

Abstract

We describe the photophysical processes that give rise to thermally activated delayed fluorescence in the excited state intramolecular proton transfer (ESIPT) molecule, triquinolonobenzene (TQB). Using transient absorption and time-resolved photoluminescence spectroscopy, we fully characterize prompt and delayed emission, phosphorescence, and oxygen quenching to reveal the reverse intersystem crossing mechanism (rISC). After photoexcitation and rapid ESIPT to the TQB-TB tautomer, emission from S1 is found to compete with thermally activated ISC to an upper triplet state, T2, very close in energy to S1 and limiting photoluminescence quantum yield. T2 slowly decays to the lowest triplet state, T1, via internal conversion. In the presence of oxygen, T2 is quenched to the ground state of the double proton transferred TQB-TC tautomer. Our measurements demonstrate that rISC in TQB occurs from T2 to S1 driven by thermally activated reverse internal conversion from T1 to T2 and support recent calculations by Cao et al. (Cao Y.; Eng J.; Penfold T. J.. Excited State Intramolecular Proton Transfer Dynamics for Triplet Harvesting in Organic Molecules. J. Phys. Chem. A 2019, 123, 2640−2649).
Thermally activated delayed fluorescence (TADF) materials have proven to be highly efficient emitters for electroluminescent organic light-emitting diodes (OLEDs).2−4 Electronic excitation in OLEDs gives rise to singlet and triplet excitonic states in a 25:75 ratio. Emissive decay from the triplet state is normally forbidden by spin conservation though, severely limiting the quantum efficiency of electroluminescence. Mechanisms through which triplet excited states can be harvested for emission and thus circumvent this limit have therefore attracted great interest. Heavy metal complex emitters enjoy rapid intersystem crossing (ISC) between the triplet and singlet states, with strong spin–orbit coupling of the heavy atom resulting in spin-mixing and emission directly from the triplet state.5−7 Purely organic emitters exhibiting TADF provide an alternate means of achieving almost 100% internal quantum efficiency, without necessitating the use of rare heavy metals.8−11 In the TADF process, triplets undergo reverse intersystem crossing (rISC) back to the singlet state with the assistance of thermal energy, followed by radiative decay back to the ground state. Recent work on TADF molecules has revealed the importance of intermediate molecular structures via conformational changes, providing an additional triplet excited state. ESIPT systems also provide the multiple intermediate structures that should accelerate RISC.
To maximize the rate of rISC (and achieve efficient triplet harvesting), the energy gap between the triplet and singlet state (ΔEST) must be minimized. Recent research has revealed that a low ΔEST and efficient rISC can be achieved by several molecular design strategies,12−14 with the most successful materials exhibiting either a donor–acceptor (D–A) or a donor–acceptor–donor (D–A–D) structure and possessing strong intramolecular charge transfer character.15−19 The mechanism underpinning high-efficiency TADF in these systems was found to be a second-order spin-vibronic coupling between the 1CT and 3CT charge transfer states, which is mediated by a locally excited 3LE triplet state.20,21 Minimizing the ΔEST between the 3LE and CT states accelerates rISC, demonstrating the potential for all energy states to impact TADF efficiency.17,22
Recently, a new category of TADF emitter has emerged in which a small ΔEST is achieved without the need for a D–A system. Here, the necessary HOMO/LUMO separation is achieved by excited state intramolecular proton transfer (ESIPT), a photoisomerization process that produces a characteristic large Stokes shift. Typically, upon photoexcitation of an enol ground state [see below for clarification of the nomenclature (structures in Figure 1)], electronic charge redistribution leads to intramolecular proton transfer over femtosecond to picosecond time scales, from the proton donor (here N–H) to the proton acceptor (here C=O), even at low temperatures. As a result, the singlet excited state keto forms contain six-membered cyclized cis-ketimine rings, stabilized by intramolecular hydrogen bonding. The molecular geometry of the keto tautomers is therefore significantly different from that of the enol form, generating the large Stokes shift.
Figure 1.

Chemical structures of hexyl-TQB (triquinolonobenzene) as well as the parent TQB ground state and excited states following ESIPT. Following naming conventions for other ESIPT materials (and focusing on the nitrogen proton donor), we call the ground state the enol form (actually enamine) and the excited states keto forms (actually ketimines).
(Here we have chosen to continue established naming conventions23−25 for the TQB proton transfer tautomers, which rely on analogy to well-studied enol–keto tautomers in organic compounds such as acetaldehyde26 and acetone.27 However, the simultaneous focus on the amine proton, the presence of a ketone acceptor, and borrowing of enol–keto nomenclature can lead to some confusion. Indeed, in Figure 1 what we classify as the “enol form” formally has three keto groups and no true enol at all. The functional group of interest is in reality an enamine. Similarly, what we classify as the “keto forms” lose their actual keto groups and gain true enol groups during ESIPT. The functional group of interest in this form is more correctly a ketimine.)
Small ESIPT molecules demonstrating TADF have been reported as attractive materials for OLED emitters, sparking new interest in this molecular design. The spectral sensitivity of ESIPT materials to changes in the surrounding medium also allows them to also be used in imaging and as fluorescent probes.23,24,32−34 In contrast to D–A(−D) materials, while rISC and TADF have been reported in several ESIPT materials no definitive experimental evidence for the underlying mechanism has been forthcoming.28−31 Additionally, the utilization of ESIPT emitters in OLEDs remains a challenge due to their limitations of low photoluminescence quantum yield and concentration quenching of the excited keto state. Understanding the rISC mechanism and factors impacting emission efficiency is crucial to enable rational design of future ESIPT emitters.
Despite these challenges, recent developments in ESIPT TADF materials have been rapid. A series of blue imidazole-based ESIPT crystal emitters have been reported with a moderate photoluminescence efficiency of 52% due to optimization of the steric configuration to minimize nonradiative relaxation.29 Novel ESIPT chromophores constituting polymeric and dendrimeric structures have also been reported with highly efficient fluorescence emission unaffected by keto form concentration quenching.35,36 A series of ESIPT molecules based on 2-(2′-hydroxyphenyl)oxazole were developed by You et al., in which TADF from the enol form was reported.37 However, in all cases, no unambiguous proof of the mechanism underpinning rISC and TADF has been given.
One particular ESIPT TADF material that has drawn great interest is triquinolonobenzene (TQB), in which the ESIPT is unhindered in the solid state because of the planarity of the molecule, resulting in small structural changes during proton transfer.38Figure 1 shows a scheme of the ESIPT process and tautomeric structures of TQB in the ground and excited states. TQB emits delayed fluorescence (DF) at room temperature with moderate total photoluminescence quantum yields of 55% in bis[2-(diphenylphosphino)phenyl]ether oxide (DPEPO) host films, while external quantum efficiencies of ≤14% have been obtained in OLEDs using 2,8-bis(diphenylphosphoryl)dibenzo[b,d]furan (PPF) or 9-(4-tert-butylphenyl)-3,6-bis(triphenylsilyl)-9H-carbazole (CzSi) host emissive layers.38 Nonetheless, it was discovered that slow rISC in these devices resulted in long exciton lifetimes, leading to quenching by singlet–triplet and triplet–triplet annihilation and large efficient roll-off. A deeper understanding of the transitions occurring between the TQB excited states is crucial to optimizing exciton dynamics, enhancing rISC, and improving OLED performance in similar ESIPT materials.
Transient absorption spectroscopy (TAS) and time-resolved photoluminescence spectroscopy have been used here to identify and monitor the evolution of excited states in TQB. In previous reports, TAS has been used to directly monitor the kinetics of TADF triplet states and correlate these to emission from singlet states.39 Elsewhere, TAS has revealed that suppressing the conformational relaxation of triplet states enhances rISC by maintaining a small ΔEST.40,41 Phosphorescence emission has also been measured at low temperatures, alongside delayed fluorescence at room temperature. These results establish the temperature dependence of intersystem crossing transitions between the excited states within the TQB molecule, and we are able to elucidate the mechanism of triplet state harvesting at room temperature as well as the quenching mechanism of the triplet excited states in the presence of oxygen. A comprehensive energy state transition diagram derived from these experiments (Figure 8) is presented as a conclusion of our work and supports recent previous computational reports of TQB by Cao et al.1
Figure 8.

Proposed energy level scheme and transitions between upper triplet states occurring in TQB.
Because the solubility of unsubstituted TQB is low, to allow preparation of low-surface roughness films (at high dye loading) as well as high-concentration solutions suitable for TAS, we designed and synthesized trihexyl-substituted TQB, with the structure shown in Figure 1. Subsequent use of “TQB” in this work refers to the hexyl-substituted version, except where explicitly stated otherwise [e.g., for zeonex films (Figure S4)]. The synthesis procedures, chemical characterization data, and details of all measurement techniques are described in the Supporting Information. The absorption and emission spectra and fluorescence lifetime (Figure S2) revealed that alkyl substitution of the TQB structure has little influence on the excited state dynamics.
Figure 2 shows the normalized absorption spectrum of TQB in toluene, representing the optical transitions from the TQB-TA ground state (before ESIPT takes place). A structured absorption band indicates strong vibronic contributions to the electronic transition. The progression spacing of 0.2 eV (1610 cm–1) corresponds to C=C vibrational modes coupled to the electronic transition from the ground state. The Stokes shift of 0.45 eV between the onsets of emission and absorption is large, which is characteristic of ESIPT. For a neat TQB film, a broad absorption band centered at 350 nm is observed, with partially resolved vibronic overtones similar to those seen in solution. A slight blue-shift in absorption is observed when TQB is incorporated into the DPEPO host (10 wt % emitter), potentially due to the increased rigidity of this host.
Figure 2.

Normalized absorption and photoluminescence emission spectra for TQB in a toluene solution (top) and for neat TQB and TQB films doped in DPEPO (bottom).
Photoluminescence emission is thought to arise from the C=N···H–O–C keto form (TQB-TB structure), due to the ESIPT process being orders of magnitude faster than fluorescence.35 An unstructured emission peak is observed in steady state measurements centered at 537 nm (2.31 eV peak, 2.71 eV onset), which agrees with previous calculations for this tautomer (2.31 eV).38 The TQB-TB tautomer has also been identified as the most stable keto form, with the lowest excited singlet state energy. Emission spectra measured in toluene and in films all show the same emission peak (Figure 2), which indicates that no charge redistribution occurs during the measurement. TQB in DPEPO displays a slight narrowing of the emission spectra, probably due to weakened TQB intermolecular interactions within the host matrix.
Picosecond to nanosecond scale TAS measurements of TQB in oxygenated and deoxygenated toluene (300 K) are shown in Figures 3 and 4. A negative induced band is initially observed at 500–600 nm (though red-shifted in the presence of oxygen), ascribed to stimulated emission (SE) from the TQB-TB singlet excited state. This is rapidly quenched, and a photoinduced absorption band (PA1) begins to grow on top of the SE. Figure 5 shows TAS spectra over the first 6 ns in degassed toluene, with an initial induced absorption band (later identified as the same feature as PA3 in Figure 4) observed from very early times (<2 ns) with an onset energy of 1.65 eV that decays with a lifetime of 5 ns. A second higher-energy band grows in over this decay time with an onset energy of 1.85 eV (PA2). PA2 overlaps with the SE band, leading to an observed isosbestic point at 580 nm (2.14 eV) and an apparent blue-shift of the SE band in Figure 5 compared to Figures 3 and 4 in the presence of oxygen, for which the PA2 signal vanishes. PA2 and PA3 are identified as triplet–triplet absorptions by noting that they are rapidly quenched in the presence of oxygen.42,43
Figure 3.

Transient absorption spectra measured from 0 to 40 ns for TQB in a toluene solution (1 mg/mL). In oxygen (top), the SE and PA3 signals are rapidly quenched while PA1 grows in. In the absence of oxygen (bottom), both PA2 and PA3 initially grow rapidly, and at later times, PA2 continues to grow while the contribution from PA3 becomes relatively smaller.
Figure 4.

Transient absorption spectra measured for TQB in a toluene solution from 0 to 1000 ns. In oxygen (top), the SE and PA3 signals are rapidly quenched while PA1 grows in. In the absence of oxygen (bottom), both PA2 and PA3 initially grow rapidly, and at later times, PA2 continues to grow while the contribution from PA3 becomes relatively smaller.
Figure 5.

Transient absorption spectra of TQB in deoxygenated toluene from 6 to 6000 ps. Both PA2 and PA3 form rapidly, with PA3 experiencing more rapid decay.
A cursory inspection of the oxygenated TAS spectra in Figure 3 reveals the presence of processes in addition to simple oxygen quenching. The strong induced absorption feature PA1 (onset of 2.0–2.1 eV) grows over the first 40 ns (eventually masking the SE, which is observed after <1 ps) and then decays. PA1 is identified as a singlet–singlet absorption due to its insensitivity to oxygen quenching and so must arise from distinct relaxation pathways caused by oxygen quenching of other excited states, discussed separately below. Nonetheless, in the absence of oxygen, the photophysics is rather clear. The tautomer TQB-TA absorbs, and following rapid ESIPT (<50 fs),1 we observe emission from the TQB-TB first excited singlet state in competition with efficient ISC. This competition limits the overall PLQY for the TQB-TB state (55% in DPEPO films, 58% in degassed toluene), which is still relatively high for ESIPT molecules but low compared to those of many high-performance D–A(−D) TADF materials in solid hosts.18,19,38
Figure 6 shows the emission decay dynamics of TQB in DPEPO at 300 and 80 K. DPEPO has an “effective” polarity similar to that of toluene, while these films are also directly representative of the emission layer in an OLED. Consistent with a TADF mechanism and previous measurements (using TCSPC and a streak camera),38 we observe millisecond scale decay kinetics and a lower overall delayed emission intensity at 80 K. Comparison of the prompt fluorescence (PF) kinetics at both temperatures shows decay with a lifetime of 6.5 ns, again in agreement with previous reports.38 After ∼100 ns, we observe a second cascade feature on the log–log plot, characteristic of TADF. Throughout the decay, we observe the same emission spectra with an onset at 2.71 eV from the TQB-TB first excited singlet state (Figure S3). Similar to previous reports, we find that the intensity of the delayed emission varies linearly with the laser excitation dose.38 This rules out biexcitonic triplet–triplet annihilation as the DF mechanism, while the preserved emission spectra between PF and DF rule out phosphorescence, leaving TADF as the only likely DF mechanism at room temperature.44
Figure 6.

Time-resolved photoluminescence for TQB films in DPEPO measured at 300 and 80 K. Representative regions of prompt fluorescence (PF) and delayed fluorescence (DF) are indicated.
Surprisingly, even at 80 K and delay times of >1 ms, we observe no unambiguously attributable phosphorescence spectrum from the DPEPO films. While previous reports of activation energies and DFT calculations can be used to estimate the lowest triplet energy of TQB at ∼2.5 eV (singlet at 2.7 eV and ΔEST ≈ EA = 200 meV), the delayed emission retains an onset of 2.7 eV throughout (Figure S3). In contrast, cryogenic measurements of very low concentration zeonex films doped with “regular” (nonhexyl) TQB [<0.5 wt % (Methods in the Supporting Information)] allow the phosphorescence to be resolved (Figure S4). In agreement with previous estimates, at long delay times a new emission band appears, onset of 2.5 eV in the polymeric host, which we ascribe to phosphorescence from the lowest-energy triplet state of the TQB-TB configuration, T1. These measurements define an experimental ΔEST of 0.18 eV for TQB-TB, in good agreement with the value estimated from previously reported activation energies determined from Arrhenius plots.38 However, we note that the rapid ISC identified as being responsible for the low PLQY of TQB seems implausible for such a large ΔEST.
Reconciling these data with TAS, we find the lower triplet state of the PA3-induced triplet–triplet absorption forms rapidly by ISC from the TQB-TB singlet state and can be rapidly quenched by oxygen. In the absence of oxygen, the PA3 band decays over several hundred nanoseconds (Figure 5) while the broader PA2 band continues to grow in (Figures 3 and 4) and has a lifetime far longer than that of PA3. Similar to PA3, the lower triplet state of the PA2 triplet–triplet band is also rapidly quenched by oxygen (Figures 3 and 4). However, as the growth and decay of PA2 and PA3 exhibit different kinetics (indeed, the decay of PA3 in Figure 5 appears to feed the later growth of PA2 in Figures 3 and 4), we can conclude that they must not share the same lower triplet state. This realization implies the involvement of a second triplet state higher in energy than the T1 state revealed by zeonex film phosphorescence, which we identify as T2.
Because they are adjacent features, we suggest that PA3 and PA2 induce absorption to the same final upper triplet state, TN. As the transition from T2 to TN involves less energy than that from T1 to TN, PA3 must arise from the T2 state while PA2 comes from the T1 state. In this case, the energy gap between T2 and T1 is found to be 0.2 eV, which places the energy of T2 at 2.73 eV, close to resonance with the TQB-TB singlet (2.71 eV, from fluorescence onset). Energy calculations for TQB also find T2 and the TQB-TB singlet state to be nearly resonant, with the T2 state 20 meV above the singlet.1 This small energy gap is thoroughly consistent with rapid ISC from the TQB-TB singlet to T2. Additionally, as PA2 grows in parasitically from PA3 and is very long-lived, we suggest this is consistent with IC from T2 to T1. This relatively slow IC suggests that the Franck–Condon factor between T2 and T1 is small, indicating they correspond to different molecular configurations and potentially different tautomers.
In the presence of O2, we see rapid quenching of the SE within 12 ns, leaving only the strong PA1 band. A contribution from PA3 is also seen at very early times (Figures 3 and 4) but is rapidly fully quenched by the oxygen. Oxygen is quenched by triplet energy transfer from T1 or T2 to 3O2, creating 1O2 and a singlet state on the TQB to preserve spin. Typically, oxygen quenching of organic triplets returns the molecule to the ground state (singlet), and while this may be true for T1, it is not a strictly necessary outcome. Instead, the strong induced absorption band PA1 (which is not quenched by oxygen and appears only in the presence of oxygen) shows that a different singlet state is reached. PA1 therefore corresponds to a singlet–singlet transition of a singlet excited state reached after O2 quenching of T2. To reveal the identity of this state, we note that, as it is an induced absorbance far from the 300–400 nm S0–S1 ground state absorption, it cannot be a transition from the TQB-TA ground state. If PA1 came from the TQB-TB state, we would expect it to have kinetics identical to that of the SE arising from the same state. We would also observe that the SE signal would be significantly quenched by reabsorption from the overlapping PA1 absorption; however, neither of these is observed. Therefore, PA1 cannot be from the S0–S1 transition of the singly proton transferred TQB-TB state.
Moving further to the left in Figure 1, we instead propose that the state formed by oxygen quenching of T2 is the ground state singlet of TQB-TC. This assignment at first appears to be consistent with TQB-TC being more conjugated than TQB-TA or TQB-TB and so possessing a smaller S0–S1 gap (2.1–2.0 eV, from the onset of PA1). The higher energy of the TQB-TC ground state is also consistent with a second endothermic proton transfer. In opposition to this assignment though, previously reported calculations suggest the specific S0–S1 gap for TQB-TC is actually very similar to the gap for the TQB-TB configuration (2.7 eV, from emission).38 However, these calculations show that the gap between TQB-TC S0 and TQB-TB S1 is 2 eV, and thus, we suggest that PA1 results from concerted TQB-TC S0 to TQB-TB S1 absorption and reverse proton transfer (rESIPT). All of the measured and proposed transitions are shown diagrammatically in Figure 8.
From calculations of the SOC matrix elements between various singlet and triplet states, the TQB-TB singlet excited state has a spin–orbit coupling strength to T2 as large as 0.8 cm–1.1 As T2 is only approximately 20 meV above this singlet state, the gap is easily overcome at 300 K to give rapid ISC. Calculations also show that the level of spin–orbit coupling between the S1 and T1 states decreased significantly (by a factor of 5, 0.2 cm–1) as a result of the proton transfer, due to the similar orbital characters of both states. This explains why direct ISC from S1 to T1 is not observed in TAS measurements.
Finally, returning to time-resolved emission measurements, Figure 6 shows that at 300 K, PF emission from TQB-TB decays faster than at 80 K due to competition with thermally activated ISC to T2. From the individual spectra in Figure 7, after 100 μs (in the DF region) we observe a combination of TADF (onset of 2.7 eV) and red-shifted emission (weakly structured), possibly due to phosphorescence coming from the large triplet population in T1 (arising from IC from T2). At 80 K, the early thermally activated ISC to T2 is greatly reduced (gap of 20 meV, and kbT ≈ 6.6 meV at 80 K), so direct ISC to T1 dominates. However, this process is very slow because of the weak SOC between the S1 and T1 states as discussed above. Thus, at 80 K, we observe relatively higher-intensity PF emission (with a slower decay time). The individual spectra at 80 K in Figure 7 no longer show a phosphorescence contribution on the red side of the emission band. This reduction in phosphorescence activity despite the lower temperatures is because ISC to T2 is suppressed and there is a much smaller total triplet population formed. We also see less TADF emission at low temperatures because there are few triplets to harvest, and because at low temperatures rIC from T1 to T2 (180 meV energy gap) must precede rISC from T2 to S1. This reveals TQB as an example of an upper state crossing or “hot exciton” TADF material, an uncommon class of emitters with particularly challenging design rules.30,45,46
Figure 7.
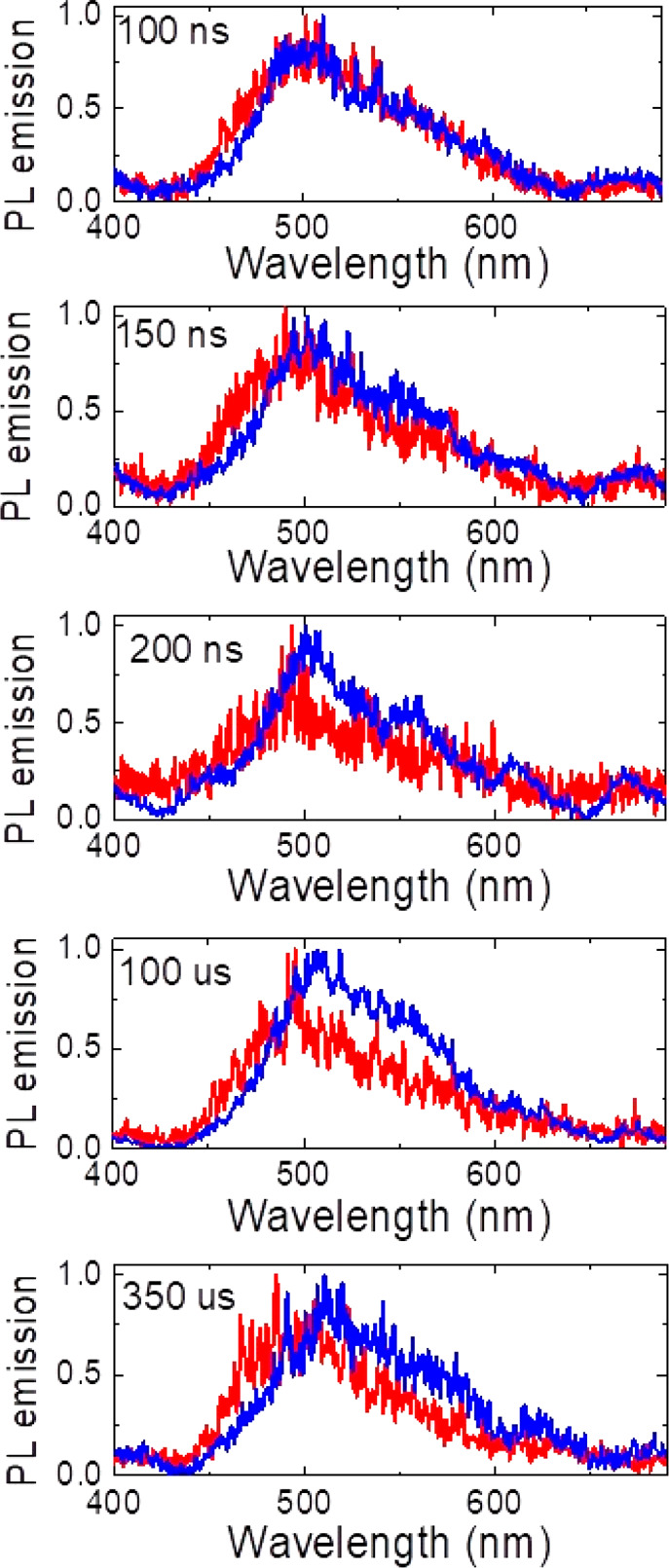
Normalized time-resolved photoluminescence spectra of TQB in DPEPO measured using gated iCCD at different time intervals, measured at 300 K (blue) and 80 K (red). Integration times are identical within each panel but different between different panels. At each delay time, the 300 K spectrum shows red-shifted structured emission features not observed at 80 K.
In conclusion, we describe the evolution of excited states within the ESIPT system TQB, including TADF and room-temperature phosphorescence (RTP) emission and employing TAS. We observe that after photoexcitation and ESIPT a rather slow emission from the first excited singlet state of TQB-TB occurs in competition with thermally activated ISC to an upper triplet state, T2, with a ΔEST of only 20 meV. Thus, the photoluminescence quantum yield of the TQB system is limited by this fast ISC. T2 excitons slowly decay to T1 via internal conversion (IC) in competition with “hot exciton” rISC and TADF emission, which is accompanied at later times by RTP from T1. At low temperatures, the ISC channel to T2 becomes energetically inaccessible and only slower ISC to T1 is active, leading to no observed phosphorescence contribution to emission despite the lower temperature, and lower TADF emission due to the larger ΔEST for T1 compared to T2.
In the presence of oxygen, both T2 and T1 are effectively quenched. However, we observe that quenching of T2 leads to a new induced absorption band that overlaps with and quenches stimulated emission. By careful measurement of the energies of all induced bands along with the energy of T1 (from zeonex film phosphorescence, found to be 0.18 eV below the TQB-TB excited singlet), we show that this new singlet–singlet induced absorption band formed by the oxygen quenching of T2 can correspond only to a 2 eV rESIPT transition from the TQB-TC ground state to the TQB-TB excited singlet. Through these assignments, we also confirm the findings of the spin–orbit coupling calculations by Cao et al.1 These results ultimately reveal that TADF in the TQB system occurs from upper triplet state rISC from T2, either before IC to or after thermally activated rIC from T1. The limited emission and triplet harvesting efficiencies reported for ESIPT emitters and OLEDs suggest that this inefficient TADF mechanism is common among them and must be avoided or engineered around in the design of future ESIPT emitters.
Acknowledgments
This work was financially supported by JST ERATO Grant JPMJER1305 and JSPS KAKENHI Grant 19H02790. The authors also thank the EPSRC (EP/P012167/1) and acknowledge the EXCILIGHT and HyperOLED projects funded by the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreements 674990 and 732013 (under Action ICT-02-2016) for funding the research in Durham. P.L.d.S. thanks the CAPES Foundation, Ministry of Education of Brazil, in particular the Science Without Borders Program for a Ph.D. studentship (Proc. 12027/13-8).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c00498.
Synthesis and structural and thermal characterization of hexyl-TQB, sample preparation and experimental methods for photophysical characterization, comparison of hexyl-TQB and bare TQB, normalized time-resolved emission spectra of hexyl-TQB in DPEPO films, and optical characterization of bare TQB in zeonex films (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cao Y.; Eng J.; Penfold T. J. Excited State Intramolecular Proton Transfer Dynamics for Triplet Harvesting in Organic Molecules. J. Phys. Chem. A 2019, 123, 2640–2649. 10.1021/acs.jpca.9b00813. [DOI] [PubMed] [Google Scholar]
- Dias F. B; Penfold T. J; Monkman A. P Photophysics of thermally activated delayed fluorescence molecules. Methods Appl. Fluoresc. 2017, 5, 012001. 10.1088/2050-6120/aa537e. [DOI] [PubMed] [Google Scholar]
- Im Y.; Kim M.; Cho Y. J.; Seo J.-A.; Yook K. S.; Lee J. Y. Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters. Chem. Mater. 2017, 29, 1946–1963. 10.1021/acs.chemmater.6b05324. [DOI] [Google Scholar]
- Zhang L.; Cheah K. W. Thermally Activated Delayed Fluorescence Host for High Performance Organic Light-Emitting Diodes. Sci. Rep. 2018, 8, 8832. 10.1038/s41598-018-27238-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi C.; Baldo M. A.; Thompson M. E.; Forrest S. R. Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 2001, 90, 5048–5051. 10.1063/1.1409582. [DOI] [Google Scholar]
- Baldo M. A.; O’Brien D. F.; You Y.; Shoustikov A.; Sibley S.; Thompson M. E.; Forrest S. R. Highly efficient phosphorescent emission from organic electroluminescent devices. Nature 1998, 395, 151–154. 10.1038/25954. [DOI] [Google Scholar]
- Minaev B.; Baryshnikov G.; Agren H. Principles of phosphorescent organic light emitting devices. Phys. Chem. Chem. Phys. 2014, 16, 1719–1758. 10.1039/C3CP53806K. [DOI] [PubMed] [Google Scholar]
- Chen X.-K.; Kim D.; Brédas J.-L. Thermally Activated Delayed Fluorescence (TADF) Path toward Efficient Electroluminescence in Purely Organic Materials: Molecular Level Insight. Acc. Chem. Res. 2018, 51, 2215–2224. 10.1021/acs.accounts.8b00174. [DOI] [PubMed] [Google Scholar]
- Cai J.-L.; Liu W.; Wang K.; Chen J.-X.; Shi Y.-Z.; Zhang M.; Zheng C.-J.; Tao S.-L.; Zhang X.-H. Highly Efficient Thermally Activated Delayed Fluorescence Emitter Developed by Replacing Carbazole With 1,3,6,8-Tetramethyl-Carbazole. Front. Chem. 2019, 7, 17. 10.3389/fchem.2019.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Data P.; Takeda Y. Recent Advancements in and the Future of Organic Emitters: TADF- and RTP-Active Multifunctional Organic Materials. Chem. - Asian J. 2019, 14, 1613–1636. 10.1002/asia.201801791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Cao X.; Wu Q.; Zhang M.; Sun N.; Zhang X.; Tao Y. Purely organic materials for extremely simple all-TADF white OLEDs: a new carbazole/oxadiazole hybrid material as a dual-role non-doped light blue emitter and highly efficient orange host. J. Mater. Chem. C 2018, 6, 3675–3682. 10.1039/C7TC04969B. [DOI] [Google Scholar]
- Hall D.; Suresh S. M.; dos Santos P. L.; Duda E.; Bagnich S.; Pershin A.; Rajamalli P.; Cordes D. B.; Slawin A. M. Z.; Beljonne D.; et al. Improving Processability and Efficiency of Resonant TADF Emitters: A Design Strategy. Adv. Opt. Mater. 2020, 8, 1901627. 10.1002/adom.201901627. [DOI] [Google Scholar]
- Romanov A. S.; Jones S. T. E.; Gu Q.; Conaghan P. J.; Drummond B. H.; Feng J.; Chotard F.; Buizza L.; Foley M.; Linnolahti M.; et al. Carbene metal amide photoemitters: tailoring conformationally flexible amides for full color range emissions including white-emitting OLED. Chem. Sci. 2020, 11, 435–446. 10.1039/C9SC04589A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella M.; Danos A.; Monkman A. P. Less Is More: Dilution Enhances Optical and Electrical Performance of a TADF Exciplex. J. Phys. Chem. Lett. 2019, 10, 793–798. 10.1021/acs.jpclett.8b03646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos P. L.; Etherington M. K.; Monkman A. P. Chemical and conformational control of the energy gaps involved in the thermally activated delayed fluorescence mechanism. J. Mater. Chem. C 2018, 6, 4842–4853. 10.1039/C8TC00991K. [DOI] [Google Scholar]
- Etherington M. K.; Franchello F.; Gibson J.; Northey T.; Santos J.; Ward J. S.; Higginbotham H. F.; Data P.; Kurowska A.; Dos Santos P. L.; et al. Regio- and conformational isomerization critical to design of efficient thermally-activated delayed fluorescence emitters. Nat. Commun. 2017, 8, 14987. 10.1038/ncomms14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherington M. K.; Gibson J.; Higginbotham H. F.; Penfold T. J.; Monkman A. P. Revealing the spin–vibronic coupling mechanism of thermally activated delayed fluorescence. Nat. Commun. 2016, 7, 13680. 10.1038/ncomms13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachelek P.; Ward J. S.; dos Santos P. L.; Danos A.; Colella M.; Haase N.; Raynes S. J.; Batsanov A. S.; Bryce M. R.; Monkman A. P. Molecular Design Strategies for Color Tuning of Blue TADF Emitters. ACS Appl. Mater. Interfaces 2019, 11, 27125–27133. 10.1021/acsami.9b06364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukhta N. A.; Higginbotham H. F.; Matulaitis T.; Danos A.; Bismillah A. N.; Haase N.; Etherington M. K.; Yufit D. S.; McGonigal P. R.; Gražulevičius J. V.; et al. Revealing resonance effects and intramolecular dipole interactions in the positional isomers of benzonitrile-core thermally activated delayed fluorescence materials. J. Mater. Chem. C 2019, 7, 9184–9194. 10.1039/C9TC02742D. [DOI] [Google Scholar]
- Gibson J.; Monkman A. P.; Penfold T. J. The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules. ChemPhysChem 2016, 17, 2956–2961. 10.1002/cphc.201600662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias F. B.; Bourdakos K. N.; Jankus V.; Moss K. C.; Kamtekar K. T.; Bhalla V.; Santos J.; Bryce M. R.; Monkman A. P. Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters. Adv. Mater. 2013, 25, 3707–3714. 10.1002/adma.201300753. [DOI] [PubMed] [Google Scholar]
- Penfold T. J.; Dias F. B.; Monkman A. P. The Theory of Thermally Activated Delayed Fluorescence for Organic Light Emitting Diodes. Chem. Commun. 2018, 54, 3926. 10.1039/C7CC09612G. [DOI] [PubMed] [Google Scholar]
- Padalkar V. S.; Seki S. Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters. Chem. Soc. Rev. 2016, 45, 169–202. 10.1039/C5CS00543D. [DOI] [PubMed] [Google Scholar]
- Sedgwick A. C.; Wu L.; Han H.-H.; Bull S. D.; He X.-P.; James T. D.; Sessler J. L.; Tang B. Z.; Tian H.; Yoon J. Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev. 2018, 47, 8842–8880. 10.1039/C8CS00185E. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Dong H.; Yang H.; Zheng Y. Solvent-Polarity-Dependent Excited-State Behavior and Thermally Active Delayed Fluorescence for Triquinolonobenzene. ACS Appl. Bio Mater. 2019, 2, 2060–2068. 10.1021/acsabm.9b00088. [DOI] [PubMed] [Google Scholar]
- Shaw M. F.; Sztáray B.; Whalley L. K.; Heard D. E.; Millet D. B.; Jordan M. J. T.; Osborn D. L.; Kable S. H. Photo-tautomerization of acetaldehyde as a photochemical source of formic acid in the troposphere. Nat. Commun. 2018, 9, 2584. 10.1038/s41467-018-04824-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack S. K.; Hehre W. J. The enol of acetone. J. Am. Chem. Soc. 1977, 99, 4845–4846. 10.1021/ja00456a062. [DOI] [Google Scholar]
- Wu K.; Zhang T.; Wang Z.; Wang L.; Zhan L.; Gong S.; Zhong C.; Lu Z.-H.; Zhang S.; Yang C. De Novo Design of Excited-State Intramolecular Proton Transfer Emitters via a Thermally Activated Delayed Fluorescence Channel. J. Am. Chem. Soc. 2018, 140, 8877–8886. 10.1021/jacs.8b04795. [DOI] [PubMed] [Google Scholar]
- Park S.; Kwon O.-H.; Lee Y.-S.; Jang D.-J.; Park S. Y. Imidazole-Based Excited-State Intramolecular Proton-Transfer (ESIPT) Materials: Observation of Thermally Activated Delayed Fluorescence (TDF). J. Phys. Chem. A 2007, 111, 9649–9653. 10.1021/jp072212p. [DOI] [PubMed] [Google Scholar]
- Hu D.; Yao L.; Yang B.; Ma Y. Reverse intersystem crossing from upper triplet levels to excited singlet: a ‘hot excition’ path for organic light-emitting diodes. Philos. Trans. R. Soc., A 2015, 373, 20140318. 10.1098/rsta.2014.0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezin A. S.; Vinogradova K. A.; Krivopalov V. P.; Nikolaenkova E. B.; Plyusnin V. F.; Kupryakov A. S.; Pervukhina N. V.; Naumov D. Y.; Bushuev M. B. Excitation-Wavelength-Dependent Emission and Delayed Fluorescence in a Proton-Transfer System. Chem. - Eur. J. 2018, 24, 12790–12795. 10.1002/chem.201802876. [DOI] [PubMed] [Google Scholar]
- Park S.; Kwon J. E.; Park S.-Y.; Kwon O.-H.; Kim J. K.; Yoon S.-J.; Chung J. W.; Whang D. R.; Park S. K.; Lee D. K.; et al. Crystallization-Induced Emission Enhancement and Amplified Spontaneous Emission from a CF3-Containing Excited-State Intramolecular-Proton-Transfer Molecule. Adv. Opt. Mater. 2017, 5, 1700353. 10.1002/adom.201700353. [DOI] [Google Scholar]
- Ernsting N. P.; Mordzinski A.; Dick B. Excited-state intramolecular proton transfer in jet-cooled 2,5-bis(2-benzothiazolyl)hydroquinone. J. Phys. Chem. 1987, 91, 1404–1407. 10.1021/j100290a026. [DOI] [Google Scholar]
- Zhou X.; Jiang Y.; Zhao X.; Guo D. ESIPT-Based Photoactivatable Fluorescent Probe for Ratiometric Spatiotemporal Bioimaging. Sensors 2016, 16, 1684. 10.3390/s16101684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Ji S.; Chen Y.; Guo H.; Yang P. Excited state intramolecular proton transfer (ESIPT): from principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. 10.1039/C2CP23144A. [DOI] [PubMed] [Google Scholar]
- Kim S.; Chang D. W.; Park S. Y.; Kawai H.; Nagamura T. Excited-State Intramolecular Proton Transfer in a Dendritic Macromolecular System: Poly(aryl ether) Dendrimers with Phototautomerizable Quinoline Core. Macromolecules 2002, 35, 2748–2753. 10.1021/ma011385o. [DOI] [Google Scholar]
- Li B.; Zhou L.; Cheng H.; Huang Q.; Lan J.; Zhou L.; You J. Dual-emissive 2-(2-hydroxyphenyl)oxazoles for high performance organic electroluminescent devices: discovery of a new equilibrium of excited state intramolecular proton transfer with a reverse intersystem crossing process. Chem. Sci. 2018, 9, 1213–1220. 10.1039/C7SC04464J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamada M.; Inada K.; Komino T.; Potscavage W. J.; Nakanotani H.; Adachi C. Highly Efficient Thermally Activated Delayed Fluorescence from an Excited-State Intramolecular Proton Transfer System. ACS Cent. Sci. 2017, 3, 769–777. 10.1021/acscentsci.7b00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase N.; Danos A.; Pflumm C.; Morherr A.; Stachelek P.; Mekic A.; Brütting W.; Monkman A. P. Kinetic Modeling of Transient Photoluminescence from Thermally Activated Delayed Fluorescence. J. Phys. Chem. C 2018, 122, 29173. 10.1021/acs.jpcc.8b11020. [DOI] [Google Scholar]
- Hosokai T.; Matsuzaki H.; Furube A.; Tokumaru K.; Tsutsui T.; Nakanotani H.; Yahiro M.; Adachi C.. Role of Intermediate State in the Excited State Dynamics of Highly Efficient TADF Molecules; SPIE, 2016; Vol. 9941, p 6. [Google Scholar]
- Kuang Z.; He G.; Song H.; Wang X.; Hu Z.; Sun H.; Wan Y.; Guo Q.; Xia A. Conformational Relaxation and Thermally Activated Delayed Fluorescence in Anthraquinone-Based Intramolecular Charge-Transfer Compound. J. Phys. Chem. C 2018, 122, 3727–3737. 10.1021/acs.jpcc.7b11411. [DOI] [Google Scholar]
- Gijzeman O. L. J.; Kaufman F.; Porter G. Oxygen quenching of aromatic triplet states in solution. Part 1. J. Chem. Soc., Faraday Trans. 2 1973, 69, 708–720. 10.1039/f29736900708. [DOI] [Google Scholar]
- Kawaoka K.; Khan A. U.; Kearns D. R. Role of Singlet Excited States of Molecular Oxygen in the Quenching of Organic Triplet States. J. Chem. Phys. 1967, 46, 1842–1853. 10.1063/1.1840943. [DOI] [Google Scholar]
- Jankus V.; Aydemir M.; Dias F. B.; Monkman A. P. Generating Light from Upper Excited Triplet States: A Contribution to the Indirect Singlet Yield of a Polymer OLED, Helping to Exceed the 25% Singlet Exciton Limit. Adv. Sci. 2016, 3, 1500221. 10.1002/advs.201500221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Kukhta N. A.; Ward J. S.; Danos A.; Batsanov A. S.; Bryce M. R.; Dias F. B. Balancing charge-transfer strength and triplet states for deep-blue thermally activated delayed fluorescence with an unconventional electron rich dibenzothiophene acceptor. J. Mater. Chem. C 2019, 7, 13224–13234. 10.1039/C9TC02175B. [DOI] [Google Scholar]
- Ward J. S.; Kukhta N. A.; dos Santos P. L.; Congrave D. G.; Batsanov A. S.; Monkman A. P.; Bryce M. R. Delayed Blue Fluorescence via Upper-Triplet State Crossing from C–C Bonded Donor–Acceptor Charge Transfer Molecules with Azatriangulene Cores. Chem. Mater. 2019, 31, 6684–6695. 10.1021/acs.chemmater.9b01184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.