

Abstract

A variety of pathogenic bacteria can infect humans, and rapid species identification is crucial for the correct treatment. However, the identification process can often be time-consuming and depend on the cultivation of the bacterial pathogen(s). Here, we present a stand-alone, enzyme-free, optical DNA mapping assay capable of species identification by matching the intensity profiles of large DNA molecules to a database of fully assembled bacterial genomes (>10 000). The assay includes a new data analysis strategy as well as a general DNA extraction protocol for both Gram-negative and Gram-positive bacteria. We demonstrate that the assay is capable of identifying bacteria directly from uncultured clinical urine samples, as well as in mixtures, with the potential to be discriminative even at the subspecies level. We foresee that the assay has applications both within research laboratories and in clinical settings, where the time-consuming step of cultivation can be minimized or even completely avoided.
Keywords: nanofluidics, optical DNA mapping, diagnostics, bacteria, UTI
Technological advances in the past decades have resulted in a variety of biodiagnostic tests that have improved the way that infectious diseases are diagnosed and treated.1 Correct pathogen identification is of great importance to improve patient outcomes and can also help in limiting the spread of disease and in infection control.2 Traditionally, the diagnosis of bacterial infections has relied on phenotypic methods or techniques such as 16S rRNA gene sequencing and MALDI-TOF mass spectrometry, both of which are either expensive and/or require pathogen cultivation before analysis.3,4 Cultivation is a time-consuming and sometimes troublesome task, as some bacteria are not easy to cultivate.5,6 Yet, most clinical laboratories still rely on phenotypic methods.
Advances in sequencing technologies have opened up for the introduction of whole-genome sequencing (WGS) in healthcare.7 In the past decade, the use of WGS has started migrating into public health practice with epidemiological associations of nosocomial infections as one of the earliest applications.8 Even if promising approaches exist,9 the extensive preparation protocols including bacterial cultivation, in combination with high costs and complex analysis, have hampered the progression of sequencing-based methods into diagnostic tools in clinical practice.10 There is, thus, a need for new, faster, and less complicated diagnostics assays for the accurate identification of bacteria.
Optical DNA mapping (ODM) is an umbrella term for methods visualizing sequence-dependent patterns along stretched, single DNA molecules, typically ranging from 100 kb to 1 Mb in size.11 Stretching of the DNA is traditionally done either on modified glass surfaces12 or in nanofluidic channels,13 where the latter allows for high throughput and uniform stretching. Contrary to many forms of DNA sequencing, ODM can analyze long, single DNA fragments without the need for any prior DNA amplification. Multiple labeling strategies for producing the sequence-specific patterns have been developed, based either on enzymatic labeling14 or modulating DNA binding affinity.15 While enzymatic labeling requires extensive labeling schemes,14,16,17 including steps to wash and remove unbound fluorophores, affinity-based methods, such as competitive binding used here,18 offer a simple approach for DNA labeling.
Even if previous efforts have been made to identify bacteria using ODM,19−27 no general approach has been reported. Overall, previous studies lack general applicability or streamlined workflows, and they rely on cultivated bacterial samples. We present here a new, fast, cultivation-free bacterial identification assay based on ODM that includes both a novel DNA extraction protocol and a new data analysis strategy. Compared to our previous study,19 the approach presented here does not require any prior knowledge about the sample content, and the new extraction protocol is designed to work for both Gram-positive and Gram-negative bacteria. The new data analysis strategy is based on assessing the uniqueness of each mapped DNA molecule, to determine the presence of a bacterial species. As a result, the ODM assay, based on the competitive binding of netropsin and YOYO-1 to DNA,18 is capable of identifying bacterial species with high precision, both in mixtures and in uncultivated urine samples. Also, because our assay is based on the analysis of single bacterial DNA molecules, we avoid potential errors induced by DNA amplification.
Results and Discussion
In this study, we demonstrate the applicability of affinity-based ODM for identifying bacterial species from clinical isolates and mixtures, as well as directly from uncultivated samples from patients with urinary tract infections (Figure 1A). A strategy, based on classic pulsed-field gel electrophoresis (PFGE) embedding of intact bacteria in agarose plugs, was developed to prepare long, intact DNA molecules from a variety of bacteria for ODM analysis. Lysis of bacterial cells in the agarose plugs was performed with a single-step combination of lysozyme and lysostaphin to ensure lysis of both Gram-positive and Gram-negative bacteria. Proteinase K treatment and washing of the plugs ensured the removal of proteins and cell debris while keeping the DNA as intact as possible. Release of the long DNA fragments from the agarose plugs was done by gentle enzymatic degradation of the agarose with agarase.28 All of the steps were optimized to reduce the time from patient sample to pure DNA; the DNA purity was verified by standard spectroscopic methods (Nanodrop and Qubit), and the quality (i.e., the size of the extracted DNA molecules) was verified during the nanofluidic experiments. In total, the incubation times were shortened from 18 to 5 h with sufficient yield, purity, and integrity of the DNA for the ODM method for all of the tested bacterial species (see below). After preparation, the principle of the DNA labeling is based on that netropsin, which is a nonfluorescent molecule that binds specifically to AT base pairs,29 blocks these sites from the fluorescent YOYO-1, which renders an emission intensity profile where AT-rich regions will appear dark and GC-rich regions will appear bright.18,19
Figure 1.

Schematic overview of the optical DNA mapping assay. (A) Experimental outline. Bacteria are isolated and then lysed in agarose plugs to extract large (>100 kb) DNA molecules. The DNA is labeled with YOYO-1 and netropsin in a single step, creating a sequence-specific intensity profile along the DNA. To record the intensity profile, the DNA is confined in a nanofluidic channel and imaged using a fluorescence microscope. The resulting experimental intensity profiles are compared to a reference database, and the bacterial species present in the sample are identified based on profiles that match discriminatively to a single species in the database. (B) Data analysis pipeline. The time-averaged kymographs are matched to the reference database of theoretical intensity profiles generated from complete bacterial genomes. For each experimental intensity profile, the database matches are filtered as follows. First, short intensity profiles are discarded (length < Lmin). Then, the highest-scoring matches are selected (Cmax within the range max(Cmax) to max(Cmax) – Cdiff), and if all of the highest-scoring matches match to a single species, the intensity profile is classified as discriminative. Lastly, discriminative intensity profiles with sufficiently high-scoring matches (max(Cmax) > Cthresh) are reported back to the user. See Methods section for details of how the parameter space of Lmin, Cdiff, and Cthresh was explored, and see Figures 2 and 3 for the results.
The method operates by classifying intensity profiles as either discriminative or nondiscriminative on the species level (Figure 1B). Discriminative profiles are experimental intensity profiles where all high-quality matches against the reference database are to a single species. The accuracy of the methods is governed by three main parameters: Cdiff, Cthresh, and Lmin. In short, Cdiff and Cthresh determine which matches against the reference database are of sufficiently high quality, while Lmin sets the minimum acceptable profile length (see Methods section for full details). A low value of both Cthresh and Cdiff will increase the fraction of intensity profiles that are classified as discriminative, reducing the amount of required data (Figure 2A). However, the fraction of correct matches, i.e., discriminative profiles matching to the correct species, will decrease, increasing the risk for identifying the incorrect species (Figure 2B). On the other hand, a high value of both Cthresh and Cdiff will increase the required amount of data, because a large fraction of profiles will be discarded. The results showed that Cthresh does not affect the performance of the method to a large extent, unless it is set very high (Cthresh > 0.6). Because the fraction of correct matches approaches 100% for Cdiff > 0.05 with Cthresh fixed to 0.5 (Figure 3A), we decided to use a Cdiff = 0.05 and Cthresh = 0.5 for all subsequent analyses in this study. This maintained a high true positive rate, while not significantly reducing the throughput of the assay. It should, however, be noted that the choice of parameter values is dependent on the type of sample analyzed. In this study, we focused on human pathogens, which have an abundance of genome sequence data available that was used to generate the reference database of theoretical profiles. If the analyzed samples contained rare or even unknown species that are not well-represented in the reference database, more conservative values of Cdiff and Cthresh would likely be necessary to avoid false positives and achieve optimal performance.
Figure 2.

Effect of Cdiff and Cthresh on data quality and quantity. Heat maps showing fraction (%) of profiles found to be discriminative out of the total number of mapped molecules (A), and the true positive rate (TPR), i.e., the fraction (%) of the experimental profiles found to be discriminative to the correct species, out of the total number of discriminative profiles (B), as a function of Cdiff and Cthresh.
Figure 3.

Effect of Cdiff and fragment size on data quality and quantity. (A) Fraction (%) of experimental profiles found to be discriminative to the correct species out of the total number of discriminative profiles (solid line, dark green), and the fraction (%) of molecules found to be discriminative out of the total number of mapped molecules (dashed line, green), as a function of Cdiff (Cthresh fixed to 0.5). (B) The fraction (%) of the experimental molecules found to be discriminative to the correct species out of the total number of discriminative molecules (solid line, dark brown), and the fraction (%) of molecules found to be discriminative out of the total number of mapped molecules (dashed line, light brown), as a function of fragment size (Cdiff = 0.05, Cthresh = 0.5). One pixel corresponds to approximately 500 bp.
The size of the DNA molecules and, accordingly, the parameter Lmin, has a significant effect on the possibility to discriminate between species. To find the lower limit of DNA fragment size for which the ODM assay still functions reliably, an in silico simulation was performed by randomly sampling and cutting experimental profiles into fragments of lengths 100–600 pixels (approximately 50–300 kb, details in Methods section). The results revealed that profiles as small as 250 pixels (approximately 125 kb) yield the same true positive rate as that of longer fragments (Figure 3B). However, at even shorter fragment lengths, the performance dropped considerably. We, therefore, set the threshold for the minimum allowed length of a profile, Lmin, to 250 pixels. Furthermore, the percentage of molecules that were discriminative increased steadily with fragment size. Hence, fewer profiles are needed to make a reliable species identification, the longer the DNA molecules are.
As a first validation of the assay, we analyzed the DNA extracted from three different Escherichia coli (E. coli) isolates. Examples of matches between individual experimental and theoretical intensity profiles with a high degree of similarity (Cmax > 0.8) are shown in Figure 4. The same three intensity profiles are compared to their respective, best matching theoretical intensity profile of a non-E. coli species in Figure S2 in the Supporting Information. For the three E. coli isolates, a majority of the intensity profiles (77%) were discriminative, and all of them matched correctly to E. coli, demonstrating a high specificity.
Figure 4.

Results for E. coli isolates. Example fits of experimental intensity profiles (green) and their respective highest-scoring theoretical intensity profile (black) for each of the three E. coli isolates (sequence types 93, 10, and 131). The inner circle in the pie charts illustrates the species distribution in the analyzed sample, and the outer circle illustrates the obtained species distribution of the discriminative profiles (the exact number of discriminative profiles specified).
To evaluate the applicability of the assay for different bacterial species, five bacterial species relevant for urinary tract infections, both Gram-negative and Gram-positive, were analyzed: Klebsiella pneumoniae, Pseudomonas aeruginosa, Proteus mirabilis, Staphylococcus aureus, and Staphylococcus saprophyticus. For all of the species except S. saprophyticus, all of the discriminative profiles identified the correct species (Figure 5A). For S. saprophyticus, one of the seven discriminative intensity profiles matched incorrectly to Vibrio parahemolyticus. However, by requiring at least three discriminative intensity profiles for a species to consider that species present (details in Methods section), only the correct species was identified for all five isolates. Importantly, the same protocol for DNA extraction was used for both Gram-positive and Gram-negative bacteria, which is very important when analyzing unknown samples. Thus, these results demonstrate that the assay is general and can be used for a wide variety of bacterial species.
Figure 5.

Results for single-species samples and bacterial mixtures. The results obtained from single-species samples (A) and mixed samples (B, ratios specified beneath each chart), where each chart represents one sample. The inner circle illustrates the species distribution in the sample, and the outer circle illustrates the obtained species distribution of the discriminative profiles (with the exact number of discriminative profiles specified). Incorrect matches, i.e., profiles matching discriminatively to a species not present in the sample, are shown in gray.
Because each DNA molecule is analyzed individually, the assay is ideal for samples where multiple bacterial species are present. To illustrate this, five different mixes of bacteria were analyzed, varying both in the number of different species, and their ratios, and in the mixtures of Gram-positive and Gram-negative bacteria. We successfully identified all of the bacterial species present in all five mixes (Figure 5B), and only three single intensity profiles were found to be discriminative to an incorrect species. In the 25/25/25/25 mixture, one profile matched discriminatively to Burkholderia stagnalis and one to Corynebacterium diphtheriae, and in the 10/20/30/40 mixture, one profile matched discriminatively to Campylobacter jejuni. All of these incorrect species had no more than a single profile that matched discriminatively to them. Hence, given the threshold of at least three matching profiles, only the correct bacterial species were reported for all of the mixed samples.
Due to multiple factors, the assay presented here is, in its current form, not well suited to determine initial concentrations of bacteria in a sample or to specify ratios of bacteria in mixtures. These factors include differences in DNA extraction efficiency and genome size (a smaller genome yields a lower relative DNA concentration), degree of AT/GC sequence variation (resolution), and relative uniqueness of sequences in the database. With this in mind, the experimental results overlapped surprisingly well with the estimated ratios of bacteria in the mixtures (Figure 5B), based on bacterial concentration (CFU/mL). The results could potentially be improved by calibrating the assay for different bacterial species.
Because the ODM assay is a single-molecule-based technique, the amount of DNA needed to perform the analysis is as low as 10 picomoles (concentration ≥500 nM (bp)), and the amount of DNA used for the actual analysis is only approximately 10 attomoles (bp). The small amount of sample needed for analysis makes the method suitable for samples with low concentrations of bacteria, such as clinical samples, without the need to first cultivate the bacteria. As proof of concept, DNA was extracted directly from three different clinical urine samples from patients suffering from urinary tract infections. Following cultivation, bacterial-species identification was conducted with MALDI-TOF (Bruker Daltronics; Bremen, Germany), and the initial bacterial concentration was confirmed to be above 105 CFU/mL, which corresponds to the limit for the significant growth of bacterial pathogens in urine. Using the ODM assay, we were able to detect the correct bacterial species in all three samples (Figure 6).
Figure 6.

Noncultured urine samples. The inner circle illustrates the expected species distribution in each sample, and the outer circle illustrates the obtained species distribution of the discriminative profiles (the exact number of discriminative profiles indicated).
Importantly, potential contamination with human DNA molecules does not affect the results, because any large fragments of human DNA are unlikely to match discriminatively to any bacterial species. With the highly sensitive ODM assay, as with any culture-based method, there is a possibility that contaminating bacteria will give rise to false positive results. This is already a problem today in the clinical setting when using urine cultures, as low-level contamination with Gram-negative bacilli can complicate interpretation, along with asymptomatic bacteriuria. The correct way of addressing this issue is to focus on correct sampling and correct indication for UTI diagnostics. Moreover, we foresee that, with further optimized DNA extraction, the method could be used, for example, to identify bacteria in positive blood culture bottles and also, potentially, directly in cerebral spinal fluid.
Summarizing the data obtained for all of the samples of this study, 36% (344 out of 944) of the mapped DNA molecules were discriminative on the species level, and the remaining data were not used for the species identification. Out of the discriminative profiles, 99% (340 out of 344) matched the correct species, and 4 matched an incorrect species. By requiring a minimum of three discriminative profiles to confidently report a species as present in a sample, we achieved an accuracy of 100% for all of the samples. Even if they are rare, it is important to understand why incorrect discriminative matches appear. The fits between the four incorrectly matched intensity profiles, and their respective highest-scoring matches, show that they all have at least one very dominating feature, combined with an overall low-intensity variation across the profile, rendering a high Cmax even if the overall fit is rather poor (Figure S3 in the Supporting Information). The dominating features might, for example, be a result of knots in the DNA molecules, leading to local compaction of DNA and, thus, a brighter signal in these areas.30 If needed, preprocessing of the experimental data could potentially remove molecules displaying such features, increasing the specificity of the assay even further.
Another possible reason for incorrect matches is errors in the reference database, such as incorrect annotations or contamination. It should be noted that, by increasing Cdiff to 0.06, all incorrect matches were removed at the cost of fewer discriminative profiles. Importantly, even if we observed incorrect matches, we never had more than a single match to an incorrect species, making the incorrect matches easy to distinguish and discard. By requiring at least three profiles for the identification of a species, we achieved a correct species identification in all of the analyzed samples.
The vast majority of all of the mapped DNA molecules were >250 kb, with an average size of ∼350 kb. The fact that DNA molecules as short as ∼125 kb can be used to identify bacteria correctly, as shown in Figure 3B, is important. This means that it will also be possible to identify bacteria in samples where the DNA is significantly more fragmented than those in this study. Increased fragmentation can occur in dead bacteria and when using more harsh extraction protocols, for example, to speed up the assay even further.
We finally investigated the potential of using the mapped intensity profiles to discriminate also at the subspecies level by identifying the sequence type (ST) of three of the previously analyzed E. coli isolates. This is of high relevance as some STs, such as E. coli ST 131,31 display epidemic occurrence and, therefore, are clinically important to detect, not the least in complex microbial communities. We used the same method to determine whether the profiles were also discriminative on the sequence type level. Using the same parameter values, we were able to indicate the correct sequence types of all of the three isolates (Figure 7). We, therefore, foresee that, in the future, it should be possible to use the mapped intensity profiles to not only resolve the species of a present bacterium but also access subspecies information, such as clonal complexes and phylogroups. Moreover, plasmids, which are already present in the DNA extraction, could be mapped in the same experiment, enabling plasmid tracing in outbreak situations or resistance genes detection, as we have previously demonstrated in several different studies.32−38
Figure 7.

Results from the subspecies identification of the E. coli isolates. The inner circle in the pie charts illustrates the expected distribution of E. coli sequence types in each sample, and the outer circle illustrates the obtained distribution of profiles discriminative on the sequence type level (with the exact number of discriminative profiles specified). Note that only one discriminative fragment was obtained for the E. coli isolate belonging to ST10. This is below the required threshold of three discriminative fragments used at the species level.
To conclude, we have developed an affinity-based ODM assay capable of identifying bacteria with very high precision, not only in single cultures but also in mixtures, as well as directly in clinical urine samples. The presented DNA extraction protocol is general and works for both Gram-negative and Gram-positive bacteria. Moreover, our results suggest that the highly specific intensity profiles generated with the ODM assay, together with our new data analysis strategy, have the potential to be discriminative even at the subspecies level. At present, the lead time from the urine sample to the result is down to 8 h, and we anticipate that this can be substantially reduced when the process is fully automated. We foresee that the assay could have applications both within research laboratories as well as in clinical settings, where this methodology could complement time-consuming, cultivation-based methods.
Methods
Bacterial Samples
The bacteria used in the study were selected based on clinical relevance; for details see Table S1 in the Supporting Information. For the cultivated bacterial samples, the strains were stored in 10% DMSO stocks at −80 °C, plated on Luria–Bertani (LB) agar plates with 1.5% agar, and later grown in LB broth at 37 °C before DNA isolation. Mixes of strains were prepared in the same manner by growing separate cultures overnight and mixing relative amounts of each strain to achieve the selected ratios before DNA isolation. The noncultivated urine samples were collected at the Karolinska University Hospital in Stockholm and used directly for DNA isolation. Pseudoanonymized samples were shared with the researchers carrying out the ODM experiments, without sharing the key making patients identifiable. No informed consent was collected from patients, as per the ethical committee assessment (recordal 2018/2735-31/2).
DNA Isolation
The method used for DNA extraction was designed to obtain large-sized (>100 kb) DNA molecules for subsequent labeling and analysis. The DNA extraction was initially performed by method i, CHEF Genomic DNA kit from BIO-RAD, and later by method ii, a tailor-made extraction protocol, inspired by the work of Matushek et al.39 In short, for method i, an overnight culture of the bacteria was diluted 100-fold and allowed to grow until it reached an OD600 of 0.8–1.0. For each milliliter of agarose plugs, 5 × 108 cells were centrifuged. For the noncultivated samples, 1–3 mL of urine was centrifuged. The bacterial pellet was resuspended in a cell suspension buffer, combined with 2% CleanCut agarose (50 °C), and cast into plug molds. The plugs were incubated in lysozyme buffer for 2 h at 37 °C, rinsed with sterile water, and incubated overnight in Proteinase K reaction buffer at 50 °C. The next day, the plugs were washed four times for 1 h in a 1× wash buffer at room temperature with gentle agitation. The plugs were stored in wash buffer at 4 °C until further use. For this method, all of the buffers used were premade by the kit manufacturer (BIO-RAD). For method ii, 250 μL of overnight culture or 1–3 mL of a noncultivated urine sample was spun down and the pellet was resuspended in 50 μL of 2× lysis buffer (1× lysis buffer = 6 mM Tris HCL pH 7.4, 1 M NaCl, 10 mM EDTA pH 7.5, 0.5% Brij, 0.2% deoxycholate, and 0.5% sodium lauryl sarcosine), with 1 mg/mL lysozyme, 20 mg/mL RNase A, and 100 μg/mL lysostaphin added fresh on the day of the experiment; this was mixed with 50 μL of 1.6% low-melting-point agarose (50 °C) and allowed to solidify in a plug mold. The plug was incubated in 300 μL of 1× lysis buffer at 37 °C for 2 h. Next, the plug was incubated in 300 μL of EPS solution (10 mM Tris HCL pH 7.4, 1 mM EDTA), including 100 μg/mL proteinase K and 1% sodium dodecyl sulfate, which was added fresh on the day of the experiment, at 50 °C for 1 h. Finally, all of the residual EPS solution was discarded, and the plug was incubated in TE buffer (10 mM Tris HCL pH 7.4, 0.1 mM EDTA) at 50 °C for 1 h before storage at 4 °C. Method ii is effective for both Gram-negative and Gram-positive bacteria and reduces the overall time for DNA extraction by almost a factor of five. There was no notable difference in the quality of the extracted DNA when using extraction methods i or ii.
The agarose plugs (100 μL) were melted in 20 μL of 10× CutSmart Buffer (New England Biolabs) and 78 μL of MQ-water at 70 °C for 10 min, followed by incubation at 42 °C for 10 min, prior to the addition of 2 μL of agarase (ThermoFisher Scientific, 0.5 U/L) and a second incubation at 42 °C for at least 1 h. The DNA concentration was determined using a Qubit Fluorometer 2.0 (ThermoFisher Scientific).
Sample Preparation and Nanofluidic Experiments
The sequence-based intensity profiles for the ODM experiments were created by the addition of YOYO-1 (excitation of 491 nm/emission of 509 nm, Invitrogen) and netropsin (Sigma-Aldrich).18 A 0.5× TBE (Tris-Borate-EDTA, Medicago, 10 μL) solution was prepared with 1 μM (base pairs) extracted bacterial DNA, 1 μM (base pairs) λ-DNA (included as an internal size reference, 48 502 bp, Roche Biochem Reagents), 0.2 μM YOYO-1 (ratio of DNA/YOYO is 10:1), and 60 μM netropsin (ratio of netropsin/YOYO is 300:1), followed by incubation at 50 °C for 30 min. Next, the DNA solution was diluted by a factor of 10 with 88 μL of MQ-water and 2 μL of β-mercaptoethanol (used to prevent photodamaging, Sigma-Aldrich), obtaining a final buffer concentration of 0.05× TBE.
To record the intensity profiles, the DNA fragments were confined in nanofluidic channels and imaged using a fluorescence microscope. The nanofluidic experiments were performed using 500 μm long nanochannels with a cross section of 100 × 150 nm2 (height × width) (see Figure S1 in the Supporting Information), fabricated in silica utilizing standard methods.40 The nanochannels were spanned by two microchannels, which were connected to two loading wells each. For each sample, 10 μL (1 picomole, 100 nM, bacterial DNA) of the prepared DNA sample was loaded onto the chip, and the DNA was forced into the nanochannels using pressure-driven N2 flow. The DNA was imaged using a fluorescence microscope (Zeiss AxioObserver.Z1) equipped with a 63× (1.6× optovar) oil immersion objective (NA = 1.46, Zeiss) and an Andor iXon EMCCD camera. For each DNA molecule, 50 frames were acquired using 100 ms exposure.
Data Analysis
The processing of output data from the nanofluidics-based ODM fluorescence imaging experiments was divided into three main parts: (i) generation and time averaging of kymographs to generate intensity profiles, (ii) comparison of the experimental intensity profiles to a reference database of theoretical intensity profiles, and (iii) identification of intensity profiles that were discriminative on the species level (Figure 1B).
The first part converts an imaging
output (movie of up to 50 time frames) to a kymograph, the steps for
which are explained in detail in the Supporting Information of a previous study.28 The kymographs were used to generate time averages (intensity profiles).
In the second part, all of the experimental intensity profiles from
a sample were compared with a reference database of theoretical intensity
profiles. The database was based on all of the complete bacterial
genomes in RefSeq (as of October 16, 2018), excluding sequences shorter
than 500 kb or with the word “plasmid” in their FASTA
headers. In total, the resulting reference database consisted of theoretical
intensity profiles based on 10 310 sequences belonging to 2355
different bacterial species. Theoretical intensity profiles were generated
as described in a previous study41 and
stretched to the measured nanometer/base pair ratio, as described
previously.28 In the comparison, each experimental
intensity profile, i, was matched against each theoretical
intensity profile, j, using every possible start
position, k, in the theoretical profile, and match
scores, Ci,j,k, were calculated using the Pearson correlation
coefficient. For each combination of experimental and theoretical
intensity profiles, the following information was saved for the highest-scoring
match: match score (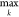 Ci,j,k = Cmax), start position
in the theoretical profile (k), length of the experimental
profile, and stretch factor.
Ci,j,k = Cmax), start position
in the theoretical profile (k), length of the experimental
profile, and stretch factor.
In the third part (Figure 1B), the Cmax scores were used to identify intensity profiles that were
discriminative on the species level in the following way. The analysis
results depend on the settings of three parameters, which are described
below: Cdiff, Lmin, and Cthresh. First, all of the experimental
intensity profiles shorter than a set threshold, Lmin, were removed from further analysis. Then, considering
one experimental intensity profile at the time, we identified high-quality
matches against the reference database by discarding all of the matches
against theoretical intensity profiles with a Cmax score more than the Cdiff value
lower than the theoretical intensity profile with the highest score
( Ci,j,k). Next, an experimental profile was classified
as discriminative at the species level if the following two criteria
were met: (a) all remaining high-quality matches were against theoretical
profiles belonging to a single species and (b) the best match had
a Cmax score above a set threshold, Cthresh. From a set of experimental profiles,
the species distribution of the discriminative profiles was reported.
All of the other profiles were discarded as they were classed as noninformative.
Ci,j,k). Next, an experimental profile was classified
as discriminative at the species level if the following two criteria
were met: (a) all remaining high-quality matches were against theoretical
profiles belonging to a single species and (b) the best match had
a Cmax score above a set threshold, Cthresh. From a set of experimental profiles,
the species distribution of the discriminative profiles was reported.
All of the other profiles were discarded as they were classed as noninformative.
Because there is a risk for false positives, i.e., intensity profiles that are discriminative but to an incorrect species, a threshold was implemented for the minimum number of intensity profiles required before confidently identifying a bacterial species as present in a sample. Out of all of the DNA molecules mapped in this study, only 0.4% were classified as false positives. By requiring at least three intensity profiles that are discriminative to the same species to identifying the species as present, the average number of mapped DNA molecules required to state the presence of an incorrect species in the sample, under the assumption of independence, is approximately 100 000. To set a strict threshold, considering that typically fewer than 100 DNA molecules were mapped per isolate in this study, any identification of a species by fewer than three discriminative profiles was deemed unreliable.
To test the effect of different parameter values, the true positive rate, i.e., the proportion of the discriminative profiles that were discriminative to the correct species, as well as the proportion of discriminative profiles out of all of the measured profiles, was tested using different values of the parameters Cdiff (range 0.01–0.1, step length 0.01) and Cthresh (range 0.3–0.7, step length 0.05). One sample for each of the species included in this study was used for the parameter evaluation to avoid any species-specific bias: isolates EC3, KP1, PA1, PM1, SA, and SS (see Table S1 in the Supporting Information).
To evaluate
the sensitivity of the assay to the size of the DNA molecules and,
by extension, the effect of the Lmin parameter,
experimental intensity profiles were randomly cut in silico into fragments of a specified length using the same samples as those
used for the parameter evaluation. We generated fragments of lengths
100–600 pixels, in 50-pixel intervals. To generate the fragments,
we used bootstrapping, i.e., random sampling with replacement, by
first counting the number of possible fragments, Ki, for each intensity profile. The probability
for selecting an experimental intensity profile i then becomes 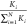 . We used MATLAB’s
command randsample() to pick an experimental
profile i from this probability. Finally, a subsample
of the specified length from the randomly drawn intensity profile
was randomly selected based on a uniform distribution [MATLAB’s randi()]. For each included sample and fragment length,
a set of 100 (not necessarily distinct) fragments was generated. The
cut fragments were analyzed in terms of the true positive rate and
the proportion of discriminative profiles using the parameters selected
after the parameter evaluation of Cdiff and Cthresh.
. We used MATLAB’s
command randsample() to pick an experimental
profile i from this probability. Finally, a subsample
of the specified length from the randomly drawn intensity profile
was randomly selected based on a uniform distribution [MATLAB’s randi()]. For each included sample and fragment length,
a set of 100 (not necessarily distinct) fragments was generated. The
cut fragments were analyzed in terms of the true positive rate and
the proportion of discriminative profiles using the parameters selected
after the parameter evaluation of Cdiff and Cthresh.
Acknowledgments
This work was supported by the EU Horizon 2020 program BeyondSeq (Grant 634890, given to F.W.), the Erling Persson Family Foundation (supporting C.G.G., T.A., L.S., F.W., and E.K.), the Swedish Research Council (Grant 2014-4305, given to T.A.), and the Crafoord Foundation (Grant 20180675, given to T.A.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.9b00464.
Overview of bacterial isolates, schematic overview of the design of the nanofluidic chip, best match of non-E. coli species, and fits of intensity profiles that match discriminatively to incorrect species (PDF)
Author Contributions
◆ V.M., M.N., and A.J. contributed equally to this work
The authors declare no competing financial interest.
Supplementary Material
References
- Giljohann D. A.; Mirkin C. A. (2009) Drivers of biodiagnostic development. Nature 462 (7272), 461–4. 10.1038/nature08605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley S. O. (2017) What Are Clinically Relevant Levels of Cellular and Biomolecular Analytes?. ACS sensors 2 (2), 193–197. 10.1021/acssensors.6b00691. [DOI] [PubMed] [Google Scholar]
- Chakravorty S.; Helb D.; Burday M.; Connell N.; Alland D. (2007) A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69 (2), 330–9. 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal N.; Kumar M.; Kanaujia P. K.; Virdi J. S. (2015) MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Front. Microbiol. 6, 791. 10.3389/fmicb.2015.00791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd K. G.; Steen A. D.; Ladau J.; Yin J.; Crosby L. (2018) Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 3 (5), e00055-18 10.1128/mSystems.00055-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenollar F.; Raoult D. (2007) Molecular diagnosis of bloodstream infections caused by non-cultivable bacteria. Int. J. Antimicrob. Agents 30, 7–15. 10.1016/j.ijantimicag.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Berg J. S.; Khoury M. J.; Evans J. P. (2011) Deploying whole genome sequencing in clinical practice and public health: meeting the challenge one bin at a time. Genet. Med. 13 (6), 499–504. 10.1097/GIM.0b013e318220aaba. [DOI] [PubMed] [Google Scholar]
- Besser J.; Carleton H. A.; Gerner-Smidt P.; Lindsey R. L.; Trees E. (2018) Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 24 (4), 335–341. 10.1016/j.cmi.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwkamp T. A.; Thair S.; Rosen M. J.; Blair L.; Lindner M. S.; Vilfan I. D.; Kawli T.; Christians F. C.; Venkatasubrahmanyam S.; Wall G. D.; Cheung A.; Rogers Z. N.; Meshulam-Simon G.; Huijse L.; Balakrishnan S.; Quinn J. V.; Hollemon D.; Hong D. K.; Vaughn M. L.; Kertesz M.; Bercovici S.; Wilber J. C.; Yang S. (2019) Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease. Nature microbiology 4 (4), 663–674. 10.1038/s41564-018-0349-6. [DOI] [PubMed] [Google Scholar]
- Rossen J. W. A.; Friedrich A. W.; Moran-Gilad J. (2018) Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect. 24 (4), 355–360. 10.1016/j.cmi.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Levy-Sakin M.; Ebenstein Y. (2013) Beyond sequencing: optical mapping of DNA in the age of nanotechnology and nanoscopy. Curr. Opin. Biotechnol. 24 (4), 690–8. 10.1016/j.copbio.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Neely R. K.; Deen J.; Hofkens J. (2011) Optical mapping of DNA: single-molecule-based methods for mapping genomes. Biopolymers 95 (5), 298–311. 10.1002/bip.21579. [DOI] [PubMed] [Google Scholar]
- Tegenfeldt J. O.; Prinz C.; Cao H.; Chou S.; Reisner W. W.; Riehn R.; Wang Y. M.; Cox E. C.; Sturm J. C.; Silberzan P.; Austin R. H. (2004) The dynamics of genomic-length DNA molecules in 100-nm channels. Proc. Natl. Acad. Sci. U. S. A. 101 (30), 10979–83. 10.1073/pnas.0403849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo K.; Dhingra D. M.; Odijk T.; de Pablo J. J.; Graham M. D.; Runnheim R.; Forrest D.; Schwartz D. C. (2007) A single-molecule barcoding system using nanoslits for DNA analysis. Proc. Natl. Acad. Sci. U. S. A. 104 (8), 2673–8. 10.1073/pnas.0611151104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller V.; Westerlund F. (2017) Optical DNA mapping in nanofluidic devices: principles and applications. Lab Chip 17 (4), 579–590. 10.1039/C6LC01439A. [DOI] [PubMed] [Google Scholar]
- Das S. K.; Austin M. D.; Akana M. C.; Deshpande P.; Cao H.; Xiao M. (2010) Single molecule linear analysis of DNA in nano-channel labeled with sequence specific fluorescent probes. Nucleic Acids Res. 38 (18), e177 10.1093/nar/gkq673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E. T.; Hastie A.; Lin C.; Ehrlich D.; Das S. K.; Austin M. D.; Deshpande P.; Cao H.; Nagarajan N.; Xiao M.; Kwok P. Y. (2012) Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 30 (8), 771–6. 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg L. K.; Persson F.; Berg J.; Bergstrom J.; Fransson E.; Olsson L.; Persson M.; Stalnacke A.; Wigenius J.; Tegenfeldt J. O.; Westerlund F. (2012) A single-step competitive binding assay for mapping of single DNA molecules. Biochem. Biophys. Res. Commun. 417 (1), 404–8. 10.1016/j.bbrc.2011.11.128. [DOI] [PubMed] [Google Scholar]
- Nilsson A. N.; Emilsson G.; Nyberg L. K.; Noble C.; Stadler L. S.; Fritzsche J.; Moore E. R.; Tegenfeldt J. O.; Ambjornsson T.; Westerlund F. (2014) Competitive binding-based optical DNA mapping for fast identification of bacteria--multi-ligand transfer matrix theory and experimental applications on Escherichia coli. Nucleic Acids Res. 42 (15), e118 10.1093/nar/gku556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand N. O.; Smith D. A.; Wilkinson A. A.; Rushton A. E.; Busby S. J. W.; Styles I. B.; Neely R. K. (2019) DNA barcodes for rapid, whole genome, single-molecule analyses. Nucleic Acids Res. 47 (12), e68 10.1093/nar/gkz212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protozanova E.; Zhang M.; White E. J.; Mollova E. T.; Broeck D. T.; Fridrikh S. V.; Cameron D. B.; Gilmanshin R. (2010) Fast high-resolution mapping of long fragments of genomic DNA based on single-molecule detection. Anal. Biochem. 402 (1), 83–90. 10.1016/j.ab.2010.03.024. [DOI] [PubMed] [Google Scholar]
- Sabirova J. S.; Xavier B. B.; Ieven M.; Goossens H.; Malhotra-Kumar S. (2014) Whole genome mapping as a fast-track tool to assess genomic stability of sequenced Staphylococcus aureus strains. BMC Res. Notes 7, 704. 10.1186/1756-0500-7-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S. K.; Pantrang M.; Stahl B.; Briska A. M.; Stemper M. E.; Wagner T. K.; Zentz E. B.; Callister S. M.; Lovrich S. D.; Henkhaus J. K.; Dykes C. W. (2012) Comparative whole-genome mapping to determine Staphylococcus aureus genome size, virulence motifs, and clonality. J. Clin Microbiol 50 (11), 3526–3533. 10.1128/JCM.01168-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan W. R.; Briska A.; Stahl B.; Wagner T. K.; Zentz E.; Henkhaus J.; Lovrich S. D.; Agger W. A.; Callister S. M.; DuChateau B.; Dykes C. W. (2010) Use of optical mapping to sort uropathogenic Escherichia coli strains into distinct subgroups. Microbiology (London, U. K.) 156 (7), 2124–35. 10.1099/mic.0.033977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotewicz M. L.; Jackson S. A.; LeClerc J. E.; Cebula T. A. (2007) Optical maps distinguish individual strains of Escherichia coli O157: H7. Microbiology (London, U. K.) 153 (6), 1720–33. 10.1099/mic.0.2006/004507-0. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Savarino S. J.; Venkatesan M. M. (2006) Subtractive hybridization and optical mapping of the enterotoxigenic Escherichia coli H10407 chromosome: isolation of unique sequences and demonstration of significant similarity to the chromosome of E. coli K-12. Microbiology (London, U. K.) 152 (4), 1041–54. 10.1099/mic.0.28648-0. [DOI] [PubMed] [Google Scholar]
- Bouwens A.; Deen J.; Vitale R.; D’Huys L.; Goyvaerts V.; Descloux A.; Borrenberghs D.; Grussmayer K.; Lukes T.; Camacho R.; Su J.; Ruckebusch C.; Lasser T.; Van De Ville D.; Hofkens J.; Radenovic A.; Frans Janssen K. P. (2020) Identifying Microbial Species by Single-Molecule DNA Optical Mapping and Resampling Statistics. NAR Genomics and Bioinformatics 2 (1), 1. 10.1093/nargab/lqz007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller V.; Dvirnas A.; Andersson J.; Singh V.; KK S.; Johansson P.; Ebenstein Y.; Ambjörnsson T.; Westerlund F. (2019) Enzyme-free optical DNA mapping of the human genome using competitive binding. Nucleic Acids Res. 47, e89. 10.1093/nar/gkz489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer C.; Marck C.; Schneider C.; Guschlbauer W. (1979) Influence of nucleotide sequence on dA.dT-specific binding of Netropsin to double stranded DNA. Nucleic Acids Res. 6 (8), 2831–2837. 10.1093/nar/6.8.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifenberger J. G.; Dorfman K. D.; Cao H. (2015) Topological events in single molecules of E. coli DNA confined in nanochannels. Analyst 140 (14), 4887–4894. 10.1039/C5AN00343A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. R.; Porter S.; Thuras P.; Castanheira M. (2017) The Pandemic H30 Subclone of Sequence Type 131 (ST131) as the Leading Cause of Multidrug-Resistant Escherichia coli Infections in the United States (2011–2012). Open Forum Infect Dis 4 (2), 089–089. 10.1093/ofid/ofx089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblom A.; Kk S.; Muller V.; Oz R.; Sandstrom H.; Ahren C.; Westerlund F.; Karami N. (2019) Interspecies plasmid transfer appears rare in sequential infections with extended-spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae. Diagn. Microbiol. Infect. Dis. 93, 380. 10.1016/j.diagmicrobio.2018.10.014. [DOI] [PubMed] [Google Scholar]
- Johnning A.; Karami N.; Tang Hallback E.; Muller V.; Nyberg L.; Buongermino Pereira M.; Stewart C.; Ambjornsson T.; Westerlund F.; Adlerberth I.; Kristiansson E. (2018) The resistomes of six carbapenem-resistant pathogens - a critical genotype-phenotype analysis. Microbial genomics 4 (11), 1. 10.1099/mgen.0.000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg L. K.; Quaderi S.; Emilsson G.; Karami N.; Lagerstedt E.; Muller V.; Noble C.; Hammarberg S.; Nilsson A. N.; Sjoberg F.; Fritzsche J.; Kristiansson E.; Sandegren L.; Ambjornsson T.; Westerlund F. (2016) Rapid identification of intact bacterial resistance plasmids via optical mapping of single DNA molecules. Sci. Rep. 6, 30410. 10.1038/srep30410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller V.; Rajer F.; Frykholm K.; Nyberg L. K.; Quaderi S.; Fritzsche J.; Kristiansson E.; Ambjörnsson T.; Sandegren L.; Westerlund F. (2016) Direct identification of antibiotic resistance genes on single plasmid molecules using CRISPR/Cas9 in combination with optical DNA mapping. Sci. Rep. 6, 37938. 10.1038/srep37938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torche P. C.; Muller V.; Westerlund F.; Ambjornsson T. (2017) Noise reduction in single time frame optical DNA maps. PLoS One 12 (6), e0179041 10.1371/journal.pone.0179041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller V.; Karami N.; Nyberg L. K.; Pichler C.; Torche Pedreschi P. C.; Quaderi S.; Fritzsche J.; Ambjörnsson T.; Åhrén C.; Westerlund F. (2016) Rapid Tracing of Resistance Plasmids in a Nosocomial Outbreak Using Optical DNA Mapping. ACS Infect. Dis. 2 (5), 322–328. 10.1021/acsinfecdis.6b00017. [DOI] [PubMed] [Google Scholar]
- Bikkarolla S. K.; Nordberg V.; Rajer F.; Muller V.; Kabir M. H.; KK S.; Dvirnas A.; Ambjornsson T.; Giske C. G.; Naver L.; Sandegren L.; Westerlund F. (2019) Optical DNA mapping combined with Cas9-targeted resistance gene identification for rapid tracking of resistance plasmids in a neonatal intensive care unit outbreak. mBio 10 (4), e00347–19 10.1128/mBio.00347-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matushek M. G.; Bonten M. J.; Hayden M. K. (1996) Rapid preparation of bacterial DNA for pulsed-field gel electrophoresis. J. Clin Microbiol 34 (10), 2598–600. 10.1128/JCM.34.10.2598-2600.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson F.; Tegenfeldt J. O. (2010) DNA in nanochannels-directly visualizing genomic information. Chem. Soc. Rev. 39 (3), 985–999. 10.1039/b912918a. [DOI] [PubMed] [Google Scholar]
- Dvirnas A.; Pichler C.; Stewart C. L.; Quaderi S.; Nyberg L. K.; Müller V.; Kumar Bikkarolla S.; Kristiansson E.; Sandegren L.; Westerlund F.; Ambjörnsson T. (2018) Facilitated sequence assembly using densely labeled optical DNA barcodes: A combinatorial auction approach. PLoS One 13 (3), e0193900 10.1371/journal.pone.0193900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.