

Abstract
Hyperpolarized water can be a valuable aid in protein NMR, leading to amide group 1H polarizations that are orders of magnitude larger than their thermal counterparts. Suitable procedures can exploit this to deliver 2D 1H–15N correlations with good resolution and enhanced sensitivity. These enhancements depend on the exchange rates between the amides and the water, thereby yielding diagnostic information about solvent accessibility. This study applied this “HyperW” method to four proteins exhibiting a gamut of exchange behaviors: PhoA(350–471), an unfolded 122-residue fragment; barstar, a fully folded ribonuclease inhibitor; R17, a 13.3 kDa system possessing folded and unfolded forms under slow interconversion; and drkN SH3, a protein domain whose folded and unfolded forms interchange rapidly and with temperature-dependent population ratios. For PhoA4(350–471) HyperW sensitivity enhancements were ≥300×, as expected for an unfolded protein sequence. Though fully folded, barstar also exhibited substantial enhancements; these, however, were not uniform and, according to CLEANEX experiments, reflected the solvent-exposed residues. R17 showed the expected superposition of ≥100-fold enhancements for its unfolded form, coexisting with more modest enhancements for their folded counterparts. Unexpected, however, was the behavior of drkN SH3, for which HyperW enhanced the unfolded but, surprisingly, enhanced even more certain folded protein sites. These preferential enhancements were repeatedly and reproducibly observed. A number of explanations—including three-site exchange magnetization transfers between water and the unfolded and folded states; cross-correlated relaxation processes from hyperpolarized “structural” waters and labile side-chain protons; and the possibility that faster solvent exchange rates characterize certain folded sites over their unfolded counterparts—are considered to account for them.
Introduction
Nuclear magnetic resonance (NMR) plays an irreplaceable role in biophysical studies. NMR can tackle complex systems such as proteins in solution under native or near-physiological conditions, and provide information about the structures and dynamics of these systems with atomic resolution. Despite this potential, NMR in general—and NMR of large biomolecules in particular—suffers from inherent sensitivity issues. Improving sensitivity and signal-to-noise ratio in NMR has therefore been the focus of extensive efforts, including the use of hyperpolarization methods that can impart orders-of-magnitude sensitivity enhancements to a variety of solutions and solids.1−5 Out of all methods for nuclear hyperpolarization, dissolution dynamic nuclear polarization (DNP) stands out in its generality to enhance the sensitivity of high-field solution-state NMR and MRI measurements.6−9 However, the ex situ nature of this approach—where the sample is hyperpolarized in one magnet under cryogenic conditions and then transferred as a liquid to another system for its eventual observation—fails when it is attempted on large biomolecules subject to very fast low-field relaxation processes. Hyperpolarized water10−12 (HyperW) NMR was recently introduced to overcome this limitation and enable the study of proteins and nucleic acids.13 HyperW NMR relies on the fact that H2O’s protons can be hyperpolarized by dissolution DNP into the tens of percent, and if suitably handled their relaxation times can reach into the tens of seconds. These protons, being labile, can then spontaneously exchange with groups in biomolecules—for instance, with amides in unfolded proteins or intrinsically disordered proteins/domains (IDPs/IDDs). This will then hyperpolarize the amide protons for long enough to enable the acquisition of 2D 1H–15N NMR correlations, particularly if the direct excitation of the water reservoir that is constantly supplying the amides with polarization is avoided. Initial HyperW biomolecular 1D and 2D NMR experiments delivered considerable sensitivity enhancements— ≥300× over their thermal counterparts—for mixtures of short peptides,14 albeit with poor spectral resolution. Hyperpolarized water has also enabled the study of weak protein interactions15 and IDPs.16 More recently,17 this method was used to achieve substantial enhancements for the Parkinson’s-disease-associated IDP α-synuclein as well as full 2D 1H–15N HMQC NMR resolution using an optimized water-injection experimental setup. With sensitivity enhancement values resolved for each amide group in the polypeptide, a simple model based on the Bloch–McConnell equations was then developed that could translate these HyperW enhancements in terms of the residue-specific dynamics characterizing amide/water exchanges for α-synuclein.
This work extends these optimized HyperW measurements to a wider variety of protein structures. These included the fully unfolded protein fragment PhoA(350–471) from alkaline phosphatase, for which the sensitivity enhancements observed were substantial and distributed randomly throughout the sequence. Also included was barstar, a protein which, although fully folded, also evidenced double-digit enhancements for certain residues—particularly solvent-exposed ones, for which ancillary CLEANEX experiments confirmed that the HyperW method acts as a kind of “exchange filter”. The third kind of system analyzed involved equilibria between coexisting unfolded and folded conformations, interconverting at different rates; these included the R17 domain of chicken-brain α-spectrin, and a terminal Src homology 3 domain from Drosophila, drkN SH3. In both cases the HyperW approach was able to light up both coexisting folded/unfolded populations, and to deliver from these enhancements site- and state-discriminated pictures of solvent accessibility for both folded and unfolded forms. For the R17 dimer these were, as expected, ca. 5–10× higher for the unfolded form. Different, however, was the behavior observed for drkN SH3, where repeated experiments consistently indicated equal or larger enhancements for residues in the folded form compared to their unfolded counterparts. Potential mechanisms and consequences of such observations are assessed.
Materials and Methods
Sample Preparation
An E. coli PhoA (residues 350–471) fragment was produced and purified as described by Saio et al.18 This PhoA(350–471) (PhoA4) was cloned into a pET-16b vector. The final gene incorporates an N-terminal Hexa-His-MBP tag followed by a Tobacco Etch Virus (TEV) protease cleavage site. A culture of BL21(DE3) harboring the PhoA4 plasmid was grown in M9 minimal medium supplemented with 1 g/L 15N-labeled ammonium chloride and ampicillin (100 μg/mL). The culture was induced at OD600 = 0.5 and overexpressed at 18 °C overnight. The protein was isolated from the lysate using a Ni-NTA column, and the His tag was removed by incubation with TEV protease overnight at 4 °C. PhoA4 was separated from the tag and TEV protease by passing it over a Ni-NTA column and further purified on a Superdex 75 size exclusion column (GE Healthcare). The samples containing PhoA4 were buffer exchanged to a concentration of 1.5 mM or 0.35 mM in a 99.9% D2O buffer (50 mM HEPES, pD 7.5, 50 mM KCl). For HyperW dissolutions, 140–150 μL aliquots of this solution were placed in a 5 mm NMR tube for their subsequent analysis. Following the hyperpolarized water injection, the sample was thus diluted to either 0.6 mM or 0.13 mM protein. For the reference, high protonated water content sample, 35 μL of the 0.35 mM PhoA4 solution was diluted with a 90% H2O buffer (50 mM HEPES, pH 7.5, 50 mM KCl) to a concentration of 0.13 mM protein in 82.5% H2O.
Barstar was produced and purified as described by Schreiber et al.19 In brief, a culture of BL21(DE3)pLysS harboring a plasmid encoding a mutated barstar (C40A, C82A) was grown in M9 minimal medium supplemented with 1 g/L 15N-labeled ammonium chloride, ampicillin (100 μg/mL), and chloramphenicol (17 μg/mL). The culture was induced at OD600 = 0.6 with 200 μM isopropyl β-d-1-thiogalactopyranoside (IPTG) and grown overnight at 30 °C. The cell pellet was resuspended in buffer (10 mM Tris pH 8, 1 mM EDTA, 100 mM NaCl, 1 mM PMSF, 50 mg/mL lysozyme, and DNase) and disrupted with a cooled cell disrupter (Constant Systems) followed by centrifugation. Barstar found in the soluble fraction was isolated by precipitation with 40–80% ammonium sulfate. After centrifugation the pellet was resuspended in a minimal volume of buffer (50 mM Tris pH 8, 100 mM NaCl), injected to a gel filtration column (Hiload_Superdex_75_26/60, GE Healthcare), and pre-equilibrated with the same buffer. Final purification on an anion exchange column (HiTrap_Q_HP, GE Healthcare) involved elution with 300 mM NaCl. The fractions containing barstar were dialyzed to DDW and lyophilized. For the HyperW experiments, lyophilized barstar was reconstituted in a D2O buffer (50 mM sodium phosphate, pD 7) at a concentration of ∼4 mM. For their subsequent analysis, 130–140 μL aliquots of this solution were placed in a 5 mm NMR tube. Following the hyperpolarized water injection, the sample was thus diluted to 1.3–1.6 mM protein. For the reference, high protonated water content sample, a post-injection sample was lyophilized to dryness and subsequently reconstituted in the same volume of 90% H2O to give rise to the same final protein concentration of 1.6 mM.
The R17 domain dimer was produced and purified as described by Sekhar et al.20 In brief, the gene encoding the L90A R17 domain of chicken-brain α-spectrin was cloned into a pET-29b(+) vector. The final gene incorporates an N-terminal hexa-His tag followed by a short linker and a TEV protease cleavage site. A culture of BL21(DE3) cells harboring the R17 plasmid was grown at 37 °C in M9 minimal medium supplemented with 1 g/L 15N-labeled ammonium chloride and kanamycin (50 μg/mL). The culture was grown to OD600 = 0.8 and overexpressed at 22 °C overnight. The protein was isolated from the lysate using a Ni-NTA column, and the His tag was removed by incubation with TEV protease overnight at 4 °C. R17 was separated from the tag and TEV protease by passing it over a Ni-NTA column and further purified on a Superdex 75 size exclusion column (GE Healthcare). The protein eluted as two peaks (monomer and dimer), and the dimer fractions were collected. The samples containing 15N-labeled R17 dimer were buffer exchanged to a concentration of 1.23 mM in a 99.9% D2O buffer (50 mM HEPES, pD 7.5, 50 mM KCl). For the HyperW dissolution experiment, a 140 μL aliquot of this solution was placed in a 5 mm NMR tube for its subsequent analysis. Following the hyperpolarized water injection, the sample was thus diluted to a protein concentration of 0.57 mM.
The drkN SH3 domain was produced and purified as described by Sekhar et al.20 The gene for the SH3 domain of Drosophila melanogaster enhancer of sevenless 2B protein (drkN SH3) was cloned into the pET-28 vector using PCR amplification (Kapa Hifi, Kapa Biosystems, MA, USA) followed by Gibson assembly (New England Biolabs, MA, USA). The final gene incorporates an N-terminal Hexa-His tag followed by a TEV protease cleavage site. A culture of BL21(DE3) cells harboring the drkN SH3 plasmid was grown at 37 °C in M9 minimal medium supplemented with 1 g/L 15N-labeled ammonium chloride and kanamycin (50 mg/L). The culture was grown to OD600 = 0.8 and overexpressed at 25 °C overnight. The protein was isolated from the lysate using a Ni-NTA column under denaturing (6 M guanidinium chloride) conditions. The unfolded protein was refolded on the column before elution by lowering the denaturant concentration stepwise from 6 to 4, 2, 1, and finally to 0 M. The His tag was removed by incubation with TEV protease overnight at 4 °C. DrkN SH3 was separated from the tag and TEV protease by passing it over a Ni-NTA column and further purified on a Superdex 75 size exclusion column (GE Healthcare). The samples containing drkN SH3 were buffer exchanged to concentrations of 0.8 mM or 1.3 mM in a 99.9% D2O buffer (50 mM HEPES, pD 7.5, 50 mM KCl). For HyperW dissolutions at 50 °C, 130 or 80 μL aliquots of the 0.8 mM solution, or 140 μL of the 1.3 mM solution, were placed in a 5 mm NMR tube for their subsequent analysis. For HyperW dissolutions at 37 °C, 145 μL of the 1.3 mM solution was placed in the 5 mm NMR tube. Following four hyperpolarized water injections, the sample was thus diluted to 0.26/0.16/0.59 mM protein (at 50 °C) or 0.51 mM (at 37 °C). For the reference, high protonated water content samples, the first two post-injection samples were lyophilized to dryness and subsequently reconstituted in the same volume with 90% H2O to give rise to the same final protein concentrations of 0.26/0.16 mM. The third high water content sample was prepared by dilution of 145 μL of 1.3 mM in a 99.9% D2O buffer with a 100% H2O buffer (50 mM HEPES, pH 7.5, 50 mM KCl) to a concentration of 0.52 mM protein and 87.4% H2O. The latter was used as a reference at both 50 and 37 °C. Further sample preparation details are given in the figure captions.
Dynamic Nuclear Polarization
Water was hyperpolarized using an Oxford Instruments Hypersense equipped with a 3.35 T magnet. The system was modified by adding to the Oxford-supplied E2M80 vacuum pump an EH-500 Edwards booster capable of taking the operating pressure to 1 Torr. Polarization was thus typically done at ∼1.05–1.30 K. DNP was achieved by irradiating at ∼94.1 GHz a 10 mM 4-amino-TEMPO (4AT) nitroxide radical, dissolved in ca. 100 μL solutions containing 15% glycerol and 85% H2O (v/v). Optimized microwave power levels and pumping time were 100 mW (nominal) and 180 min, respectively. Following this irradiation, samples were dissolved with a 99.9% D2O buffer; approximately 300 μL of a melted, hyperpolarized sample was then transferred into the NMR magnet using a pre-heated (60 °C) tubing line and injected into a 5 mm tube containing the targeted biomolecules dissolved in buffered D2O.
Injection Setup
Sample injections were carried out on an automated pressurized system, achieving robust, reproducible transfers with minimum bubble formation. The system and its design have been described elsewhere.14,17 In brief, it relies on a two-state valve operation,21−23 controlling the filling of the NMR tube using a three-port accessory involving both forward and backward gas pressures and controlled by Arduino-based software.23 Following previous optimization of the injection setup for obtaining high-resolution two-dimensional (2D) protein spectra,17 the injection system driving pressure was set to a gradient between 17 and 3.5 bar.
NMR Spectroscopy
Post-dissolution NMR experiments were conducted using a 5 mm liquid-nitrogen-cooled “Prodigy” probe in a 14.1 T Bruker magnet interfaced to an Avance III console. These experiments included 2D NMR acquisitions, which were triggered upon injecting the hyperpolarized water sample into the NMR tubes waiting with their samples inside the magnet bore. Experiments were carried out at nominal temperatures of either 37 or 50 °C, as detailed below. In view of the claims made below for the case of co-existing folded/unfolded protein states, particular attention was paid to the thermal reliability and uniformity of the sample temperatures resulting upon co-mixing the pre-heated hyperpolarized water with the pre-heated protein solution waiting inside the NMR tube. An idea of the thermal gradients and thermal stabilization of the ensuing mix is presented in the Supporting Information (SI), Figure S1, which analyzes the stabilization of the NMR signal throughout a 2D HyperW NMR acquisition performed at 50 °C, on the basis of two water-enhanced residues with temperature-sensitive resonances. It follows from this analysis that the temperature stabilizes to within one degree of the target value, within ∼10 s into the signal acquisition. 2D HyperW NMR spectra were acquired using the 1H–15N HMQC sequence given in the SI, Figure S2.14,17 This sequence fully excites and echoes the downfield amide region selectively and employs minimal recycle delays,24,25 in order to maximize the signal from the hyperpolarized exchangeable sites while minimizing the water depolarization losses. Unless otherwise noted, thermal equilibrium measurements were carried out on the same sample with the same hardware and using the same pulse sequence but with longer recycle delays, to obtain reliable measures of the HyperW site-specific enhancements. Ancillary CLEANEX-PM26 experiments were collected on the same spectrometer and probe at 50 or 37 °C. ZZ-exchange and methyl-TROSY experiments were measured on 5 mm cryogenically cooled probes in 14.1 or 18.8 T Bruker magnets interfaced to Bruker AvanceNeo or Avance III consoles, respectively, at 50 or 37 °C. All NMR data were processed using the Bruker Topspin software and subsequently plotted and analyzed using Matlab. Non-uniform sampling (NUS) using a Poisson-gap sampling schedule and spectral reconstructions was implemented using the hmsIST software,27 in combination with Topspin.
Results and Discussion
HyperW on a Disordered Peptide: Alkaline Phosphatase 350–471 Fragment PhoA4
Disordered proteins are natural candidates for water-based hyperpolarization enhancements, since their amide protons are exposed to the solvent, and the ensuing rapid amide/water exchange rates should facilitate substantial enhancements.17,28 An example of this is provided by the fully disordered protein fragment PhoA(350–471) (PhoA4). This 122-residue polypeptide is completely unfolded under reducing conditions.18,29−31 Consistent with this, the NMR chemical shifts of the PhoA4 fragment match the values known for the same residues in the full-length protein.18Figure 1 compares a representative 2D 1H–15N HMQC spectrum measured at 50 °C for this unfolded 15N-labeled protein upon injection of hyperpolarized water against that of a thermal counterpart, both containing only ca. 2% protonated H2O. Notice that in this conventional spectrum, measured using the same sample at the same temperature, most peaks broaden beyond detection due to fast exchanges with the solvent. While this exposure conspires against normal 2D NMR, it facilitates the magnetization transfer from the hyperpolarized water, leading to strongly enhanced peaks. This evidences a certain complementarity between HyperW-based and conventional HMQC acquisitions. While enhancements can be calculated only with relatively large uncertainties when the hyperpolarized spectrum is compared against a thermal spectrum collected from the dissolution DNP sample, peaks emerge from the noise if the PhoA4 HMQC spectrum is measured with the same sequence in a fully protonated H2O buffer at 50 °C (SI, Figure S3). The average sensitivity enhancement that can be then calculated for the unfolded PhoA4 fragment is ∼260× when considering all peaks in the spectrum. This high enhancement is typical of what we have obtained in unfolded protein injections, using our 14.1 T NMR and hyperpolarization setup.
Figure 1.
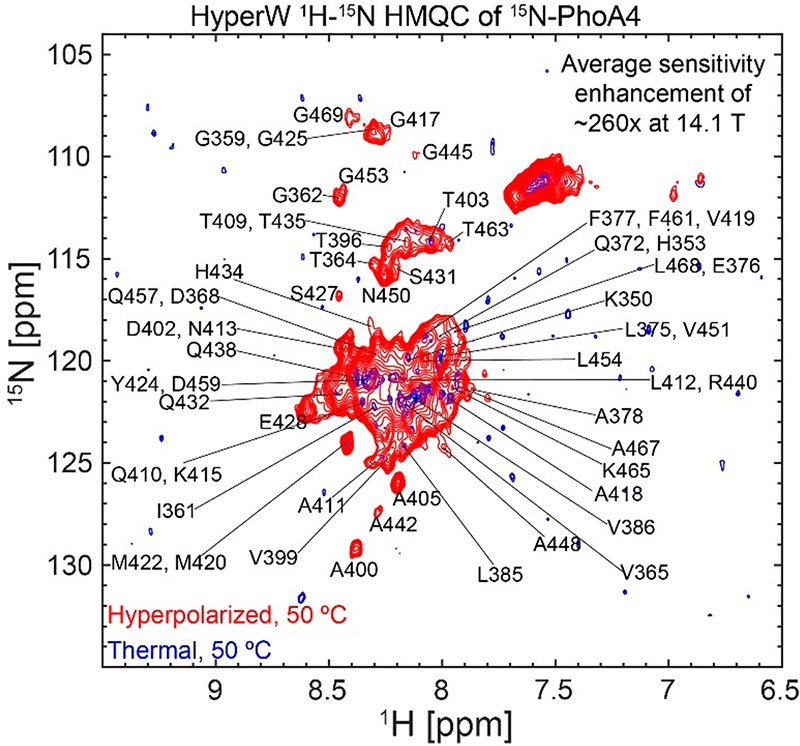
Comparison between 2D HyperW (red) and conventional (blue) 1H–15N HMQC spectra measured on 15N-PhoA4. 2.8 mL of super-heated buffered D2O (50 mM HEPES, pD 7.5, 50 mM KCl) was used to dissolve an 85/15 water/glycerol pellet containing 10 mM 4-amino-TEMPO. This pellet had been polarized at 1.12 K for ∼3 h 30 min using 100 mW of microwave irradiation at 94.195 GHz. ∼240 μL of the resulting hyperpolarized water solutions were injected into a 5 mm NMR tube containing ∼140 μL of a 1.5 mM 15N-labeled PhoA4 solution. Partial tentative assignment of residues indicated by single-letter amino acid codes is done based on the BMRB entry of the full-length PhoA.32 Both spectra were recorded at 50 °C using 64 hypercomplex t1 increments and hypercomplex34 acquisition covering indirect- and direct-domain spectral widths of 6009.6 and 1825.8 Hz. The HyperW spectrum was recorded using two phase-cycled scans per t1 increment. Additional experimental parameters: 14.1 T Prodigy-equipped NMR; total acquisition times of 73 s for the hyperpolarized spectrum (acquisition time of 213.0 ms, repetition delay of 0.037 s) and 14 h 12 min for the thermal spectrum (320 scans per t1 increment, acquisition time of 213.0 ms, and a repetition delay of 1 s).
By comparing to the BMRB entry of the full-length PhoA32 and extrapolating according to the changes that peaks undergo with temperature and pH, several peaks in the HyperW spectrum (Figure 1) can be tentatively assigned. With these assignments, enhancements can be calculated for specific residues; the average enhancement for these resolved residues (Figure 2) is ∼130×, substantially lower than what arises by considering the overall peak volume of the spectra. It is also clear that within this assignable set there are sites which get enhanced much more than others, a heterogeneity that could reflect water accessibility and/or local residue charges. To evaluate the influence of the former, we relied on secondary structure propensity (SSP) scores, which can range from +1 for a completely structured α-helix, through 0 in a disordered structure, to −1 for a β-sheet.33 Saio et al.18 calculated SSPs for this protein fragment; the gray bars in Figure 2 illustrate these parameters as a function of the primary sequence. Also added to Figure 2 are orange and red squares indicating positively and negatively charged residues, respectively. Unlike what had been previously observed for α-synuclein, the sensitivity enhancements evidenced by HyperW HMQC do not appear to correlate with these electrostatic charges in the sequence; the correlation arising between the enhancements and the SSP values is also questionable—if present at all (SI, Figure S4).
Figure 2.
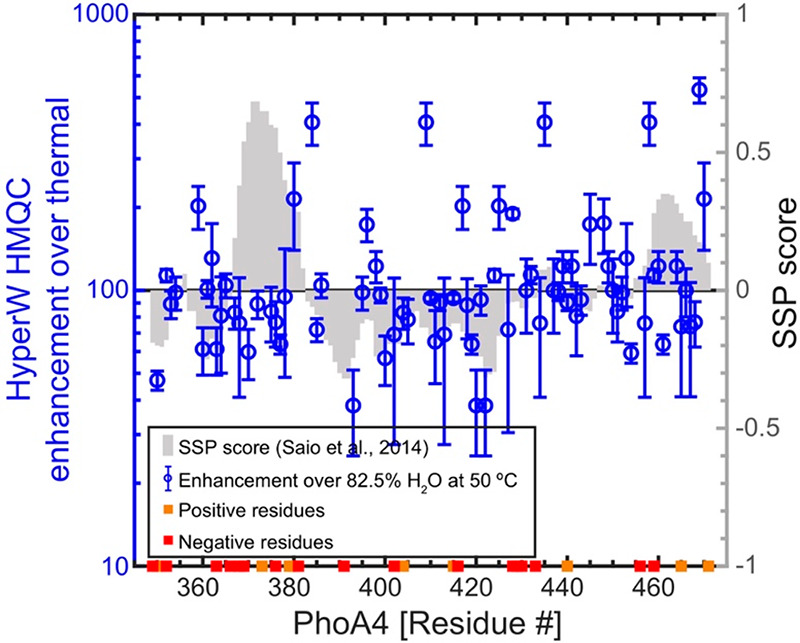
HyperW HMQC sensitivity enhancements calculated for resolved residues in the 15N-labeled PhoA4 protein fragment. The sensitivity enhancements were extracted by comparing peak volumes between the HyperW HMQC spectrum (e.g., Figure 1, red) and thermal equilibrium spectra measured in an 82.5% H2O buffer (Figure S3). The values are averaged for three HyperW HMQC experiments, after normalizing to the H2O proton enhancement in each experiment; the “error bars” reflect the scattering obtained over the course of these repeated injections for each residue. Sensitivity enhancements compared against SSP scores (gray bars) given in the literature18 based on NMR 13Cα and 13Cβ chemical shifts. Charged residues are also mapped on the sequence with orange (positively charged) and red (negatively charged) squares.
HyperW NMR on a Fully Structured Peptide: Barstar
Barstar is an 89-residue protein from Bacillus amyloliquefaciens bacteria with a well-defined, folded structure.35,36 Extensive work has been done on this protein as a model of folding,37−42 with most crystallographic and folding studies centering on the C40/82A mutant. We thus chose this well-studied construct to test the outcome of HyperW HMQC experiments on a well-folded paradigm. Figure 3 shows the sensitivity enhancements that HyperW HMQC NMR at 50 °C imparts on this double C40/C82A barstar mutant (note that the protein is stable at this temperature, as its Tm is ∼70–75 °C at the pH ∼7–8 used in this study43−45). The excellent volume repeatability delivered by the post-DNP rapid injection system allows one to achieve very good shimming conditions, leading to clearly resolved resonances with chemical shifts that are characteristic of well-folded structures (this is consistent with what has been recently reported by Kadeřávek et al. for ubiquitin,46 regarding the compatibility of water-derived hyperpolarization with studies of folded biopolymers). In fact, after taking into account the changes in chemical shifts with temperature, it was possible to assign most of the peaks in the HyperW HMQC (80 out of 89) based on literature data;47 these are annotated in Figure 3A. Despite the good site resolution achieved, it is also clear that peaks along the indirect dimension of the HyperW experiments are broader than in their thermally collected counterpart spectra. This reflects the limited lifetime of the water hyperpolarization, which, driven by T1, by chemical exchange with the biomolecule, and by decays induced by pulse non-idealities (even though pulses were tuned to avoid touching the water resonance), put an upper bound on the number of points that can be conventionally sampled along the t1 domain. For the kind of systems hereby analyzed, ca. 30–60 s is the time available for probing the indirect dimension of the 2D NMR spectra. Non-uniform sampling (NUS)48,49 should be able improve this resolution further while retaining the same overall experimental time. Figure 3B illustrates this with HyperW and thermal spectra recorded and processed on the same sample with NUS, where the effective t1 evolution time was increased 4-fold and an improvement in resolution along the indirect dimension for both experiments (thermal and hyperpolarized) is evident. Overall the average sensitivity enhancements in both regularly and non-uniformly sampled experiments are comparable, as the longer evolution times employed in the latter are offset by the smaller number of points (and hence fewer pulses) employed.
Figure 3.

(A) Comparison between 2D HyperW (red) and conventional (blue) 1H–15N HMQC spectra measured on 15N-labeled barstar C40/82A double mutant. 2.8 mL of super-heated buffered D2O (50 mM sodium phosphate, pD 7) was used to dissolve an 85/15 water/glycerol pellet containing 10 mM 4-amino-TEMPO. The pellet was polarized at 1.20 K for ∼3 h using 100 mW nominal microwave irradiation at 94.195 GHz. ∼215 μL of the resulting hyperpolarized water solutions was injected into a 5 mm NMR tube containing ∼140 μL of a ∼4 mM 15N-labeled barstar mutant solution. Partial assignment of 80 (out of 89) residues is indicated here by single-letter amino acid code, on the basis of Wong et al.47 The three peaks marked x, y, and z are unassigned and are attributed to free amino acids in the sample. Both spectra were recorded at 50 °C using 64 hypercomplex t1 increments34 covering indirect- and direct-domain spectral widths of 7211.5 and 1825.8 Hz. The HyperW spectrum was recorded using two phase-cycled scans per t1 increment. Total experimental times were 72 s for the hyperpolarized spectrum (acquisition time of 213.0 ms, repetition delay of 0.037 s) and 2 h 51 min for the thermal spectrum (64 scans and an acquisition time of 213.0 ms per t1 increment, repetition delay of 1 s). (B) Non-uniform sampling improves HyperW resolution. Non-uniformly sampled 2D HyperW (red) and non-uniformly sampled conventional (blue) 1H–15N HMQC spectra were measured on the 15N-labeled barstar C40/82A double mutant. 2.8 mL of super-heated buffered D2O (50 mM sodium phosphate, pD 7) was used to dissolve the 85/15 water/glycerol pellet containing 10 mM 4-amino TEMPO. The pellet was polarized at ∼1.18 K for ∼3 h 03 min using microwave irradiation of 100 mW, 94.195 GHz. ∼250 μL of the resulting hyperpolarized water solution was injected into a 5 mm NMR tube containing ∼130 μL of a ∼4 mM 15N-labeled barstar mutant solution. Both spectra were recorded at 50 °C, sampling 25% of 256 hypercomplex t1 increments34 covering indirect- and direct-domain spectral widths of 7211.5 and 1825.8 Hz, leading to a 4-fold increase in maximum effective t1 evolution. The HyperW spectrum was recorded using two phase-cycled scans per t1 increment. Total experiment times were ∼80 s for the hyperpolarized spectrum (acquisition time of 213.0 ms, repetition delay of 0.037 s) and 11 h 50 min for the thermal spectrum (256 scans recorded and 213.0 ms acquisition time per t1 increment, with a repetition delay of 1 s).
Identification of the individual peaks reveals a remarkably heterogeneous picture for the HyperW enhancements characterizing barstar, which range from <1× for some residues to >300× for others (Figure 4). These sensitivity enhancements are calculated by comparing peak volumes between the HyperW HMQC spectrum (such as in Figure 3A, red) and the thermal equilibrium spectrum measured for the same sample in 90% H2O buffer. In general, residues in loops and otherwise disordered regions of the folded conformation appeared enhanced to a greater extent than those in the structured segments, highlighting again the relation between HyperW signal enhancements and accessibility to the hyperpolarized solvent. However, for other residues, including I13 and amides in helix-3 and helix-4 in the protein, the measured enhancements are also high. The close connection between these enhancements and water/amide exchange rates is further confirmed by CLEANEX-PM NMR,26 an experiment designed to highlight water-exposed residues. In these experiments the water resonance is selectively excited and allowed to exchange over a variable mixing period τm with the amide proton spins. At the end of these mixing periods a fast HSQC sequence is used for detection, and the amide resonances’ peak intensities are monitored for every τm on a per-residue basis. Using short mixing times, only the fast-exchanging amides will have enough magnetization coming from water. The longer the amides’ protons are allowed to exchange with the water, the higher their magnetization will be. Figure 5A illustrates the close match between long-τm CLEANEX-PM experiments and the HyperW data.
Figure 4.
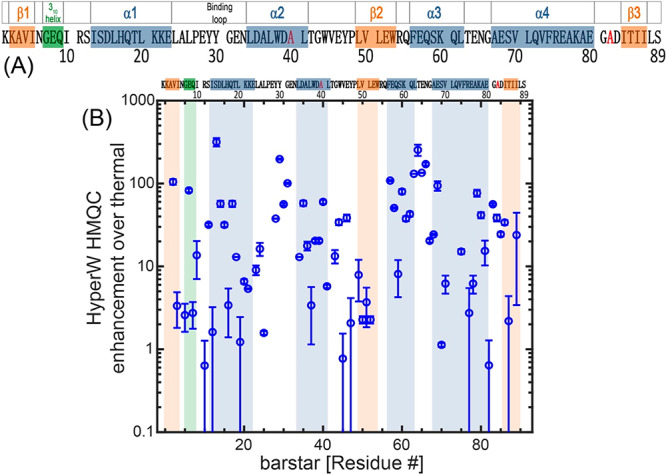
(A) 89-residue barstar C40/82A sequence analyzed in this study. Secondary structure elements47 are denoted above the sequence and shaded in blue (α-helices), orange (β-strands), and green (310 helix). A flexible loop, which plays an important role in binding barnase,50,51 is also noted. The C40/82A mutations are shown in red. (B) HyperW HMQC sensitivity enhancements observed for the assigned residues of the 15N-labeled mutant. The sensitivity enhancements were calculated by comparing peak volumes between the HyperW HMQC spectrum (such as in Figure 3A, red) and the thermal equilibrium spectrum measured for the same sample in 90% H2O buffer. The values are averaged for two HyperW HMQC experiments, after normalizing to the H2O proton enhancement in each experiment; “error bars” reflect the scattering of these experiments. Blue, orange, and green shaded areas are drawn on the regions corresponding to the secondary structure elements in (A).
Figure 5.

HyperW method correlates well with CLEANEX measurements for barstar. (A) 1H–15N CLEANEX Fast-HSQC spectrum with τm = 40 ms (black) and HyperW 1 H–15N HMQC (red, taken from Figure 3A) measured on 15N-labeled barstar. The post-dissolution ∼355 μL sample, which contained ∼1.6 mM barstar and ∼1.8% H 2O buffer (50 mM sodium phosphate, pH 7), was lyophilized and subsequently reconstituted in the same volume of 90% H2O. For the CLEANEX measurements, indirect- and direct-domain spectral widths of 7812.5 and 2130.1 Hz were covered, using 64 t1 hypercomplex increments.34 64 scans were collected using a 131.1 ms acquisition time and a relaxation delay of 2 s. Total experimental time was ∼5 h for each different mixing time τm. For HyperW HMQC, the acquisition parameters were as in Figure 3. All measurements were done at 50 °C on a 14.1 T Prodigy-equipped NMR spectrometer. (B) Comparing the amide proton exchange rates kHN arising for different barstar residues as extracted from CLEANEX experiments26 at 14.1 T (black squares), with the corresponding HyperW HMQC sensitivity enhancements (blue circles). The sensitivity enhancements were calculated by comparing peak volumes between the HyperW HMQC spectrum (such as in Figure 3A, red) and the thermal equilibrium spectrum measured for the corresponding sample in 90% H2O buffer. The values are averaged for two HyperW HMQC experiments after normalizing to the H2O proton enhancement in each experiment. All measurements were done at 50 °C.
The theory for extracting exchange rates k from CLEANEX is well established26,52−55 and is based on the equation
| 1 |
where V is the CLEANEX peak volume and V0 the corresponding peak volume in a reference HSQC spectrum. RHN,app is the apparent relaxation rate for the amides, containing contributions from the longitudinal relaxation rate 1/T1HN and from the transverse relaxation rate 1/T2HN, while the apparent relaxation rate for water is its longitudinal relaxation rate RH2O,app = 1/T1W. The rate constant k is related to the amide–water exchange rate kHN used in our previous analyses of HyperW signal enhancements17 by kHN = XH2Ok; since XH2O (the molar fraction of H2O) ≈ 1, kHN ≈ k. CLEANEX-derived rates should thus be, within the uncertainty limits of the relaxation and overall DNP enhancement (ε) parameters, similar to those arising from HyperW methods. Figure 5B shows that there is indeed a relatively good correlation (r = 0.63, calculated in a linear enhancement vs kHN plot) between the measurements.
HyperW NMR on R17: Highlighting the Unfolded State in a Folded/Unfolded Coexisting System
Chicken brain α-spectrin repeat 17 (R17) is a 118-residue domain, which exists in equilibrium between a well-folded state (F) and an unfolded state (U).20 The exchange dynamics between these states is very slow on the NMR time scale, with an exchange rate kex = kF→U + kU→F, which has an upper limit of 0.01 s–1 at 37 °C.20 This provides an interesting platform for assessing the “exchange filter” model put forward for barstar: as individual resonances should be observable for each of these forms, one expects that the HyperW enhancement will highlight the unfolded, exposed residues over their folded, protected counterparts. Figure 6A,B demonstrates that this is indeed the case, by comparing hyperpolarized and thermal data recorded at 37 °C and 2% H2O on this 13.3 kDa polypeptide, where the [U]/[F] equilibrium constant is ∼1. Even a cursory investigation of the spectra shows that the HyperW procedure enhances the disordered residues appearing in the central 8–9 ppm/118–128 ppm 1H/15N amide region more strongly than the well-resolved peaks arising from the folded form and appearing in the periphery of this “box”. The relatively good HyperW HMQC line shapes allow us to use literature data56 in order to assign individual peaks—but only for the folded form. The majority of the unfolded peaks, unfortunately, overlap and prevent us from performing a similar assignment. HyperW enhancements measured for the assigned folded and the partly assigned unfolded peaks are summarized in Figure 6C,D. These data confirm that the method preferentially enhances the signals from residues in the unfolded conformation over those in the folded one—the average enhancement for the unfolded form is ∼100×, while for the folded one it is ∼25×. These unfolded and folded case values are comparable to those observed for the PhoA4 and barstar cases, respectively. For specific residues such as the indole group of W26 that can be identified in both unfolded and folded resonances, the enhancements are 35× and 10×, respectively. As enhancements are influenced by the rates of exchange and in unfolded forms these exchanges are facilitated, this is in good accord with typical amide/solvent exposure expectations.
Figure 6.

HyperW vs thermal HMQC results for R17, a protein possessing unfolded and folded conformations in slow U⇋F exchange. (A, B) Comparisons between 2D HyperW (red) and the thermal (blue) 1H–15N HMQC spectra measured for a 15N-R17 dimer at 37 °C. 2.8 mL of super-heated buffered D2O (50 mM HEPES, pD 7.5, 50 mM KCl) was used to dissolve an 85/15 water/glycerol pellet containing 10 mM 4-amino-TEMPO. The pellet was polarized at 1.20 K for 3 h using microwave irradiation of 100 mW, 94.195 GHz. ∼160 μL of the resulting hyperpolarized water solution was injected into a 5 mm NMR tube containing ∼140 μL of a ∼1.2 mM 15N-R17 solution. Partial assignment of residues indicated by single-letter amino acid codes is done based on the BMRB entry of R17.56 Resonances of the folded conformation are labeled with their respective assignments, resonances of the unfolded form are marked with an asterisk, and unassigned peaks are either overlapped folded and unfolded conformation residues or residues belonging to the latter. The indole peak of W26 is assigned with a prime (′) for the unfolded conformation and without a prime for the folded one. The full spectrum is shown in (A), and a zoomed-in view (highlighted square) in (B). This spectrum was recorded at 37 °C using 64 hypercomplex t1 increments34 covering indirect- and direct-domain spectral widths of 7211.5 and 1947.5 Hz. The HyperW spectrum was recorded using two phase-cycled scans per t1 increment. Total experimental time was 63 s for the hyperpolarized spectrum (acquisition time of 177.5 ms, repetition delay of 0.037 s) and 20 h 07 min for the thermal spectrum (256 scans with an acquisition time of 177.5 ms and repetition delay of 2 s per t1 increment). (C, D) HyperW HMQC sensitivity enhancements for residues of the 15N-labeled R17 domain at 37 °C. The sensitivity enhancements were measured by comparing peak volumes between the HyperW HMQC spectrum and the thermal equilibrium spectrum measured for the same sample as in (A). Sensitivity enhancements for the folded state are marked with blue circles (C), and those of the unfolded state are marked with red circles (D). Note that there is no assignment available for the unfolded state; therefore, the enhancements in (D) are plotted against sequential peak numbers.
Paradigm Broken: HyperW Differential Enhancements of the Folded and Unfolded drkN SH3 Domain are Biased toward the Former
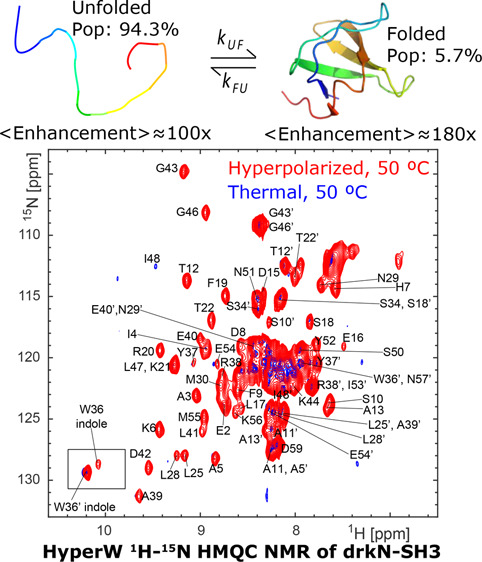
SH3 is a small protein domain, found as a modular entity in a variety of eukaryotic and viral proteins.57,58 The SH3 domain from the Drosophila signal transduction protein, drkN, has an important role in behavioral neuroplasticity, in activation-dependent learning, and in memory formation.59 It also has an interesting dynamics that was targeted by several investigations,60−64 which showed that this 6.9 kDa polypeptide exists in equilibrium between a well-folded ground state (F) and an unfolded excited state (U).65,66 These equilibrium dynamics are slow, and thus in a simple 1H–15N HSQC spectrum one can distinguish and assign peaks which belong to both states. Figure 7A shows a set of 1H–15N HMQC spectra measured at different temperatures on the 15N-labeled SH3 domain from drkN, collected without hyperpolarization. These data illustrate a shift in populations in favor of the unfolded state as temperature is gradually increased; Figure 7B highlights this with an enlargement focusing on the indole peak from the Trp36 side chain, where resonances arising from F and U states at each temperature are clearly resolved, and their changing intensities can be well quantified. To further characterize this folded/unfolded equilibrium under the conditions of our study, we implemented a series of ZZ-exchange NMR measurements67 (SI, Figure S5) that quantify both the kinetics and thermodynamics of slow conformational exchanges such as SH3’s U⇋F process. SI, Table S1 summarizes these kinetic and population values, as derived by these measurements on SH3 at the three temperatures that we explored.
Figure 7.

SH3 folded and unfolded states visualized by HyperW HMQC. (A) 2D 1H–15N HMQC spectra measured for a ∼520 μM 15N-drkN-SH3 domain in an 87.4% H2O buffer (50 mM HEPES buffer, 50 mM KCl, pH 7.5) at 27 °C (blue), 37 °C (green), and 50 °C (black). Indirect- and direct-domain spectral widths of 9014.4 and 2312.7 Hz were covered, using 64 hypercomplex t1 increments.34 The flip angle of the selective excitation was 90°, and 16 scans were collected using a 56.8 ms acquisition time and a relaxation delay of 2 s. Total experimental time was 1 h 12 min. (B) Enlarged region of Trp36 indole peak (marked with a black rectangle in (A)) showing the thermally driven rise of the unfolded state. (C) Comparison between 2D HyperW (red) and thermally polarized (blue) 1 H–15N HMQC spectra measured for the 15N-drkN-SH3 domain. 2.8 mL of buffered D2O (50 mM HEPES, pD 7.5, 50 mM KCl) was used to dissolve an 85/15 water/glycerol pellet containing 10 mM 4-amino-TEMPO. The pellet was polarized at 1.17 K for 3 h 30 min using 100 mW microwave irradiation at 94.195 GHz. ∼180 μL of the resulting hyperpolarized water solution was injected into a 5 mm NMR tube containing ∼140 μL of a ∼1.3 mM 15N-drkN-SH3 solution. Partial assignment of the various residues indicated by single-letter amino acid codes is done on the basis of Zhang et al.65 Resonances of the folded conformation are labeled with these assignments, and resonances of the unfolded form are marked with an added prime (′). These spectra were recorded at 50 °C using 64 hypercomplex t1 increments covering indirect- and direct-domain spectral widths of 7211.5 and 1947.5 Hz. The HyperW spectrum was recorded using two phase-cycled scans per t1 increment. Total experimental time was 63 s for the hyperpolarized spectrum (acquisition time of 177.5 ms, repetition delay of 0.037 s) and 13 h 50 min for the thermal spectrum (176 scans, acquisition time of 177.5 ms and a repetition delay of 2 s per t1 increment). (D) Enlarged region of the Trp36 indole peak (marked with a black rectangle in (C)).
Figure 7C compares 2D HyperW vs thermal 1H–15N HMQC spectra measured for the same post-dissolution SH3 sample at 50 °C and 2% H2O. A mostly unfolded state dominates these spectra, whose residues (indicated by primes added to the single-letter amino acid codes) are once again significantly enhanced by the injection of hyperpolarized water. Interestingly, however, one can also observe a significant enhancement of the folded state peaks; see, for instance, Figure 7D, zoomed in on the Trp36 indole peak from the folded (F) and unfolded (U) states. The reported sensitivity enhancements (Figure 8) are calculated by comparing peak volumes between the HyperW HMQC spectrum (such as in Figure 7C, red) and the thermal equilibrium spectrum measured for the same sample in 90% H2O buffer, after suitable rescaling to equate the proton concentrations. The degree of enhancement of these F-derived peaks is not easy to quantify from the thermal counterpart, as at an abundance of ∼5.7% their visibility is limited. Furthermore, the reproducibility of hyperpolarized water injections is not perfect. Still, after n = 3 injections performed under a priori identical conditions and after extensive signal averaging of the thermal samples, systematically higher enhancements are revealed at 50 °C for this and many other F-state residues, than for their U-state counterparts. This is illustrated in Figure 8 in a number of different representations, which aim at conveying the extensive experimental data that indicates that in this system, at 50 °C, water hyperpolarization enhances the majority of the assignable residues in the folded form of drkN SH3 more than in its unfolded counterpart. This anomalous behavior is observed to a smaller extent at 37 °C, even if the folded residue enhancements are then still considerably higher than in any of the other folded proteins examined in this study.
Figure 8.

(A) 59-residue drkN-SH3 domain sequence analyzed in this study. Secondary structure elements in the folded state (as measured at 20 °C62) are denoted above the sequence and shaded in orange (β-strands) and green (310 helix). Three β-sheets are formed in this small protein, and their β-strands are connected by straight lines in the cartoon. (B, C) HyperW HMQC sensitivity enhancements for assigned residues of the 15N-labeled drkN SH3 domain at 50 °C (full symbols) and 37 °C (open symbols). The sensitivity enhancements were calculated by comparing peak volumes between the HyperW HMQC spectrum (such as in Figure 7, red) and the thermal equilibrium spectrum measured for the same sample in ∼90% H2O buffer. The values at 50 °C are averages for three nominally identical HyperW HMQC experiments after normalizing to the H2O proton enhancement in each experiment, and the “error bars” denote the spreads observed in these experiments; only residues whose identity could be verified were included in the analysis (see SI Table S3 for further information). Sensitivity enhancements for the folded state are marked with blue circles (B), and those of the unfolded state are marked with red circles (C). (D, E) Different renderings of the observed experiments, showing the relative enhancement ratio of folded vs unfolded peaks in all the experiments recorded (D), and as correlations between the folded and unfolded enhancements observed in all the experiments at 37 and 50 °C (E). Orange and green shaded areas are drawn in (B, D) for regions which correspond to the secondary structure elements in (A).
As the results shown in Figure 8 depart from the standard paradigm according to which unfolding should promote a more facile water/amide exchange process and hence a higher HyperW enhancement, numerous ancillary tests were performed to corroborate and further understand these findings. The simplest of them, repeated injections, gave fairly reproducible results—at least within the limits of our HyperW NMR setup, and within the resolution constraints imposed by the relatively broad unfolded spectral patterns (SI, Table S3). CLEANEX-PM experiments were also undertaken on the post-dissolution samples, but at 50 °C they failed to provide sufficient sensitivity to measure the exchange rates of either the folded (minority) or unfolded (broadened) sites. Samples that had been analyzed by HyperW were thus lyophilized, resuspended in 90% H2O/D2O buffer, and subjected to CLEANEX-PM analyses at 50 °C. Figure 9 summarizes representative findings of these experiments. As can be appreciated from the CLEANEX-PM spectrum measured with a mixing time comparable to the HyperW recycling delay, the buildup process only highlighted the more abundant U-derived resonances; peaks belonging to the folded state are not observed in these experiments—primarily due to their low populations. Furthermore, a relatively weak correlation (r ≈ 0.50) was found between the unfolded state exchange rates measured in these CLEANEX-PM studies and the corresponding sensitivity enhancements observed in the HyperW HMQC for the unfolded resonances (Figure 9B).
Figure 9.

(A) 1H–15N CLEANEX fast-HSQC spectrum with τm = 40 ms (black) and HyperW 1 H– 15N HMQC (red, taken from Figure 7C) measured on 15N-labeled drkN SH3. Notice how HyperW enhancements appear to correlate with CLEANEX-PM measurements for the unfolded state of the drkN SH3 domain. For the CLEANEX-PM measurements (black), one of the post-dissolution samples containing 160 μM drkN SH3 and ∼2.4% H2O buffer (50 mM HEPES, 50 mM KCl, pH 7.5) was lyophilized and subsequently reconstituted in the same volume with 90% H2O. For CLEANEX the indirect- and direct-domain spectral widths were 7211.5 and 2069.2 Hz, covered using 64 t1 hypercomplex increments and STATES acquisition.34NS = 128 scans were collected using a 142.0 ms acquisition time and a relaxation delay of d1 = 2 s. Total experimental time was ∼10 h for each different mixing time τm. For the HyperW HMQC, acquisition parameters were as in Figure 7C. All measurements were done at 50 °C on a 14.1 T Prodigy-equipped NMR spectrometer. (B) Amide proton exchange rates arising for different drkN SH3 residues in the unfolded state as extracted from CLEANEX-PM experiments at 14.1 T, 50 °C (black squares).
HyperW HMQC measurements were repeated at 37 °C, where the folded state is more abundant and the rates of U⇋F interconversion are, according to ancillary ZZ-exchange and methyl-TROSY experiments (see SI, Figure S5 and Tables S1 and S2), slower. Figure 8 summarizes these results (open symbols). As can be seen, both folded and unfolded peaks are now enhanced systematically less than at 50 °C; this is as expected, given the decrease in the solvent exchange rates occurring upon lowering the temperature, and a decrease in the water T1 that will lead to shorter hyperpolarization lifetimes. Furthermore, individual residues are now enhanced to comparable degrees in their folded and unfolded forms. CLEANEX-PM measurements were repeated for SH3 under these conditions to measure amide exchange rates for the resonances of the unfolded and folded forms (SI, Figure S6). The measured signal enhancements for the folded state of drkN-SH3 at 37 °C correlate well (r = 0.85) with solvent–amide exchange rates measured by CLEANEX-PM at this temperature (Figure S6A), while for the unfolded state this correlation is weaker (r = 0.49, see Figure S6B). The enhancements observed at 37 °C are in agreement with the expectations deriving from the HyperW examples discussed above, as the enhancements for folded state residues at this temperature are not larger than for their unfolded counterparts. This lifts the need for an explanation of anomalous folded-vs-unfolded enhancements at this lower temperature but does not shed light on the behavior observed at 50 °C.
A feature that distinguishes SH3’s 50 °C behavior both from its behavior at 37 °C, and from the R17 case, concerns the presence of a relatively fast U⇋F interconversion between a dominant U and a minority F state. At these conditions the folded form corresponds to what is normally considered to be an “invisible” state,68 which is only made visible here by the unusually large enhancements brought to its amide peaks by the hyperpolarized water injection. This suggests the possibility of an alternate route to the water(H) ⇋ amide(H) exchange facilitating the HyperW HMQC enhancement, along the lines shown in Scheme 1. In this case the folded form is hyperpolarized by two concurrent processes: one where the water protons undergo direct chemical exchange with the amides of the folded state, and another where this exchange happens with the protons of the highly populated unfolded state—and then this unfolded state undergoes a conformational conversion into the low-populated folded form.
Scheme 1. Potential Exchange Processes Defining HyperW Experiments on drkN SH3 Domain.
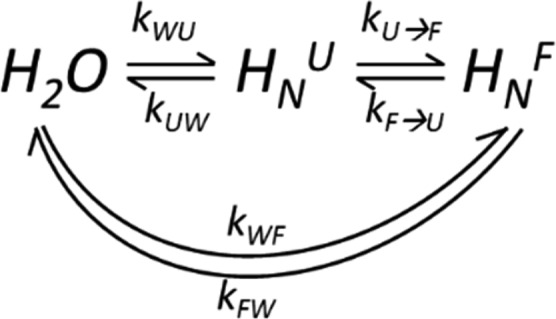
U and F denote a residue’s unfolded and folded conformations; kWU, kWU are the exchange rates of the water protons with the amides in the unfolded and folded states, and kXW (X = U,F) are the rates of the backward reactions. kU→F and kF→U are the rates of the U⇋F protein interconversion.
A theoretical Bloch–McConnell exchange model was developed to test whether these additional dynamics could explain the anomalous enhancement of the folded over the unfolded residues; calculations showed that the enhancements measured for the folded state residues could then indeed be larger than for the unfolded state—but only if the solvent exchange rates for these folded residues are faster than for their unfolded counterparts (Figure 10A,B). In search for an alternative that would demand less radical assumptions, the exchange model was expanded to include potential effects of different cross-relaxation processes. In order to account for these, a system of Bloch−McConnell−Solomon equations was set up, accounting for possible transfers among water, amide, and aliphatic magnetizations, ⟨H2O⟩z, ⟨HNU⟩z and ⟨HNF⟩z, ⟨HCU⟩z and ⟨HCF⟩z, the latter present in both the folded and unfolded states. For the sake of completeness, we included in this model the possibility that the 33 labile side-chain sites in this 59-residue peptide—representing hydroxyls, guanidinium and amines, and summarized by a magnetization ⟨HX⟩—might also be enhanced by exchanges with the hyperpolarized water, and transfer their hyperpolarization via cross-relaxation to the targeted amide sites. While a more complete account of this model is given in the SI, the overall system of equations that we considered was28
 |
2 |
Diagonal elements in the matrix above are given by rF = kFW + kFU + R1HNF + 1/T1F, rU = kUW + kUF + R1HNU + 1/T1U, R1HCF, R1HCU, R1HXF, R1HXU, and rW = 1/T1W + kWU + kWF + 2kWX. In these expressions the rates kFU and kUF represent the exchange rates between the folded and unfolded states (Scheme 1); R1HNF, R1HNU, R1HXF, R1HXU, R1HCF, and R1HCU are the auto-relaxation rates of the amide, the labile side chain, and the aliphatic protons in folded and unfolded states (including dipolar interactions between the 15N and HN, HN and two HC’s, HN and a suitably weighted HX, and HN and the hyperpolarized water, as well as between pairs HC–HC of aliphatic protons). Notice that an additional rate 1/T1 was added to the relaxation terms of each amide proton and of the water to account for other potential effects—arising, for instance, from the residual radical. The various σ’s in eq 2 represent in turn the cross-relaxation rates among the various proton populations. ⟨H2O⟩z(eq), ⟨HN⟩z(eq), ⟨HX⟩z(eq), and ⟨HC⟩z(eq) are the water, the amide (N), the labile side chain (X), and the aliphatic (C) proton magnetizations at thermal equilibrium. For both hyperpolarized and thermal calculations, the equilibrium polarizations were scaled according to a priori known molar fractions:
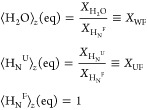 |
3 |
The population of the exchangeable side-chain protons was reduced to match the ratio between these nuclei and the amides in the protein. Exchange rates are also related to each other by the water and protein molar fractions
Water relaxation times were estimated from
independent experiments, while kinetic parameters for the U⇋F
interconversion process were extracted from the ZZ-exchange experiment
shown in the SI and recorded at 50 °C
(SI Figure S5 and Table S1). With all this
information, and using additional known parameters and standard assumptions
(delay between scans, number of t1 increments,
number of signals averaged scans, coherence transfer efficiencies,
etc.; see SI for a full derivation of this
model and the assumptions involved), the relative enhancement of the
HyperW vs the thermal HMQC experiments was cast in terms of three
variables: the initial enhancement factor ε =  of the injected hyperpolarized water over
its thermal magnetization—a parameter that affected the enhancement
of all residues, in both the folded and unfolded states, homogeneously; kUW, the rate of exchange between water and an
unfolded residue; and kFW, the rate of
exchange between water and a folded residue. Numerical calculations
based on eq 2 were carried
out for hyperpolarized and for thermally polarized HMQC acquisitions
for sets of exchange rates kUW and kFW, and the ensuing signal enhancement (Enh)
was determined for each pair of residues in the set as
of the injected hyperpolarized water over
its thermal magnetization—a parameter that affected the enhancement
of all residues, in both the folded and unfolded states, homogeneously; kUW, the rate of exchange between water and an
unfolded residue; and kFW, the rate of
exchange between water and a folded residue. Numerical calculations
based on eq 2 were carried
out for hyperpolarized and for thermally polarized HMQC acquisitions
for sets of exchange rates kUW and kFW, and the ensuing signal enhancement (Enh)
was determined for each pair of residues in the set as
Figure 10.

Relative HyperW/thermal enhancement per scan predicted by the numerical solutions of SI eqs S6–S12 for a protein residue subject to the 2D 1H–15N HMQC sequence depicted in Figure S2. Calculations were repeated for thermal (ε = 1) and hyperpolarized (ε = 500) water scenarios as a function of exchange rates kUW and kFW. Additional assumptions included T1W = 15 s, [H2O] = 0.92 M (to account for a dilution to 1.7% after dissolution), [protein] = 0.59 mM, pU = 94.3% in the hyperpolarized experiment and 96% in the thermal (to account for equilibrium differences in protonated and deuterated solvents), pF = 5.7% in the hyperpolarized experiment and 4% in the thermal one (Table S2), kUF = 1.9 s–1, and kFU = 31.4 s–1. The correlation times τc for the folded and unfolded states were assumed to be equal to 3.4 and 0.8 ns, respectively. The number of scans per increment were 2 and 128 for the hyperpolarized and thermal experiments, and N1 = 128 increments for both cases. Other considerations regarding the auto-relaxation and cross-relaxation are as detailed in the SI. Enhancements were calculated by taking the ratio of the expected HyperW and thermal equilibrium signals recorded with fixed repetition times tR,Hyp = 0.24 s and tR,TE = 1.21 s. Numerically simulated per-scan enhancements for the unfolded and folded conformations are plotted as 3D surfaces and as a function of exchange rates kUW and kFW for relevant ≤60 s–1 values. (A, B) Results expected for different intrinsic relaxation times of the folded (T1F) and unfolded (T1U) states (indicated on the top of each panel), assuming that cross-relaxation processes occur solely within the HN, NH, and two aliphatic side-chain protons, Hc1 and Hc. Note that when a larger intrinsic relaxation rate 1/T1F is assumed, the per-scan enhancements for the folded state will be larger for slightly slower kFW, but these effects are small. (C, D) Effects introduced when the possibility of cross-relaxation from the hyperpolarized water is added to the model in (A), assuming the indicated correlation times τc of the folded and unfolded states. The only significant bias of the HyperW enhancements provided by the exchange processes (A, B) arises when assuming particularly suitable correlation times and short interatomic water–amide distances (panel D). (E, F) Same as models (C, D), but now incorporating the possibility of having cross-relaxation between the amides and a labile 1H (X), which could be part of an hydroxyl, amino, or guanidinio side chain. For simplicity the τc’s used to model these additional relaxation processes were assumed as for the structural waters, and the rates of exchange with the solvent were assumed 30 Hz for all forms. Again, notice that very short internuclear distances would be required for these cross-relaxation processes to have a noticeable effect in biasing the F/U enhancements.
Figure 10 shows a summary of these calculations, which focuses on illustrating how the solvent exchange rates kUW and kFW will affect the per-scan enhancement of different sites in drkN SH3’s unfolded and folded conformations. For a range of intrinsic relaxation times T1F and T1U and for typical water enhancement factors (ε ≈ 500), these plots show two surfaces that intersect when kUW ≈ kFW—with some parameters being fixed as per the SH3 experiments and the ancillary independently measured data, and others varied so as to illustrate their effects. It follows from this model that the enhancements measured on the folded state residues could indeed be very large—even larger than for the unfolded state—but in the absence of water-derived cross-relaxation effects, this would require that the solvent exchange rates for these folded residues be faster than for their unfolded counterparts. Indeed, the U⇋F interconversion, intraresidue cross-relaxation effects and ad hoc 1/T1 rates will affect the symmetry of the folded and unfolded state enhancements slightly, but for the values measured independently for kUF and kFU, this asymmetry is relatively small (Figure 10A,B). Only if the T1F value is for some reason much larger than T1U will a slight bias toward the folded-form enhancement arise (as a result of the partial saturation of the thermal signal used as reference, a condition that was not met in our experiments), and hence the apparent enhancement of the folded site will look larger than that of its unfolded counterpart. When strong water-associated cross-relaxation effects deriving either from the hyperpolarized water itself or from labile side-chain protons that have been hyperpolarized by the water are included, however (e.g., Figure 10D,F), the experimental data can also be reproduced if it is assumed that kFW < kUW. Notice, however, that even under these assumptions—which bias cross-relaxation enhancements toward the folded form by virtue of having assumed relatively long correlation times and short internuclear distances between the hyperpolarization sources and the targeted protons—the maximal F/U ratios reached under the kFW < kUW condition amount to tens of percent.
Discussion and Conclusions
The injection of hyperpolarized water in precise aliquots into a regular NMR setting followed by the acquisition of high-resolution 2D data was applied to a wide range of protein structures and shown to be a technique that can serve two main purposes. On one hand it can help to sensitize 2D HMQC NMR experiments, to the point of highlighting low-populated “invisible” states that would be hard to observe in equilibrium with their more populated states.68,69 On the other hand, the experiment affords enhancements that can in general be translated into insight about relative solvent exchange for different residues within the same sample/protein. This could be important, as given reasonably well-known parameters including the hyperpolarized water enhancement and the effective T1 relaxation decays, absolute values of water/amide exchange rates could also be derived. These features were explored here using an array of representative protein systems, chosen to illustrate a variety of scenarios. The largest enhancements were observed, as could have been expected, for the case of disordered proteins like the PhoA4 fragment, for which nearly all residues exhibited enhancements ≥100×—and several residues exceeded 500-fold enhancement values. Also in agreement with the aforementioned exchange-dominated model was the behavior of barstar, a well-folded protein that exhibited correspondingly smaller enhancements. Notable heterogeneities in the enhancement of the different barstar residues were noted, yet these correlated well with their readiness for water exchanges, as evidenced by CLEANEX-PM measurements. Previous reports suggested that even though barstar holds a well-defined three-dimensional structure, it is still dynamic and flexible;37,45,51,70,71 this could help to rationalize the observed HyperW/CLEANEX NMR behavior in terms of local disorder. In fact, previous H/D exchange studies investigated amide/water exchange rates for different barstar residues and found that these exchange rates correlate with calculated relative surface accessibility.71 It is generally accepted that, in folded proteins, protons residing in flexible loops will be the most surface-exposed, while protons in secondary structure elements will be involved in hydrogen bonding or buried in the protein core, and hence their exchange rates would be slower.72 While the HyperW enhancements observed for barstar are higher for loop regions and exposed amides (Figure 4), several residues do not follow this correlation: most of these belong to an α-helix and are apparently involved in hydrogen bonds, yet still exhibit high enhancements. This could, however, still be explained in terms of the solvent accessibility of these residues, as they might reside on a more surface-exposed side of the α-helix.
Attention was then turned to two proteins featuring coexisting folded and unfolded states. One of these, R17, behaved within expectations: The folded peaks of R17 showed enhancements in the 1–100× range, while the same residues in the unfolded form showed enhancements in the 10–500-fold range (Figure 6). While the resolution of this U form in the HMQC experiment was not sufficiently high to permit residue-specific analyses, the trends respected the behavior described above for amide exchange in unfolded and folded proteins; this was as expected, given the relatively slow interconversion between R17’s F and U forms. By contrast, the second system analyzed, drkN SH3, revealed an anomaly: for the majority of the assignable residues, consistently larger HyperW enhancements were observed in the folded than in the unfolded states at 50 °C. Just as the residues’ enhancements were heterogeneous in each of the previously discussed systems, a distribution again characterized the individual residues’ enhancements in both U and F states—in SH3 this anomaly arises from the fact that the HyperW enhancements at 50 °C were in general larger for the folded state of the same amino acid than for its unfolded counterpart. These anomalous trends consistently emerged when examining the post-dissolution samples as well as lyophilized post-dissolution samples that had been reconstituted in per-protio solvents for the sake of improving the sensitivity, even after their populations had been suitably corrected to account for solvent differences. CLEANEX measurements shed little light on the origin of this behavior: for the 50 °C case, [F] ≪ [U]; this, plus CLEANEX’s limited sensitivity, prevented the characterization of the minority, “invisible” F-state behavior (while, however, still allowing measurements of the unfolded state’s behavior; see Figure 9).
In an effort to explain how hyperpolarized water could enhance certain residues more in their folded than in their unfolded states, a model based on Bloch–McConnell’s and Solomon’s equations was developed. This relied on independently measured relaxation times, on U⇋F kinetic and thermodynamic equilibrium parameters that were also independently measured, and on a variety of potentially concurrent self- and cross-relaxation phenomena. The correlation times of folded and unfolded proteins were estimated based on values for chains of similar size; the main unknowns in this model were thus the rates of folded- and unfolded-state exchanges with water and the extent of water–protein and intraprotein cross-relaxations. With this model we explored whether an amide proton in the U-form could gain magnetization from the hyperpolarized water but then “lose it” rapidly to a minority F-state that would then display unusually large enhancements as a result of combining multiple sources of hyperpolarization. These effects (Figure 10A,B) were not significant. Our model then considered whether cross-relaxation of the amides to other, non-exchangeable (and therefore not hyperpolarized) protons in the protein could bias measurements and result in an artificially higher F-form enhancement. These effects, however, ended up leading to bigger losses for the more structured folded form than for the more mobile unfolded form; if there is any bias derived from these effects, it should thus be working against the apparent enhancements observed for the F residues. Inclusion of ancillary ad hocT1 terms did not have much influence, either. The model was therefore expanded to allow for drkN SH3 amide proton enhancements to arise from other sources, including the possibility of differential folded/unfolded cross-relaxations between the amide groups and the hyperpolarized solvent, as well as between the amide groups and labile side-chain protons. The former, in particular, might lead to sizable contributions if structural-like hydration waters are involved.76,77 When assuming that correlation times were sufficiently short for the unfolded and long for the folded forms, and that the intermolecular 1H–1H distances were sufficiently short to ensure a strong Overhauser interaction, these additions predicted that HyperW enhancements could indeed be larger for the folded than for the unfolded forms—while still respecting the kFW ≤ kUW condition (Figure 10D–F). The resulting enhancement differences, however, were still relatively small: ≤50% for the best kFW = kUW case, compared to the differential enhancement factors of ca. 200–400% that are observed for numerous residues at 50 °C (Table S3).
In view of this, other potentially confounding factors were explored. One of them concerned the possibility of thermally induced drkN SH3 degradation and/or aggregation, which were found to occur at 50 °C but only over 48 h; these, however, are not relevant time scales for the ca. minute long times involved in our NMR measurements. Another potentially important factor that was considered concerned potential miscalibrations of the temperatures assumed in the HyperW experiment: as lower HyperW measurement temperatures would mean larger-than-assumed folded/unfolded drkN SH3 ratios in the sample, this could lead, after normalizing by intensities measured on a correctly set, thermally polarized 50 °C sample, to a bias in the ensuing folded/unfolded enhancement calculations. While no such artifacts were observed in calibration measurements (data not shown), we also relied on SH3’s own high temperature dependence to evaluate what the effects of dealing with lower-than-expected post-mixing temperature would be. Comparisons against variable-temperature drkN SH3 HMQC data showed that, post-injection, HyperW sample temperatures reached the targeted 49–50 °C within ca. 10 s (SI Figure S1). The various HyperW data sets collected in this study were still re-evaluated under the possibility that the sudden injection process dropped the sample’s temperature to 47 °C, but as shown by Figure S7, this would still leave, within experimental errors, the majority of assignable folded peaks in the HyperW spectra equally or more enhanced than their unfolded counterparts. As mentioned earlier, the population imbalances that may arise upon comparing folded/unfolded equilibria in mostly deuterated (e.g., HyperW) and mostly protonated (thermal) water were also considered; these were also measured via ancillary ZZ-exchange and methyl-TROSY experiments (SI, Tables S1 and S2), and their effects were included in all our enhancement estimations.
When examining which folded-form drkN SH3 sites showed the largest HyperW enhancements (Figure 8), residues at or near disordered loops stood out: for these cases nearly 300× enhancements could be measured, vis-à-vis ∼100-fold enhancements for their unfolded counterparts (see SI Table S3 for a summary of drkN SH3’s 50 °C results). This might explain why these residues are enhanced more than other amides in better folded regions—or in other folded systems we have examined. It still leaves the question, however, of how the same residue can be more readily enhanced by hyperpolarized water in a folded than in an unfolded form. Although the solvent/amide and labile side chain/amide cross-relaxation arguments made above could partly explain this behavior, it is hard to discard completely the role that amide–solvent exchange rates could play in this anomaly. Solvent–protein exchange measurements have been the focus of decades of systematic studies,73−75 with NMR- and mass-spectrometry-based H/D exchange measurements being the most established methods for measuring them.76−81 These solvent/amide exchange measurements, which are clearly related to the HyperW NMR measurement, have in turn been intimately linked with the degree of folding (or intermolecular binding) of a protein.77,82 This derives from the reasonable assumption that the more easily that water can access a specific amide moiety, the faster the rate of exchange with water will be.83,84 A change in the rate of solvent exchange will thus reflect a change in the solvent accessibility that the residue in question experiences—up to a maximum rate given by the exchange of the isolated amide (for instance, in a model dipeptide structure). Decades of H/D exchange studies have also revealed that many factors beyond solvent accessibility may influence a particular amide’s solvent exchange rate and change it by factors of up to a billion-fold. Foremost among these factors are the group’s local acidity,85−90 the effective electrostatic charge of the residue involved,87−89,91−96 and the electrostatic shielding imposed by a residue’s neighbors.89,97,98 On the basis of these very strong influences, it has been hypothesized, and even predicted by numerical methods,89,99 that anomalous cases may arise where rates of H/D exchange do not correlate with exposure to the solvent—and hence with a residue’s degree of folding. To the best of our knowledge, however, such predictions have not heretofore been experimentally detected. In this respect, the HyperW method provides a unique experimental window that could enable the discovery of such instances: by its very nature it probes the solvent accessibility directly and in very short time scales; it does so in a residue-by-residue fashion; it provides the ability to discriminate between peaks arising from coexisting folded and unfolded forms; and by virtue of its enhanced sensitivity it enables one to see minority states that under normal conditions would be invisible. As such, it allowed us to monitor enhancements of SH3’s folded and unfolded states under conditions that are at the threshold of total unfolding. It remains to be seen whether additional experiments can be devised that shed further light on the origins of the unusually high HyperW enhancements displayed by the folded SH3 residues over their unfolded counterparts.
The present study presented some of the promising avenues opened by HyperW NMR in protein research. The observations verified that even in its present form it can be used to sensitize the spectra of IDPs by several hundred-fold. The findings showed that even proteins like barstar, which are typically considered to be essentially folded, can also experience substantial enhancements that inform about the local structure and dynamics of the protein. Most intriguingly, this work also provided a new experimental tool to examine coexisting folded and unfolded protein states—even when one of these is present at what are normally “invisible” concentrations. Still, numerous additions could further extend the analytical power of this approach to solution-state protein NMR spectroscopy. Aspects in need of improvements from the DNP standpoint include increasing the volume and the hyperpolarization of the water,100 eliminating the polarizing radical,101,102 and—foremost of all—reducing the dilution experienced by the hyperpolarized water. Additional improvements investigated partially in this work, like the reliance on NUS schemes, could also facilitate higher sensitivity, higher resolution,48,49 and extensions to higher dimensionalities.103−105 Several of these advances are currently in the making, in the hope of revisiting the behavior of multiple protein unfolded/folded equilibria and of probing solvent accessibility in more complex interacting systems.
Acknowledgments
This work would not have been possible without the technical assistance of the late Koby Zibzener. We are also grateful to Dr. S. F. Cousin for assistance in the cross-relaxation calculations, to Dr. Shira Albeck (ISPC, Weizmann Institute of Science) for the barstar protein, and to Ms. Ivana Petrovic for expression of 13C-labeled drkN-SH3. This work was supported by the Kimmel Institute for Magnetic Resonance (Weizmann Institute), the EU Horizon 2020 program (Marie Sklodowska-Curie Grant 642773 and FET-OPEN Grant 828946, PATHOS), Israel Science Foundation Grant 965/18, and the Perlman Family Foundation. R.R. was also supported by the Israel Science Foundation Grant 1889/18 and the Minerva Foundation Research Grant.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00807.
Additional details about the experimental procedures and pulse sequences used; controls on the postinjection temperatures; additional measurements on PhoA4; additional ZZ-exchange, CLEANEX-PM, and methyl-TROSY measurements on SH3 at 37 and 50 °C; site-specific enhancements for SH3 at 50 °C; and details on the model accounting for multi-site exchanges with cross-relaxation (PDF)
Author Present Address
§ O.S.: Department of Biochemistry, Duke University School of Medicine, 307 Research Dr., Durham NC 27710, USA
Author Present Address
∥ G.L.O.: Institute of Biological Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 38, 1090 Vienna, Austria
The authors declare no competing financial interest.
Supplementary Material
References
- Koers E. J.; van der Cruijsen E. A. W.; Rosay M.; Weingarth M.; Prokofyev A.; Sauvée C.; Ouari O.; van der Zwan J.; Pongs O.; Tordo P.; Maas W. E.; Baldus M. NMR-based structural biology enhanced by dynamic nuclear polarization at high magnetic field. J. Biomol. NMR 2014, 60 (2), 157–168. 10.1007/s10858-014-9865-8. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Okuno Y.; Cavagnero S. Sensitivity enhancement in solution NMR: emerging ideas and new frontiers. J. Magn. Reson. 2014, 241, 18–31. 10.1016/j.jmr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Andreas L.; Griffin R. G. Magic Angle Spinning NMR of Proteins: High-Frequency Dynamic Nuclear Polarization and 1H Detection. Annu. Rev. Biochem. 2015, 84 (1), 465–497. 10.1146/annurev-biochem-060614-034206. [DOI] [PubMed] [Google Scholar]
- Akbey Ü.; Oschkinat H. Structural biology applications of solid state MAS DNP NMR. J. Magn. Reson. 2016, 269, 213–224. 10.1016/j.jmr.2016.04.003. [DOI] [PubMed] [Google Scholar]
- Jaudzems K.; Polenova T.; Pintacuda G.; Oschkinat H.; Lesage A. DNP NMR of biomolecular assemblies. J. Struct. Biol. 2019, 206 (1), 90–98. 10.1016/j.jsb.2018.09.011. [DOI] [PubMed] [Google Scholar]
- Ardenkjær-Larsen J. H.; Fridlund B.; Gram A.; Hansson G.; Hansson L.; Lerche M. H.; Servin R.; Thaning M.; Golman K. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (18), 10158–10163. 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragavan M.; Chen H.-Y.; Sekar G.; Hilty C. Solution NMR of Polypeptides Hyperpolarized by Dynamic Nuclear Polarization. Anal. Chem. 2011, 83 (15), 6054–6059. 10.1021/ac201122k. [DOI] [PubMed] [Google Scholar]
- Chen H.-Y.; Ragavan M.; Hilty C. Protein Folding Studied by Dissolution Dynamic Nuclear Polarization. Angew. Chem. 2013, 125 (35), 9362–9365. 10.1002/ange.201301851. [DOI] [PubMed] [Google Scholar]
- Ardenkjær-Larsen J.-H.; Boebinger G. S.; Comment A.; Duckett S.; Edison A. S.; Engelke F.; Griesinger C.; Griffin R. G.; Hilty C.; Maeda H.; Parigi G.; Prisner T.; Ravera E.; van Bentum J.; Vega S.; Webb A.; Luchinat C.; Schwalbe H.; Frydman L. Facing and Overcoming Sensitivity Challenges in Biomolecular NMR Spectroscopy. Angew. Chem., Int. Ed. 2015, 54 (32), 9162–9185. 10.1002/anie.201410653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardenkjær-Larsen J. H.; Laustsen C.; Pullinger B.; Kadlecek S.; Emami K.; Rizi R. Hyperpolarized Water for Interventional Angiography. Proc. Int. Soc. Magn. Reson. Med. 2011, 19, 3534. [Google Scholar]
- Harris T.; Bretschneider C.; Frydman L. Dissolution DNP NMR with solvent mixtures: Substrate concentration and radical extraction. J. Magn. Reson. 2011, 211 (1), 96–100. 10.1016/j.jmr.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris T.; Szekely O.; Frydman L. On the Potential of Hyperpolarized Water in Biomolecular NMR Studies. J. Phys. Chem. B 2014, 118 (12), 3281–3290. 10.1021/jp4102916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic M.; Olsen G. L.; Pinter G.; Hymon D.; Furtig B.; Schwalbe H.; Frydman L.. Hyperpolarized water (HyperW) NMR of imino resonances in nucleic acids: ≈400x signal enhancements and 1D/2D NMR probing of fast reaction kinetics. 60th Experimental Nuclear Magnetic Resonance Conference, Asilomar, Pacific Grove, CA, 2019.
- Olsen G.; Markhasin E.; Szekely O.; Bretschneider C.; Frydman L. Optimizing water hyperpolarization and dissolution for sensitivity-enhanced 2D biomolecular NMR. J. Magn. Reson. 2016, 264, 49–58. 10.1016/j.jmr.2016.01.005. [DOI] [PubMed] [Google Scholar]
- Chappuis Q.; Milani J.; Vuichoud B.; Bornet A.; Gossert A. D.; Bodenhausen G.; Jannin S. Hyperpolarized Water to Study Protein–Ligand Interactions. J. Phys. Chem. Lett. 2015, 6 (9), 1674–1678. 10.1021/acs.jpclett.5b00403. [DOI] [PubMed] [Google Scholar]
- Kurzbach D.; Canet E.; Flamm A. G.; Jhajharia A.; Weber E. M. M.; Konrat R.; Bodenhausen G. Investigation of Intrinsically Disordered Proteins through Exchange with Hyperpolarized Water. Angew. Chem., Int. Ed. 2017, 56 (1), 389–392. 10.1002/anie.201608903. [DOI] [PubMed] [Google Scholar]
- Szekely O.; Olsen G. L.; Felli I. C.; Frydman L. High-Resolution 2D NMR of Disordered Proteins Enhanced by Hyperpolarized Water. Anal. Chem. 2018, 90 (10), 6169–6177. 10.1021/acs.analchem.8b00585. [DOI] [PubMed] [Google Scholar]
- Saio T.; Guan X.; Rossi P.; Economou A.; Kalodimos C. G. Structural Basis for Protein Antiaggregation Activity of the Trigger Factor Chaperone. Science 2014, 344 (6184), 1250494. 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G.; Fersht A. R. The refolding of cis- and trans-peptidylprolyl isomers of barstar. Biochemistry 1993, 32 (41), 11195–11203. 10.1021/bi00092a032. [DOI] [PubMed] [Google Scholar]
- Sekhar A.; Velyvis A.; Zoltsman G.; Rosenzweig R.; Bouvignies G.; Kay L. E. Conserved conformational selection mechanism of Hsp70 chaperone-substrate interactions. eLife 2018, 7, e32764 10.7554/eLife.32764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen S.; Hilty C. Time-Resolved Dynamic Nuclear Polarization Enhanced NMR Spectroscopy. Angew. Chem., Int. Ed. 2008, 47 (28), 5235–5237. 10.1002/anie.200801492. [DOI] [PubMed] [Google Scholar]
- Bowen S.; Hilty C. Rapid sample injection for hyperpolarized NMR spectroscopy. Phys. Chem. Chem. Phys. 2010, 12 (22), 5766–5770. 10.1039/c002316g. [DOI] [PubMed] [Google Scholar]
- Katsikis S.; Marin-Montesinos I.; Pons M.; Ludwig C.; Günther U. L. Improved Stability and Spectral Quality in Ex Situ Dissolution DNP Using an Improved Transfer Device. Appl. Magn. Reson. 2015, 46 (7), 723–729. 10.1007/s00723-015-0680-5. [DOI] [Google Scholar]
- Schanda P.; Brutscher B. Very Fast Two-Dimensional NMR Spectroscopy for Real-Time Investigation of Dynamic Events in Proteins on the Time Scale of Seconds. J. Am. Chem. Soc. 2005, 127 (22), 8014–8015. 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Kupče E̅.; Brutscher B. SOFAST-HMQC Experiments for Recording Two-dimensional Heteronuclear Correlation Spectra of Proteins within a Few Seconds. J. Biomol. NMR 2005, 33 (4), 199–211. 10.1007/s10858-005-4425-x. [DOI] [PubMed] [Google Scholar]
- Hwang T.-L.; van Zijl P. C. M.; Mori S. Accurate Quantitation of Water-amide Proton Exchange Rates Using the Phase-Modulated CLEAN Chemical EXchange (CLEANEX-PM) Approach with a Fast-HSQC (FHSQC) Detection Scheme. J. Biomol. NMR 1998, 11 (2), 221–226. 10.1023/A:1008276004875. [DOI] [PubMed] [Google Scholar]
- Hyberts S. G.; Milbradt A. G.; Wagner A. B.; Arthanari H.; Wagner G. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 2012, 52 (4), 315–327. 10.1007/s10858-012-9611-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell H. M. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958, 28 (3), 430–431. 10.1063/1.1744152. [DOI] [Google Scholar]
- Akiyama Y.; Ito K. Export of Escherichia coli alkaline phosphatase attached to an integral membrane protein, SecY. J. Biol. Chem. 1989, 264 (1), 437–442. [PubMed] [Google Scholar]
- Bardwell J. C. A.; McGovern K.; Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell 1991, 67 (3), 581–589. 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- Kamitani S.; Akiyama Y.; Ito K. Identification and characterization of an Escherichia coli gene required for the formation of correctly folded alkaline phosphatase, a periplasmic enzyme. EMBO J. 1992, 11 (1), 57–62. 10.1002/j.1460-2075.1992.tb05027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Rossi P.; Saio T.; Kalodimos C. G. Structural basis for the antifolding activity of a molecular chaperone. Nature 2016, 537 (7619), 202–206. 10.1038/nature18965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J. A.; Singh V. K.; Jia Z.; Forman-Kay J. D. Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: Implications for fibrillation. Protein Sci. 2006, 15 (12), 2795–2804. 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- States D. J.; Haberkorn R. A.; Ruben D. J. A two-dimensional nuclear overhauser experiment with pure absorption phase in four quadrants. J. Magn. Reson. (1969-1992) 1982, 48 (2), 286–292. 10.1016/0022-2364(82)90279-7. [DOI] [Google Scholar]
- Hartley R. W. Barnase and barstar: two small proteins to fold and fit together. Trends Biochem. Sci. 1989, 14 (11), 450–454. 10.1016/0968-0004(89)90104-7. [DOI] [PubMed] [Google Scholar]
- Schreiber G.; Fersht A. R. Interaction of barnase with its polypeptide inhibitor barstar studied by protein engineering. Biochemistry 1993, 32 (19), 5145–5150. 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- Agashe V. R.; Shastry M. C. R.; Udgaonkar J. B. Initial hydrophobic collapse in the folding of barstar. Nature 1995, 377, 754. 10.1038/377754a0. [DOI] [PubMed] [Google Scholar]
- Shastry M. C. R.; Udgaonkar J. B. The Folding Mechanism of Barstar: Evidence for Multiple Pathways and Multiple Intermediates. J. Mol. Biol. 1995, 247 (5), 1013–1027. 10.1006/jmbi.1994.0196. [DOI] [PubMed] [Google Scholar]
- Rami B. R.; Udgaonkar J. B. Mechanism of Formation of a Productive Molten Globule Form of Barstar. Biochemistry 2002, 41 (6), 1710–1716. 10.1021/bi0120300. [DOI] [PubMed] [Google Scholar]
- Li H.; Frieden C. Comparison of C40/82A and P27A C40/82A Barstar Mutants Using 19F NMR. Biochemistry 2007, 46 (14), 4337–4347. 10.1021/bi6026083. [DOI] [PubMed] [Google Scholar]
- Sinha K. K.; Udgaonkar J. B. Barrierless evolution of structure during the submillisecond refolding reaction of a small protein. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (23), 7998–8003. 10.1073/pnas.0803193105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Chakraborty K.; Khatua P.; Bandyopadhyay S. Microscopic dynamics of water around unfolded structures of barstar at room temperature. J. Chem. Phys. 2015, 142 (5), 055102 10.1063/1.4907007. [DOI] [PubMed] [Google Scholar]
- Agashe V. R.; Udgaonkar J. B. Thermodynamics of Denaturation of Barstar: Evidence for Cold Denaturation and Evaluation of the Interaction with Guanidine Hydrochloride. Biochemistry 1995, 34 (10), 3286–3299. 10.1021/bi00010a019. [DOI] [PubMed] [Google Scholar]
- Khurana R.; Hate A. T.; Nath U.; Udgaonkar J. B. pH dependence of the stability of barstar to chemical and thermal denaturation. Protein Science: A Publication of the Protein Society 1995, 4 (6), 1133–1144. 10.1002/pro.5560040612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintrode P. L.; Griko Y. V.; Privalov P. L. Structural energetics of barstar studied by differential scanning microcalorimetry. Protein Sci. 1995, 4 (8), 1528–1534. 10.1002/pro.5560040810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadeřávek P.; Ferrage F.; Bodenhausen G.; Kurzbach D. High-Resolution NMR of Folded Proteins in Hyperpolarized Physiological Solvents. Chem. - Eur. J. 2018, 24 (51), 13418–13423. 10.1002/chem.201802885. [DOI] [PubMed] [Google Scholar]
- Wong K.-B.; Fersht A. R.; Freund S. M. V. NMR 15N relaxation and structural studies reveal slow conformational exchange in barstar C40/82A. J. Mol. Biol. 1997, 268 (2), 494–511. 10.1006/jmbi.1997.0989. [DOI] [PubMed] [Google Scholar]
- Mobli M.; Hoch J. C. Nonuniform sampling and non-Fourier signal processing methods in multidimensional NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 83 (Suppl. C), 21–41. 10.1016/j.pnmrs.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billeter M. Non-uniform sampling in biomolecular NMR. J. Biomol. NMR 2017, 68 (2), 65–66. 10.1007/s10858-017-0116-7. [DOI] [PubMed] [Google Scholar]
- Buckle A. M.; Schreiber G.; Fersht A. R. Protein-protein recognition: Crystal structural analysis of a barnase-barstar complex at 2.0-Å resolution. Biochemistry 1994, 33 (30), 8878–8889. 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- Lubienski M. J.; Bycroft M.; Freund S. M. V.; Fersht A. R. Three-dimensional solution structure and 13C assignments of barstar using nuclear magnetic resonance spectroscopy. Biochemistry 1994, 33 (30), 8866–8877. 10.1021/bi00196a003. [DOI] [PubMed] [Google Scholar]
- Jeener J.; Meier B. H.; Bachmann P.; Ernst R. R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71 (11), 4546–4553. 10.1063/1.438208. [DOI] [Google Scholar]
- Schwartz A. L.; Cutnell J. D. One- and two-dimensional NMR studies of exchanging amide protons in glutathione. J. Magn. Reson. (1969-1992) 1983, 53 (3), 398–411. 10.1016/0022-2364(83)90213-5. [DOI] [Google Scholar]
- Dobson C. M.; Lian L.-Y.; Redfield C.; Topping K. D. Measurement of hydrogen exchange rates using 2D NMR spectroscopy. J. Magn. Reson. (1969-1992) 1986, 69 (2), 201–209. 10.1016/0022-2364(86)90072-7. [DOI] [Google Scholar]
- Mori S.; Abeygunawardana C.; van Zijl P. C. M.; Berg J. M. Water Exchange Filter with Improved Sensitivity (WEX II) to Study Solvent-Exchangeable Protons. Application to the Consensus Zinc Finger Peptide CP-1. J. Magn. Reson., Ser. B 1996, 110 (1), 96–101. 10.1006/jmrb.1996.0015. [DOI] [PubMed] [Google Scholar]
- Brenner A. K.; Kieffer B.; Travé G.; Frøystein N. Å.; Raae A. J. Thermal stability of chicken brain α-spectrin repeat 17: a spectroscopic study. J. Biomol. NMR 2012, 53 (2), 71–83. 10.1007/s10858-012-9620-y. [DOI] [PubMed] [Google Scholar]
- Cohen G. B.; Ren R.; Baltimore D. Modular binding domains in signal transduction proteins. Cell 1995, 80 (2), 237–248. 10.1016/0092-8674(95)90406-9. [DOI] [PubMed] [Google Scholar]
- Pawson T. Protein modules and signalling networks. Nature 1995, 373 (6515), 573–580. 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- Moressis A.; Friedrich A. R.; Pavlopoulos E.; Davis R. L.; Skoulakis E. M. C. A Dual Role for the Adaptor Protein DRK in Drosophila Olfactory Learning and Memory. J. Neurosci. 2009, 29 (8), 2611–2625. 10.1523/JNEUROSCI.3670-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow N. A.; Zhang O.; Forman-Kay J. D.; Kay L. E. Comparison of the Backbone Dynamics of a Folded and an Unfolded SH3 Domain Existing in Equilibrium in Aqueous Buffer. Biochemistry 1995, 34 (3), 868–878. 10.1021/bi00003a021. [DOI] [PubMed] [Google Scholar]
- Tollinger M.; Skrynnikov N. R.; Mulder F. A. A.; Forman-Kay J. D.; Kay L. E. Slow Dynamics in Folded and Unfolded States of an SH3 Domain. J. Am. Chem. Soc. 2001, 123 (46), 11341–11352. 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- Bezsonova I.; Singer A.; Choy W.-Y.; Tollinger M.; Forman-Kay J. D. Structural Comparison of the Unstable drkN SH3 Domain and a Stable Mutant. Biochemistry 2005, 44 (47), 15550–15560. 10.1021/bi0512795. [DOI] [PubMed] [Google Scholar]
- Marsh J. A.; Neale C.; Jack F. E.; Choy W.-Y.; Lee A. Y.; Crowhurst K. A.; Forman-Kay J. D. Improved Structural Characterizations of the drkN SH3 Domain Unfolded State Suggest a Compact Ensemble with Native-like and Non-native Structure. J. Mol. Biol. 2007, 367 (5), 1494–1510. 10.1016/j.jmb.2007.01.038. [DOI] [PubMed] [Google Scholar]
- Mazouchi A.; Zhang Z.; Bahram A.; Gomes G.-N.; Lin H.; Song J.; Chan H. S.; Forman-Kay J. D.; Gradinaru C. C. Conformations of a Metastable SH3 Domain Characterized by smFRET and an Excluded-Volume Polymer Model. Biophys. J. 2016, 110 (7), 1510–1522. 10.1016/j.bpj.2016.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang O.; Kay L. E.; Olivier J. P.; Forman-Kay J. D. Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 1994, 4 (6), 845–858. 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- Zhang O.; Forman-Kay J. D. Structural Characterization of Folded and Unfolded States of an SH3 Domain in Equilibrium in Aqueous Buffer. Biochemistry 1995, 34 (20), 6784–6794. 10.1021/bi00020a025. [DOI] [PubMed] [Google Scholar]
- Farrow N. A.; Zhang O.; Forman-Kay J. D.; Kay L. E. A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J. Biomol. NMR 1994, 4 (5), 727–734. 10.1007/BF00404280. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Kay L. E. NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol. 2009, 5 (11), 808–814. 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Bouvignies G.; Kay L. E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134 (19), 8148–8161. 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- Ramachandran S.; Udgaonkar J. B. Stabilization of Barstar by Chemical Modification of the Buried Cysteines. Biochemistry 1996, 35 (26), 8776–8785. 10.1021/bi9600759. [DOI] [PubMed] [Google Scholar]
- Bhuyan A. K.; Udgaonkar J. B. Two structural subdomains of barstar detected by rapid mixing NMR measurement of amide hydrogen exchange. Proteins: Struct., Funct., Genet. 1998, 30 (3), 295–308. . [DOI] [PubMed] [Google Scholar]
- Gregory R. B.; Rosenberg A.. Protein conformational dynamics measured by hydrogen isotope exchange techniques. Methods in Enzymology; Academic Press, 1986; Vol. 131, pp 448–508. [DOI] [PubMed] [Google Scholar]
- Hvidt A.; Nielsen S. O.. Hydrogen Exchange in Proteins. In Advances in Protein Chemistry; Anfinsen C. B., Anson M. L., Edsall J. T., Richards F. M., Eds.; Academic Press, 1966; Vol. 21, pp 287–386. [DOI] [PubMed] [Google Scholar]
- Englander S. W.; Kallenbach N. R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983, 16 (4), 521–655. 10.1017/S0033583500005217. [DOI] [PubMed] [Google Scholar]
- Maier C. S.; Deinzer M. L.. Protein Conformations, Interactions, and H/D Exchange. In Methods in Enzymology; Academic Press, 2005; Vol. 402, pp 312–360. [DOI] [PubMed] [Google Scholar]
- Halle B. Cross-relaxation between macromolecular and solvent spins: The role of long-range dipole couplings. J. Chem. Phys. 2003, 119, 12372–85. 10.1063/1.1625632. [DOI] [Google Scholar]
- Persson F.; Halle B. How amide hydrogens exchange in native proteins. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10383–10388. 10.1073/pnas.1506079112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander S. W. Protein Folding Intermediates and Pathways Studied by Hydrogen Exchange. Annu. Rev. Biophys. Biomol. Struct. 2000, 29 (1), 213–238. 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- Hoofnagle A. N.; Resing K. A.; Ahn N. G. Protein Analysis by Hydrogen Exchange Mass Spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003, 32 (1), 1–25. 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- Krishna M. M. G.; Hoang L.; Lin Y.; Englander S. W. Hydrogen exchange methods to study protein folding. Methods 2004, 34 (1), 51–64. 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Jaswal S. S. Biological insights from hydrogen exchange mass spectrometry. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834 (6), 1188–1201. 10.1016/j.bbapap.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Pan J.; Liu Y.-H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40 (3), 1224–1234. 10.1039/C0CS00113A. [DOI] [PubMed] [Google Scholar]
- Woodward C.; Li R. The slow-exchange core and protein folding. Trends Biochem. Sci. 1998, 23 (10), 379. 10.1016/S0968-0004(98)01282-1. [DOI] [PubMed] [Google Scholar]
- Truhlar S. M. E.; Croy C. H.; Torpey J. W.; Koeppe J. R.; Komives E. A. Solvent Accessibility of Protein Surfaces by Amide H/2H Exchange MALDI-TOF Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17 (11), 1490–1497. 10.1016/j.jasms.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Eigen M. Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Part I: Elementary Processes. Angew. Chem., Int. Ed. Engl. 1964, 3 (1), 1–19. 10.1002/anie.196400011. [DOI] [Google Scholar]
- Molday R. S.; Kallen R. G. Substituent effects on amide hydrogen exchange rates in aqueous solution. J. Am. Chem. Soc. 1972, 94 (19), 6739–6745. 10.1021/ja00774a029. [DOI] [Google Scholar]
- Matthew J. B.; Richards F. M. The pH dependence of hydrogen exchange in proteins. J. Biol. Chem. 1983, 258 (5), 3039–3044. [PubMed] [Google Scholar]
- Anderson J. S.; Hernández G.; LeMaster D. M. A Billion-fold Range in Acidity for the Solvent-Exposed Amides of Pyrococcus furiosus Rubredoxin. Biochemistry 2008, 47 (23), 6178–6188. 10.1021/bi800284y. [DOI] [PubMed] [Google Scholar]
- LeMaster D. M.; Anderson J. S.; Hernández G. Peptide conformer acidity analysis of protein flexibility monitored by hydrogen exchange. Biochemistry 2009, 48 (39), 9256–9265. 10.1021/bi901219x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMaster D. M.; Anderson J. S.; Hernández G. Normal carbon acid referencing for protein amide hydrogen exchange. Magn. Reson. Chem. 2007, 45 (7), 601–604. 10.1002/mrc.2003. [DOI] [PubMed] [Google Scholar]
- Kim P. S.; Baldwin R. L. Influence of charge on the rate of amide proton exchange. Biochemistry 1982, 21 (1), 1–5. 10.1021/bi00530a001. [DOI] [PubMed] [Google Scholar]
- Tüchsen E.; Woodward C. Hydrogen kinetics of peptide amide protons at the bovine pancreatic trypsin inhibitor protein-solvent interface. J. Mol. Biol. 1985, 185 (2), 405–419. 10.1016/0022-2836(85)90412-7. [DOI] [PubMed] [Google Scholar]
- Delepierre M.; Dobson C. M.; Karplus M.; Poulsen F. M.; States D. J.; Wedin R. E. Electrostatic effects and hydrogen exchange behaviour in proteins: The pH dependence of exchange rates in lysozyme. J. Mol. Biol. 1987, 197 (1), 111–122. 10.1016/0022-2836(87)90613-9. [DOI] [PubMed] [Google Scholar]
- Fogolari F.; Esposito G.; Viglino P.; Briggs J. M.; McCammon J. A. pKa Shift Effects on Backbone Amide Base-Catalyzed Hydrogen Exchange Rates in Peptides. J. Am. Chem. Soc. 1998, 120 (15), 3735–3738. 10.1021/ja963133m. [DOI] [Google Scholar]
- Anderson J. S.; LeMaster D. M.; Hernández G. Electrostatic Potential Energy within a Protein Monitored by Metal Charge-Dependent Hydrogen Exchange. Biophys. J. 2006, 91 (11), L93–L95. 10.1529/biophysj.106.094219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández G.; Anderson J. S.; LeMaster D. M.. Electrostatics of Hydrogen Exchange for Analyzing Protein Flexibility. In Protein NMR Techniques; Shekhtman A., Burz D. S., Eds.; Humana Press: Totowa, NJ, 2012; pp 369–405. [DOI] [PubMed] [Google Scholar]
- Hernández G.; LeMaster D. M.. NMR Analysis of Native-State Protein Conformational Flexibility by Hydrogen Exchange. In Protein Structure, Stability, and Interactions; Shriver J. W., Ed.; Humana Press: Totowa, NJ, 2009; pp 285–310. [DOI] [PubMed] [Google Scholar]
- Anderson J. S.; Hernández G.; LeMaster D. M. Sidechain conformational dependence of hydrogen exchange in model peptides. Biophys. Chem. 2010, 151 (1), 61–70. 10.1016/j.bpc.2010.05.006. [DOI] [PubMed] [Google Scholar]
- McAllister R. G.; Konermann L. Challenges in the Interpretation of Protein H/D Exchange Data: A Molecular Dynamics Simulation Perspective. Biochemistry 2015, 54 (16), 2683–2692. 10.1021/acs.biochem.5b00215. [DOI] [PubMed] [Google Scholar]
- Bowen S.; Ardenkjær-Larsen J. H. Enhanced performance large volume dissolution-DNP. J. Magn. Reson. 2014, 240, 90–94. 10.1016/j.jmr.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Capozzi A.; Cheng T.; Boero G.; Roussel C.; Comment A. Thermal annihilation of photo-induced radicals following dynamic nuclear polarization to produce transportable frozen hyperpolarized 13C-substrates. Nat. Commun. 2017, 8, 15757. 10.1038/ncomms15757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi A.; Coi A.; Karlsson M.; Lerche M. H.; Ardenkjær-Larsen J. H.. Preparation of Radical-Free Hyperpolarized Water using Photo-induced non-persistent Radicals on a “SpinLab-like” dissolution-DNP Polarizer. 58th Experimental Nuclear Magnetic Resonance, Asilomar, US, 2017.
- Bermel W.; Bertini I.; Felli I. C.; Lee Y.-M.; Luchinat C.; Pierattelli R. Protonless NMR Experiments for Sequence-Specific Assignment of Backbone Nuclei in Unfolded Proteins. J. Am. Chem. Soc. 2006, 128 (12), 3918–3919. 10.1021/ja0582206. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Van Melckebeke H.; Brutscher B. Speeding Up Three-Dimensional Protein NMR Experiments to a Few Minutes. J. Am. Chem. Soc. 2006, 128 (28), 9042–9043. 10.1021/ja062025p. [DOI] [PubMed] [Google Scholar]
- Bermel W.; Bertini I.; Chill J.; Felli I. C.; Haba N.; Kumar M. V. V.; Pierattelli R. Exclusively Heteronuclear 13C-Detected Amino-Acid-Selective NMR Experiments for the Study of Intrinsically Disordered Proteins (IDPs). ChemBioChem 2012, 13 (16), 2425–2432. 10.1002/cbic.201200447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.