

Abstract
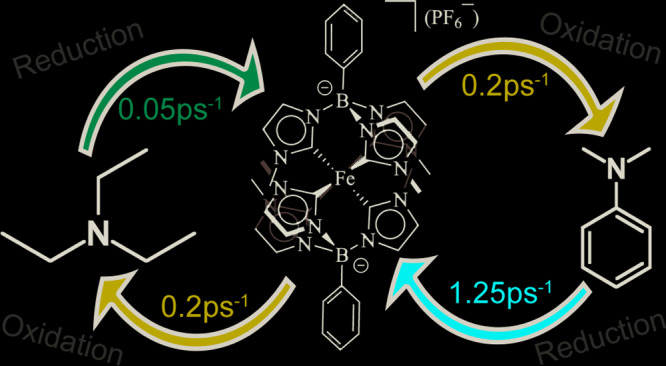
Photoinduced bimolecular charge transfer processes involving the iron(III) N-heterocyclic carbene (FeNHC) photosensitizer [Fe(phtmeimb)2]+ (phtmeimb = phenyltris(3-methyl-imidazolin-2-ylidene)borate) and triethylamine as well as N,N-dimethylaniline donors have been studied using optical spectroscopy. The full photocycle of charge separation and recombination down to ultrashort time scales was studied by investigating the excited-state dynamics up to high quencher concentrations. The unconventional doublet ligand-to-metal charge transfer (2LMCT) photoactive excited state exhibits donor-dependent charge separation rates of up to 1.25 ps–1 that exceed the rates found for typical ruthenium-based systems and are instead more similar to results reported for organic sensitizers. The ultrafast charge transfer probed at high electron donor concentrations outpaces the solvent dynamics and goes beyond the classical Marcus electron transfer regime. Poor photoproduct yields are explained by donor-independent, fast charge recombination with rates of ∼0.2 ps–1, thus inhibiting cage escape and photoproduct formation. This study thus shows that the ultimate bottlenecks for bimolecular photoredox processes involving these FeNHC photosensitizers can only be determined from the ultrafast dynamics of the full photocycle, which is of particular importance when the bimolecular charge transfer processes are not limited by the intrinsic excited-state lifetime of the photosensitizer.
There have been long-standing efforts to replace rare and expensive transition metals like ruthenium1−3 by Earth-abundant metals such as iron in photosensitizers for molecular-based technologies such as dye-sensitized solar cells and photocatalysis.4,5 Iron is of particular interest in this regard because it is the most abundant transition metal on earth. Recent studies on Fe light-harvesting complexes6 have demonstrated significant progress in terms of efficient interfacial electron transfer,7,8 photoluminescence,9,10 and record excited-state lifetimes into the nanosecond regime.10,11
The Fe(III) N-heterocyclic carbene (NHC) complex [Fe(phtmeimb)2]PF6 (1) (phtmeimb = phenyltris(3-methyl-imidazolin-2-ylidene)borate) (see Scheme 1A), with a lifetime of ∼2 ns, was furthermore found to be capable of driving bimolecular photoredox reactions.10 Despite efficient bimolecular quenching of 1, the formation of long-lived photoproducts was limited to yields of about 5%. A Stern–Volmer analysis suggested typical diffusion-limited kinetics, raising significant questions about what limits the product formation.10 Furthermore, the Fe(III) hexacarbene complexes exhibit a rare type of photophysics involving a low-spin (doublet) 3d5 ground state and a doublet ligand-to-metal charge transfer (2LMCT) excited state. This contributes to a broader current interest to develop earth-abundant photoactive complexes featuring LMCT excited states.1,3,12
Scheme 1. (A) Structures of [Fe(phtmeimb)2]+, N,N-Dimethylaniline (DMA), and Triethylamine (TEA) (Hydrogen Atoms Omitted for Clarity); (B) Schematic Bimolecular Photocycle Involving the FeIII Sensitizer, Quencher (Q), and Photoproduct (PP) Undergoing Charge Separation (CS) and Charge Recombination (CR).

A typical bimolecular photocycle is presented in Scheme 1B. Upon irradiation the Fe(III) sensitizer is excited to the 2LMCT state. Subsequent charge separation (CS) reduces the sensitizer while oxidizing the quencher. From there the constituents can separate, resulting in photoproduct (PP) formation, or the photocycle can be completed by charge recombination (CR). Experimentally, the photocycle is typically traced by (i) quenching of the photoluminescence (PL) intensity of the excited sensitizer and (ii) emerging absorption or emission features from the PP. However, a comprehensive elucidation of all of the elementary steps in the full photocycle can be achieved only by studying the ultrafast photoinduced dynamics at the pico- or femtosecond time scale.13−16
As a first step for the present type of Fe(III) photosensitizer, we investigate the dynamics using two types of quencher molecules: (I) aliphatic triethylamine (TEA) and (II) aromatic N,N-dimethylaniline (DMA). The oxidation potentials of these electron donors differ slightly, i.e. 0.960 and 0.756 V vs SCE in acetonitrile for TEA and DMA, respectively.17 This yields free energies of −0.396 and −0.600 eV for the reduction of 1 and oxidation of TEA and DMA, respectively, based on the excited-state redox potential of 1.10
For comparative purposes, we performed UV/vis, steady-state emission, and time-resolved absorption spectroscopy on solutions of 1 in acetonitrile (MeCN) with added quenchers. Both the absorption and emission spectra (see Figure 1) exhibit virtually unchanged characteristics of 1, and additional absorbance of the quenchers is observed only for wavelengths below 400 nm (see the Supporting Information). For both quenchers, the slopes of the intensity and the pseudo-first-order lifetime curves as a function of quencher concentration match well, indicating that for low to intermediate quencher concentrations (<800 mM) pure dynamic quenching is observed (see Figure 1). These measurements also indicate that quenching by DMA is more efficient than quenching by TEA, as reflected in the dynamic quenching rates for TEA and DMA, respectively (for details of the underlying Stern–Volmer analysis, see the Supporting Information). This agrees with the free energy values given above. In particular, the dynamic quenching rate of 1 in DMA is more than 2 orders of magnitude higher than in the prototype case of tris(bipyridine)ruthenium(II)18,19 ([Ru(bpy)3]2+), indicating fundamental differences from the processes found for 1.
Figure 1.

Normalized luminescence intensities (solid symbols) and excited-state lifetimes (open symbols) of 1 in MeCN upon addition of TEA (orange) or DMA (blue) on a double logarithmic scale vs normalized quencher concentration. The inset shows the normalized absorbance and emission spectra of 1 in MeCN (black dashed) with added TEA (orange) or DMA (blue).
Next, we investigate the time-resolved dynamics with a particular focus on high quencher concentrations. When the average distance between quencher molecules becomes smaller than the size of the sensitizer, it is reasonable to assume that the sensitizer and quencher are always in close contact. In this static quenching regime, intrinsic charge transfer rates become accessible, as previously applied in ultrafast dynamics studies of organic sensitizers.17,20−22 Transient absorption (TA) spectroscopy with ∼100 fs resolution was employed for these measurements (see the Supporting Information).
First we summarize the TA signature in pure MeCN (Figure 2 inset).10 In general, a positive photoinduced absorption (PIA) reflects absorption of a multitude of excited states (ESA) and may include contributions of photoproducts, e.g., oxidized donor. A negative signal corresponds to either ground-state bleach (GSB) or stimulated emission (SE). In the case of 1 we observe several features: A negative SE band at ∼680 nm and several ESA bands. The latter are separated into (i) a band rising toward the long-wavelength part of the visible spectrum, (ii) a band at ∼575 nm, and (iii) a band rising toward the near-ultraviolet. The dip between the last two ESA bands corresponds to the GSB, which is expected to peak at ∼505 nm but is overwhelmed by the stronger ESA. These features all decay single-exponentially with a lifetime ∼2 ns.10 This is also reflected in two isosbestic points at zero differential absorption, between ESA and SE. An isosbestic point corresponds to a state-to-state transition. Here, this is the transition from the excited state to the ground state. This underlines that the ESA and SE correspond to the same excited state.
Figure 2.

(A) Normalized differential absorption spectra at 1 ps time delay in pure MeCN (blue) and with 4200 mM TEA (orange). (B) Normalized differential absorption transients in the stimulated emission (SE) region (700 nm) of 1 in MeCN for various concentrations of TEA from 0 mM (blue) to 5040 mM (yellow). (C) Differential absorption transients of 1 with TEA at a concentration of 4254 mM. Each transient has been divided by the stimulated emission intensity at the respective step. The GSB region was fit by a double-exponential function (blue line).
Next we focus on the influence of TEA on the dynamics. The differential absorption spectrum and transients in the SE region are given in Figure 2 (for a set of full differential absorption spectra, see the Supporting Information). Even for very high TEA concentrations the spectral shape does not change. In agreement with the observed steady-state emission quenching, the lifetime of ESA and SE shortens by 2 orders of magnitude to ∼20 ps (see Figure 2) at the maximum TEA concentration (5040 mM). We associate this shortening of excited-state lifetime with the photoinduced CS, corresponding to a rate constant of 0.05 ps–1. This is consistent with the substantial negative free energy for reduction of the excited complex 1 by TEA. As we do not expect any significant TEA cation signal (see the Supporting Information), the CS process should also be observable in either reduced species of 1 or a stoichiometric amount of GSB that persists until the CR process occurs. The signature of reduced 1 in the short-wavelength spectral region is, however, not covered by our experiment. Additionally, we do not observe any long-lived GSB. The absence of both TEA cation and a long-lived GSB signal suggests that either the CR quickly follows the CS and no long-lived product is formed or, contrary to our expectation, the presence of TEA facilitates 1 to undergo internal conversion to the ground state without CS occurring at all. To unambiguously verify CS, we have divided the signals at all times by the corresponding amplitude of the SE signal (see the Supporting Information). In the case of internal conversion, these normalized transients should become time-independent constants in all wavelength regions. At high TEA concentration, we obtain a nonconstant time dependence in the GSB spectral region (see Figure 2). This deviation directly reflects the relation of the CS and CR rates. Fitting of the obtained dynamics reveals a rate of 0.25 ps–1 for CR (see the Supporting Information).
Next we study the dynamics with DMA. At low DMA concentrations, the TA spectra resemble those of MeCN/TEA mixtures. For higher DMA concentrations, the GSB becomes clearly pronounced, the ESA decreases, and at ∼450 nm another feature emerges (see Figure 3). The latter agrees with the expected absorption of DMA cation.20 Analyzing the transients in the selected spectral regions yields the decrease of the SE as expected from the reported emission quenching. More informative are the blue ESA and GSB regions. At low DMA concentration, i.e., in the diffusion-limited regime, the signal is dominated by the ESA dynamics. At high DMA concentration, a subpicosecond buildup of additional signal appears that is positive in the ESA region and negative in the GSB region. We interpret this as a fast decay of the initially dominating ESA of 1 without recovery of the ground-state absorption, which thus reveals the negative GSB signal. With the same dynamics, an additional but weak ESA component assigned to DMA cation arises toward the blue spectral region. Importantly, both the rise and decay dynamics of the negative GSB do not change above a DMA concentration of ∼800 mM but clearly differ from the SE and ESA decay at corresponding DMA concentrations. SE and ESA continuously speed up with increasing DMA concentration, but the GSB only changes its amplitude. We associate this behavior with the vanishing contribution of diffusion as the average distance between 1 and the DMA molecules decreases with increasing concentration. For the highest DMA concentration, the rise and decay of the GSB have negligible diffusion contributions and thus reflect the CS and CR processes between 1 and DMA molecules in close contact. Consequently, by fitting the GSB observed for the highest DMA concentration with a double-exponential function, we extract a CS rate of 1.25 ps–1 and a CR rate of 0.17 ps–1.
Figure 3.

(A) Differential absorption spectra of 1 in MeCN at time delay of 1 ps for DMA concentrations from 9 mM (blue) to 5000 mM (yellow). For comparison, the absorbances of DMA+ (green) and 1 (light blue) are shown with the stimulated emission of 1 (red) calculated from the steady-state emission spectrum (sign reversed), all on an arbitrary scale. (B–D) Differential absorption transients around 450 nm (ESA), 520 nm (GSB), and 660 nm (SE), with quencher concentrations as for the spectra above.
In summary, we have investigated the full bimolecular photocycle between the excited state of the Fe(III)–NHC complex [Fe(phtmeimb)2]+ and common sacrificial donor molecules (TEA and DMA). The ultrafast dynamics at high DMA concentration is assigned to charge separation and recombination due to faster separation than recombination. This is in clear contrast to the case of TEA, where charge recombination is significantly faster than charge separation. Although the CR rate is ∼0.2 ps–1 for both quenchers, the CS rate differs drastically: 0.05 ps–1 in TEA and 1.25 ps–1 in DMA. This trend agrees with, but is too large to be dominated by, differences in the driving force. Similar to studies of coumarins, we assign this difference to enhanced coupling of the sensitizer to aromatic DMA compared with aliphatic TEA.17 Furthermore, the charge transfer in DMA appears to be independent of the slower solvation dynamics.21 Following studies on organic light harvesters and DMA, we associate the ultrafast charge transfer with nuclear reorganization as described by the Sumi–Marcus model.24,25 Additionally, the observed rates are significantly higher than the reported values for standard Ru sensitizers.18,26 Our observed CS rates put this iron-based complex in the same category as organic molecules like oxazines20 and coumarins.17 Despite very efficient charge separation, formation of the photoproduct is not favored. This is attributed to very fast spin-allowed charge recombination. The spin-allowed CR rates in this study are comparable to reported rates in Ru and Os sensitizers, where the formal spin-forbidden nature of the transitions is largely lifted by strong spin–orbit coupling.27 One strategy to explore further would be to utilize the intrinsically smaller spin–orbit coupling in Fe(III)–NHC complexes to achieve higher photoproduct yields in photocycles where spin-forbidden CR is suppressed.
Overall, these results provide key insights to guide further efforts to better utilize the full potential of the promising excited-state properties of novel Fe(III)–NHC and other 2LMCT photosensitizers.
Acknowledgments
The Swedish Research Council (VR), the Swedish Foundation for Strategic Research (SSF), the Knut and Alice Wallenberg Foundation (KAW), and the Swedish Energy Agency (Energimyndigheten) are acknowledged for financial support. N.W.R. acknowledges funding from the Alexander von Humboldt Foundation within the Feodor-Lynen Fellowship Program. K.W. thanks the LMK Foundation for support. O.P. thanks the Carl Tryggers Stiftelse for support. S.K. thanks the Wenner-Gren Foundation for support. We thank Linnea Lindh and Jens Uhlig for contributing to experiments and discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00755.
Description of experimental setups and sample preparation, determination of quenching rates, normalization procedure for transients obtained for the TEA quencher series, and full series of transient absorption spectra for both quencher series (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- McCusker J. K. Electronic Structure in the Transition Metal Block and Its Implications for Light Harvesting. Science 2019, 363 (6426), 484–488. 10.1126/science.aav9104. [DOI] [PubMed] [Google Scholar]
- Campagna S.; Puntoriero F.; Nastasi F.; Bergamini G.; Balzani V.. Photochemistry and Photophysics of Coordination Compounds: Ruthenium. In Photochemistry and Photophysics of Coordination Compounds I; Springer: Berlin, 2007; pp 117–214. [Google Scholar]
- Wenger O. S. Is Iron the New Ruthenium?. Chem. - Eur. J. 2019, 25 (24), 6043–6052. 10.1002/chem.201806148. [DOI] [PubMed] [Google Scholar]
- Bozic-Weber B.; Constable E. C.; Housecroft C. E. Light Harvesting with Earth Abundant D-Block Metals: Development of Sensitizers in Dye-Sensitized Solar Cells (DSCs). Coord. Chem. Rev. 2013, 257 (21–22), 3089–3106. 10.1016/j.ccr.2013.05.019. [DOI] [Google Scholar]
- Förster C.; Heinze K. Photophysics and Photochemistry with Earth-Abundant Metals—Fundamentals and Concepts. Chem. Soc. Rev. 2020, 49 (4), 1057–1070. 10.1039/C9CS00573K. [DOI] [PubMed] [Google Scholar]
- Lindh L.; Chábera P.; Rosemann N. W.; Uhlig J.; Wärnmark K.; Yartsev A.; Sundström V.; Persson P. Photophysics and Photochemistry of Iron Carbene Complexes for Solar Energy Conversion and Photocatalysis. Catalysts 2020, 10 (3), 315. 10.3390/catal10030315. [DOI] [Google Scholar]
- Harlang T. C. B.; Liu Y.; Gordivska O.; Fredin L. A.; Ponseca C. S.; Huang P.; Chábera P.; Kjaer K. S.; Mateos H.; Uhlig J.; Lomoth R.; Wallenberg R.; Styring S.; Persson P.; Sundström V.; Wärnmark K. Iron Sensitizer Converts Light to Electrons with 92% Yield. Nat. Chem. 2015, 7 (11), 883–889. 10.1038/nchem.2365. [DOI] [PubMed] [Google Scholar]
- Duchanois T.; Liu L.; Pastore M.; Monari A.; Cebrián C.; Trolez Y.; Darari M.; Magra K.; Francés-Monerris A.; Domenichini E.; Beley M.; Assfeld X.; Haacke S.; Gros P. NHC-Based Iron Sensitizers for DSSCs. Inorganics 2018, 6, 63. 10.3390/inorganics6020063. [DOI] [Google Scholar]
- Chábera P.; Liu Y.; Prakash O.; Thyrhaug E.; Nahhas A. El; Honarfar A.; Essén S.; Fredin L. A.; Harlang T. C. B.; Kjær K. S.; Handrup K.; Ericson F.; Tatsuno H.; Morgan K.; Schnadt J.; Häggström L.; Ericsson T.; Sobkowiak A.; Lidin S.; Huang P.; Styring S.; Uhlig J.; Bendix J.; Lomoth R.; Sundström V.; Persson P.; Wärnmark K. A Low-Spin Fe(III) Complex with 100-ps Ligand-to-Metal Charge Transfer Photoluminescence. Nature 2017, 543 (7647), 695–699. 10.1038/nature21430. [DOI] [PubMed] [Google Scholar]
- Kjær K. S.; Kaul N.; Prakash O.; Chábera P.; Rosemann N. W.; Honarfar A.; Gordivska O.; Fredin L. A.; Bergquist K.-E.; Häggström L.; Ericsson T.; Lindh L.; Yartsev A.; Styring S.; Huang P.; Uhlig J.; Bendix J.; Strand D.; Sundström V.; Persson P.; Lomoth R.; Wärnmark K. Luminescence and Reactivity of a Charge-Transfer Excited Iron Complex with Nanosecond Lifetime. Science 2019, 363 (6424), 249–253. 10.1126/science.aau7160. [DOI] [PubMed] [Google Scholar]
- Braun J. D.; Lozada I. B.; Kolodziej C.; Burda C.; Newman K. M. E.; van Lierop J.; Davis R. L.; Herbert D. E. Iron(II) Coordination Complexes with Panchromatic Absorption and Nanosecond Charge-Transfer Excited State Lifetimes. Nat. Chem. 2019, 11 (12), 1144–1150. 10.1038/s41557-019-0357-z. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lee T. S.; Favale J. M.; Leary D. C.; Petersen J. L.; Scholes G. D.; Castellano F. N.; Milsmann C. Delayed Fluorescence from a Zirconium(IV) Photosensitizer with Ligand-to-Metal Charge-Transfer Excited States. Nat. Chem. 2020, 12, 345–352. 10.1038/s41557-020-0430-7. [DOI] [PubMed] [Google Scholar]
- Ponseca C. S.; Chábera P.; Uhlig J.; Persson P.; Sundström V. Ultrafast Electron Dynamics in Solar Energy Conversion. Chem. Rev. 2017, 117 (16), 10940–11024. 10.1021/acs.chemrev.6b00807. [DOI] [PubMed] [Google Scholar]
- Kumpulainen T.; Lang B.; Rosspeintner A.; Vauthey E. Ultrafast Elementary Photochemical Processes of Organic Molecules in Liquid Solution. Chem. Rev. 2017, 117 (16), 10826–10939. 10.1021/acs.chemrev.6b00491. [DOI] [PubMed] [Google Scholar]
- Kiefer L. M.; Kubarych K. J. Solvent Exchange in Preformed Photocatalyst-Donor Precursor Complexes Determines Efficiency. Chem. Sci. 2018, 9 (6), 1527–1533. 10.1039/C7SC04533F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen P. N.; Watanabe H.; Tamaki Y.; Ishitani O.; Kimura S. Relaxation Dynamics of [Re(CO)2(Bpy){P(OEt)3}2](PF6) in TEOA Solvent Measured by Time-Resolved Attenuated Total Reflection Terahertz Spectroscopy. Sci. Rep. 2019, 9 (1), 11772. 10.1038/s41598-019-48191-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castner E. W.; Kennedy D.; Cave R. J. Solvent as Electron Donor: Donor/Acceptor Electronic Coupling Is a Dynamical Variable. J. Phys. Chem. A 2000, 104 (13), 2869–2885. 10.1021/jp9936852. [DOI] [Google Scholar]
- Kavarnos G. J.; Turro N. J. Photosensitization by Reversible Electron Transfer: Theories, Experimental Evidence, and Examples. Chem. Rev. 1986, 86 (2), 401–449. 10.1021/cr00072a005. [DOI] [Google Scholar]
- Ballardini R.; Varani G.; Indelli M. T.; Scandola F.; Balzani V. Free Energy Correlation of Rate Constants for Electron Transfer Quenching of Excited Transition Metal Complexes. J. Am. Chem. Soc. 1978, 100 (23), 7219–7223. 10.1021/ja00491a017. [DOI] [Google Scholar]
- Kandori H.; Kemnitz K.; Yoshihara K. Subpicosecond Transient Absorption Study of Intermolecular Electron Transfer between Solute and Electron-Donating Solvents. J. Phys. Chem. 1992, 96 (20), 8042–8048. 10.1021/j100199a039. [DOI] [Google Scholar]
- Nagasawa Y.; Yartsev A. P.; Tominaga K.; Johnson A. E.; Yoshihara K. Temperature Dependence of Ultrafast Intermolecular Electron Transfer Faster than Solvation Process. J. Chem. Phys. 1994, 101 (7), 5717–5726. 10.1063/1.467357. [DOI] [Google Scholar]
- Vauthey E. Direct Measurements of the Charge-Recombination Dynamics of Geminate Ion Pairs Formed upon Electron-Transfer Quenching at High Donor Concentration. J. Phys. Chem. A 2001, 105 (2), 340–348. 10.1021/jp0023260. [DOI] [Google Scholar]
- Sumi H.; Marcus R. A. Dynamical Effects in Electron Transfer Reactions. J. Chem. Phys. 1986, 84 (9), 4894–4914. 10.1063/1.449978. [DOI] [Google Scholar]
- Nadler W.; Marcus R. A. Dynamical Effects in Electron Transfer Reactions. II. Numerical Solution. J. Chem. Phys. 1987, 86 (7), 3906–3924. 10.1063/1.451951. [DOI] [Google Scholar]
- Haga M.; Dodsworth E. S.; Eryavec G.; Seymour P.; Lever A. B. P. Luminescence Quenching of the Tris(2,2′-bipyrazine)ruthenium(II) Cation and Its Monoprotonated Complex. Inorg. Chem. 1985, 24 (12), 1901–1906. 10.1021/ic00206a041. [DOI] [Google Scholar]
- Olmsted J.; Meyer T. J. Factors Affecting Cage Escape Yields Following Electron-Transfer Quenching. J. Phys. Chem. 1987, 91 (6), 1649–1655. 10.1021/j100290a071. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.