

Abstract
Bruton’s tyrosine kinase (BTK) is a major drug target for B-cell related malignancies; however, existing BTK inhibitors approved for cancer treatment have significant off-targets that limit their use for autoimmune and inflammatory diseases. Remibrutinib (LOU064) is a novel covalent BTK inhibitor that binds an inactive BTK conformation, which affords it unprecedented selectivity. Its optimization led to rapid BTK engagement in vivo and fast clearance, further limiting systemic exposure. Remibrutinib is currently in phase 2 clinical trials for treatment of chronic urticaria and Sjoegren’s syndrome.
BTK is an essential protein for signaling of the B cell receptor.1 As such, it plays an important role in B cell differentiation and proliferation. This has made BTK a highly attractive drug target for B cell malignancies such as chronic lymphocytic leukemia and mantle cell lymphoma, as well as autoimmune and inflammatory diseases. Covalent BTK inhibitors such as ibrutinib and acalabrutinib exploit the presence of a nucleophilic cysteine residue in position 481 in BTK in order to achieve very high (sub-nM) binding affinities that result in potent BTK inhibition, and they were approved for treatment of several B-cell cancers. However, these compounds suffer from limited selectivity, as they react with other kinases that bear a cysteine at the same position and also reversibly inhibit additional kinases, resulting in serious side effects.2 This off-target activity may be acceptable for cancer treatment, due to the severe nature of the disease and limited time frame of the treatment. However, for chronic treatment of inflammatory or autoimmune diseases and in order to increase patient compliance, much higher selectivity is necessary. In this issue, Angst et al. report such a selective covalent BTK inhibitor: remibrutinib (LOU064).3
The need for potent and highly selective inhibition of BTK had previously prompted investigators to search for molecules with alternative binding modes for BTK. A milestone in the search was the development of CGI1746, a reversible and highly selective BTK inhibitor.4 CGI1746 binds BTK in a modified, inactive conformation (Figure 1A), in which the regulatory Y551 is rotated and forms a new binding pocket (H3 pocket). This binding mode offers two benefits: first, it dramatically enhances the selectivity of CGI1746 toward BTK due to the sequence variability around the binding pocket. Second, it inhibits phosphorylation of Y551 by upstream kinases, thus enhancing the inhibition of BTK. Fenebrutinib, a selective reversible BTK inhibitor optimized from CGI1746, is currently in clinical trials.
Figure 1.

Remibrutinib achieves selectivity by binding an inactive conformation of BTK. (A) Structure of BTK-remibrutinib complex (PDB code 6TFP). In the complex Tyr551 (white) is rotated inward and interacts with the cyclopropylphenyl group and is unavailable for phosphorylation. The active conformation of Tyr551 in the complex of BTK with ibrutinib is depicted in green (PDB code 5P9J). (B) Relative binding of ibrutinib and remibrutinib to various kinases, illustrating the dramatic differences in selectivity.
Angst et al. developed remibrutinib based on this new binding mode (Figure 1A). The covalent binding enables remibrutinib to reach high and sustained occupancy of BTK without the need for sustained systemic exposure typically required for reversible inhibitors. The authors performed an exhaustive campaign to optimize BTK binding and PK/PD properties culminating with a cyclopropylphenyl group as the H3 pocket binding motif, an aminopyrimidine scaffold as the hinge binding motif, and terminal acrylamide group for covalent binding to C481. The resulting compound exhibited potent BTK inhibition (IC50 = 1.3 nM) with excellent PK/PD properties (>120 min stability in human plasma, 94% BTK occupancy at a 3 mg/kg dose).
It is interesting to note that the acrylamide electrophile was installed on the core recognition element through a fairly flexible linker, containing four rotatable bonds. This is unusual for potent covalent inhibitors and challenges the notion that rigid and exact placement of the electrophile is a necessary requirement to achieve optimal covalent binding. The authors evaluated more rigid linkers which resulted in similar IC50 values.
Irreversible covalent binding of remibrutinib to BTK was confirmed by jump dilution kinetic studies, intact protein LC/MS, and a crystal structure. Comparison of remibrutinib to existing BTK inhibitors indicated an excellent selectivity profile among kinases, with off-target activity measured only against BMX and TEC (Figure 1B), as well as negligible off-target activity against other targets such as GPCRs and nuclear receptors.
An important benefit in the use of covalent inhibitors is the ability to achieve high sustained occupancy of the target without the need for extended exposure to the compound. Remibrutinib showed an excellent pharmacokinetic profile in animal models, with high permeability and rapid elimination, enabling rapid and sustained BTK engagement after dosing, followed by fast clearance of excess drug from the body. To test how quickly remibrutinib engages BTK, the authors used an ELISA assay that quantifies the total amount of BTK using an anti-BTK antibody, and the free (nonbound) BTK using a biotinylated covalent probe. Using this assay, the authors tested the binding kinetics of remibrutinib in human blood and in rats, showing highly potent and sustained inhibition of BTK with transient exposures to the drug (Figure 2).
Figure 2.
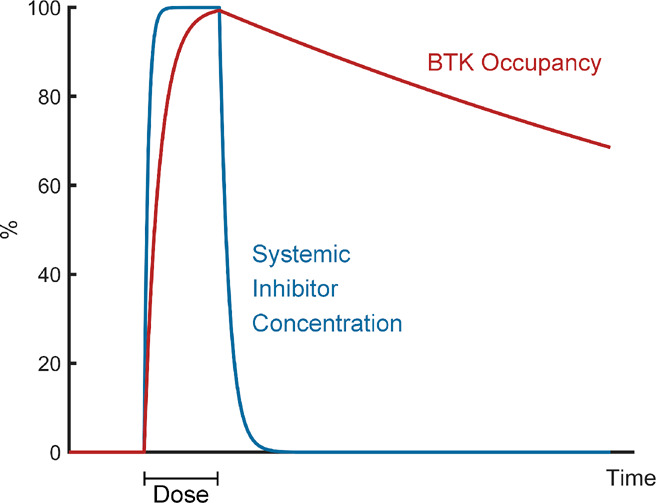
Rapid binding and fast clearance limit systemic exposure. Shown is a schematic illustration of remibrutinib binding and clearance dynamics. Rapid, irreversible binding to BTK, coupled with fast clearance, enables achievement of high BTK occupancy for extended periods of time with a transient dose of the inhibitor.
Finally, the authors tested the effect of remibrutinib on immune response in animal models. Remibrutinib displayed potent inhibition of antibody production in rats following immunization by sheep red blood cells and also inhibited inflammation in a rat model of collagen-induced arthritis. These results place remibrutinib as a promising clinical candidate for the treatment of inflammatory and autoimmune diseases via selective BTK inhibition.
Remibrutinib is currently in phase 2 clinical studies for chronic spontaneous urticaria and Sjoegren’s syndrome. Other covalent BTK inhibitors also progress in the race for chronic treatment. Evobrutinib has started phase 3 trials for relapsing multiple sclerosis; PRN1008, a reversible covalent BTK inhibitor, is in phase 3 trials for treatment of pemphigus; SAR442168 has successfully completed phase 2 trials for multiple sclerosis, and additional compounds are progressing through the pipeline.5 We wish great success to all of these efforts and hope they will support further rational design of safe covalent inhibitors for chronic indications, evolving the field of covalent drug discovery beyond oncology.
The authors declare no competing financial interest.
References
- Pal Singh S.; Dammeijer F.; Hendriks R. W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17 (1), 57. 10.1186/s12943-018-0779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigberg L. A.; Smith A. M.; Sirisawad M.; Verner E.; Loury D.; Chang B.; Li S.; Pan Z.; Thamm D. H.; Miller R. A.; Buggy J. J. The Bruton Tyrosine Kinase Inhibitor PCI-32765 Blocks B-Cell Activation and Is Efficacious in Models of Autoimmune Disease and B-Cell Malignancy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (29), 13075–13080. 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angst D.; Gessier F.; Janser P.; Vulpetti A.; Wälchli R.; Beerli C.; Littlewood-Evans A.; Dawson J.; Nuesslein-Hildesheim B.; Wieczorek G.; Gutmann S.; Scheufler C.; Hinniger A.; Zimmerlin A.; Funhoff E. G.; Pulz R.; Cenni B. Discovery of LOU064 (Remibrutinib), a Potent and Highly Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2020, 10.1021/acs.jmedchem.9b01916. [DOI] [PubMed] [Google Scholar]
- Di Paolo J. A.; Huang T.; Balazs M.; Barbosa J.; Barck K. H.; Bravo B. J.; Carano R. A. D.; Darrow J.; Davies D. R.; DeForge L. E.; Diehl L.; Ferrando R.; Gallion S. L.; Giannetti A. M.; Gribling P.; Hurez V.; Hymowitz S. G.; Jones R.; Kropf J. E.; Lee W. P.; Maciejewski P. M.; Mitchell S. A.; Rong H.; Staker B. L.; Whitney J. A.; Yeh S.; Young W. B.; Yu C.; Zhang J.; Reif K.; Currie K. S. Specific Btk Inhibition Suppresses B Cell- and Myeloid Cell-Mediated Arthritis. Nat. Chem. Biol. 2011, 7 (1), 41–50. 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Duan W.; Cu X.; Liang C.; Xin M. Bruton’s Tyrosine Kinase (BTK) Inhibitors in Treating Cancer: A Patent Review (2010-2018). Expert Opin. Ther. Pat. 2019, 29 (4), 217–241. 10.1080/13543776.2019.1594777. [DOI] [PubMed] [Google Scholar]