nervous systems, fuel metabolic derangements are relevant to the pathogenesis of HF. The “energy-starvation” hypothesis is now well established and has led some to characterize the failing heart as “an engine out of fuel”.2 As a metabolically demanding organ, the heart consumes more than seven times its weight in adenosine triphosphate (ATP) daily, with limited reserves available.3 Therefore, augmenting cardiac ATP production in a bioenergetically efficient manner is of significant interest to the HF field.
Specific fuel metabolic derangements during the pathophysiologic transition to HF include early impairments in fatty acid oxidation (FAO) followed by alterations in glucose oxidation.4 Given that FAs are the chief source of fuel for the normal heart, these metabolic re-programming events set the stage for reduced ability to regenerate ATP. In this setting, animal and human models have demonstrated increased ketone body utilization in the failing heart.3, 5 Very recently, pre-clinical and clinical studies suggest that exogenous delivery of ketones may improve cardiovascular function as well as prevent the development of pathological remodeling en-route to HF.6, 7 These small, early studies have offered a glimpse into a promising new target for pharmacologic treatment of HF. The potential significance of ketosis associated with sodium glucose cotransporter 2 (SGLT2) inhibitors and reductions in HF morbidity and cardiovascular mortality observed in patients with HF (irrespective of diabetes status) further motivates investigation of the potential benefits of ketones in patients with HF.8, 9 Accordingly, the goals of this review are to characterize normal myocardial ketone metabolism, describe metabolic reprogramming and shifts in substrate utilization in patients with HF, and review recent studies of therapeutic ketosis both in humans and animal models.
KETONE METABOLISM: A BRIEF PRIMER
The energetic requirements of the human myocardium are robust due to the persistent pumping of approximately 7200 liters of blood per day against systemic vascular resistance, utilizing 35 liters of oxygen to generate the requisite kilogram quantities of ATP daily.10 Given limited capacity to store energy, the heart turns over ATP stores at the remarkable rate of every 10 seconds to accomplish these tasks. FAs provides the bulk of energetic substrate (60-90%) under normal conditions. However, the heart demonstrates metabolic flexibility to adapt to changing substrate availability, utilizing glucose (through glycolysis and glucose oxidation), ketone bodies, lactate, and branched chain amino acids (BCAA) as necessary. Mitochondrial β-oxidation supplies the vast majority of ATP substrate.4 Comprehensive review of cardiovascular metabolic substrates have been discussed elsewhere.4, 11 However, for the purposes of this article, we will review the role and physiology of ketone metabolism in greater detail.
Hepatic Production and Myocardial Metabolism of Ketone Bodies.
Normally, the myocardium minimally extracts ketone bodies under non-fasting conditions,12 as ketones are not normally present in great quantity. Teleologically, ketosis has been hypothesized as an evolutionary mechanism to preserve carbohydrate stores in periods of stress and starvation. Since the brain is largely incapable of oxidizing FA and predominantly relies on glucose, ketones bodies conserve glucose since ketones can be used in the periphery (and can themselves constitute neurologic fuel). In addition, ketones reduce proteolysis that would be required to support gluconeogenesis from amino acids. Ketogenesis is therefore a critical part of maintaining metabolic homeostasis during periods of fasting or stress.13
Ketone bodies are produced mainly in the liver from mobilized FA, though other fuel sources (ketogenic amino acids leucine and lysine) may contribute to limited ketogenesis.14, 15 During states of FA catabolism, such as fasting (Figure 1), mobilized FAs enter hepatocytes and ultimately undergo complete mitochondrial β-oxidation to form several molecules of acetyl-CoA. Several enzymatic reactions catalyze the conversion of acetyl-CoA to acetoacetate, the first ketone body able to be delivered in the circulation. Acetoacetate can also be reduced to β-hydroxybutyrate (BHB) for systemic transport. Therefore, BHB, acetoacetic acid, and the breakdown product acetone are the three ketone bodies endogenously produced. Levels of approximately 2–3 mmol/L of BHB can be reached after a few days of fasting.16 Ketogenesis is influenced by hormonal signaling, transcriptional regulation, and post-transcriptional modification.13 Fates of ketone bodies include urinary or lung elimination, lipid synthesis, or transport to extra-hepatic tissue for utilization.
Figure 1: Pathways of Hepatic Ketogenesis and Myocardial Ketolysis.
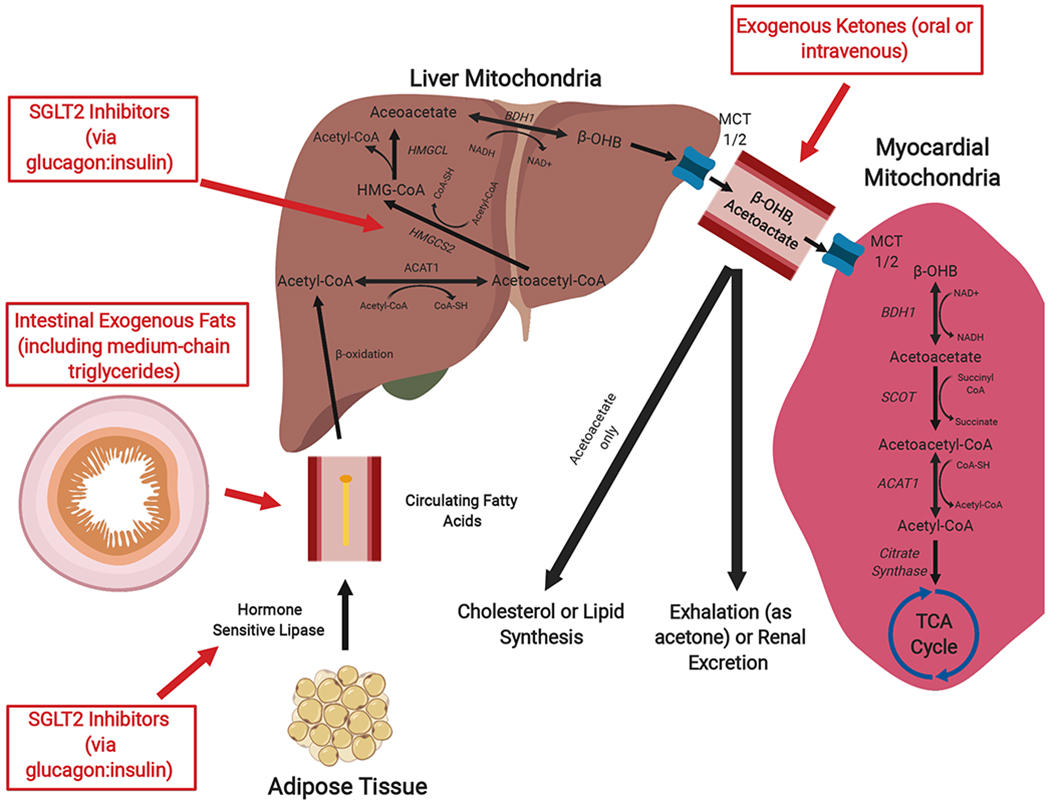
Exogenous fats are absorbed through the gastrointestinal tract (specifically, medium chain triglycerides can be hydrolyzed by lipases to generate medium chain fatty acids). In addition, free fatty acids generated by adipolysis are circulated to the liver, where they undergo fatty acid oxidation in the hepatic mitochondria to produce acetyl CoA. Subsequent ketogenesis ultimately produces two mature ketone bodies: acetoacetate and β-hydroxybutyrate (β-OHB). Export from the hepatocytes and import into the cardiomyocyte is accomplished via monocarboxylate transporters. In cardiomyocyte mitochondria, ketolysis produces acetyl CoA that can be used in the tricyclic acid cycle. Notably, circulating ketones that are not oxidized by tissue can be 1) used for lipid or cholesterol synthesis (acetoacetate only) or 2) eliminated through the urine or exhaled as acetone (derived from decarboxylation of acetoacetate). Therapies used to achieve ketosis are noted in red. Abbreviations: ACAT, acetyl-CoA acetyltransferase; BHB, β-hydroxybutyrate; BDH, BHB dehydrogenase; HMGCL, HMG-CoA lyase; HMGCS2, HMG-CoA synthase; MCT, monocarboxylate transporter; SCOT, succinyl-CoA:3 oxoacid-CoA transferase; TCA; tricyclic acid.
Circulating ketone bodies are taken up by tissues via monocarboxylate transporters (MCTs) for use in organs capable of ketone oxidation. In myocardial mitochondria, BHB dehydrogenase 1 (BDH1) oxidizes BHB to acetoacetate. Acetoacetate is then activated to acetoacetyl CoA by the CoA transferase succinyl-CoA:3 oxoacid-CoA transferase (SCOT), which then undergoes a thiolysis reaction by acetyl-CoA acetyltransferase (ACAT) to produce two molecules of acetyl CoA that can enter the tricyclic acid (TCA) cycle. These enzymes are critical to ketolysis, consistent with observations that genetic defects in SCOT, ACAT, and MCT1 cause recurrent ketoacidosis.17 The myocardium has among the highest levels of ketolytic enzyme activity in the body.18 Indeed, states associated with physiological increases in circulating ketones increase myocardial glycogen content in contrast to decreases observed in skeletal muscle.19
In contrast to glucose, an anaplerotic fuel source which replenishes TCA cycle intermediates, ketones are cataplerotic and deplete TCA intermediates. Consequently, in rat hearts, ketones as a sole source of fuel led to impairments in contractile function, while delivery of anaplerotic substrates such as pyruvate reversed the pathology.20–22 These studies highlight the importance of ensuring an anaplerotic input to the TCA cycle in the context of increasing ketone body metabolism. Thus, some degree of glucose metabolism is required to provide functional benefits from ketone therapies.
Loss of ketone metabolic capability can be functionally significant in settings of myocardial stress. This is illustrated in a cardiomyocyte specific SCOT knockout mice model, resulting in impaired terminal ketone body oxidation. After eight weeks of transaortic constriction (TAC), left ventricular (LV) volume is markedly increased and EF decreased in SCOT knock-out mice compared to control mice.23 Similarly, mice with BDH1 gene knockout exhibit worsened pathological remodeling in response to combined TAC and myocardial infarction.5 BDH1 knockout mice also exhibit cardiac dysfunction with prolonged fasting.7
Impact of Ketone Bodies on Cardiac Energetic Properties and Efficiency.
Beyond their contribution to energy generation, ketones oxidation is relatively efficient in terms of oxygen utilization.4 Fatty acid oxidation (FAO) using a substrate such as palmitate renders 105 ATP molecules at the expense of 23 molecules of oxygen per two carbon moiety, with a phosphate to oxygen (P/O) ratio of 2.3.4 Glucose oxidation generates only 31 ATP molecules while consuming 6 molecule of oxygen: despite a lower ATP yield, glucose oxidation is more efficient than FAO with a higher P/O ratio (2.6).4 In this context, ketone bodies strike a balance between energetic production and efficiency. Ketone oxidation has a P/O ratio of 2.5, and thus are energetically more efficient than FAO, and generates more ATP per 2 carbon moiety than glucose. Indeed, the addition of ketone bodies to working rat hearts perfused with glucose buffer significantly increases cardiac output and efficiency in a manner similar to insulin.24
Preferential Utilization of Ketones in the Normal Heart.
Under normal conditions, oxygen consumption is not limiting, and therefore the myocardium primarily utilizes FAO and secondarily glucose oxidation. A formal recognition of the substrate hierarchy of mitochondrial fuel selection (FAO at the expense of glucose utilization) has been termed the “Randle Effect”.25 In a like manner, several studies have demonstrated preferential utilization of ketones in normal hearts, such that in the presence of increased ketones, both FA and glucose metabolism appear to be reduced.26–29
KETONE METABOLISM IN HEART FAILURE
Impaired Fatty Acid Oxidation.
The transition from normal myocardium to pathologic remodeling and ultimately heart failure consists of well-described fuel metabolic alterations, including a reduction in capacity for, and rates of, FAO.30, 31 Indeed, FAO impairment is observed in early compensated cardiac hypertrophy32 and with more severe FAO dysfunction.30 A case-control lipidomic analysis of myocardial tissues from end-stage HF patients showed significantly decreased myocardial concentrations of the majority of lipids, including long-chain acylcarnitines.3 Analogous substrate shifts can occur rapidly during induction of HF in animal models. Acute pressure overload models of HF (TAC) engender a prompt switch from FAO to glucose utilization,33, 34 and deactivation of the upstream transcriptional regulator PPAR-alpha has been implicated as a regulator of these shifts.33 In chronic pressure overload models of HF, FAO mRNA levels were markedly reduced in LVH rats compared with controls, whereas protein levels and enzyme activities were not significantly different until HF develops.30 Similar shifts away from FAO to glucose oxidation are observed early in a canine chronic tachypacing model of HF.7 This impairment in FAO combined with increased reliance on glucose oxidation resembles the fetal metabolic program.7 Overall, these shifts in metabolic programming diminish the capacity of the mitochondria to generate high quantities of ATP through FAO.
Alterations in Glucose Metabolism.
As an efficient metabolic substrate, glucose is theoretically well positioned to support the high ATP requirements of the failing heart and is generally consumed to a greater degree in early stages of HF.35 However, progressive deficits in glucose oxidation lead to greater flux through the glycolytic pathway, a much less efficient process for generation of ATP. In hypertrophied hearts, the reduced contribution of FAO to ATP generation at low workloads is matched by an increase in glycolysis.36–38 Moreover, the increase in glucose utilization in early stage HF and/or hypertrophy may deteriorate by later stages of HF,39, 40 potentially mediated by the development of insulin resistance or other unknown mechanisms.41 Myocardial tissue obtained at the time of LV assist device implantation demonstrated decreased transcript levels encoding enzymes involved in glucose and pyruvate oxidation.42 These metabolic deficits in both FAO and glucose metabolism in HF conspire to produce an “energy starved” heart that is primed to use alternative ATP-generating fuels.
Switch to Ketone Fuel Utilization.
As the failing heart progressively loses its capacity to oxidize FA and glucose, ketone bodies provide a potential alternative substrate since ketone oxidation bypasses the complex dysregulation of the beta-oxidation pathway and pyruvate dehydrogenase complex in HF. In fact, one recent study of end-stage HF patients found an abundance of a ketogenic derivative, β-hydroxybutyryl-CoA, along with decreased BHB in myocardial tissue in association with increased expression of ketolytic enzymes such as BDH1.3 Extending these findings, a mouse model of progressive HF, induced by TAC and distal coronary ligation (TAC/myocardial infarction), likewise demonstrated increased expression of BDH1 along with increased expression of genes encoding ketone body transporters (monocarboxylate transporter 1 [MCT1] and MCT2) and increased 13C-BHB utilization based on nuclear magnetic resonance studies.5 Thus, despite well-known mitochondrial dysfunction that occurs during the progression to HF and in contrast to FAO, ketones can be completely oxidized given the metabolic pathway is upregulated in the failing heart and utilization can occur via a shorter number of steps.3, 5 Reports that cardiac-specific BDH1 knockout mice exhibit more severe ventricular modeling and dysfunction following TAC/myocardial infarction while BDH1 overexpression attenuates cardiac remodeling and DNA damage in pressure overloaded model43 suggest that increased ketone utilization is adaptive.7
Ketones as a Biomarker in HF.
Several studies have reported increases in peripheral ketone levels in HF.44, 45 Exhaled acetone, for example, was able to identify HF patients with similar predictive value to B-type natriuretic peptide, and exhaled acetone levels positively correlated to New York Heart Association class.45 Among 45 patients with predominantly systolic HF, higher circulating levels of ketones were associated with lower EF, worse New York Heart Association Class, and elevated filling pressures.44 Increases in peripheral ketone levels do not themselves demonstrate the mechanism of their presence, as such elevation could reflect a decrease in utilization or an increase in production of ketones. In addition, increased ketone levels may be physiologic and compensatory in HF. In a related example, natriuretic peptides are elevated in HF and strongly prognostic for adverse cardiovascular events, though they serve necessary physiologic roles in natriuresis and sympathetic inhibition in HF. Accordingly, though elevated levels of ketone track with HF diagnosis and severity, this observation does not necessarily indicate that elevated ketones are maladaptive.
Ketone Metabolism in HFpEF vs. HFrEF.
In an investigation using arterial and coronary sinus sampling of patients with aortic stenosis with LV hypertrophy and preserved LVEF, both ketones and FA uptake increased compared to controls, while only ketone uptake increased in patients with HFrEF.46 Thus, ketone uptake appears to be utilized as metabolic fuel in both HFpEF and HFrEF. Studies assessing peripheral blood ketone levels showed that HFrEF patients had lower,47 or similar,48 levels compared to HFpEF.
EFFECTS OF THERAPEUTIC KETOSIS
As highlighted in Table 1, recent studies have investigated the role of therapeutic ketosis in animal and human studies in both normal and HF states with promising results. The method of inducing ketosis (ketogenic diet, oral therapy, and/or intravenous infusion) as well as the target population may account for distinctions observed among these studies.
Table 1.
Studies Relevant to Augmenting Ketosis in Human Heart Failure
| First Author and Year | Population | Intervention | Main Findings |
|---|---|---|---|
| Human studies | |||
| Cox, 201649 | 8 endurance athletes | Crossover study of ketone monoester and dextrose vs. carbohydrate drink | Athletes after ingesting drink containing ketone monoester cycled on average 411 m further over 30 minutes. |
| Verma, 201650 | 10 patients with diabetes and established cardiovascular disease | Before/after study with empagliflozin | Empagliflozin reduced LV mass and improved lateral E’ velocity. |
| Gormsen, 201751 | 8 healthy subjects | Randomized, crossover trial of sodium-3-OHB infusion vs. saline | Ketone infusion decreased myocardial glucose uptake and increased myocardial blood flow by 75%. |
| Nielsen, 20196 | 16 HFrEF patients (EF≤40%) | Randomized, crossover trial of 3-OHB infusion vs. isotonic saline | 3-OHB infusion increased cardiac output, reduced systemic and pulmonary vascular resistance, and marginally decreased biventricular filling pressures. |
| McMurray, 20199 | 4744 HFrEF patients (EF≤40%) | Randomized, crossover trial of dapagliflozin vs. placebo | Dapagliflozin reduced risk of worsening heart failure or cardiovascular death, irrespective of underlying diabetes mellitus. |
| Animal studies | |||
| Joubert, 201752 | Lipodystrophic (seipin knockout) mice | Dapagliflozin only | Dapagliflozin treatment significant improved diastolic function in the hypertrophic heart. |
| Lee, 201753 | Normoglycemic male Wistar rats after coronary ligation | Dapagliflozin therapy and control arm | Dapagliflozin reduced cardiac fibrosis. |
| Verma, 201854 | Mice with diabetes | Empagliflozin and control arms | Empagliflozin increased cardiac ATP production, but not through ketone oxidation. |
| Abdurrachim, 201955 | Rats with obesity, diabetes, hypertension and HF | Empagliflozin and control arms | Empagliflozin lowered myocardial ketone utilization; no effect observed on LV hypertrophy or fibrosis. |
| Horton, 20197 | TAC/MI mice and tachycardia-induced cardiomyopathy dogs | Ketogenic and normal chow for mice; 3-OHB infusion vs. no infusion in dogs with HF | Ketogenic chow improved LV remodeling in mice; 3-OHB infusion significantly improved systolic dysfunction and LV remodeling in the canine model. |
| Santos-Gallego, 201956 | HF after left anterior descending artery ligation in nondiabetic pigs | Empagliflozin versus control | Empagliflozin improved systolic function and cardiac remodeling. Empagliflozin-treated pigs switched substrate utilization to ketones, FAs, and branched chain amino acids. |
| Yurista, 201957 | Rat models without diabetes after MI | Empagliflozin and control chow arms | Empagliflozin significantly improved EF, attenuated hypertrophy, diminished fibrosis, and reduced oxidative stress after MI. Empagliflozin increased myocardial expression of the ketone body transporters and ketogenic enzymes. |
3-OHB, 3-hydroxybutyrate; EF, ejection fraction; FA, fatty acid; g, gram; h, hour; HF, heart failure; kg, kilogram; LV, left ventricle; MI, myocardial infarction; TAC, transaortic constriction.
Endogenous Ketosis in Healthy Humans.
In humans, the optimal mode to increase systemic ketone levels is unclear. The ketogenic diet (KD) is intended to mimic the biochemical effects of starvation and consists of high fat with low carbohydrate intake, a regimen that can engender difficulty with long-term adherence. The KD lowers blood glucose (through reduced glucose availability and lower gluconeogenesis) and elevates FA concentrations. Endogenous ketosis occurs in states of low insulin and elevated glucagon levels which in turn drive lipolysis, releasing FA as a substrate for hepatic ketogenesis. The lipolytic component of the ketogenic diet forms part of the basis of its popularity as a weight loss regimen.58 KDs typically result in mild to moderate ketosis with BHB levels of roughly 0.5-1.5 mmol/L, which is currently considered an accepted definition of ketosis (≥ 0.3 mmol/L).59 This level of ketosis may also reduce appetite.
Classical ketogenic diets focus on augmenting intake of long-chain fats (approximately 55-75% of daily caloric intake) and significantly reducing carbohydrate intake (5-10% of daily caloric intake, typically less than 30-50 grams per day). However, an alternative diet consisting of medium-chain triglycerides (with corresponding fatty acids of 6-12 carbon atoms) has been used to allow greater carbohydrate intake. Shorter FAs can rapidly generate ketones and have been studied in in epilepsy, inherited disorders of metabolism, and diabetes mellitus.60 Potential benefits of medium-chain triglycerides include modest weight loss and favorable changes in body composition compared to long-chain triglycerides, with similar effects on lipids.61
Limited data are available regarding cardiovascular effects of the KD. Short term KD induces a decrease in high-energy phosphate metabolism by cardiac magnetic resonance imaging spectroscopy, without echocardiographic changes in function.62 It has been shown that increases in serum free fatty acids (FFAs) favor mitochondrial uncoupling, potentially accounting for the spectroscopic findings.63 Observational data further suggest that increasing FFA concentration is associated with diastolic dysfunction.64
Chronic use of a KD raises several other long-term concerns. First, the KD can induce dyslipidemia with increased low density lipoprotein cholesterol, which may be problematic in patients with, or prone to, atherosclerotic cardiovascular disease.58 Secondly, it reduces peripheral glycogen stores, a strong determinant of exercise capacity.65 In addition, there are concerns regarding anaplerosis and depletion of TCA intermediates with the KD (depending on the strictness of conformity to the low carbohydrate portion of the diet), and whether anaplerotic substrates should be incorporated into diets would be important to investigate. Finally, population level studies suggest low carbohydrate diets are associated with increased mortality, albeit with effect modification by the source of fat or protein used to supplement the diet.66 Thus, endogenous ketosis by dietary modification may not be an ideal long term solution to maintain ketosis. Future studies of the ketogenic diet should elucidate long term atherosclerotic cardiovascular consequences, the impact of supplementation with medium-chain triglycerides and/or anaplerotic substrates, and impact on myocardial metabolism using cardiac magnetic resonance imaging and nuclear metabolic tracers.
Effects of Ketone Supplementation in Healthy Humans.
Exogenous delivery of ketones induces a unique state, since ketosis can be achieved in the presence of normal carbohydrate and FA levels. Ketone supplementation demonstrates consistent improvements in glucose control as well as reduced circulating FFAs. Intravenous infusions of sodium BHB (5 mmol/kg/h for 1.5 hours67 and 4.7/9.4/18.8 μmol/kg/min each for 1 hour68) significantly reduces glucose and FFA concentration. The latter regimen has achieved BHB levels of approximately 0.5-2 mmol/L.68 Glucose reduction is likely related to limiting hepatic gluconeogenesis and increasing peripheral glucose uptake, as it is not mediated by pancreatic stimulation.67 FFA reduction may be due to exogenous ketones negatively feeding back on their own production through BHB-mediated agonism of the PUMA-G receptor in adipose tissue, ultimately suppressing lipolysis.69
The myocardial effects of relatively high doses of exogenous therapy (to a BHB level of nearly 4 mmol/L), are substantial under carefully controlled circumstances. One study demonstrated increase in heart rate, an increase in pulse pressure (mediated by lowering of diastolic blood pressure), a 75% increase myocardial blood flow, and near halving of glucose uptake.51 There was little change in FFA, insulin, and glucose levels, though these assessments were made in the presence of a hyperinsulinemic-euglycemic clamp which may complicate the assessment. The mechanism of the increased heart rate is not clear, but may be a compensatory response to vasodilation rather than a sympathetic response, which is typically downregulated by ketones.70
Recognizing that intravenous infusion of ketones is not practical for therapeutic ketosis among outpatients, different formulations of oral ketone therapy have been studied, including ketone salts and ketone esters. Unfortunately, oral ketone salts result in a very high sodium intake and half the plasma concentration of the D-stereoisomer of BHB compared to ketone ester. The differences in isomers are important to consider: the L-isoform is much less readily oxidized, is more likely to accumulate, and is less well-studied regarding its signaling effects.71 One ketone monoester, (R)-3-hydroxybutyl (R)-3-hydroxybutyrate, has been studied for the purposes of therapeutic exogenous ketosis.65, 71, 72 This compound produces a rapid rise of blood levels of BHB in humans to nearly 3 mmol/L within 60 minutes of consumption with 282 mg/kg of KE.71 No significant cardiovascular effects have been reported at rest in healthy volunteers.72
In contrast to ketone salts,73 benefits of ketone ester therapy among athletes have been demonstrated during exercise. Early studies indicate that athletes, in particular, appear to undergo metabolic reprogramming that result in favorable and matched ketone utilization during exercise.74, 75 In a recent blinded, crossover study of endurance athletes, ketone ester therapy increased exercise distance by an average of 411 meters over 30 minutes cycling duration, demonstrating that ketone therapy improves exercise capacity among highly trained athletes, though a ceiling effect may be reached in that population.49 However, pathologic conditions that cause metabolic dysregulation and where incremental improvements in energy transduction may translate to increases in exercise capacity, such as HF, may afford the greatest clinical benefit.49
Effects of Therapeutic Ketosis in Animals with Cardiovascular Disease.
Models of therapeutic ketosis in animals tend to support a cardioprotective effect following ischemic-reperfusion injury. Ketonemia induced by 3-day fasting in mice reduced myocardial damage and arrhythmias.76 However, studies of KD following ischemia-reperfusion conflict as to whether the dietary intervention is protective77 or increases myocardial injury76. Further, the possible protective effect of KD may be enhanced by exogenous ketone therapy: fasting in addition to ketone infusion led to the least myocardial damage relative to fasting without ketone infusion or a fed state with ketone infusion in rat hearts after coronary occlusion.78 In hypertensive heart disease models, low carbohydrate, high fat (ketogenic) diets in Dahl salt-sensitive hypertensive rats and rats exposed to pressure-overload banding models results in attenuated ventricular remodeling and dysfunction compared to high carbohydrate and/or low fat diets.79–81
Increased myocardial delivery of ketone bodies has been shown to ameliorate HF in mice and in large animals. A KD that doubled levels of ketones led to significant reduction in ventricular remodeling caused by TAC/myocardial infarction in wild-type mice compared to mice fed normal chow.7 A complementary study in a canine tachypacing HF model demonstrated that a 2.5-fold increase in plasma ketones achieved via continuous delivery of ketones to the right ventricle markedly reduces cardiac dilatation and reduced cardiac output along with blunting decreases in LVEF compared with tachypacing and vehicle infusion.7 BHB delivery also decreases total peripheral resistance raising the question of whether ketones are direct vasodilators or act indirectly such as via lowering sympathetic tone.70 In canines, BHB infusion reduces glucose oxidation and lactate production, without altering FFA oxidation, consistent with a Randle effect (increased acetyl-CoA levels decrease pyruvate dehydrogenase activity).25 The corresponding decrease in glucose oxidation could be beneficial in cardiometabolic disease states in order to limit glucotoxicity. While ketone infusion augmented myocardial oxygen consumption, myocardial mechanic energetic efficiency actually improved, implying the increased oxygen consumption was not pathologic.
Notably, the direct impact of ketone bodies on cardiac energetics (phosphocreatine/ATP ratio) in the failing heart is not established. With that said, data suggest that infused ketones are oxidized, along with improvement in cardiac function, in the canine tachypacing model of HF.7 Moreover, infusion of ketones competes with glucose oxidation in this setting. Finally, mitochondria isolated from the normal mouse heart exhibit increased NADH/NAD levels and enhanced defense of the mitochondrial membrane potential when supplemented with ketones in the setting of limited supply of FA to mimic the substrate utilization of the failing heart.7
While the energetic properties of ketones are important, it is not clear that efficiency as defined by phosphate/oxygen ratio accounts for their beneficial effect in HF. In mice, ketone infusion significantly increases the failing heart’s utilization of ketones (to over 20% from <10%), but cardiac efficiency fails to improve and is similar to FA metabolic efficiency.12 Nevertheless, studies using isolated mitochondrial in the context of limiting levels of FA input into the TCA cycle report that BHB maintains the mitochondrial membrane gradient to a greater extent than other substrates.7 Thus, the “respiratory efficiency” may be improved by ketone bodies. Future animal studies should investigate relevance of exogenous ketosis in acute versus chronic models of HF as well as in HFpEF, given these studies largely investigate models of HFrEF.
Effects of Ketone Supplementation in Human HF.
Limited data in humans suggest that ketone supplementation has therapeutic potential in HF. In a randomized, crossover study in patients with compensated HFrEF, a 3-hour infusion of a racemic mixture of 7.5% 3-OHB (at 0.18 g/kg/h) achieved serum levels of 3.3 mmol/L and increased cardiac output by 40% in association with decreases in systemic and pulmonary vascular resistances, a modest increase in heart rate and stroke volume, and mildly reduced cardiac filling pressures, which were not observed with saline infusion.6 These hemodynamic changes were dose-dependent and cardiac effects were seen with even mild ketosis. Laboratory analysis revealed little difference in insulin levels, B-type natriuretic peptide, or norepinephrine levels. Myocardial external efficiency (defined as the ratio between stroke work and myocardial oxygen consumption) was unchanged with ketone infusion compared to isotonic saline. Similar hemodynamic effects of the ketone infusion were observed in an-age matched control group who were largely hypertensive.
While important, this study generates several additional questions and considerations. A key unanswered question with this study is whether the increases in cardiac output were solely due to peripheral vasodilation or due to the use of ketones as a metabolic substrate. Given that vasodilators are already a mainstay of HFrEF treatment, clarifying this distinction is key to understanding whether exogenous ketones add unique benefits in HF. The comparison with normal saline, as opposed to an alternative energetic fuel, likewise leaves unanswered questions about how the ketone infusion affected hemodynamics. In addition, favorable short-term effects (as the ketone washes out very quickly)6 may not translate to long term benefits due to concerns related to the high sodium load, increased heart rate (which may be detrimental in HFrEF), and depletion of TCA intermediates. Further, the small sample size precludes further assessment of subgroups that might differentially respond to ketone infusions (e.g. patients with diabetes mellitus). Finally, understanding how the degree of mitochondrial dysfunction affects the response to ketone infusion is an important clinical consideration.
The effects of ketone supplementation in HFpEF have not yet been assessed. However, because many of the control patients in the aforementioned study had hypertension (a common comorbidity in those with HFpEF) and achieved similar hemodynamic findings with ketone infusion to HFrEF, the same benefit may be seen in HFpEF. It is also tempting to speculate that improvement in systolic strain82 or vasodilatory effect of ketone therapy83 could be beneficial in patients with HFpEF.
Thus, ketone supplementation is therapeutically promising in human HF. However, questions of mechanism of benefit, impact on peripheral muscles and glycogen content, comparisons to other metabolic substrates, relevance in end-stage HF, and effect of anaplerotic substrate supplementation are important to investigate further.
Non-Metabolic Benefits of Ketone Bodies.
The benefits observed with ketone therapy in the aforementioned studies might also relate to the pleiotropic roles of ketone bodies in cellular signaling and modulating oxidative stress and inflammation, which recently have been comprehensively reviewed.13 In particular, ketone bodies regulate both lipolysis and sympathetic activity through their interactions with specific G-protein coupled receptors. For example, BHB binds G-protein coupled receptor GPR109A and provides negative feedback to lipolysis in adipoycytes through hormone-sensitive triglyceride lipase.69 Additionally, ketones interact with GPR41 to suppress sympathetic nervous system activity in the sympathetic ganglion.70 This latter effect may be particularly relevant in HF, given the benefits of sympathetic antagonism. In addition, BHB has anti-inflammatory activity by suppressing activation of the Nod-like receptor protein 3 (NLRP3) inflammasome.84 However, the inflammatory response may not be uniform among ketones, with studies supporting both pro-inflammatory and anti-inflammatory effects of acetoacetate.85, 86 Further, ketone bodies regulate fibroblastic growth factor 21 (FGF21), which plays an important paracrine role in the heart and favorably influences myocardial remodeling, as Fgf21−/− mice demonstrate greater cardiac hypertrophy and more deleterious responses to isoproterenol infusion.87, 88 Finally, the role of ketones in regulating appetite and promoting satiety are complex and in need of further study. Though speculative, the differential milieu of endogenous versus exogenous ketosis may be important in evoking a specific response. Since endogenous ketosis often occurs during sympathetic states, the anti-lipolytic, anti-hyperlipidemic, anti-sympathetic, and anti-inflammatory effects may be muted. Exogenous ketosis, however, might uniquely elicit these responses without requiring the sympathetic state to promote ketosis.
Epigenetic effects of BHB have also been observed. Ketone bodies inhibit class I histone deacetylases and increase the activity of two promoters involved in protecting against oxidative stress (FOXO3A and MT2).89, 90 Notably, increased histone acetylation was observed both with endogenous and exogenous means of inducing ketosis.90 These globally beneficial ketotic effects may be relevant to reversing the pathophysiologic remodeling induced by reactive oxygen species that contribute directly and indirectly to worsening HF.91 A ketone derived histone modification, β-hydroxybutyrylation, is another recently discovered epigenetic regulatory mark, though its significance needs further elucidation.92
Thus, the relevance of therapeutic ketosis in HF goes significantly beyond ATP generation. Understanding the non-metabolic benefits of ketone bodies in HF, such as the effects on inflammatory, oxidative stress, and neurohormonal pathways, is critical. Further defining these pathways in HFpEF vs. HFrEF and method of ketosis (exogenous versus endogenous) will be necessary in advancing the treatment paradigm.
Relationship between SGLT2 Inhibitors, Systemic Ketosis, and HF.
Very recently, administration of SGLT2 inhibitors as a primary HF therapeutic demonstrated a striking reduction in HF hospitalization and all-cause mortality.9 The mechanisms responsible for the salutary impact of SGLT2 inhibitors in HF are unknown. Intriguingly, SGLT2 inhibitors induce systemic ketosis but it is currently unclear whether this is a mechanism of benefit.8 Since SGLT2 receptors are not located in cardiomyocytes, indirect mechanisms, including ketosis, are likely to account for the benefit.93 SGLT2 inhibitors decrease the insulin to glucagon ratio and increase lipolysis, both of which favor formation of ketones (Figure 1).94 In fact, empagliflozin treatment mildly increases BHB levels (0.56 mmol/L in patients with diabetes, 0.27 mmol/L in patient without diabetes) with chronic dosing (28 days).95 Because SGLT2 inhibitor therapy increases ketone metabolism in animal models of HF,56, 57 increased circulating levels of ketones may plausibly increase myocardial utilization of ketones. Indeed, the systemic ketosis achieved with SGLT2 inhibitors that decrease HF hospitalization is similar to levels shown to be beneficial in HF.6
Apart from ketosis, SGLT2 therapy may have other cardioprotective effects in HF. SGLT2 inhibitors may engender favorable changes in blood pressure, weight loss, uricosuria, volume control through osmotic diuresis, natriuresis, and oxygen carrying capacity with hemoconcentration.93 In animal and human hearts with pathologic hypertrophy or diabetes, treatment with SGLT2 inhibitors improved diastolic function and reduced cardiac fibrosis, but whether these are ketone-dependent benefits is unknown.50, 52, 53 As SGLT2 inhibitors are employed to treat patients with HF, better understanding of whether ketosis is central to their mechanism of action will have implications for their target population, potential risks, use of dietary adjuvants, and safety monitoring.
CONCLUSION
Emerging evidence suggests that therapeutic ketosis may be a viable treatment target in patients with HF (Figure 2). Striking short-term cardiovascular benefits observed with exogenous ketosis, highlight the ketone metabolic axis as a promising frontier in novel HF treatment. Interest in capitalizing on the unique metabolic effects of ketone utilization is growing but begs for greater understanding of the mechanisms through which ketones alter cardiovascular function. For example, do ketone bodies work as alternate cardiac fuels, vasodilators, and/or via other signaling pathways? Will they prove useful for both chronic and acute (boosting short-term activity tolerance or in decompensated heart failure) indications? Could peripheral effects of ketosis contribute to improved outcomes in cardiomyopathies? How does ketone body supplementation interact with myocardial branched-chain amino acid metabolism (a recently discovered pathway in HF genesis)? Several other questions remain to be answered. Future studies should seek to clarify efficacy and safety (particularly hepatic effects and acid-based disturbances) of long-term ketone administration, optimal methods of achieving ketosis in HF (oral versus intravenous administration), effect on patient centered outcomes, and differential effects in HFrEF and HFpEF. For example, peripheral glycogen preserving effects may be important to both subsets of HF, though may be particularly relevant to patients with HFpEF in whom improvements in systolic function might be less relevant. It is an exciting time to investigate the potential of ketone metabolic modulation as a therapeutic strategy for HF.
Figure 2: Methods of Achieving Ketosis and Related Potential Cardiovascular Benefits.
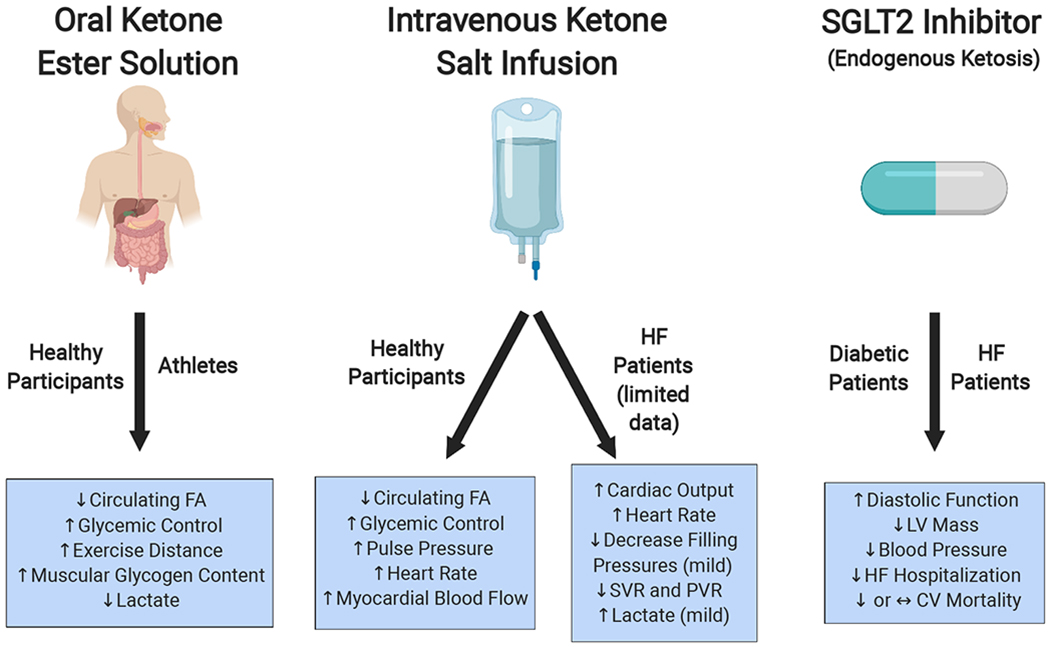
Popular, non-dietary ways of achieving ketosis include oral ketone ester drinks, intravenous ketone salt infusions, and SGLT2 inhibitors. Reported cardiovascular benefits related to the ketosis achieved are listed underneath with the corresponding patient population studied. Note that the role of ketosis as a mediator of the cardiovascular benefits observed with SGLT-2 inhibitors is speculative at this point. CV, cardiovascular; FA, fatty acid; HF, heart failure; LV, left ventricular; PVR, peripheral vascular resistance; SVR, systemic vascular resistance.
ACKNOWLEDGEMENTS
Images were created with the support of Biorender.
FUNDING
Dr. Selvaraj is supported by the National Institutes of Health (Training Grant 5-T32HL007843-23). Dr. Kelly is supported by R01 HL058493 and R01 HL128349. Dr. Margulies is supported by U10 HL110338.
DISCLOSURES
Dr. Kelly reports a significant consulting relationship with Pfizer, and a modest consulting relationship with Amgen. Dr. Margulies serves as a consultant for MyoKardia and Pfizer and receives research funding from Sanofi-Aventis and GlaxoSmithKline. No other relevant disclosures are reported.
ABBREVIATIONS AND ACRONYMS
- ACAT
acetyl-CoA acetyltransferase
- ATP
adenosine triphosphate
- BCAA
branched chain amino acids
- BHD1
β-hydroxybutyrate dehydrogenase 1
- BHB
β-hydroxybutyrate
- FAO
fatty acid oxidation
- FFA
free fatty acid
- FGF21
fibroblastic growth factor 21
- KD
ketogenic diet
- LV
left ventricular
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- SCOT
succinyl-CoA:3 oxoacid-CoA transferase (SCOT)
- SGLT2
sodium glucose cotransporter 2
- TAC
transaortic constriction (TAC)
- TCA
tricyclic acid
REFERENCES
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, American Heart Association Council on E, Prevention Statistics C, Stroke Statistics S. Heart disease and stroke statistics-2019 update: A report from the american heart association. Circulation. 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 3.Bedi KC Jr., Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, Rame JE. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Jong KA, Lopaschuk GD. Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Can J Cardiol. 2017;33:860–871. [DOI] [PubMed] [Google Scholar]
- 5.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nielsen R, Moller N, Gormsen LC, Tolbod LP, Hansson NH, Sorensen J, Harms HJ, Frokiaer J, Eiskjaer H, Jespersen NR, Mellemkjaer S, Lassen TR, Pryds K, Botker HE, Wiggers H. Cardiovascular effects of treatment with the ketone body 3-hydroxybutyrate in chronic heart failure patients. Circulation. 2019;139:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA, Kelly DP. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4: e124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrannini E, Mark M, Mayoux E. Cv protection in the empa-reg outcome trial: A “thrifty substrate” hypothesis. Diabetes Care. 2016;39:1108–1114. [DOI] [PubMed] [Google Scholar]
- 9.McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM, Committees D-HT, Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. [DOI] [PubMed] [Google Scholar]
- 10.Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, Gropler RJ, Holzhuetter HG, Kizer JR, Lewandowski ED, Malloy CR, Neubauer S, Peterson LR, Portman MA, Recchia FA, Van Eyk JE, Wang TJ, American Heart Association Council on Basic Cardiovascular S. Assessing cardiac metabolism: A scientific statement from the american heart association. Circ Res. 2016;118:1659–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGarrah RW, Crown SB, Zhang GF, Shah SH, Newgard CB. Cardiovascular metabolomics. Circ Res. 2018;122:1238–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho KL, Zhang L, Wagg C, Al Batran R, Gopal K, Levasseur J, Leone T, Dyck JRB, Ussher JR, Muoio DM, Kelly DP, Lopaschuk GD. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc Res. 2019;115:1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25:262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas LK, Ittmann M, Cooper C. The role of leucine in ketogenesis in starved rats. Biochem J. 1982;204:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang D, Yang H, Kong X, Wang K, Mao X, Yan X, Wang Y, Liu S, Zhang X, Li J, Chen L, Wu J, Wei M, Yang J, Guan Y. Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am J Physiol Endocrinol Metab. 2011;300:E287–295. [DOI] [PubMed] [Google Scholar]
- 16.Cahill GF Jr., Herrera MG, Morgan AP, Soeldner JS, Steinke J, Levy PL, Reichard GA Jr., Kipnis DM. Hormone-fuel interrelationships during fasting. J Clin Invest. 1966;45:1751–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Hasselt PM, Ferdinandusse S, Monroe GR, Ruiter JP, Turkenburg M, Geerlings MJ, Duran K, Harakalova M, van der Zwaag B, Monavari AA, Okur I, Sharrard MJ, Cleary M, O’Connell N, Walker V, Rubio-Gozalbo ME, de Vries MC, Visser G, Houwen RH, van der Smagt JJ, Verhoeven-Duif NM, Wanders RJ, van Haaften G. Monocarboxylate transporter 1 deficiency and ketone utilization. N Engl J Med. 2014;371:1900–1907. [DOI] [PubMed] [Google Scholar]
- 18.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60:143–187. [DOI] [PubMed] [Google Scholar]
- 19.Zorzano A, Balon TW, Brady LJ, Rivera P, Garetto LP, Young JC, Goodman MN, Ruderman NB. Effects of starvation and exercise on concentrations of citrate, hexose phosphates and glycogen in skeletal muscle and heart. Evidence for selective operation of the glucose-fatty acid cycle. Biochem J. 1985;232:585–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell RR 3rd, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest. 1991;87:384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taegtmeyer H On the inability of ketone bodies to serve as the only energy providing substrate for rat heart at physiological work load. Basic Res Cardiol. 1983;78:435–450. [DOI] [PubMed] [Google Scholar]
- 22.Russell RR 3rd, Taegtmeyer H. Pyruvate carboxylation prevents the decline in contractile function of rat hearts oxidizing acetoacetate. Am J Physiol. 1991;261:H1756–1762. [DOI] [PubMed] [Google Scholar]
- 23.Schugar RC, Moll AR, Andre d’Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab. 2014;3:754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keon CA, Tuschiya N, Kashiwaya Y, Sato K, Clarke K, Radda GK, Veech RL. Substrate dependence of the mitochondrial energy status in the isolated working rat heart. Biochem Soc Trans. 1995;23:307S. [DOI] [PubMed] [Google Scholar]
- 25.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. [DOI] [PubMed] [Google Scholar]
- 26.Bassenge E, Wendt VE, Schollmeyer P, Bluemchen G, Gudbjarnason S, Bing RJ. Effect of ketone bodies on cardiac metabolism. Am J Physiol. 1965;208:162–168. [DOI] [PubMed] [Google Scholar]
- 27.Jeffrey FM, Diczku V, Sherry AD, Malloy CR. Substrate selection in the isolated working rat heart: Effects of reperfusion, afterload, and concentration. Basic Res Cardiol. 1995;90:388–396. [DOI] [PubMed] [Google Scholar]
- 28.Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. Beta-hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-coa content. Am J Physiol Heart Circ Physiol. 2003;285:H1626–1631. [DOI] [PubMed] [Google Scholar]
- 29.Ziegler A, Zaugg CE, Buser PT, Seelig J, Kunnecke B. Non-invasive measurements of myocardial carbon metabolism using in vivo 13c nmr spectroscopy. NMR Biomed. 2002;15:222–234. [DOI] [PubMed] [Google Scholar]
- 30.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. [DOI] [PubMed] [Google Scholar]
- 31.Aubert G, Vega RB, Kelly DP. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta. 2013;1833:840–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akki A, Smith K, Seymour AM. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem. 2008;311:215–224. [DOI] [PubMed] [Google Scholar]
- 33.Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sack MN, Disch DL, Rockman HA, Kelly DP. A role for sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci U S A. 1997;94:6438–6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271–277. [DOI] [PubMed] [Google Scholar]
- 36.Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to atp production in hypertrophied hearts. Am J Physiol. 1994;267:H742–750. [DOI] [PubMed] [Google Scholar]
- 37.Leong HS, Grist M, Parsons H, Wambolt RB, Lopaschuk GD, Brownsey R, Allard MF. Accelerated rates of glycolysis in the hypertrophied heart: Are they a methodological artifact? Am J Physiol Endocrinol Metab. 2002;282:E1039–1045. [DOI] [PubMed] [Google Scholar]
- 38.Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. 2004;44:662–667. [DOI] [PubMed] [Google Scholar]
- 39.Zhabyeyev P, Gandhi M, Mori J, Basu R, Kassiri Z, Clanachan A, Lopaschuk GD, Oudit GY. Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res. 2013;97:676–685. [DOI] [PubMed] [Google Scholar]
- 40.Schroeder MA, Lau AZ, Chen AP, Gu Y, Nagendran J, Barry J, Hu X, Dyck JR, Tyler DJ, Clarke K, Connelly KA, Wright GA, Cunningham CH. Hyperpolarized (13)c magnetic resonance reveals early- and late-onset changes to in vivo pyruvate metabolism in the failing heart. Eur J Heart Fail. 2013;15:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res. 2016;118:1151–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, Peterson LE, Lyon CJ, Wong ST, Newgard CB, Torre-Amione G, Taegtmeyer H, Hsueh WA. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet. 2014;7:266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uchihashi M, Hoshino A, Okawa Y, Ariyoshi M, Kaimoto S, Tateishi S, Ono K, Yamanaka R, Hato D, Fushimura Y, Honda S, Fukai K, Higuchi Y, Ogata T, Iwai-Kanai E, Matoba S. Cardiac-specific bdh1 overexpression ameliorates oxidative stress and cardiac remodeling in pressure overload-induced heart failure. Circ Heart Fail. 2017;10: e004417. [DOI] [PubMed] [Google Scholar]
- 44.Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M. Blood ketone bodies in congestive heart failure. J Am Coll Cardiol. 1996;28:665–672. [DOI] [PubMed] [Google Scholar]
- 45.Marcondes-Braga FG, Gutz IGR, Batista GL, Saldiva PHN, Ayub-Ferreira SM, Issa VS, Mangini S, Bocchi EA, Bacal F. Exhaled acetone as a new biomaker of heart failure severity. Chest. 2012;142:457–466. [DOI] [PubMed] [Google Scholar]
- 46.Voros G, Ector J, Garweg C, Droogne W, Van Cleemput J, Peersman N, Vermeersch P, Janssens S. Increased cardiac uptake of ketone bodies and free fatty acids in human heart failure and hypertrophic left ventricular remodeling. Circ Heart Fail. 2018;11:e004953. [DOI] [PubMed] [Google Scholar]
- 47.Zordoky BN, Sung MM, Ezekowitz J, Mandal R, Han B, Bjorndahl TC, Bouatra S, Anderson T, Oudit GY, Wishart DS, Dyck JR, Alberta H. Metabolomic fingerprint of heart failure with preserved ejection fraction. PLoS One. 2015;10:e0124844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunter WG, Kelly JP, McGarrah RW 3rd, Khouri MG, Craig D, Haynes C, Ilkayeva O, Stevens RD, Bain JR, Muehlbauer MJ, Newgard CB, Felker GM, Hernandez AF, Velazquez EJ, Kraus WE, Shah SH. Metabolomic profiling identifies novel circulating biomarkers of mitochondrial dysfunction differentially elevated in heart failure with preserved versus reduced ejection fraction: Evidence for shared metabolic impairments in clinical heart failure. J Am Heart Assoc. 2016;5:e003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, King MT, Dodd MS, Holloway C, Neubauer S, Drawer S, Veech RL, Griffin JL, Clarke K. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016;24:256–268. [DOI] [PubMed] [Google Scholar]
- 50.Verma S, Garg A, Yan AT, Gupta AK, Al-Omran M, Sabongui A, Teoh H, Mazer CD, Connelly KA. Effect of empagliflozin on left ventricular mass and diastolic function in individuals with diabetes: An important clue to the empa-reg outcome trial? Diabetes Care. 2016;39:e212–e213. [DOI] [PubMed] [Google Scholar]
- 51.Gormsen LC, Svart M, Thomsen HH, Sondergaard E, Vendelbo MH, Christensen N, Tolbod LP, Harms HJ, Nielsen R, Wiggers H, Jessen N, Hansen J, Botker HE, Moller N. Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: A positron emission tomography study. J Am Heart Assoc. 2017;6:e005066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joubert M, Jagu B, Montaigne D, Marechal X, Tesse A, Ayer A, Dollet L, Le May C, Toumaniantz G, Manrique A, Charpentier F, Staels B, Magre J, Cariou B, Prieur X. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes. 2017;66:1030–1040. [DOI] [PubMed] [Google Scholar]
- 53.Lee TM, Chang NC, Lin SZ. Dapagliflozin, a selective sglt2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via stat3 signaling in infarcted rat hearts. Free Radic Biol Med. 2017;104:298–310. [DOI] [PubMed] [Google Scholar]
- 54.Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, Dyck JE, Uddin GM, Oudit GY, Mayoux E, Lehrke M, Marx N, Lopaschuk GD. Empagliflozin increases cardiac energy production in diabetes: Novel translational insights into the heart failure benefits of sglt2 inhibitors. JACC Basic Transl Sci. 2018;3:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abdurrachim D, Teo XQ, Woo CC, Chan WX, Lalic J, Lam CSP, Lee PTH. Empagliflozin reduces myocardial ketone utilization while preserving glucose utilization in diabetic hypertensive heart disease: A hyperpolarized (13) c magnetic resonance spectroscopy study. Diabetes Obes Metab. 2019;21:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, Flores E, Garcia-Ropero A, Sanz J, Hajjar RJ, Fuster V, Badimon JJ. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. 2019;73:1931–1944. [DOI] [PubMed] [Google Scholar]
- 57.Yurista SR, Sillje HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, van Goor H, van Veldhuisen DJ, de Boer RA, Westenbrink BD. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. 2019;21:862–873. [DOI] [PubMed] [Google Scholar]
- 58.Nordmann AJ, Nordmann A, Briel M, Keller U, Yancy WS Jr., Brehm BJ, Bucher HC. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: A meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166:285–293. [DOI] [PubMed] [Google Scholar]
- 59.Gibson AA, Seimon RV, Lee CMY, Ayre J, Franklin J, Markovic TP, Caterson ID, Sainsbury A. Do ketogenic diets really suppress appetite? A systematic review and meta-analysis. Obes Rev. 2015;16:64–76. [DOI] [PubMed] [Google Scholar]
- 60.Augustin K, Khabbush A, Williams S, Eaton S, Orford M, Cross JH, Heales SJR, Walker MC, Williams RSB. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018;17:84–93. [DOI] [PubMed] [Google Scholar]
- 61.Mumme K, Stonehouse W. Effects of medium-chain triglycerides on weight loss and body composition: A meta-analysis of randomized controlled trials. J Acad Nutr Diet. 2015;115:249–263. [DOI] [PubMed] [Google Scholar]
- 62.Holloway CJ, Cochlin LE, Emmanuel Y, Murray A, Codreanu I, Edwards LM, Szmigielski C, Tyler DJ, Knight NS, Saxby BK, Lambert B, Thompson C, Neubauer S, Clarke K. A high-fat diet impairs cardiac high-energy phosphate metabolism and cognitive function in healthy human subjects. Am J Clin Nutr. 2011;93:748–755. [DOI] [PubMed] [Google Scholar]
- 63.Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004;364:1786–1788. [DOI] [PubMed] [Google Scholar]
- 64.Leichman JG, Aguilar D, King TM, Vlada A, Reyes M, Taegtmeyer H. Association of plasma free fatty acids and left ventricular diastolic function in patients with clinically severe obesity. Am J Clin Nutr. 2006;84:336–341. [DOI] [PubMed] [Google Scholar]
- 65.Cox PJ, Clarke K. Acute nutritional ketosis: Implications for exercise performance and metabolism. Extrem Physiol Med. 2014;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seidelmann SB, Claggett B, Cheng S, Henglin M, Shah A, Steffen LM, Folsom AR, Rimm EB, Willett WC, Solomon SD. Dietary carbohydrate intake and mortality: A prospective cohort study and meta-analysis. Lancet Public Health. 2018;3:e419–e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balasse E, Ooms HA. Changes in the concentrations of glucose, free fatty acids, insulin and ketone bodies in the blood during sodium beta-hydroxybutyrate infusions in man. Diabetologia. 1968;4:133–135. [DOI] [PubMed] [Google Scholar]
- 68.Mikkelsen KH, Seifert T, Secher NH, Grondal T, van Hall G. Systemic, cerebral and skeletal muscle ketone body and energy metabolism during acute hyper-d-beta-hydroxybutyratemia in post-absorptive healthy males. J Clin Endocrinol Metab. 2015;100:636–643. [DOI] [PubMed] [Google Scholar]
- 69.Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, Jin L, Liaw C, Chen R, Richman J, Connolly D, Offermanns S, Wright SD, Waters MG. (d)-beta-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor puma-g. J Biol Chem. 2005;280:26649–26652. [DOI] [PubMed] [Google Scholar]
- 70.Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, Kobayashi M, Hirasawa A, Tsujimoto G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via g protein-coupled receptor 41 (gpr41). Proc Natl Acad Sci U S A. 2011;108:8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, Faull OK, Magor-Elliott S, Hiyama S, Stirling M, Clarke K. On the metabolism of exogenous ketones in humans. Front Physiol. 2017;8:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, Ho M, Roberts A, Robertson J, Vanitallie TB, Veech RL. Kinetics, safety and tolerability of (r)-3-hydroxybutyl (r)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol. 2012;63:401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Malley T, Myette-Cote E, Durrer C, Little JP. Nutritional ketone salts increase fat oxidation but impair high-intensity exercise performance in healthy adult males. Appl Physiol Nutr Metab. 2017;42:1031–1035. [DOI] [PubMed] [Google Scholar]
- 74.Johnson RH, Walton JL, Krebs HA, Williamson DH. Metabolic fuels during and after severe exercise in athletes and non-athletes. Lancet. 1969;2:452–455. [DOI] [PubMed] [Google Scholar]
- 75.Johnson RH, Walton JL. The effect of exercise upon acetoacetate metabolism in athletes and non-athletes. Q J Exp Physiol Cogn Med Sci. 1972;57:73–79. [DOI] [PubMed] [Google Scholar]
- 76.Wang P, Tate JM, Lloyd SG. Low carbohydrate diet decreases myocardial insulin signaling and increases susceptibility to myocardial ischemia. Life Sci. 2008;83:836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Al-Zaid NS, Dashti HM, Mathew TC, Juggi JS. Low carbohydrate ketogenic diet enhances cardiac tolerance to global ischaemia. Acta Cardiol. 2007;62:381–389. [DOI] [PubMed] [Google Scholar]
- 78.Zou Z, Sasaguri S, Rajesh KG, Suzuki R. Dl-3-hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1968–1974. [DOI] [PubMed] [Google Scholar]
- 79.Sharma N, Okere IC, Barrows BR, Lei B, Duda MK, Yuan CL, Previs SF, Sharov VG, Azimzadeh AM, Ernsberger P, Hoit BD, Sabbah H, Stanley WC. High-sugar diets increase cardiac dysfunction and mortality in hypertension compared to low-carbohydrate or high-starch diets. J Hypertens. 2008;26:1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duda MK, O’Shea KM, Lei B, Barrows BR, Azimzadeh AM, McElfresh TE, Hoit BD, Kop WJ, Stanley WC. Low-carbohydrate/high-fat diet attenuates pressure overload-induced ventricular remodeling and dysfunction. J Card Fail. 2008;14:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Okere IC, Young ME, McElfresh TA, Chess DJ, Sharov VG, Sabbah HN, Hoit BD, Ernsberger P, Chandler MP, Stanley WC. Low carbohydrate/high-fat diet attenuates cardiac hypertrophy, remodeling, and altered gene expression in hypertension. Hypertension. 2006;48:1116–1123. [DOI] [PubMed] [Google Scholar]
- 82.Shah AM, Claggett B, Sweitzer NK, Shah SJ, Anand IS, Liu L, Pitt B, Pfeffer MA, Solomon SD. Prognostic importance of impaired systolic function in heart failure with preserved ejection fraction and the impact of spironolactone. Circulation. 2015;132:402–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weber T, Chirinos JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J. 2018;39:3847–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E, Dixit VD. The ketone metabolite beta-hydroxybutyrate blocks nlrp3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanikarla-Marie P, Jain SK. Hyperketonemia (acetoacetate) upregulates nadph oxidase 4 and elevates oxidative stress, icam-1, and monocyte adhesivity in endothelial cells. Cell Physiol Biochem. 2015;35:364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jain SK, Kannan K, Lim G, McVie R, Bocchini JA Jr. Hyperketonemia increases tumor necrosis factor-alpha secretion in cultured u937 monocytes and type 1 diabetic patients and is apparently mediated by oxidative stress and camp deficiency. Diabetes. 2002;51:2287–2293. [DOI] [PubMed] [Google Scholar]
- 87.Fisher FM, Maratos-Flier E. Understanding the physiology of fgf21. Annu Rev Physiol. 2016;78:223–241. [DOI] [PubMed] [Google Scholar]
- 88.Planavila A, Redondo I, Hondares E, Vinciguerra M, Munts C, Iglesias R, Gabrielli LA, Sitges M, Giralt M, van Bilsen M, Villarroya F. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat Commun. 2013;4:2019. [DOI] [PubMed] [Google Scholar]
- 89.Nagao M, Toh R, Irino Y, Mori T, Nakajima H, Hara T, Honjo T, Satomi-Kobayashi S, Shinke T, Tanaka H, Ishida T, Hirata K. Beta-hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem Biophys Res Commun. 2016;475:322–328. [DOI] [PubMed] [Google Scholar]
- 90.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr., de Cabo, Ulrich S, Akassoglou, Verdin E. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–2190. [DOI] [PubMed] [Google Scholar]
- 92.Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, Xia C, Peng C, Ruan H, Kirkey M, Wang D, Jensen LM, Kwon OK, Lee S, Pletcher SD, Tan M, Lombard DB, White KP, Zhao H, Li J, Roeder RG, Yang X, Zhao Y. Metabolic regulation of gene expression by histone lysine beta-hydroxybutyrylation. Mol Cell. 2016;62:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lytvyn Y, Bjornstad P, Udell JA, Lovshin JA, Cherney DZI. Sodium glucose cotransporter-2 inhibition in heart failure: Potential mechanisms, clinical applications, and summary of clinical trials. Circulation. 2017;136:1643–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonner C, Kerr-Conte J, Gmyr V, Queniat G, Moerman E, Thevenet J, Beaucamps C, Delalleau N, Popescu I, Malaisse WJ, Sener A, Deprez B, Abderrahmani A, Staels B, Pattou F. Inhibition of the glucose transporter sglt2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med. 2015;21:512–517. [DOI] [PubMed] [Google Scholar]
- 95.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, Mari A, Pieber TR, Muscelli E. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. 2016;65:1190–1195. [DOI] [PubMed] [Google Scholar]