

Abstract
While numerous studies have confirmed ATP’s importance in bladder physiology/pathophysiology, the literature is still conflicted regarding the mechanism of ATP release from the urothelium. Multiple mechanisms have been identified including non-vesicular release via pannexin channels as well as vesicular release via a mechanism blocked by botulinum toxin. Recently, it has been shown that lysosomes contain significant stores of ATP which can be released extracellularly in response to Toll-like receptor (TLR) stimulation. The goal of the current study was to determine if lysosomal exocytosis occurs in urothelial cells in response to TLR4 stimulation by its agonist, bacterial lipopolysaccharide (LPS). Stimulation of TRT-HU1 human urothelial cells with 100μg/ml LPS significantly increased ATP release, which was inhibited by the destruction of lysosomes using glycyl-L-phenylalanine-β-naphthylamide, but not by the pannexin channel antagonist Brilliant Blue FCF. Conversely, stimulation with the nicotinic agonist cytisine induced ATP release that was sensitive to Brilliant Blue FCF but not glycyl-L-phenylalanine-β-naphthylamide. LPS stimulation also induced the release of the lysosomal acid phosphatases. LPS increased lysosomal pH and direct alkalization of lysosomal pH using chloroquine or bafilomycin A1 induced ATP and acid phosphatase release, indicating an important role for pH in lysosomal exocytosis. Additionally, stimulation of lysosomal Transient Receptor Potential Mucolipin 1 calcium channels evoked intracellular calcium transients as well as ATP release. These data indicate that LPS-induced ATP release from urothelial cells is mediated by lysosomal exocytosis, a vesicular mechanism distinctly separate from non-vesicular release via pannexin channels.
Keywords: adenosine triphosphate, urinary bladder, lower urinary tract, purinergic, epithelial, lysosome, lysosomal pH, chloroquine, bafilomycin A1
Introduction
ATP is an important transmitter in the sensory limb of the micturition reflex. For example, distension of the bladder during filling causes a release of ATP from the urothelium (1), which is thought to act in a paracrine manner on afferent nerves present in the bladder wall to increase their firing (2). Increased urothelial ATP release is also thought to play a significant role in pathological bladder disorders, as urinary ATP is significantly increased in patients suffering from urinary tract infections (3), overactive bladder (4) or Bladder Pain Syndrome / Interstitial Cystitis (5). Given this link to bladder pathology, there has been interest in targeting purinergic signaling between the urothelium and sensory nerves as a treatment for bladder pathology, however progress has been hampered due to an incomplete understanding of the mechanisms controlling urothelial ATP release.
Multiple mechanisms of urothelial ATP release have been studied over the past decade. Our lab and others have demonstrated that stretch or chemical stimulation of the urothelium releases ATP through pannexin channels, large-pore ion channels permeable to purines (1). However, there is also considerable evidence that urothelial cells release ATP via a vesicular mechanism. Chief among this evidence is the expression of the vesicular nucleotide transporter (VNUT, gene name: Slc17A9) in the urothelium and that ATP is stored in discrete vesicles inside urothelial cells (6). Additionally, our own experiments demonstrated that ATP released by the urothelium into the lumen of the rat bladder in response to bacterial lipopolysaccharides (LPS) was not completely blocked by the pannexin antagonist Brilliant Blue FCF (BB-FCF), indicating a contribution of another, possibly vesicular, mechanism of ATP release (1). For example, retinal pigmented epithelial cells exhibit both vesicular and non-vesicular (pannexin-mediated) ATP release (7,8). Mechanical stimulation of these cells releases ATP through pannexin channels, but release triggered by stimulation of the Toll-like receptor (TLR) is independent of pannexin channels but is suppressed after cells are treated with glycyl-l-phenylalanine 2-naphthylamide (GPN), a cathepsin C substrate that destroys lysosomes (8,9). Furthermore, TLR-mediated ATP release is accompanied by the release of lysosomal enzymes, such as acid phosphatase, indicating that lysosomes can store ATP and that stimulation of the cells can lead to release of lysosomal contents into the extracellular space (i.e. exocytosis). Our present study was undertaken to determine if LPS-mediated ATP release from the urothelium occurs through lysosomal exocytosis.
Methods and Materials
Drugs, Reagents and Solutions:
All reagents used in this study were obtained from Sigma-Aldrich Inc. or Tocris, Inc. (Denver, CO). The serotype of LPS used in our studies was 0111:B4 (Sigma-Aldrich). The extracellular solution used to maintain cells in our ATP release studies (hereafter called “isotonic solution”) consisted of (in mM): NaCl 105, KCl 5, HEPES Acid 6, Na HEPES 4, NaHCO3 5, glucose 5, CaCl2 1.3, MgCl2 0.5, Mannitol 60; pH 7.4. The osmolarity of this solution was measured at ~300 mOsmol/L, which matched the measured osmolarity of the keratinocyte media used to grow the cells. For our calcium imaging studies, cells were maintained in Hanks’ Balanced Saline Solution (HBSS), which consisted of (in mM): NaCl 138, KCl 5, KH2P04 0.03, Na2HPO4 0.03, NaHCO3 4, Glucose 5.6, Na HEPES 10, MgCl2 1, CaCl2 2; pH 7.4. All drug stocks were made in isotonic solution/HBSS with the exception of glycyl-L-phenylalanine-β-naphthylamide, ML-SA1 and bafilomycin A1, which were initially dissolved in DMSO and diluted in isotonic solution/HBSS to a final DMSO concentration of 0.1%.
TRT-HU1 cell culture:
TRT-HU1 cells, an hTERT-immortalized human urothelial cell line, were obtained from Drs. Raymond Rackley and Joeseph DiDonato of the Cleveland Clinic (10). These cells were grown in serum-free keratinocyte medium containing 1% Penicillin-Streptomycin-Amphotericin B and 10μg/ml Blasticidin (as a selection marker for hTERT). Keratinocyte medium was purchased from Invitrogen and contained epidermal growth factor and bovine pituitary extract. Cells were grown in T25 cell culture flasks (Fisher Scientific) and passaged when they reached 75–80% confluency. Cells used for this study were from passage 30–40.
ATP Release:
TRT-HU1 cells were passaged, plated on cell-culture treated 96-well plates with white opaque walls and clear bottoms (Fisher Scientific) and allowed to grow until 50–75% confluent (~2 days). At the time of the experiment, the medium in the plate was carefully aspirated and the cells washed with 100μl of isotonic solution. This solution was aspirated again and the cells covered in either 50μl of isotonic solution or 50μl of isotonic solution containing a 2X concentration of one of the antagonists used in the study. This plate was returned to the incubator for 20 minutes to allow any ATP released by mechanical stimulation when switching solutions to be broken down by ATPases. After the incubation, 50μl of either isotonic solution alone (for a negative control) or 50μl of isotonic solution containing a 2X concentration of one of the agonists used in the study (such as LPS) was added to the volume already in the plate (for a final 1X concentration of agonists/antagonists) and the plate returned to the incubator for 20 minutes. Following this second incubation, 50μl of luciferin/luciferase assay mix (Sigma-Aldrich) was added to each well. A piece of white adhesive contact paper was placed on the bottom of the plate to make it opaque before immediately reading in a plate-reader capable of luminescence readings (Victor3, PerkinElmer) with an integration time of 100 milliseconds. Relative luminescence readings were converted to ATP concentrations using a standard curve with known concentrations of ATP and containing the agonists/antagonists used in the experiment to correct for any interference they may have on the luciferin/luciferase assay.
Acid Phosphatase Measurements:
TRT-HU1 cells were passaged, plated on cell-culture treated 24-well plates and allowed to grow until 50–75% confluent. At the time of the experiment, the medium was replaced for 60 minutes with 500μl of either isotonic solution or 20μM glycyl-l-phenylalanine 2-naphthylamide (GPN). The cells were then stimulated by pipetting 50μl of 1mg/ml LPS, 1mM cytisine or 100μM chloroquine into the appropriate wells for a final concentration of 100μg/ml, 100μM or 10μM, respectively. Control (unstimulated) cells received 50μl of isotonic solution. The cells were incubated in the drug solutions for 2 hours at 37°C, then the extracellular solution was collected from each well and immediately frozen at −80 °C for acid phosphatase assays later. Acid phosphatase activity was determined using a colormetric assay kit (Sigma-Aldrich, Inc) using the manufacturer’s instructions. Briefly, 50μl of each sample (in triplicate) was incubated at 37°C with 50μl of a 4-nitrophenyl phosphate solution in citrate buffer for 30 min and then 200μl of a stop solution (0.5 N NaOH) was added to each reaction. Blank samples containing only citrate buffer and standard samples containing a known quantity of p-nitrophenol enzyme substrate were also added to the plate. The absorbance of the plate was read at 405nm using the Tecan Spark 20M plate reader. Acid phosphatase activity was determined using the following equation:
where “A405[x]” represents the absorbance readings at 405nm for the listed samples, “0.05μmol/ml” equals the concentration of 4-nitophenol in the standard solution, “0.3ml” equals the total volume of the unknown samples, “30 min” is the time samples were incubated with 4-nitrophenol and “50μl” equals the volume of each unknown sample in the assay. These readings were then normalized by dividing them by the average of the control (unstimulated) samples (% control).
Lysosomal pH measurements:
Lysosomal pH was measured as described previously (11). In brief, cells were grown in black 96-well plates, and allowed to grow until 50–75% confluent. At the time of the experiment, the media was removed and replaced with 50μl of either isotonic solution alone (control) or drugs dissolved in isotonic solution. After a 2 hour incubation, the extracellular solution was aspirated and the cells rinsed and incubated with 5 μM LysoSensor Yellow/Blue DND 160 (Invitrogen Corp.) for 3 min followed by a 15 min post-incubation in isotonic solution. Lysosomal pH was determined from the ratio of light excited at 340 nm vs. 380 nm (520 nm) using a plate reader (Tecan Spark 20M) and calibrated by exposing cells to 10μM H+/Na+ ionophore monensin and 20 μM H+/K+ ionophore nigericin in a solution containing (in mM) 20 MES, 110 KCl and 20 NaCl at pH 4.0–6.0 for 15 min.
Calcium Imaging (Fura-2):
TRT-HU1 cells grown on collagen-coated, glass coverslips were loaded with Fura-2 AM by incubating the cells in 5uM Fura-2AM in HBSS containing 0.2% (w/v) Pluronic F-127 detergent for 20 minutes at room temperature. Following loading, the coverslips were placed into a flow chamber on an upright microscope (Olympus BX61WI) with a Hamamatsu CCD camera (ORCA-ER) and a data analyzation computer running image analysis software (Simple-PCI, Compix). Cells were maintained in HBSS throughout the experiment by a gravity-fed perfusion system. To record changes in [Ca2+]i the cells were alternatively illuminated at 340 and 380 nm by using a xenon lamp and imaged at 510 nm. Increases in intracellular calcium were detected as a change in the ratio of emission at each excitation wavelength (340/380). Data reported is the peak increase in Fura-2 ratio over baseline during a 5-minute perfusion of agonist (ML-SA1).
Data Analysis:
All data are expressed as mean ± standard error of the mean. Significance was defined as p<0.05 and was determined using an appropriate statistical test after testing if the data set conformed to a normal distribution. Normality and statistical significance were determined using Prism 8.3 statistical software (GraphPad Software, LLC). The particular test performed for each experiment is denoted in the figure legend. ATP/acid phosphatase levels were normalized to the mean value of control cells for each plate to correct for any variations in cell density and activity of the assay between experiments.
Results:
Pannexin channels do not mediate urothelial release of ATP in response to lipopolysaccharides:
To examine the pathway(s) responsible for controlling LPS-mediated ATP release from the urothelium, we stimulated the TRT-HU1 human urothelial cell line with LPS. Consistent with our previous experiments in the rat (1), LPS (100μg/ml) significantly increased extracellular ATP (increase of 24.3 ± 2.2% over unstimulated controls, Figure 1A). However, pre-incubation of the cells with the pannexin channel antagonist BB-FCF (100μM) did not decrease this release (30.8 ± 2.1 % increase over control, Figure 1A). BB-FCF alone had no significant effect on ATP release (Figure 1A). Stimulation of α3 nicotinic receptors using cytisine (100μM), also significantly increased basal ATP release (38.0 ± 3.7% over control, Figure 1A) and this release was inhibited by BB-FCF (7.1 ± 1.7% increase over control, Figure 1A) or by the α3 nAChR antagonist 2,2,6,6-Tetramethylpiperidin-4-yl heptanoate (TMPH, 6.6 ± 2.0% increase over control, Figure 1A). These data suggest that a non-pannexin related, mechanism is responsible for ATP release in response to LPS.
Figure 1: LPS-mediated ATP release involves lysosomes, but not pannexin channels.

(A) ATP release assay in human urothelial TRT-HU1 cells, expressed as a percent change from baseline (Isotonic solution only control). Brilliant Blue FCF (BB-FCF) inhibits ATP release induced by the nicotinic receptor agonist cytisine, but not by the TLR4 agonist lipopolysaccharide (LPS). ** p<0.01 as compared to isotonic only control by one-way ANOVA with Holm-Sidak multiple comparison test. ++ p<0.01 as compared to cytisine by one-way ANOVA with Holm-Sidak multiple comparison test. (B) Glycyl-L-phenylalanine-β-naphthylamide (GPN) inhibits LPS-induced ATP release, but not cytisine-induced release. ** p<0.01 as compared to isotonic only control by one-way ANOVA with Holm-Sidak multiple comparison test. ++ p<0.01 as compared to LPS by one-way ANOVA with Holm-Sidak multiple comparison test. Numbers on each bar represent the number of wells in 96-well plates where the indicated conditions were tested.
To test if the lysosome could also be playing a role in TLR-mediated ATP release in urothelial cells, we pretreated TRT-HU1 cells with GPN (20μM, 30 min) before stimulating with LPS. GPN is a cell-permeable substrate for the lysosomal enzyme Cathepsin C. When cleaved by cathepsin, GPN exerts an osmotic effect that disrupts the membrane of the lysosome, leading to its destruction (9). GPN pre-treatment completely blocked LPS-mediated ATP release in TRT-HU1 cells (1.2 ± 1.9% increase over control, Figure 1B). Conversely, cytisine-induced ATP release was not diminished after GPN pretreatment (28.3 ± 5.7% increase over control). These data suggest that LPS-mediated ATP release is mediated through lysosomal exocytosis.
LPS stimulation of TRT-HU1 cells also causes the release of lysosomal acid phosphatases:
To confirm that ATP is being released by lysosomal exocytosis, we tested the extracellular fluid for acid phosphatase activity (12). Acid phosphatases are highly concentrated in lysosomes, where they are responsible for catalyzing the hydrolysis of phosphate monoesters under acidic conditions. Therefore, their presence in the extracellular fluid would indicate lysosomal exocytosis. Stimulation of TRT-HU1 cells with LPS (100μg/ml) significantly increased extracellular acid phosphatase activity (126.5 ± 7.8 % change from control, p<0.0001, Figure 2). Pretreatment of cells with GPN prevented this increase (−12.2 ± 4.6% change from control, p=0.1560, Figure 2). Additionally, treatment of cells with cytisine, which activates ATP release through pannexin channels and not by lysosomal exocytosis, did not increase extracellular phosphatase activity (3.1±3.4%, p=0.9697, Figure 2).
Figure 2: Stimulation with LPS also increases extracellular acid phosphatase activity.
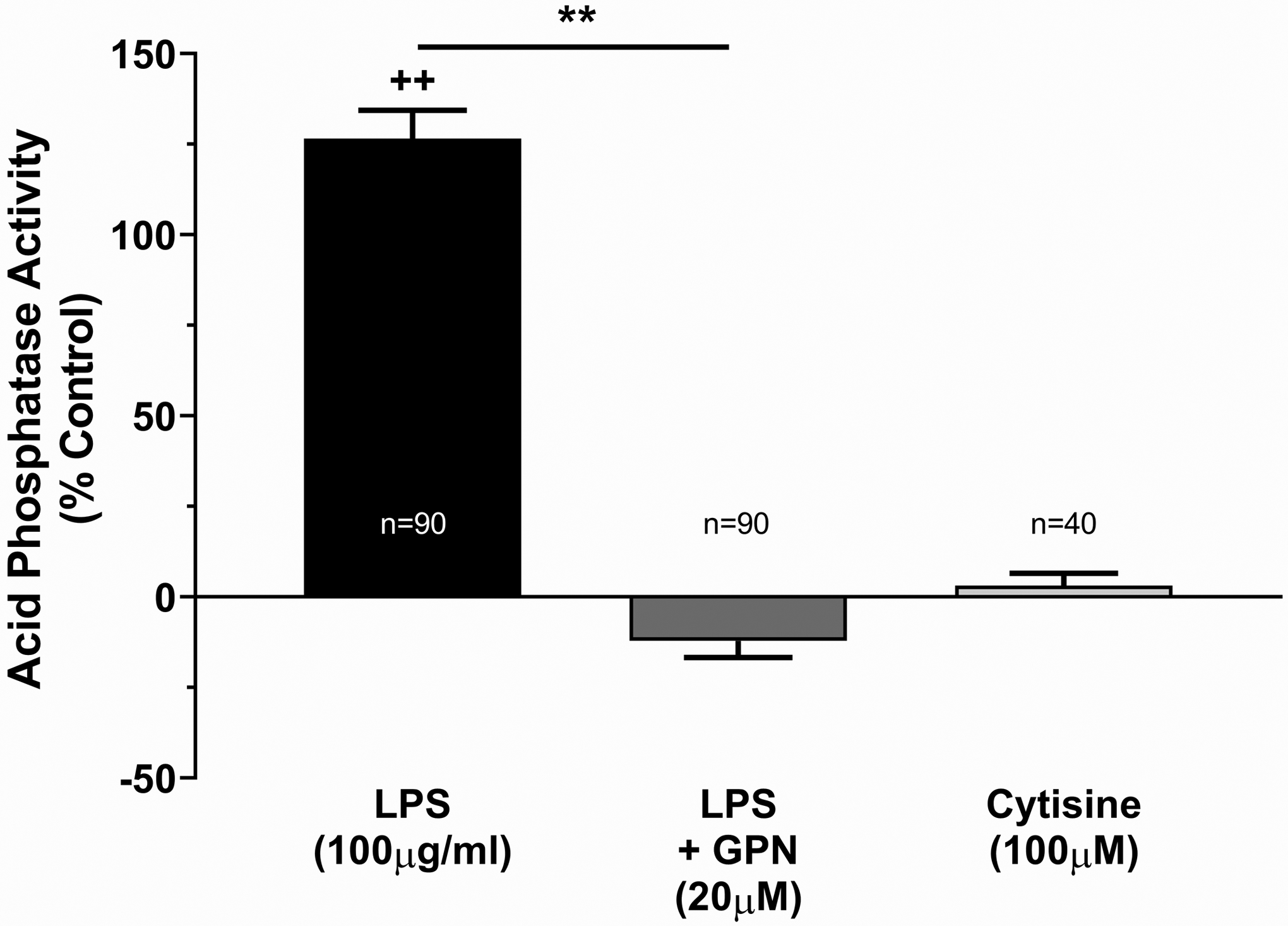
Acid phosphatase activity in the extracellular fluid (isotonic solution) of TRT-HU1 cells after stimulation with LPS, expressed as a percent of control. ** p<0.01 by one-way ANOVA with Holm-Sidak multiple comparison test. ++ p<0.01 as compared to isotonic only control by one-way ANOVA with Holm-Sidak multiple comparison test. Numbers on each bar represent the number of wells in 96-well plates where the indicated conditions were tested.
LPS stimulation of TRT-HU1 cells increases lysosomal pH:
It has been previously shown that TLRs can control autophagy and that stimulation of these receptors can alkalinize lysosomes and lead to their exocytosis (8). To confirm a role for lysosomal alkalization in LPS-induced ATP release from TRT-HU1 cells, we directly measured lysosomal pH using the Lysosensor Yellow/Blue DND 160 fluorescent dye. Cells stimulated with LPS for 1 hour showed a significant increase in lysosomal pH (from 4.5 to 5.2, p<0.001, Figure 3A). This increase was similar to increases elicited by chloroquine (pH 5.0) and bafilomycin A1 (pH 5.4), agents known for their ability to increase lysosomal pH (Figure 3A)(13,14). Chloroquine and bafilomycin A1 also significantly increased ATP release (13.6 ± 2.9% and 33.2 ± 4.8%, respectively), indicating that lysosomal alkalization is sufficient to induce lysosomal exocytosis (Figure 3B).
Figure 3: Lysosomal ATP release depends on lysosomal pH.

(A) Changes in lysosomal pH in TRT-HU1 cell induced by various agents. The pH was measured using LysoSensor Yellow/Blue DND 160 in TRT-HU1 cells. ** p<0.01 as compared to control by a Kruskal-Wallace nonparametric test with Dunn’s multiple comparison test. (B) ATP release from TRT-HU1 cells after alkalinizing lysosomal pH with chloroquine or bafilomycin A1. ** p<0.01 as compared to control by a Kruskal-Wallace nonparametric test with Dunn’s multiple comparison test.
Activation of lysosomal TRPML1 channels induces ATP release:
Transient Receptor Potential Mucolipin 1 (TRPML1) is a calcium-permeable ion channel predominately located on the lysosomal membrane. It is known that the lysosome is a significant calcium store and it is thought that TRPML1 mediates the release of calcium from the lysosome which in turn regulates fusion of the lysosome with the plasma membrane (15). One possible trigger for TRPML1 channels is a decrease in the hydrogen ion concentration in the lumen of the lysosome (i.e. lysosomal alkalization), suggesting that this receptor may play a role in the lysosomally-mediated ATP release in response to LPS that we have described above.
Stimulation of TRT-HU1 cells with the TRPML1 agonist ML-SA1 elicited a calcium transient, consistent with the previously discussed role of TRPML1 as a lysosomal calcium channel (Figure 4A). ML-SA1 stimulation also increased extracellular ATP (31.8 ± 2.4% over control, Figure 4B) and acid phosphatase concentrations (30.7 ± 5.5% over control, Figure 4C). Pre-incubation with GPN prevented this release of ATP and acid phosphatase (Figures 4B & 4C).
Figure 4: Activation of the lysosomal calcium channel TRPML1 induces ATP release through lysosomal exocytosis.

(A) Fura-2 calcium measurements in TRT-HU1 cells after stimulation with the TRPML1 agonist ML-SA1. **p<0.05 as compared to vehicle (DMSO) control by paired t-test. (B) Representative traces depicting average Fura-2 signals for one experiment (n=12 cells). Black bars indicate when 100μM ML-SA1 was applied. (C) ATP release from TRT-HU1 cells in response to ML-SA1. ** p<0.01 by one-way ANOVA with Holm-Sidak multiple comparison test. ++ p<0.01 as compared to ML-SA1. (D) Extracellular acid phosphatase activity following ML-SA1 stimulation. ** p<0.01 by one-way ANOVA with Holm-Sidak multiple comparison test. ++ p<0.01 as compared to ML-SA1.
Discussion:
It has been hypothesized that ATP released from the urothelium plays an important role in the pathophysiology of the urinary bladder. For example, it has been demonstrated that urinary ATP levels are significantly increased in patients diagnosed with Overactive Bladder (OAB) or Bladder Pain Syndrome/Interstitial Cystitis (BPS/IC) (3–5). Additionally, manipulation of urinary ATP levels in anesthetized rats through intravesical administration of apyrase or an inhibitor of endogenous ATPases can alter reflex bladder reflexes (1). A long list of stimuli induces the release of ATP from the urothelium (16), but there is uncertainty regarding the mechanism of ATP release. Early studies focused on vesicular release based on the dependence of ATP release on extracellular calcium as well as the ability of inhibitors of vesicular trafficking and fusion such as brefeldin A or botulinum toxin to block urothelial ATP release (17,18). More recently, however, the discovery of non-vesicular mechanisms of ATP release such as pannexin channels has reshaped our understanding of purinergic signaling in the urothelium (1,19). Nonetheless, sufficient evidence still exists to support the presence of a vesicular ATP storage and release mechanism in the urothelium. The most significant of this evidence has been the discovery of the vesicular nucleotide transporter (VNUT or SLC17A9) and its localization to the urothelium (6). There is also growing evidence that more than one mechanism of ATP release exists in the urothelium. For example, Nakagomi, et. al. demonstrated that urothelial cells from VNUT knockout mice showed diminished ATP release in response to small amounts of physical stretch, but ATP release in response to larger amounts of stretch were resistant to VNUT knockout (6). Our own research showed that distension evoked ATP release in vivo into the rat bladder lumen is completely blocked by antagonists or siRNA knockdown of pannexin, however ATP release is response to LPS was incompletely blocked by pannexin channel inhibition (1). Thus it is becoming clear that the urothelium has multiple mechanisms of ATP release, which may be differentially activated by distinct stimuli.
Our current research revealed that human urothelial cells have at least two separate mechanisms for ATP release, pannexin channels and lysosomal exocytosis (Figure 5). Additionally, we have shown that pannexin channels mediate ATP release induced by nicotinic receptor stimulation, while lysosomal exocytosis accounts for LPS-induced ATP release. ATP released in response to LPS stimulation is blocked by GPN, a cathepsin C substrate that causes lysosomal disruption, but not by the pannexin antagonist Brilliant Blue FCF. LPS stimulation also increased extracellular concentrations of acid phosphatase, a lysosomally resident protein also released by lysosomal exocytosis. This release of ATP appears to be driven by an increase in lysosomal pH because LPS increases lysosomal pH and direct alkalization of lysosomes using chloroquine or bafilomycin A1 is sufficient to induce ATP release. Stimulation of TRPML1 channels, believed to be lysosomal calcium channels activated by increased lysosomal pH, is also sufficient to induce ATP and acid phosphatase release. Taken together, our data implicate lysosomal exocytosis as the mechanism of LPS-induced ATP release in the urothelium. It is unclear, however, whether ATP released by lysosomal exocytosis has a physiological role in bladder function. Our research did not examine whether lysosomal ATP release resulted in altered bladder activity, thus we cannot yet say that it important for the increased bladder inflammation and detrusor overactivity induced in the rat by LPS. Our previous research, however, demonstrated that intravesical instillation of LPS, which we now know induces lysosomal ATP release, increased ATP concentrations in the lumen of the bladder by roughly 2-fold in non-distended bladders and over 5-fold in bladders distended to a pressure of 15 cm of water (1). This suggests that lysosomal exocytosis can induce ATP release from the urothelium in vivo in a significant enough concentration to play a physiological/pathophysiological role.
Figure 5: Hypothetical model of urothelial ATP release mechanisms.

(Left) Proposed mechanism of pannexin-mediated ATP release from the urothelium. Activation of α3 nAChRs (presented in this study) or stretch-activated mechanosensors (presented in our previous study (1)) induce ATP release through pannexin channels, which we hypothesize involves an influx of extracellular calcium (25). (Right) Stimulation of TLR4 by LPS or bacteria increases lysosomal pH through an, as yet, unknown pathway. This alkalization of the lysosomal lumen activates TRPML1 receptors on the lysosomal membrane, releasing calcium from lysosomal stores and promoting fusion of the lysosome with the plasma membrane.
Aside from our current research, there is growing evidence supporting a role for lysosomes as an ATP store and release mechanism in other cell types. It has been shown that VNUT completely co-localizes with lysosomal markers in epithelial cells such as retinal pigmented epithelia and HEK293 cells (8,20). Additionally, purified lysosomes actively take up ATP and uptake is blocked in lysosomes purified from cells treated with VNUT siRNA (20). Regarding the release of lysosomally stored ATP, it has been shown that increasing lysosomal pH, either through direct means such as treatment of cells with the lysosomotropic agent CHQ, or through indirect means by activating receptor pathways that result in lysosomal alkalization (i.e. TLR stimulation), that the lysosome will fuse with the plasma membrane and release its contents, including ATP (8,21). It has been hypothesized that this mechanism represents an important defense response against bacterial infection; some bacteria can prevent lysosomal degradation by neutralizing the acidic environment required for proper lysosomal enzyme function (21). In this case, increased lysosomal pH leads to activation of lysosomal calcium channels and fusion of the lysosome to the plasma membrane, discharging the undigested bacteria as well as the lysosomal store of ATP. This ATP then acts as a pathogen associated molecular pattern (PAMP) that induces the activation of the innate immune system to clear the bacterial infection (22). It is also possible that lysosomal alkalization as a result of age (23) or mutations in lysosomal proton pumps (vH+-ATPases) (24) can lead to lysosomal exocytosis and may help drive sterile inflammation. Hence, the lysosome may be an attractive target for treating bladder pathology involving inflammation, such as Interstitial Cystitis/Bladder Pain Syndrome (IC/PBS), a disorder also associated with increased urinary ATP levels. One possible target for inhibiting lysosomal exocytosis could be lysosomal TRPML1 channels, as our current research has demonstrated an important role for these channels in activating lysosomal exocytosis.
References
- 1.Beckel JM, Daugherty SL, Tyagi P, Wolf-Johnston AS, Birder LA, Mitchell CH, de Groat WC. Pannexin 1 channels mediate the release of ATP into the lumen of the rat urinary bladder. J Physiol 2015; 593: 1857–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birder LA. More than just a barrier: urothelium as a drug target for urinary bladder pain. Am J Physiol Renal Physiol 2005; 289: F489–495. [DOI] [PubMed] [Google Scholar]
- 3.Gill K, Horsley H, Kupelian AS, Baio G, De Iorio M, Sathiananamoorthy S, Khasriya R, Rohn JL, Wildman SS, Malone-Lee J. Urinary ATP as an indicator of infection and inflammation of the urinary tract in patients with lower urinary tract symptoms. BMC Urol 2015; 15: 7–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Contreras-Sanz A, Krska L, Balachandran AA, Curtiss NL, Khasriya R, Kelley S, Strutt M, Gill HS, Taylor KM, Mansfield KJ, Wu C, Peppiatt-Wildman CM, Malone-Lee J, Duckett J, Wildman SS. Altered urothelial ATP signaling in a major subset of human overactive bladder patients with pyuria. Am J Physiol Renal Physiol 2016; 311: F805–f816. [DOI] [PubMed] [Google Scholar]
- 5.Sun Y, Chai TC. Augmented extracellular ATP signaling in bladder urothelial cells from patients with interstitial cystitis. American Journal of Physiology - Cell Physiology 2006; 290: C27–C34. [DOI] [PubMed] [Google Scholar]
- 6.Nakagomi H, Yoshiyama M, Mochizuki T, Miyamoto T, Komatsu R, Imura Y, Morizawa Y, Hiasa M, Miyaji T, Kira S, Araki I, Fujishita K, Shibata K, Shigetomi E, Shinozaki Y, Ichikawa R, Uneyama H, Iwatsuki K, Nomura M, de Groat WC, Moriyama Y, Takeda M, Koizumi S. Urothelial ATP exocytosis: regulation of bladder compliance in the urine storage phase. Sci Rep 2016; 6: 29761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reigada D, Lu W, Zhang M, Mitchell CH. Elevated pressure triggers a physiological release of ATP from the retina: Possible role for pannexin hemichannels. Neuroscience 2008; 157: 396–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beckel JM, Gómez NM, Lu W, Campagno KE, Nabet B, Albalawi F, Lim JC, Boesze-Battaglia K, Mitchell CH. Stimulation of TLR3 triggers release of lysosomal ATP in astrocytes and epithelial cells that requires TRPML1 channels. Scientific Reports 2018; 8: 5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berg TO, Strømhaug E, Løvdal T, Seglen O, Berg T. Use of glycyl-L-phenylalanine 2-naphthylamide, a lysosome-disrupting cathepsin C substrate, to distinguish between lysosomes and prelysosomal endocytic vacuoles. Biochem J 1994; 300 (Pt 1): 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Ji M, DiDonato JA, Rackley RR, Kuang M, Sadhukhan PC, Mauney JR, Keay SK, Freeman MR, Liou LS, Adam RM. An hTERT-immortalized human urothelial cell line that responds to anti-proliferative factor. In Vitro Cell Dev Biol Anim 2011; 47: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guha S, Coffey EE, Lu W, Lim JC, Beckel JM, Laties AM, Boesze-Battaglia K, Mitchell CH. Approaches for detecting lysosomal alkalinization and impaired degradation in fresh and cultured RPE cells: evidence for a role in retinal degenerations. Experimental eye research 2014; 126: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 1997; 137: 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 1991; 266: 17707–17712. [PubMed] [Google Scholar]
- 14.Homewood CA, Warhurst DC, Peters W, Baggaley VC. Lysosomes, pH and the anti-malarial action of chloroquine. Nature 1972; 235: 50–52. [DOI] [PubMed] [Google Scholar]
- 15.Gomez NM, Lu W, Lim JC, Kiselyov K, Campagno KE, Grishchuk Y, Slaugenhaupt SA, Pfeffer BA, Fliesler SJ, Mitchell CH. Robust lysosomal calcium signaling through channel TRPML1 is impaired by lysosomal lipid accumulation. FASEB J 2018; 32: 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burnstock G Purinergic signalling in the urinary tract in health and disease. Purinergic Signal 2014; 10: 103–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sui G, Fry CH, Montgomery B, Roberts M, Wu R, Wu C. Purinergic and muscarinic modulation of ATP release from the urothelium and its paracrine actions. Am J Physiol Renal Physiol 2013; 306: F286–F298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith CP, Vemulakonda VM, Kiss S, Boone TB, Somogyi GT. Enhanced ATP release from rat bladder urothelium during chronic bladder inflammation: effect of botulinum toxin A. Neurochem Int 2005; 47: 291–297. [DOI] [PubMed] [Google Scholar]
- 19.Timoteo MA, Carneiro I, Silva I, Noronha-Matos JB, Ferreirinha F, Silva-Ramos M, Correia-de-Sa P. ATP released via pannexin-1 hemichannels mediates bladder overactivity triggered by urothelial P2Y6 receptors. Biochem Pharmacol 2014; 87: 371–379. [DOI] [PubMed] [Google Scholar]
- 20.Cao Q, Zhao K, Zhong XZ, Zou Y, Yu H, Huang P, Xu TL, Dong XP. SLC17A9 protein functions as a lysosomal ATP transporter and regulates cell viability. J Biol Chem 2014; 289: 23189–23199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao Y, Li G, Zhang X, Xu H, Abraham SN. A TRP Channel Senses Lysosome Neutralization by Pathogens to Trigger Their Expulsion. Cell 2015; 161: 1306–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol 2013; 3: 414–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Truschel ST, Clayton DR, Beckel JM, Yabes JG, Yao Y, Wolf-Johnston A, Birder LA, Apodaca G. Age-related endolysosome dysfunction in the rat urothelium. PLoS ONE 2018; 13: e0198817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing research reviews 2016; 32: 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beckel JM, Birder LA. Differential expression and function of nicotinic acetylcholine receptors in the urinary bladder epithelium of the rat. The Journal of Physiology 2012; 590: 1465–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]