

Abstract
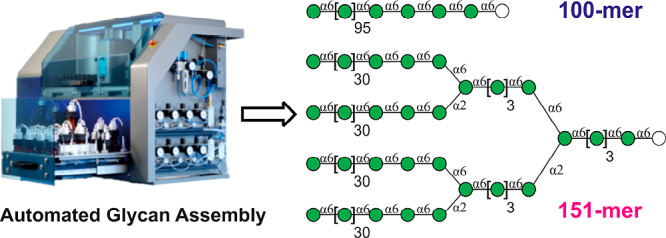
Polysaccharides are the most abundant biopolymers on earth that serve various structural and modulatory functions. Pure, completely defined linear and branched polysaccharides are essential to understand carbohydrate structure and function. Polysaccharide isolation provides heterogeneous mixtures, while heroic efforts were required to complete chemical and/or enzymatic syntheses of polysaccharides as long 92-mers. Here, we show that automated glycan assembly (AGA) enables access to a 100-mer polysaccharide via a 201-step synthesis within 188 h. Convergent block coupling of 30- and 31-mer oligosaccharide fragments, prepared by AGA, yielded a multiple-branched 151-mer polymannoside. Quick access to polysaccharides provides the basis for future material science applications of carbohydrates.
Automated solid-phase syntheses of peptides,1 DNA,2 and RNA3 were key to a molecular understanding of the structure and function of these biopolymers (Table 1). Rapid access to defined oligomers and polymers enabled applications such as DNA amplification4 and next-generation sequencing.5 Detailed molecular insights have remained difficult for carbohydrates that are isolated as heterogeneous mixtures and have been challenging to synthesize. Polysaccharides, in contrast to polypeptides and polynucleotides, are branched, and each glycosidic linkage constitutes a new stereogenic center that allows for a myriad of different polymers to be formed from just a few monosaccharides. Oligosaccharide construction by chemical,6 enzymatic,7 or chemoenzymatic8 synthesis mandates stereo- and regiocontrol at every step. Typically, labor-intensive total syntheses focused on the preparation of single molecules. Efforts to create large, branched polysaccharides have resorted to block couplings using oligosaccharides prepared by total synthesis, as was the case for a 92-mer polysaccharide, the largest synthetic carbohydrate made to date.9 The automated glycan assembly (AGA), based on the solid-phase synthesis paradigm like automated polynucleotide and polypeptide synthesis, utilizes iterative cycles consisting of coupling, capping, and deprotection steps that are executed by an automated instrument.10 Coupling efficiencies determine the length of biopolymers that can be prepared without intermediate purification. For polysaccharides, that limit was reached at 50-mer polysaccharides.11,12 Inspired by polynucleotide syntheses where 200-mers are routinely prepared,13 we developed automated synthesis protocols that ensure high coupling efficiencies while minimizing cycle times. Polymannosides serve to illustrate the concept as they do not form rigid tertiary structures that complicate the assembly of cellulose and chitin oligomers.14 The automated synthesis of 100-mer polymannoside 3, the largest polysaccharide prepared by any synthetic method to date, served as an initial goal to benchmark the methodological advances. Using a recently developed linker15 and improved AGA protocols, a linear 100-mer polymannoside was assembled from differentially protected monosaccharide building blocks. Convergent [31 + 30 + 30 + 30 + 30] block coupling of fully and partially protected polysaccharides, prepared by AGA, furnished a branched 151-mer polymannoside.
Table 1. Comparison of Automated Solid-Phase Syntheses of Biopolymers.
| biopolymer | peptides | DNA | RNA | polysaccharides |
| introduced | 19631 | 19812 | 19983 | 200110 |
| structure | linear | linear | linear | branched; regiocontrol required! |
| linkage | amide | phosphodiester | phosphodiester | glycosidic bond |
| stereogenic center | no | no | no | yes; stereocontrol required! |
| capping | for selected sequences | routinely | routinely | recently added |
| coupling yield | 99.5 | >99.99 | 98.5 | 98.75 |
| length | 50–100 mer | ≈200 | 120 | 100 mer (this work) |
Polystyrene Merrifield resin equipped with a photocleavable linker 1 was placed in the reaction vessel of an automated synthesizer, and mannose thioglycoside building block 2 was employed for AGA on a 19 μmol scale using a four-step cycle. The incorporation of each monomer consisted of an acidic washing step to prevent quenching by any remaining base (20 min), followed by the coupling step (20–60 min depending on polymer length) using 5–6.5 equiv of building block 2. Next, a capping step (30 min) prevented any unreacted nucleophiles from engaging in subsequent couplings to produce (n – 1)-polysaccharides that are difficult to separate from the desired product. Removal of the temporary Fmoc carbonate protective group (14 min) exposes the nucleophile for the subsequent coupling step (Figure 1). After every 20 coupling cycles, ten beads of resin were removed from the reaction vessel, and the product was cleaved for HPLC and MALDI mass spectrometry analysis to ascertain synthesis success. At the 40-mer stage, the glycosylation time was doubled from 20 to 40 min to ensure quantitative reactions as failure sequences were increasingly detected in the crude products (see the Supporting Information). Beyond the 80-mer stage, all glycosylations were allowed to proceed for 60 min to ensure highest possible yields. The 100 coupling cycles were executed automatically within 188 h and consumed 9 g of building block 2 to yield 39 mg of 100-mer α-(1–6)-polymannoside 3 after photocleavage from the resin and normal-phase HPLC purification (5% yield). The main byproduct (26 mg, 3% yield) of the 100-mer missing three benzyl ether protective groups that were lost during photochemical release from the resin was obtained (see the SI). Global deprotection of this byproduct yields the same final product (8% overall yield, 98.75% stepwise yield).
Figure 1.

Automated glycan assembly of polysaccharides. (A) Concept: four-step synthesis cycle consisting of an acidic wash, the glycosylation, capping to mask any unreacted nucleophiles, and cleavage of Fmoc carbonate for the next glycosylation. LG, leaving group; pPG, permanent protecting group; tPG, temporary protecting group. (B) AGA of 100-mer α-(1–6)-polymannoside 4. Merrifield resin 1 with a photolabile linker was placed in the reaction vessel of an automated synthesizer that executed coupling cycles consisting of an acidic wash, a glycosylation employing mannose thioglycoside building block 2, capping to block any unreacted nucleophile, and cleavage of the temporary Fmoc protective group (see the Supporting Information). Finally, protected 100-mer α-(1–6)-polymannoside 3 is released from the solid support by photocleavage. Two-step global deprotection yields 100-mer α-(1–6)-polymannoside 4.
Cleavage of all ester protective groups by treatment with sodium methoxide, followed by palladium-catalyzed hydrogenolysis under pressure to release all benzyl ether protective groups, produced 100-mer polymannoside 4 after preparative reverse phase HPLC. 100-mer polymannoside 4 (1 mg) was characterized by 1H and 13C NMR as well as MALDI-TOF analysis. AGA of the largest polysaccharide assembled to date illustrates the power of the solid phase synthesis to drastically reduce the synthesis time while relying exclusively on commercially available reagents.
Even larger and more complex polysaccharides would be accessible by block couplings of polysaccharides prepared by AGA. Two polysaccharide blocks, linear 30-mer α-(1–6)-polymannoside 7 and branched 31-mer polymannoside 10 when unified in a 31 + 30 + 30 + 30 + 30 coupling, will create branched 151-mer 11, to illustrate the approach. Mannose building block 2 was incorporated into linear α-(1–6)-polymannoside 30-mer 6 within 45 h by AGA using resin 5 that, upon photocleavage, reveals an oligosaccharide with a free reducing end. The hemiacetal form can be readily equipped with different anomeric leaving groups to produce glycosylating agents such as 30-mer glycosyl trichloroacetimidate 7 or 30-mer glycosyl fluoride 8 (Figure 2A). Branched 31-mer polymannoside 10 that will serve as acceptor in the subsequent block coupling was prepared by AGA using α-(1–6) mannose building block 2 and differentially protected mannose building block 9 for the branching positions. Merrifield resin 1 equipped with an n-alkyl amine linker that remains at the reducing end was the starting point to prepare 31-mer 10 in 50 h. Photocleavage of the crude oligosaccharide product, followed by normal-phase HPLC purification, yielded 30 mg of pure 31-mer 10 (12% yield over 66 steps) (Figure 2B).
Figure 2.

Synthesis of branched 151-mer polymannoside 12 via a 31 + 30 + 30 + 30 + 30 block coupling of 30-mer polymannoside glycosylating agents (7 and 8) as well as branched 31-mer polymannoside acceptor 10 prepared by AGA. (A) AGA of 30-mer glycosylating agents (7 and 8) by AGA. Thirty consecutive coupling cycles using mannose thioglycoside building block 2 were performed on Merrifield resin 5, followed by photocleavage which yielded partially protected 30-mer 6. Polymannoside 6 was converted to the corresponding 30-mer α-(1–6)-polymannoside donors 7 and 8 by treatment with trichloroacetonitrile or deoxo-fluor. (B) 31-mer polymannoside acceptor 10 was assembled. After incorporating 4 times mannose thioglycoside building block 2 (6.5 equiv), branching building block 9 followed. The first two parallel couplings with building block 2 required 10 equiv to glycosylate the secondary C2-hydroxyl group and install an α-1,2 linkage. After the next three parallel glycosylations with building block 2, this process was repeated following incorporation of two further branching points with building block 8, in order to rapidly grow 31-mer polymannoside. Photocleavage and subsequent purification yielded 30 mg of 31-mer polymannoside acceptor 10. (C) Union of 30-mer glycosylating agent 8 and branched 31-mer 10 yielded 78% of fully protected 151-mer 11. Removal of all protective groups by methanolysis followed by hydrogenolysis yielded 1.8 mg of branched 151-mer polymannoside 12.
The convergent assembly of 151-mer polysaccharide 11 relied on the 4-fold simultaneous glycosylation of branched 31-mer 10 using a 30-mer glycosylating agent. 30-mer trichloroacetimidate 7 failed to couple to 31-mer 10 at −20 °C upon activation with TMSOTf acid but rather hydrolyzed rapidly as no product was formed. Release of a leaving group that can act as a nucleophile upon activation of glycosyl trichloroacetimidates renders these glycosylating agents problematic for challenging glycosylations that involve sterically demanding nucleophiles.16
Therefore, 30-mer 6 was converted to the corresponding glycosyl fluoride 8 to determine whether a different anomeric leaving group and activator system would result in a successful 31 + 30 + 30 + 30 + 30 coupling to furnish 151-mer 11. The use of 6 equiv of 30-mer glycosyl fluoride 8 and 31-mer acceptor 10 in the presence of a silver triflate–hafnocene dichloride mixture as activator furnished mainly the desired 151-mer 11 accompanied by about 20% 121-mer byproduct(s) as judged by HPLC and MALDI mass spectrometry (Figure 2C). Using silver perchlorate as a promotor,17 complete glycosylation without the formation of significant amounts of byproducts was achieved in just 30 min (see the SI). Purification by normal-phase HPLC furnished a fully protected 27 mg (78% isolated yield) of 151-mer 11. To remove 302 benzyl ether protective groups, 144 benzoate, and four acetate esters as well as one Cbz amine protective group, fully protected 151-mer 11 (11 mg) was treated with sodium methoxide for 16 h to cleave all 148 ester groups, followed by hydrogenation in the presence of palladium on carbon under hydrogen pressure for 72 h to cleave all benzyl ethers and the Cbz group. Fully deprotected 151-mer 12 was obtained in 41% yield (1.8 mg). Analysis by 1H, 13C NMR, as well as MALDI-mass spectrometry unequivocally ascertained the synthesis of branched 151-mer 12 as the largest synthetic polysaccharide to date.
The rapid construction of linear and branched polysaccharides up to 100-mers using monosaccharides on an automated synthesizer provided the basis for the construction of polysaccharides as large as 151-mers by a 31 + 30 + 30 + 30 + 30 block coupling. Fully and partially protected polysaccharides prepared by AGA can serve as glycosylating agents and glycosyl acceptors and open many possibilities to prepare carbohydrate materials containing natural and non-natural monomers. Like automated polynucleotide and polypeptide synthesis, biological and material science applications are almost limitless considering the many roles polysaccharides play in nature.
Acknowledgments
We gratefully acknowledge the generous financial support of the Max-Planck Society. This project received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement 642870. We sincerely thank Dr. Kim Le Mai Hoang for providing the linker-equipped solid support and suggestions and Eva Settels for helping in HPLCs and other analytical devices.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00751.
Synthesis procedures, AGA modules, and characterization data of polysaccharides, including HPLC chromatograms and mass and NMR spectra (PDF)
The authors declare the following competing financial interest(s): P.H.S. holds a significant financial interest in GlycoUniverse GmbH and CoKGaA the company that commercializes the automated glycan synthesizer described in this manuscript. The other authors declare no competing financial interests.
Supplementary Material
References
- Merrifield R. B. Automated Synthesis of Peptides. Science 1965, 150, 178–185. 10.1126/science.150.3693.178. [DOI] [PubMed] [Google Scholar]
- Caruthers M. Gene Synthesis Machines: DNA Chemistry and Its Uses. Science 1985, 230, 281–285. 10.1126/science.3863253. [DOI] [PubMed] [Google Scholar]
- Scaringe S. A.; Wincott F. E.; Caruthers M. H. Novel RNA Synthesis Method Using 5‘-O-Silyl-2‘-O-orthoester Protecting Groups. J. Am. Chem. Soc. 1998, 120, 11820–11821. 10.1021/ja980730v. [DOI] [Google Scholar]
- Lashkari D. A.; Hunicke-Smith S. P.; Norgren R. M.; Davis R. W.; Brennan T. An Automated Multiplex Oligonucleotide Synthesizer: Development of High-Throughput, Low-cost DNA Synthesis. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 7912–7915. 10.1073/pnas.92.17.7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin S.; McPherson J. D.; McCombie W. R. Coming of Age: Ten Years of Next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Schmidt R. R. New Principles for Glycoside-Bond Formation. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- Wen L.; Edmunds G.; Gibbons C.; Zhang J.; Gadi M. R.; Zhu H.; Fang J.; Liu X.; Kong Y.; Wang P. G. Toward Automated Enzymatic Synthesis of Oligosaccharides. Chem. Rev. 2018, 118, 8151–8187. 10.1021/acs.chemrev.8b00066. [DOI] [PubMed] [Google Scholar]
- Yu H.; Chen X. One-Pot Multienzyme (OPME) Systems for Chemoenzymatic Synthesis of Carbohydrates. Org. Biomol. Chem. 2016, 14, 2809–2818. 10.1039/C6OB00058D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Xiong D.-C.; Chen S.-C.; Wang Y.-S.; Ye X.-S. Total synthesis of Mycobacterial Arabinogalactan Containing 92 Monosaccharide Units. Nat. Commun. 2017, 8, 14851. 10.1038/ncomms14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante O. J.; Palmacci E. R.; Seeberger P. H. Automated Solid-Phase Synthesis of Oligosaccharides. Science 2001, 291, 1523–1527. 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- Naresh K.; Schumacher F.; Hahm H. S.; Seeberger P. H. Pushing the Limits of Automated Glycan Assembly: Synthesis of a 50 mer Polymannoside. Chem. Commun. 2017, 53, 9085–9088. 10.1039/C7CC04380E. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Kononov A.; Delbianco M.; Seeberger P. H. A Capping Step During Automated Glycan Assembly Enables Access to Complex Glycans in High Yield. Chem. - Eur. J. 2018, 24, 6075–6078. 10.1002/chem.201801023. [DOI] [PubMed] [Google Scholar]
- Ma S.; Tang N.; Tian J. DNA Synthesis, Asembly and Applications in Synthetic Biology. Curr. Opin. Chem. Biol. 2012, 16, 260–267. 10.1016/j.cbpa.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbianco M.; Kononov A.; Poveda A.; Yu Y.; Diercks T.; Jiménez-Barbero J.; Seeberger P. H. Well-Defined Oligo- and Polysaccharides as Ideal Probes for Structural Studies. J. Am. Chem. Soc. 2018, 140, 5421–5426. 10.1021/jacs.8b00254. [DOI] [PubMed] [Google Scholar]
- Le Mai Hoang K.; Pardo-Vargas A.; Zhu Y.; Yu Y.; Loria M.; Delbianco M.; Seeberger P. H. Traceless Photolabile Linker Expedites the Chemical Synthesis of Complex Oligosaccharides by Automated Glycan Assembly. J. Am. Chem. Soc. 2019, 141, 9079–9086. 10.1021/jacs.9b03769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vorm S.; Hansen T.; van Hengst J. M. A.; Overkleeft H. S.; van der Marel G. A.; Codee J. D. C. Acceptor Reactivity in Glycosylation Reactions. Chem. Soc. Rev. 2019, 48, 4688–4706. 10.1039/C8CS00369F. [DOI] [PubMed] [Google Scholar]
- Matsumoto T.; Maeta H.; Suzuki K.; Tsuchihashi l. G.-i. New Glycosidation Reaction 1: Combinational use of Cp2ZrCl2-AgClO4 for Activation of Glycosyl Fluorides and Application to Highly β-selective Gylcosidation of D-mycinose. Tetrahedron Lett. 1988, 29, 3567–3570. 10.1016/0040-4039(88)85294-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.