

Subcellular environments control how signaling sensors respond to allosteric effectors by remodeling the free-energy landscape.
Abstract
The functional response of a signaling system to an allosteric stimulus often depends on subcellular conditions, a phenomenon known as pluripotent allostery. For example, a single allosteric modulator, Rp-cAMPS, of the prototypical protein kinase A (PKA) switches from antagonist to agonist depending on MgATP levels. However, the mechanism underlying such pluripotent allostery has remained elusive for decades. Using nuclear magnetic resonance spectroscopy, ensemble models, kinase assays, and molecular dynamics simulations, we show that allosteric pluripotency arises from surprisingly divergent responses of highly homologous tandem domains. The differential responses perturb domain-domain interactions and remodel the free-energy landscape of inhibitory excited states sampled by the regulatory subunit of PKA. The resulting activation threshold values are comparable to the effective free energy of regulatory and catalytic subunit binding, which depends on metabolites, substrates, and mutations, explaining pluripotent allostery and warranting a general redefinition of allosteric targets to include specific subcellular environments.
INTRODUCTION
Allosteric modulation is a proven approach to increase selectivity for target receptors (1–6). Allosteric modulators are typically more selective than orthosteric ligands, which target highly conserved active sites (3). However, the functional response of an allosteric system to an allosteric stimulus may vary depending on the experimental conditions, a phenomenon known as pluripotent allostery (7–10). For example, the breast cancer drug tamoxifen serves its intended antagonist role for estrogen receptors (ERs) in breast tissue, but it acts as an agonist for uterus ERs, enhancing the risk of endometrial cancer (8). These undesired side effects are rationalized through a three ligand-binding trimer model, which effectively explains the therapeutically relevant agonism-antagonism switch in ERs (7). However, pluripotent allostery in other systems, such as kinases, remains poorly understood. For example, the Rp-cAMPS phosphorothioate analog of cyclic adenosine monophosphate (cAMP) (Rp; Fig. 1A) has been known for decades to act as an effective allosteric inhibitor of isoform 1a protein kinase A (PKA) in the presence of excess MgATP, but as an allosteric agonist in the absence of excess MgATP (Fig. 1, C and D) (11). Yet, the mechanism underlying the MgATP-dependent allosteric switch of Rp is currently still elusive.
Fig. 1. Rp-cAMPS as prototype of allosteric pluripotency.
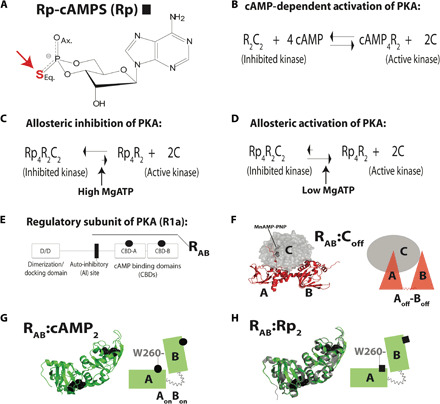
(A) The Rp-cAMPS cAMP analog. (B) Activation equilibrium of PKA. (C and D) Pluripotent allostery of Rp, which functions as an allosteric antagonist or agonist in the presence or absence of excess MgATP, respectively (11). (E) Domain organization of the regulatory (R)–subunit R1a and of the RAB construct (91 to 379). (F and G) Crystal structures and respective cartoons of C:RAB (PDB: 2QCS) (18) and RAB:cAMP2 (PDB: 1RGS) (31), with the cAMP-binding domains (CBDs) bridged by W260 of CBD-B interacting with cAMP in CBD-A. Bound nucleotides are shown as stick or sphere representation. (H) As (G), but for the crystal structure of the RAB:Rp2 complex (PDB: 1NE4, gray ribbon) (23) superimposed to the structure of RAB:cAMP2 (PDB: 1RGS, green ribbon). The two structures are virtually identical (RMSD = 0.47 Å).
Understanding pluripotent allostery in PKA is a central unmet challenge in the kinase field not only because PKA serves as a prototype for the protein kinase superfamily, but also because PKA is a cancer driver (12–14). Given the cancer predisposition caused by defective PKA inhibition, hundreds of cAMP analogs have been screened for PKA antagonism (15, 16). Yet, to date, the only known cAMP antagonist of PKA is Rp (Fig. 1A), which remains the best currently available lead for allosteric inhibition of PKA (15, 16). Hence, it is essential to understand the Rp mechanism of action, including pluripotent allostery.
The PKA regulatory subunit (R) functions as a cAMP-dependent competitive inhibitor of the PKA catalytic subunit (C) (17–19). In the inhibited PKA holoenzyme, two catalytic subunit molecules bind to a dimeric regulatory subunit (R2) (Fig. 1B) (17). Upon cAMP binding, each inhibitory R-subunit undergoes a conformational change, unleashing the catalytic activity of the C-subunit to phosphorylate substrate proteins (Fig. 1B) (17). The full-length R-subunit, i.e., R1a (1 to 379), includes an N-terminal dimerization domain, followed by a linker and two cAMP-binding domains (CBDs; CBD-A and CBD-B; Fig. 1E) (17). The linker includes an auto-inhibitory region (Fig. 1E), and the monomeric R-subunit construct spanning this region and CBD-A and CBD-B, i.e., R1a (91 to 379) or RAB (Fig. 1E), is sufficient for full inhibition and cAMP-dependent activation of PKA (18–20).
In the inhibitory C:RAB complex, both CBDs adopt a conformation denoted as “off” and interact with the C-subunit, although the CBDs are not in close contact with one another (red triangles; Fig. 1F). This inhibitory-competent CBD topology is denoted here as Aoff-Boff, with the hyphen between A and B indicating that CBD-A and CBD-B are not interacting (18, 20). In the noninhibitory RAB:cAMP2 complex (Fig. 1G), both CBDs switch to a conformation denoted as “on” and come into contact with one another (green rectangles; Fig. 1G). This inhibitory-incompetent CBD topology is denoted as AonBon, with the absence of the hyphen indicating that CBD-A and CBD-B are interacting (18–21). Hence, the terms “on/off” refer to the inability/ability of each CBD to promote inhibitory interactions of R with C that make C inaccessible to the substrate to be phosphorylated. Allosteric effectors typically act by controlling the on/off transitions of each CBD, which modulate the binding affinity of R for C and, consequently, the inhibition of PKA.
Rp binds both CBDs of R1a but, unlike cAMP, forms a stable inhibitory complex in the presence of MgATP, i.e., Rp4:R2:C2 (Fig. 1C) (15, 16, 22, 23). Despite extensive crystallization attempts, the structure of this inhibitory complex is currently unknown. The closest system amenable to crystallization so far has been RAB:Rp2 without C, but the structure of RAB:Rp2 is surprisingly very similar to that of RAB:cAMP2, which cannot bind C [root mean square deviation (RMSD) = 0.47 Å; Fig. 1, G and H] (23). Therefore, the structure of RAB:Rp2 may explain the agonism observed for Rp in the absence of excess MgATP (Fig. 1D), but alone is not sufficient to rationalize the antagonism of Rp in the presence of excess MgATP. Why does replacing cAMP with Rp in RAB:cAMP2 make RAB competent to bind and inhibit C without changing the structure of RAB? Without an answer to this question, the mechanism of action for Rp remains unclear, limiting further development of allosteric inhibitors for PKA.
We hypothesize that the primary difference between the inhibitory RAB:Rp2 and the noninhibitory RAB:cAMP2 complexes is in the conformational dynamics. Here, we test this hypothesis by investigating the interactions of Rp with RAB and C:RAB by nuclear magnetic resonance (NMR). Our comparative NMR analyses reveal how Rp remodels the conformational ensemble accessible to PKA R1a and provide a foundation to build an ensemble allosteric model (EAM) for PKA. We show how to measure input EAM parameters by NMR (24–27) and how, based on the PKA EAM, it is possible to relate microscopic dynamics to kinase function as measured by enzymatic assays, explaining allosteric pluripotency.
RESULTS
Rp-cAMPS acts as an antagonist for CBD-A and as an agonist for the homologous CBD-B in the absence of interdomain interactions
As a first step to understand the effect of Rp on the off versus on conformational equilibria of RAB, we analyzed the W260A RAB mutant, which selectively silences the interdomain interactions without substantially perturbing the allosteric network within each domain (20). The analysis of the W260A RAB:Rp2 complex is expected to reveal how Rp controls the off versus on conformational equilibrium of each separate domain. For this purpose, we compared the 1H,15N transverse-relaxation optimized spectroscopy (TROSY) spectrum of W260A RAB:Rp2 to three key reference 1H,15N TROSY spectra, i.e., RAB:C [molecular weight (MW) = ~73 kDa], which traps the conformation of RAB domains in the off state; RAB:cAMP2 (MW = ~33 kDa), which is assumed to represent the on state of each CBD; and the fully unbound apo RAB spectrum. Our comparative analyses focused on cross-peaks arising from RAB residues that are sufficiently distant from the C- and cAMP-binding interfaces to report primarily on the off versus on equilibria. For example, the cross-peaks of L221 and S297 specifically report on the conformational equilibria of CBD-A and CBD-B, respectively, and exhibit a linear pattern for the aforementioned reference states (i.e., RAB:C, RAB:cAMP2, and apo RAB), as shown in Fig. 2 (A and B). This linearity points to a fast off versus on exchange regime, in which linear chemical shift averaging implies that cross-peak positions reflect the relative populations of off and on states within each domain. The intermediate position of the apo RAB cross-peaks relative to those of RAB:C and RAB:cAMP2 (Fig. 2, A and B) means that each apo domain samples a nearly degenerate equilibrium with comparable populations of on and off CBD conformers.
Fig. 2. Identifying the states accessible to the RAB ensemble in the presence of Rp-cAMPS.
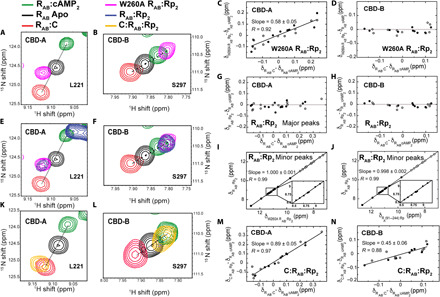
(A and B) Representative TROSY cross-peaks of 15N,2H-labeled W260A RAB:Rp2 (pink) for residues sensing primarily the “on” versus “off” equilibria of CBD-A and CBD-B. The corresponding cross-peaks in apo RAB (black), RAB:cAMP2 (green), and WT RAB:C (red) are also shown as comparison benchmarks. (C) Fractional inhibition of CBD-A in W260A RAB:Rp2 through the chemical shift (δ) correlation slope. Closed and open circles in (C) to (N) indicate 1H and 15N chemical shifts downscaled by a 0.2 factor, respectively. (D) As (C) for CBD-B. (E and F) As (A) and (B) including WT RAB:Rp2 (blue). (G and H) As (C) and (D) for the major cross-peaks of WT RAB:Rp2. (I) Correlation between the chemical shifts of the minor peaks in CBD-A of WT RAB:Rp2 and the W260A RAB:Rp2 mutant. (J) As (I) but versus the isolated CBD-A construct R1a (91 to 244) bound to Rp-cAMPS. (K and L) As (A) and (B) replacing W260A RAB:Rp2 with the WT C: RAB:Rp2 quaternary complex (orange). (M and N) As (C) and (D) for the WT C:RAB:Rp2 quaternary complex.
The comparative TROSY analysis of W260A RAB:Rp2 relative to the three reference states reveals that in the absence of interdomain interactions, Rp shifts the conformational equilibrium of apo CBD-A slightly to the off state (Fig. 2A). In notable contrast to CBD-A, CBD-B samples mostly the on state in W260A RAB:Rp2, similar to RAB:cAMP2 (Fig. 2B). Given the homology between the two tandem domains, the differential CBD-A versus CBD-B response to Rp is unexpected, and therefore, we extended our analysis of W260A RAB:Rp2 to a broader set of conformational equilibria–sensing residues (Fig. 2, C and D). This broader analysis provides a more quantitative measurement of the off versus on state populations, ensuring that they are not biased by the choice of a single residue within each domain. Specifically, the slopes of the (δW260A R:Rp2 − δR:cAMP2) versus (δR:C − δR:cAMP2) chemical shift correlation plots for CBD-A (Fig. 2C) and CBD-B (Fig. 2D) indicate that the average fraction of the off state sampled by W260A RAB:Rp2 is ~60% for CBD-A and ~5% for CBD-B. Overall, the chemical shift analyses of W260A RAB:Rp2 unexpectedly reveal that, in the absence of interdomain interactions, Rp acts as an antagonist for CBD-A but as an agonist for the homologous CBD-B. We then examined how the two CBDs respond to Rp in wild-type (WT) RAB.
WT RAB:Rp2 samples a conformational ensemble with a ground state in a closed AonBon topology similar to WT RAB:cAMP2 but excited states in an open topology lacking interdomain interactions
To probe the contribution of interdomain interactions to the conformational equilibria sampled by RAB:Rp2, we also acquired an HN TROSY spectrum of WT RAB:Rp2 (Fig. 2, E and F). We observed that CBD-B samples mostly the on state, similar to W260A RAB:Rp2, where the interdomain interactions are negligible (Fig. 2, F and H). Unlike CBD-B, CBD-A residues unexpectedly exhibit two sets of peaks in slow exchange and with different intensities (Fig. 2, E and G). The major and minor peaks are at positions similar to WT RAB:cAMP2 and W260A RAB:Rp2, respectively. This unexpected observation indicates that in WT RAB:Rp2, CBD-A samples a conformational ensemble with ground and excited (i.e., higher energy) states. In the ground state, CBD-A is in the on conformation similar to WT RAB:cAMP2, in which CBD-A interacts with the on CBD-B through the stacking of the W260 indole and the adenine base. This suggests that the interdomain interactions selectively stabilize the on state of CBD-A.
In the excited state, the interdomain interactions that stabilize the ground state are lost and CBD-A samples a fast-exchanging equilibrium between on and off conformers. This result is supported by the excellent match between the chemical shifts of the minor peaks in WT RAB:Rp2 and of W260A RAB:Rp2 (Fig. 2, E and I). Furthermore, the chemical shifts of the minor peaks of CBD-A in WT RAB:Rp2 match also those of the isolated CBD-A:Rp (Fig. 2J), confirming that the minor peaks of CBD-A in WT RAB:Rp2 arise from open topologies, where CBD-A does not interact with CBD-B. The overall population of the open topology excited state corresponding to the minor peaks is 17 ± 5%, after correcting for the differential relaxation of open versus closed topologies (fig. S1A). Considering that the population of off state in the open topology is ~60% (Fig. 2C), the excited state of WT RAB:Rp2 includes 10 ± 3% and 7 ± 2% of CBD-A in the off and on conformations, respectively, while CBD-B remains in the on conformer in all the detectable states of the ensemble accessed by WT RAB:Rp2 (Fig. 3C).
Fig. 3. Conformational ensembles accessible to apo and C-bound RAB as a function of Rp-cAMPS binding.
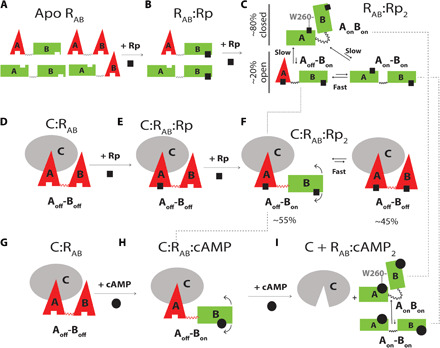
(A) States accessible within the degenerate free-energy landscape of apo RAB (20). Red triangles and green rectangles denote off and on conformations of each CBD, respectively. The on/off notation refers to the inability/ability of each CBD to promote inhibitory interactions of R with C. (B) States accessible to RAB:Rp. No interdomain interactions occur when CBD-A is apo. (C) States accessible to RAB:Rp2. The dashed lines indicate that the Aoff-Bon excited open state sampled by RAB:Rp2 is also sampled by C:RAB:Rp2 and is a transient intermediate along the cAMP-dependent activation pathway. (D and E) Rp binds first preferentially to CBD-A of C-bound RAB further stabilizing the inhibitory conformation of CBD-A. (F) States accessible to the quaternary complex C:RAB:Rp2, showing that when CBD-B is bound to Rp, it samples the “on” conformation as well. (G to I) Traditional sequential binding of cAMP to C:RAB, resulting in PKA activation. Unlike the case of Rp binding to C:RAB, cAMP binds first to CBD-B and then CBD-A (18). The dashed lines illustrate that the inhibition-incompetent RAB:cAMP2 complex samples the same ground states as RAB:Rp2 as well as one of the excited states.
When the C-subunit of PKA is added to WT RAB:Rp2 in the presence of excess MgCl2 and adenylyl-imidodiphosphate (AMP-PNP), a non-hydrolyzable analog of ATP, it selectively binds to and stabilizes the off conformer of CBD-A (~90% off; Fig. 2, K and M, and fig. S2A). However, the C-subunit only partially shifts the conformational equilibrium of CBD-B to yield a ~45% off state (Fig. 2, L and N, and fig. S2A), resulting in a two-state conformational ensemble for C:RAB:Rp2 (Fig. 3F). Hence, the excited state in the ternary RAB:Rp2 complex with an open topology and CBD-A and CBD-B in the off and on conformers, respectively, is inhibition competent because it becomes a ground state in the inhibited quaternary C:RAB:Rp2 complex (Fig. 3, C and F).
To better understand how these two complexes form upon Rp binding, we titrated Rp into the apo RAB and C:RAB samples and monitored the titration through HN TROSY spectra to measure domain-specific binding affinities for Rp (fig. S3). The results show that Rp binds C:RAB with an “A-to-B” order, which is opposite to cAMP binding, and Rp binds to apo RAB with a “B-to-A” order, similar to cAMP (fig. S3 and text S1). On the basis of these results, it is possible to propose a qualitative mechanism for the Rp antagonism-agonism switch in PKA, i.e., for pluripotent allostery (Fig. 3, A to F).
A qualitative mechanism of pluripotent allostery in PKA
In the absence of the C-subunit, apo RAB samples an ensemble of states that are accessible within a nearly degenerate free-energy landscape (20). This consists of four states with each noninteracting domain in fast exchange between comparable populations of off and on conformations (Fig. 3A) (20). Upon addition of Rp, it preferentially occupies CBD-B first, selectively stabilizing the on conformation of CBD-B, while CBD-A remains unoccupied, and thus, interdomain interactions are still negligible (Fig. 3B). As the concentration of Rp increases further, Rp also binds to CBD-A, facilitating interdomain interactions that partially stabilize a closed topology, where both domains of RAB:Rp2 are in the on conformation (AonBon; Fig. 3C). However, RAB:Rp2 also samples substantial populations (~20%) of open topologies, where CBD-A is in fast exchange between the off and the on conformations, i.e., Aoff-Bon and Aon-Bon, respectively (Fig. 3C).
When Rp is added to C:RAB, it preferentially binds to CBD-A first, stabilizing it in the off state, and then to CBD-B, promoting a partial off-to-on transition (Fig. 3, D to F). When the quaternary C:RAB:Rp2 complex forms, RAB samples the Aoff-Bon and Aoff-Boff states (Fig. 3F). In marked contrast to Rp, when cAMP is added to C:RAB, it preferentially binds to CBD-B first, transiently sampling the C:RAB:cAMP-binding intermediate with RAB in the Aoff-Bon state (Fig. 3, G and H). Then, cAMP binds also to CBD-A, stabilizing both domains in an on conformation and releasing the C-subunit (Fig. 3I) (18). Hence, the Rp antagonism arises from the stabilization by Rp of two inhibitory states of C:RAB:Rp2, i.e., Aoff-Boff as in the inhibited C:RAB complex and Aoff-Bon, which is a transient intermediate along the cAMP-dependent activation pathway, but is stably trapped by C:RAB:Rp2 (Fig. 3, D to I).
The model of Fig. 3 offers a qualitative rationalization of the Rp agonism-antagonism switch in PKA. The ensemble of RAB:Rp2 includes three main conformations: a ground closed state, AonBon, and two excited open states: Aon-Bon and Aoff-Bon (Fig. 3C). The first two states are also sampled by the RAB:cAMP2 complex, albeit with different populations (Fig. 3I), and are inhibitory incompetent given their negligible affinity for the C-subunit. The third state (Aoff-Bon) represents instead a unique feature of RAB:Rp2 that differentiates it from RAB:cAMP2, and is inhibitory competent because it is populated also in the stable C:RAB:Rp2 inhibited complex (Fig. 3F). As a result, the mixed Aoff-Bon excited state sampled by RAB:Rp2 drives Rp to act as an antagonist when C binds RAB with sufficiently high affinity to selectively stabilize Aoff-Bon relative to AonBon and Aon-Bon, resulting in a stable C:RAB:Rp2 complex and PKA inhibition. However, if the affinity of C for RAB is reduced, for example, due to lower MgATP levels (28–30), the inhibitory Aoff-Bon state remains excited in the presence of C, while the ground, most populated state is still AonBon, which, being inhibition incompetent, leads to PKA activation. Hence, the model of Fig. 3 (A to F) provides a viable framework to explain the MgATP-dependent Rp agonism-antagonism switch observed for PKA (11).
Given the potential of the model in Fig. 3 to account for the Rp agonism-antagonism switch, we sought to validate it further by additional investigations of how interdomain interactions are coupled to the on-off equilibria within each domain. Such coupling is a central feature of our model (Fig. 3), as it suggests that when both domains are bound to cyclic nucleotides, interdomain interactions occur only if both CBDs are in the on state (Fig. 3). When either CBD adopts the off state, domain-domain interactions are weakened, and conversely, when interdomain interactions are present, the conformational equilibrium of each CBD shifts to the on state. To refine this hypothesis, we performed molecular dynamics (MD) simulations of both RAB:Rp2 and C:RAB:Rp2 (Fig. 4, A, D, and F, and figs. S4 to S6).
Fig. 4. RAB interdomain interactions probed through MD simulations and PREs.
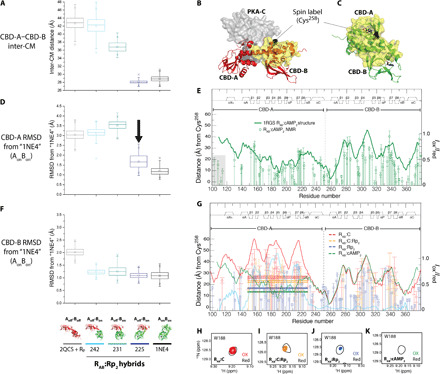
(A) Simulated CM distances between CBD-A and CBD-B for RAB:Rp2 in the Aoff-Boff, AonBon, and hybrid Aoff-Bon models with three different A/B boundaries (i.e., residues 242, 231, and 225, as shown in the bottom of F). (B and C) PRE design. The spin label is at Cys258 in CBD-B. Yellow surfaces are within 25 Å of the spin label. In C-bound (cAMP2-bound) RAB, interdomain interactions are minimal (maximal). (D) CBD-A RMSD versus AonBon. The closed topology induces the Aoff-Bon ➔ AonBon transition (black arrow). (E) PRE control. The Iox/Ired ratios (green circles) track the distance from the spin label (dark green line). Deviations occur in dynamic regions, e.g., flexible linker (gray highlight) (20, 38). (F) As (D), but for CBD-B. (G) Iox/Ired ratios for the RAB:C (red), RAB:C:Rp2 (orange), and RAB:Rp2 (blue) complexes. Dashed lines indicate the average Iox/Ired ratio for the region with the largest 2QCS (red) versus 1RGS (green) difference (cyan) in distance from the spin label. Solid lines mark the respective average ±1 SD. (H to K) Representative HN TROSY cross-peaks of oxidized and reduced samples. Contour levels were adjusted according to the intensity normalization as per Materials and Methods to account for aggregation.
The allosteric pluripotency mechanism predicts that domain-domain interactions are coupled to intradomain on-off equilibria
The MD simulations of RAB:Rp2 were executed starting from the AonBon and Aoff-Boff reference states, as well as the mixed Aoff-Bon state. The AonBon and Aoff-Boff states are based on the crystal structures of RAB:Rp2 [Protein Data Bank (PDB) code: 1NE4] (23) and C:RAB (PDB code: 2QCS) (18), respectively, while the hybrid Aoff-Bon states were generated by combining CBD-A from the crystal structure of C:RAB and CBD-B from the crystal structure of RAB:Rp2 with three different A/B boundaries within the helices that connect the two domains, i.e., residues 242, 231, and 225 (Fig. 4, A, D, and F and fig. S4, A to E), which reside close to the hinge points for the closed-to-open transition (18). The A/B boundary dictates whether the center-of-mass (CM) distance between the two domains approaches the closed versus open topologies of the AonBon versus Aoff-Boff structures, with 242 and 231 sampling the open topology, and 225 approaching the closed topology (Fig. 4A).
To assess the structural propensities of each domain within the hybrid RAB structures, RMSDs of the CBD-A and CBD-B from the RAB:Rp2 and C:RAB x-ray structures were computed (Fig. 4, D and F, and fig. S5, A, D, F, and G). As expected, CBD-B remains close to the conformation in the 1NE4 control structure for all three hybrid structures (Fig. 4F and fig. S5G). Meanwhile, the CBD-A domain remains close to the conformation in the C:RAB control structure for the 231 and 242 hybrid structures, but shifts to the conformation in the RAB:Rp2 control structure for the 225 hybrid structure (Fig. 4D and fig. S5F). Therefore, a shift of the R-subunit to its closed conformation, as in the 225 hybrid, appears to promote a shift of CBD-A to its on conformation, further corroborating our hypotheses based on the chemical shift analysis of RAB:Rp2 (Fig. 3C).
The MD simulations outlined above were also repeated in the presence of C-subunit (figs. S4, F to I; S5, B, C, and E; and S6). As in the case of the hybrid 225 RAB:Rp2 complex, the hybrid 225 Aoff-Bon structure shifts to the AonBon state (fig. S6, E, G, I, and J) with a closed topology similar to cAMP-bound RAB (figs. S5C and S6, A and C) and dissociates from the C-subunit (figs. S5, B and E, and S6, B, D, and F). In contrast, the more open hybrid 231 and 242 Aoff-Bon structures do not shift to the AonBon closed state (fig. S6, E, G, I, and J) and preserve an open topology (figs. S5C and S6, A and C), enabling CBD-A to remain in its off state and bound to the C-subunit (figs. S5B and S6, B and D). However, CBD-B in the hybrid 231 and 242 Aoff-Bon structures is displaced away from the C-subunit more than in the Aoff-Boff case (fig. S5E and S6F), and as a result, the A-B distance remains consistently shorter than in the canonical C:RAB complex (i.e., Aoff-Boff; fig. S5C) yet larger than in the closed AonBon topology (fig. S5C) because CBD-A is still stabilized in the off conformer by its binding interaction with the C-subunit. Overall, on the basis of the MD simulations, the A-B interdomain distances are predicted to decrease in the following order: C:RAB > C:RAB:Rp2 > RAB:Rp2 > RAB:cAMP2. We tested this prediction through paramagnetic relaxation enhancement (PRE) NMR measurements (Fig. 4, B, C, E, and G to K).
PRE experiments confirm the coupling of domain-domain interactions and intradomain on-off equilibria
PRE measurements are ideally suited to probe interdomain distances by engineering a spin label in one domain and observing the relaxation enhancement induced in the other domain. In the case of RAB, we introduced a cysteine mutation at residue 258 to covalently link the spin label to CBD-B and observe the relaxation enhancement in CBD-A, with the goal of probing the transition between open and closed domain topologies (Fig. 4, B and C).
The PRE data were acquired for four different samples: RAB:cAMP2, RAB:Rp2, C:RAB, and C:RAB:Rp2 (Fig. 4, E and G to K). The Iox/Ired PRE ratios for RAB:cAMP2 correlate well with the distances from residue 258 predicted on the basis of the crystal structure of RAB:cAMP2 (PDB code 1RGS; Fig. 4E) (31), confirming the correct positioning of the spin label at residue 258 and its efficacy in inducing relaxation enhancements. To probe the interdomain separation, we focused on the region of CBD-A subject to the largest variations in the distance from residue 258 when going from the C-bound (Aoff-Boff) to the cAMP-bound structure (AonBon), i.e., residues 150 to 230 (Fig. 4G, cyan trace). We then calculated the average Iox/Ired PRE ratios within this region for the RAB:cAMP2, RAB:Rp2, C:RAB, and C:RAB:Rp2 samples (Fig. 4G, dashed bars).
The average PRE ratios scale in the order C:RAB > C:RAB:Rp2 > RAB:Rp2 > RAB:cAMP2, as expected on the basis of the proposed chemical shift–based Rp antagonism-agonism model and MD simulations (Figs. 3 and 4, A, D, and F, and figs. S4 to S6). Overall, the PRE data consistently confirm the dependence of domain-domain interactions on the intradomain on-off equilibria predicted by the Rp antagonism-agonism switch mechanism (Fig. 3, A to F); i.e., they confirm that the probability of having CBD-A and/or CBD-B in the off state scales with the weakening of the interdomain interactions. We then used the validated Rp antagonism-agonism switch mechanism (Fig. 3, A to F) to build and parameterize a general EAM (32, 33) of pluripotent allostery for PKA, which enables quantitative predictions of kinase activity and, therefore, stringent comparisons to data from kinase assays.
A quantitative EAM of pluripotent allostery in PKA
To quantitatively relate the RAB dynamics (Fig. 3) to the regulation of kinase function, we built an EAM of PKA inhibition and activation, as explained in text S2. The EAM relies on four sets of input parameters (table S1): (i) the on versus off free-energy difference for each apo CBD, denoted as ΔGA and ΔGB; (ii) the state-specific association constants of Rp for each isolated domain; (iii) the free energy of interaction between the two CBDs both in the on state with CBD-A bound to the cyclic nucleotide, denoted as ΔGAB; and (iv) the state-specific affinities of the C-subunit for RAB. The key assumptions of our EAM are that (i) ΔGA and ΔGB are not affected by C or cNMP binding; (ii) ΔGAB is not affected by binding of C or a second cNMP molecule to CBD-B; (iii) the inhibition of PKA arises uniquely from R binding C and competing with the kinase substrates; and (iv) allosteric effectors act by controlling the affinity of R for C.
The EAM input parameters were measured on the basis of the populations and free energies determined by NMR chemical shift analyses and by hydrogen/deuterium (H/D) exchange protection factors for WT versus W260A, as explained in the Supplementary Materials (text S3, table S1, and figs. S1 and S2). In addition, a single scaling factor (γ) was introduced for the state-specific association constants of the C-subunit (table S1), to account for the dependence of the R:C affinity on the concentration of MgATP (28). For example, at low [MgATP], the effective R:C affinity decreases, requiring γ values <1. On the basis of input parameters (i) to (iv) (table S1), we modeled how cyclic nucleotide binding rescales the normalized thermodynamic statistical weights for each of the 18 possible states of the RAB ensemble, resulting from the combination of on/off, open/close, and apo/holo equilibria for the two domains in RAB (table S2). Starting from the ensembles of both apo and cyclic nucleotide-bound RAB, we used binding polynomials to compute R:C affinities in both scenarios, which were, in turn, used to calculate the change in kinase activity occurring upon addition of cyclic nucleotides (Supplementary Text S2 and S3 and fig. S7).
To illustrate how the EAM explains the pluripotent allostery of PKA, we simulated how the free-energy hierarchy of the states accessible to the apo RAB ensemble is remodeled by Rp and C-subunit binding (Fig. 5). When excess Rp binds to apo RAB in the absence of C, the AonBon closed state becomes the ground state, while the remaining states, which include the inhibitory-competent states (i.e., the Aoff-Boff and Aoff-Bon states), are at excited free-energy levels (Fig. 5A). When the C-subunit is introduced, the RAB:Rp2 free-energy level hierarchy is further remodeled according to the effective affinity of C for RAB.
Fig. 5. Remodeling of the free-energy landscape of apo RAB by Rp and the PKA C-subunit explains allosteric pluripotency.
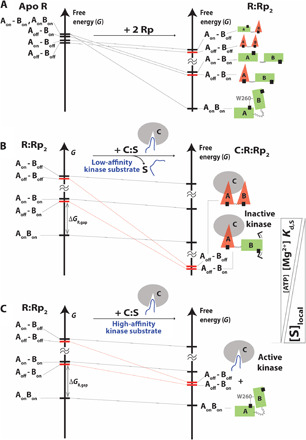
(A) Effect of Rp binding on the ensemble of states accessible to apo RAB. As the Boltzmann populations of the state decrease, the size of the cartoon is reduced. The inhibitory-competent conformations of RAB:Rp2 (red bars) are excited states. (B) Effect of PKA C-subunit binding on the ensemble of states accessible to RAB:Rp2. If the binding of the substrate to the kinase is sufficiently weak, and the MgATP level is sufficiently high, the inhibitory states “Aoff-Boff” and “Aoff-Bon” become the most stable conformations of RAB in the C:RAB:Rp2 ensemble. The kinase is inactive. ΔGR,gap is the inhibitory excited versus noninhibitory ground state free-energy difference. (C) In the case of substrates that bind C with sufficiently high affinity to compete with RAB or when the MgATP level decreases, due to the lower effective affinity of RAB:Rp2 for C, the most stable conformation of RAB accessible to the C:RAB:Rp2 ensemble is the “AonBon” state, which is noninhibitory. Hence, the kinase is active.
When the effective RAB:C affinity is high, e.g., presence of excess [MgATP] and a competing kinase substrate with low affinity and/or concentration, the free-energy levels of the excited inhibitory states are lowered to the ground level by C binding, resulting in the formation of a stable C:RAB:Rp2 complex and PKA inhibition (Fig. 5B). When the effective RAB:C affinity is low, e.g., lower [MgATP] or presence of a competing substrate with high affinity and/or concentration, then the inhibitory states are not sufficiently stabilized by C binding to reach the ground level. In this case, the C:RAB:Rp2 inhibitory complex is unstable, resulting in PKA activation (Fig. 5C). Hence, we hypothesize that not only the concentration of MgATP but also the affinity and concentration of the kinase substrate modulate the effect of Rp on PKA and trigger the Rp antagonism-agonism switch. To test this EAM-based prediction on the determinants of pluripotent allostery and further validate the model, we compared the fractional changes in kinase activity (ϕ) computed through the EAM to experimental data from kinase assays.
The MgATP concentration and kinase substrate affinity control the pluripotent allostery of PKA by remodeling the free-energy landscape of the R-subunit
To test our predictions on how the MgATP concentration and the affinity of competing kinase substrates modulate the activity of Rp-bound PKA, we first computed the Rp-dependent kinase activity (ϕ) in the presence (γ = 1) and in the absence of high [MgATP] (γ = 10−3) using the EAM and the measured input parameters (table S1). The predicted dose-dependent kinase activity profiles clearly reflect the antagonism-agonism switch of PKA by Rp-cAMPS (Fig. 6A), in agreement with previous observations (11). The activation constant (Ka) values and the Hill coefficient predicted from the EAM are in excellent agreement with the experimental values measured for the full-length R1a subunit (Fig. 6A) (11), validating the EAM. Furthermore, on the basis of the EAM, it is possible to dissect the determinants of pluripotent allostery. For example, the EAM model reveals that when the effective R:C affinity is weak (γ = 10−3), partial activation unexpectedly remains even in the absence of interdomain interaction (ΔGAB = 0 RT). The fact that CBD-A is in the off state when the two tandem domains do not interact (Fig. 3, A to F) means that the main drivers of the antagonism-to-agonism switch observed for the PKA-Rp system are both domain-domain interactions and the ability of Rp to shift CBD-B to the on state. The notable contribution of CBD-B to the activation of PKA was largely unexpected given the homology between the two tandem domains (31).
Fig. 6. Changes in MgATP concentration and/or substrate affinity trigger the Rp antagonism-to-agonism switch for PKA R1a.
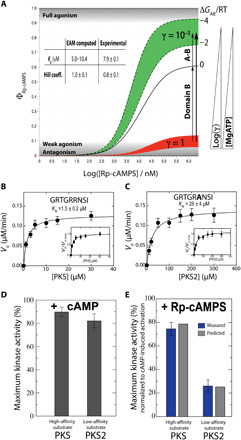
(A) Antagonism-to-agonism switch triggered by [MgATP] reduction. Dose-dependent kinase activation (ϕ) in the presence (red, γ = 1) and absence of MgATP (green, γ = 10−3). The γ scaling factor models the reduction in R:C affinity occurring upon removal of MgATP (table S1). (B to E) Antagonism-to-agonism switch triggered by substrate affinity increases. The substrates PKS and PKS2 exhibit Km values different by approximately one order of magnitude (B and C), and the increase in apparent affinity causes a switch to almost full agonism for Rp-cAMPS (E), while cAMP acts as an activator for both substrates, irrespective of the apparent affinity (D). In (E), blue bars are from kinase assays, while gray bars are computed on the basis of the EAM (see Materials and Methods). Error bars reflect the SD from triplicate measurements.
We also hypothesized that increasing the apparent affinity of the kinase substrates triggers the antagonism-to-agonism switch of Rp, as kinase substrates and the inhibitory linker of R compete for the active site of C, thus weakening the effective R:C affinity. To test this hypothesis, we used two substrates with different apparent affinities: PKS (Km = 1.5 ± 0.2 μM; Fig. 6B) and PKS2 (Km = 26 ± 4 μM; Fig. 6C), and we compared the respective experimental kinase activities to the EAM predictions (Fig. 6E). As expected from our hypothesis, upon binding of excess Rp, almost full agonism is observed with the high-affinity substrate (i.e., PKS), while only partial agonism is observed with the low-affinity substrate (i.e., PKS2). Moreover, the predicted kinase activities from the EAM are in agreement with the experimental values measured for full-length R1a subunit, further validating the relevance of our model of pluripotent allostery for the full-length integral PKA system.
DISCUSSION
An EAM of pluripotent allostery in PKA
On the basis of our comparative NMR analysis of C:RAB:Rp2, RAB:Rp2, W260A RAB:Rp2, RAB:cAMP2, and apo RAB, we propose an ensemble model of pluripotent allostery for PKA (Fig. 5). The proposed model addresses the long-standing question of why Rp functions as an antagonist under some conditions but as an agonist under others. The main difference between RAB:cAMP2 and RAB:Rp2 lies in the free-energy gap between the inhibitory-competent excited states (Aoff-Bon and Aoff-Boff) and the inhibitory-incompetent ground state (AonBon), which is common to both ternary complexes. This free-energy gap is denoted here as ΔGR,gap. The ΔGR,gap is lower in RAB:Rp2 than in RAB:cAMP2, and when the C-subunit binds the Aoff-Bon and Aoff-Boff states of RAB with high affinity (e.g., in the presence of excess MgATP), they become more stable than the AonBon state, resulting in the formation of the C:RAB:Rp2 inhibited complex and in Rp antagonism (Fig. 5B). When the affinity of the C-subunit for the excited inhibitory states is reduced (e.g., in the absence of excess MgATP), the AonBon state remains the ground state even in the presence of C and a stable inhibited complex cannot form, leading to Rp agonism (Fig. 5C). In the case of RAB:cAMP2, the free-energy gap between the inhibitory-competent excited states and the inhibitory-incompetent ground state is too large to result in antagonism, even in the presence of excess MgATP.
The proposed model not only provides a simple but general rationalization of pluripotent allostery, but also offers an unprecedented view of the previously elusive C:RAB:Rp2 quaternary inhibitory complex, resolving prior apparent discrepancies in the available RAB crystal structures. We show that while the C:RAB:Rp2 complex locks CBD-A in its off state, CBD-B samples a degenerate free-energy landscape with comparable populations of on and off states (Figs. 3F and 5B). When in the on state, CBD-B is disengaged from both the C-subunit and CBD-A and becomes inherently dynamic, explaining why the C:RAB:Rp2 complex is refractory to crystallization attempts. Interestingly, the Aoff-Bon RAB state in the C:RAB:Rp2 complex (Fig. 3F) resembles a transient cAMP-binding intermediate (Fig. 3H), given the B-to-A binding order of cAMP, which is opposite to Rp. Our model also explains why the crystal structures of RAB:cAMP2 and RAB:Rp2 are virtually identical (Fig. 1H) and why, in the absence of CBD-B, CBD-A adopts similar conformations when bound to C or 8-Br-Rp-cAMPS (34). The RAB:cAMP2 and RAB:Rp2 complexes share similar ground states with a closed topology, stabilized by interdomain interactions that selectively favor the AonBon state (Fig. 5A). However, upon deletion of CBD-B, the CBD-A/B contacts are lost and Rp-bound CBD-A samples the off state, as in the open topology of RAB:Rp2. Hence, our model is fully consistent with the previous crystallographic data. While these structures offer an essential framework to understand the interactions between Rp and the ground states of the R-subunit, it is clear that dynamics of the excited states sampled by R must be factored in to explain the inhibitory mechanism of Rp and pluripotent allostery in general (Fig. 5).
Another unique aspect of the model of Fig. 5 is that it unveils the key drivers of pluripotent allostery in PKA. The proposed EAM shows that agonism is driven by two key factors. First, Rp acts as an agonist for the isolated CBD-B but as an antagonist for the isolated CBD-A. The CBD-B agonism was unexpected, given the homology between the two tandem CBDs, but it is a primary driver of PKA activation because it reduces the affinity of R for C (Fig. 6A). Second, the A/B interdomain interactions promote agonism as they selectively stabilize the inhibitory-incompetent AonBon ground state with a closed interdomain topology relative to the excited states with open topologies (Fig. 5A), as consistently shown by both MD simulations and PRE experiments (Fig. 4 and figs. S4 to S6). Together, the Rp-induced stabilization of the on state of CBD-B and the CBD-A/B interactions favor agonism, while the Rp-induced stabilization of the off state of CBD-A promotes antagonism. The balance between these agonism- and antagonism-driving interactions controls the free-energy gap that separates the excited inhibitory states from the ground noninhibitory state of RAB:Rp2 (ΔGR,gap). The ΔGR,gap defines a critical threshold that must be overcome by the R:C affinity in order for antagonism to prevail over agonism (Fig. 5, B and C).
Metabolomic, proteomic, and genomic triggers of pluripotent allostery in PKA
On the basis of the proposed threshold model of pluripotent allostery, the antagonism-agonism switch occurs whenever the effective binding free energy for the association of the C-subunit with the R-subunit inhibitory excited states exceeds the ΔGR,gap (Fig. 5, B and C). This simple notion not only addresses the long-standing question of why the allosteric response of PKA to Rp is MgATP-dependent (Fig. 6A), but also reveals that the pluripotent allostery of PKA is a general phenomenon, with the antagonism-agonism switch controlled by other factors as well. These include the concentration and apparent affinity of kinase substrates (Fig. 6, B to E) and disease-related genetic mutations that weaken the R:C affinity. For example, Cushing’s syndrome driver mutations reduce the affinity of C for R, leading to aberrant constitutive activation of PKA (35). The Rp antagonism observed for WT PKA may suggest that Rp offers a viable means to reduce the overactivation of PKA typical of Cushing’s disease. However, given the marginal binding free energy between the mutated C-subunit and the R-subunit, our pluripotent allosteric model indicates that Rp is more likely to function as an agonist for Cushing’s patients.
The Cushing’s example illustrates the need to redefine the concept of allosteric drug target beyond the allosteric receptor to also account for conditions. The definition of allosteric target should also include the local metabolomic (e.g., MgATP), proteomic (e.g., kinase substrates), and genomic (e.g., disease-related mutations) contexts, as they serve as critical determinants of pluripotent allostery. An important implication of this broader redefinition of allosteric drug target is that the response to a given stimulus by a given receptor may vary depending on subcellular localization and on the specific signalosome in which PKA is integrated. Subcellular localization and signalosomes dictate the effective local concentrations and types of substrates and metabolites to which PKA is exposed (36, 37), potentially affecting pluripotent allostery.
Concluding remarks
We have shown that the allosteric response of the prototypical cAMP-dependent kinase cannot be recapitulated by a simple on-off switch as in traditional allostery, but is more similar to a three-way switch. In the latter, the response to one on-off switch is contingent upon another switch, as expected for pluripotent allostery. We show that one of the simplest but most effective explanations of such pluripotent allostery is a threshold response model in which the outcome of a given allosteric stimulus depends primarily on the free-energy gap that separates the excited inhibitory states from the ground noninhibitory state of the cyclic nucleotide monophosphate (cNMP)–bound R-subunit (ΔGR,gap). Notably, if both tandem domains responded similarly to Rp, ΔGR,gap would be either too low or too high relative to the free energy of R:C binding, resulting in either antagonism or agonism irrespective of experimental conditions. However, the differential response of the two CBDs to Rp ensures that ΔGR,gap is tuned to values more similar to the effective free energy of R:C association, leading to pluripotent allostery.
As part of the pluripotent allosteric model of PKA, we have mapped through NMR the sought-after dynamic conformational ensembles of the RAB:Rp2 and C:RAB:Rp2 complexes. These ensembles serve as the foundation for a model of kinase pluripotent allostery, which, in turn, enables quantitative predictions of kinase activity based on EAM input parameters measured through NMR. Hence, the approaches used here illustrate a general strategy, in which ensemble allosteric models bridge between dynamics as revealed by NMR and function as measured by enzyme assays. We anticipate the results and the experimental design proposed here to be transferable to other multidomain proteins functioning as molecular switches and signal transducers.
MATERIALS AND METHODS
Materials and protein expression and purification
Rp-cAMPS (>99% purity) was purchased from Biolog, while cAMP (>98.5% purity) was purchased from Sigma-Aldrich and S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanesulfonothioate (MTSL) was purchased from Toronto Research Chemicals. PKA R1a (91 to 379, 33 kDa), (119 to 379, 29 kDa), and (91 to 244, 17 kDa) constructs were expressed and purified according to previously published protocols (38, 39). PKA C-subunit (40 kDa) was expressed (38) or purchased (P2645, Sigma-Aldrich). AMP-PNP was purchased from Sigma-Aldrich. The kinase substrates, PKS (GRTGRRNSI) and PKS2 (GRTGRANSI), were synthesized and purchased from GenScript. The Kinase-Glo reagents were purchased from Promega.
General NMR spectroscopy
NMR data were acquired on a Bruker AVANCE 700-MHz spectrometer equipped with a 5-mm TCI cryoprobe. Unless otherwise specified, we processed the NMR data using either NMRpipe or Bruker’s Topspin with linear prediction and a resolution-enhancing 60° shifted sine-squared bell window function. Spectral analysis was implemented in NMRFAM-SPARKY using Gaussian line fitting. All experiments were acquired at 306 K in the NMR buffer [50 mM MOPS (pH 7.0), 100 mM NaCl, 10 mM MgCl2, 5 mM dithiothreitol (DTT), and 0.02% sodium azide] with 5% 2H2O, unless otherwise specified. TROSY triple-resonance three-dimensional (3D) experiments [i.e., CBCA(CO)NH, HNCACB, HNCA, and HN(CO)CA] and/or spectral comparisons, if no ambiguities were present, were used for spectral assignments.
Chemical shift analyses
Uniformly 2H,15N-labeled PKA R1a (91 to 379) WT, W260A, and (91 to 244) WT were concentrated to 50 or 100 μM in the NMR buffer [50 mM MOPS (pH 7.0), 100 mM NaCl, 10 mM MgCl2, 5 mM DTT, with or without 1 mM cAMP or 2 mM Rp-cAMPS, 0.02% sodium azide, and 5% 2H2O]. The C-subunit–bound R1a (91 to 379) complex was prepared with 1 mM AMP-PNP (Sigma-Aldrich) and with or without 2 mM Rp-cAMPS in the NMR buffer, as previously described (38). TROSY 2D experiments were recorded using 12 or 16 scans, a recycle delay of 1.7 s with 80 and 1024 complex points, and spectral widths of 31.8 and 15.9 ppm for the 15N and 1H dimensions, respectively. Chemical shift projection analyses (CHESPA) were implemented as previously described (38). The positions of the on versus off equilibria of W260A R1a (91 to 379), WT R1a (91 to 379), and C-bound WT R1a (91 to 379) were evaluated through chemical shift correlation slope analyses in the presence of 2 mM Rp-cAMPS to ensure both Rp-cAMPS binding sites were saturated. Residues 156, 157, 159, 162, 179, 215, 216, 217, 220, 221, and 223 were used for the CBD-A chemical shift correlation analyses, and residues 293, 296, 297, 317, 320, 339, 340, 343, 345, and 347 were used for CBD-B.
Measurement of R1a domain-specific Kd constants for Rp-cAMPS
Binding of Rp-cAMPS to the apo and C-bound R1a (91 to 379) was monitored by titration of Rp-cAMPS into 30 μM apo 2H, 15N-labeled R1a (91 to 379) or 50 μM 2H, 15N-labeled R1a (91 to 379):C samples and acquiring for each titration point 15N-1H 2D TROSY spectra using 128 scans and a recycle delay of 1.7 s with 80 and 1024 complex points and spectral widths of 31.8 and 15.9 ppm for the 15N and 1H dimensions, respectively. Peak intensity or chemical shifts were measured for the probe residues N186, G193, and S297 to extract the dissociation constant (Kd) values of Rp-cAMPS for CBD-A and CBD-B. The Kd values of Rp-cAMPS for R1a (91 to 379):C were determined from binding isotherms built through chemical shift changes, as the bound/unbound exchange is fast in the Rp-cAMPS titration into R1a (91 to 379):C (fig. S3). The Kd values of Rp-cAMPS for apo R1a (91 to 379) were determined from binding isotherms built through cross-peak intensities, as the bound/unbound exchange is slow in the Rp-cAMPS titration into apo R1a (91 to 379; fig. S3). Dilution effects for the titration of the apo R1a (91 to 379) sample were corrected by using a peak of a residue in the β-barrel that did not undergo notable chemical shift change upon binding of Rp-cAMPS (i.e., R340). The intensity or chemical shift change of the last titration point was used for normalization to calculate the domain-specific fraction bound (〈v〉). The domain-specific dissociation constants and respective errors were estimated through linear Scatchard regressions.
H/D exchange experiments
Protection factor (PF) values were measured by NMR-monitored H/D amide exchange for the WT and the W260A mutant of the R1a (119 to 379) construct. For this purpose, the protein was concentrated to 100 μM in 50 mM MES (pH 6.5), 100 mM NaCl, 5 mM DTT, 2 mM EDTA, 2 mM EGTA, 0.02% sodium azide, and 5% 2H2O. The cAMP-bound sample was prepared by adding excess 100 μM cAMP. Heteronuclear single-quantum coherence (HSQC) spectra using a recycle delay of 1.0 s with 128 and 1024 complex points and spectral widths of 31.8 and 15.9 ppm for the 15N and 1H dimensions, respectively, were recorded. For the first 30 HSQC spectra, four scans were acquired, while for the remaining 60 HSQC spectra, eight scans were used. To provide data points for the slow decaying peaks, additional HSQC spectra were acquired once per week. H/D exchange rates were measured using Levenberg-Marquardt nonlinear least square exponential fitting, as previously described (39). The maximal PF value for domain A (i.e., residues 119 to 244) was determined by identifying first the maximum logPF value and then selecting other CBD-A logPF values that fell within error from the maximum. The average and the SD of the logPF values selected according to this protocol were used as the logPFmax and the related error, respectively, in both WT and W260A R1a (119 to 379) constructs. The WT versus W260A difference in the average logPFmax values resulted in the ΔlogPFmax used for eq. S56 (text S3).
PRE analysis
Position 258 was selected as spin-label attachment point as it meets multiple requirements for optimal PRE measurements. Residue 258 is sufficiently removed from the C and cAMP binding sites and the interdomain interfaces to avoid substantial perturbations of these interactions by the introduction of the mutation and of the spin-labeled cysteine (Fig. 4B). Residue 258 is also sufficiently distant from CBD-A in the absence of interdomain interaction (>30 Å; Fig. 4B) but sufficiently close to CBD-A in the presence of interdomain interaction (<30 Å; Fig. 4C) to efficiently probe by PRE the transition between open and closed domain topologies. R1a (91 to 379) includes two cysteines, and both are located in CBD-B (i.e., residues 345 and 360). These cysteines are shielded from solvent in both the cAMP- and C-bound structures (PDB: 1RGS and 2QCS, respectively), exhibiting SASA values of 1.73 Å2 (residue 345) and 1.96 Å2 (residue 360) in the 1RGS structure and 1.23 Å2 (residue 345) and 1.17 Å2 (residue 360) in the 2QCS structure. Thus, we expect minimal or negligible attachment of MTSL to these cysteines.
The Asp258Cys PKA R1a (91 to 379) mutant was prepared according to the protocol used for WT R1a 91 to 379. The reducing reagent in the buffer was removed using a PD10 column before the addition of the MTSL spin label. The reduced Asp258Cys PKA R1a (91 to 379) was incubated with threefold molar excess of MTSL for 3 hours at room temperature. Excess MTSL was then removed with a PD10 column. The protein sample was then concentrated to 30 μM, and 2 mM cAMP or Rp-cAMPS were added. For the R1a (91 to 379):C and R1a (91 to 379):C:Rp2 samples, the MTSL was first covalently attached to the Asp258Cys R1a (91 to 379) mutant, as described above. After removing the excess MTSL with a PD10 column, the R:C complex was formed by incubating the Asp258Cys R1a (91 to 379) attached to MTSL with 1.5 molar excess of C-subunit overnight at 4°C in the presence of 1 mM AMP-PNP and then performing size exclusion chromatography to isolate the R:C complex. 15N-1H 2D TROSY spectra using a recycle delay of 1.7 s with 80 and 1024 complex points and spectral widths of 31.8 and 15.9 ppm for the 15N and 1H dimensions, respectively, were acquired for the paramagnetic samples. To obtain the diamagnetic control, 5 mM DTT was added and incubated for 1 hour at room temperature, after which we reacquired TROSY spectra. The total data acquisition time for PRE experiments was 30 hours for each complex, resulting in partial signal losses, presumably from partial aggregation of proteins, that needed to be corrected for a reliable analysis of PRE experiments. For this purpose, two independent approaches were taken. The first approach was to quantify the extent of sample decay by monitoring the residue-specific cross-peak intensities over time through the acquisition of multiple replicate TROSY spectra for each sample (i.e., oxidized or reduced). The average intensity loss was then calculated for both the oxidized (paramagnetic) and the reduced (diamagnetic) samples. On the basis of the average intensity losses, the sample decay occurring during PRE data acquisition was quantified and used to correct the PRE intensities accordingly. The second approach was taken to validate the first method and relies on the residues in β7 of CBD-B, which do not undergo changes in distance from 258 in the off and the on conformations. These residues were used as reference to normalize all other residues. For all four complexes analyzed by PRE (R:C, R:C:Rp2, C:Rp2, and R:cAMP2), the two approaches resulted in oxidized versus reduced ratios within experimental error. The analysis resulting from the first approach was used for the PRE plots in Fig. 4 (E and G to K). The error bars were generated starting from the errors on the intensities estimated using the signal-to-noise ratios of each peak and propagating these errors to the PRE intensity ratios (Iox/Ired).
Measurement of Michaelis-Menten constants through kinase assay
Phosphorylation in the presence of 10 nM PKA C-subunit (P2645, Sigma-Aldrich) and increasing concentration of PKS (GRTGRRNSI; 0 to 30 μM; GenScript) or PKS2 (GRTGRANSI; 0 to 300 μM; GenScript) was allowed to progress for 25 min in 50 μl of the assay buffer [40 mM tris (pH 7.5), 20 mM MgCl2, 10 μM adenosine 5′-triphosphate (ATP), and bovine serum albumin (BSA; 0.1 mg/ml)]. To reach the final reacting concentrations stated above, we diluted the stock concentrations of PKA C-subunit and the substrates 10-fold. The kinase-catalyzed reaction was terminated by adding 50 μl of the Kinase-Glo Luciferase Reagent (Promega) and was incubated for 10 min at room temperature before measuring the luminescence with a BioTek Cytation 5 spectrophotometer in triplicate. The Michaelis-Menten constant (Km) was determined through a nonlinear fitting of the Vo versus [S] plot using the Michaelis-Menten equation (Vo = Vo.max [S]/(Km + [S]).
Measurement of PKA holoenzyme activation by cAMP and Rp-cAMPS
Full-length PKA R1a (1 to 379) was purified following a protocol previously described (40). The activity of PKA C-subunit (P2645, Sigma-Aldrich) was determined by the luminescent kinase assay (Kinase Glo, Promega). The reaction mixture of 10 nM PKA C-subunit, 12 nM R1a (1 to 379), and 50 μM cAMP or 100 μM Rp-cAMPS was prepared in 40 mM tris (pH 7.5), 20 mM MgCl2, 10 μM ATP, and BSA (0.1 mg/ml). The reaction mixture was incubated for 15 min at room temperature, after which 50 μM substrate (PKS or PKS2) was added and allowed to react for 2 hours in 50 μl of the reaction mixture. To reach the final reacting concentrations stated above, we diluted the stock concentrations of PKA C-subunit, PKA R1a, cyclic nucleotides, and the substrates 10-fold. The kinase-catalyzed reaction was terminated by adding 50 μl of Kinase-Glo Luciferase Reagent (Promega) and was incubated for 10 min at room temperature before measuring the luminescence with a BioTek Cytation 5 spectrophotometer in triplicate. The relative kinase activity was calculated by normalizing based on the luminescence values corresponding to the maximum activity of C (i.e., in the absence of R1a and cNMP) and the maximum inhibition of C (i.e., in the presence of R1a but in the absence of cNMP), as per eq. S41 (text S2). The kinase activation data measured under these experimental conditions are consistent with ΔGAB and γ values of −4 (−2) RT and 0.4 (0.07), respectively.
MD simulations
MD simulations in explicit solvent were performed starting from several “hybrid” structures of the PKA R1a (91 to 379) construct containing both CBDs of PKA R1a, either unbound or in complex with the PKA 1a catalytic subunit (C-subunit; fig. S4 and table S3). The initial conformations for the simulations were constructed on the basis of x-ray crystal structures of the R-subunit with Rp-cAMPS ligands bound to both CBD domains (representing the on state of each CBD; PDB ID: 1NE4) and the R-subunit/C-subunit complex without bound Rp-cAMPS (representing the off state of each CBD; PDB ID: 2QCS). The simulations starting from the 1NE4 and 2QCS x-ray structures served as controls during analysis (fig. S4 and table S3). With the exception of the 2QCS control structure, all structures were simulated with Rp-cAMPS ligands bound to both CBD domains (table S3), and when Rp-cAMPS needed to be added to the 2QCS-derived CBD-A and CBD-B domains, the addition was achieved via overlay of the CBD-A and CBD-B domains with PDB structures 3PLQ and 1NE4, respectively (23, 34).
Initial structure preparation for MD simulations
A construct spanning residues 91 to 379 of R1a was used for the Rp-cAMPS–bound R-subunit simulations, and an additional construct spanning residues 13 to 350 of the PKA 1a C-subunit was included for all C-subunit–bound R-subunit simulations (fig. S4 and table S3). Initial structures for the 1NE4 and 2QCS structure control simulations (fig. S4, A and F) were obtained by first deleting all water molecules, and any R-subunit residues N-terminal to residue 91 or C-terminal to residue 379, from the respective PDB structures, and using Swiss PDB Viewer to reconstruct partially missing side chains on the protein surface (41). In addition, other nonprotein components besides bound Rp-cAMPS were deleted from the structures, with the exception of bound AMP-PNP and Mn2+ ligands found in the C-subunit of the 2QCS PDB structure, which were modified to ATP and Mg2+ by editing the PBD text file, in accordance with the conditions used in kinase assays. An Rp-cAMPS–bound version of the 2QCS structure was then obtained by docking Rp-cAMPS ligands into both CBD domains (table S3). The docking of Rp-cAMPS into the CBD-A domain was achieved by overlaying the x-ray structure of the 8-Br-Rp-cAMPS–bound CBD-A domain (PDB ID: 3PLQ) onto the CBD-A domain of the 2QCS structure at residues 152 to 226 (i.e., the CBD-A β-core), deleting the 8-Br substituent to obtain bound Rp-cAMPS, while docking of Rp-cAMPS into the CBD-B domain was achieved by overlaying the 1NE4 x-ray structure onto the CBD-B domain of the 2QCS structure at the CBD-B base-binding region (i.e., residues 297 to 302 and 311 to 316) and phosphate-binding cassette (i.e., residues 323 to 335) regions. An Rp-cAMPS–bound version of the 2QCS structure R-subunit lacking the bound C-subunit was also constructed, by deleting the atomic coordinates for the C-subunit (and its bound ATP and Mg2+ ligands) from the Rp-cAMPS–bound 2QCS structure (fig. S4C and table S3).
Initial structures for the hybrid structure simulations were obtained from the 1NE4 control and Rp-cAMPS–bound 2QCS structures by grafting the CBD-B domain from the 1NE4 control structure onto the CBD-A domain of the Rp-cAMPS–bound 2QCS structure via backbone superimposition at three different locations along the intervening α-helical region between the two CBD domains, saving the atomic coordinates for the 2QCS-derived CBD-A domain residues and 1NE4-derived CBD-B domain residues as a new structure (fig. S4, C to E and G to I, and table S3). The three graft locations were selected in accordance with kinks that were observed in the intervening α-helical region of the 1NE4 structure, but not in the 2QCS structure, resulting in three distinct CBD-A/CBD-B grafts (fig. S4, C to E and G to I, and table S3). The atomic coordinates for the 2QCS-derived C-subunit (and its bound ATP and Mg2+ ligands) were deleted from the grafted structures to obtain the three hybrid R-subunit structures lacking the bound C-subunit (fig. S4, C to E, and table S3) but were retained to achieve the respective C-subunit–bound hybrid structures (fig. S4, G to I, and table S3). Molecular structure topology and CHARMM parameters for Rp-cAMPS were calculated using the SwissParam software, as previously described (42).
MD simulation protocol
All MD simulations were performed using a previously described protocol (42) with box dimensions of 108 Å for the 1NE4 control structure, 116 Å for the three hybrid R-subunit structures, or 134 Å for the C-subunit–bound structures and used 2.1-GHz 32-core Broadwell compute nodes accelerated with two NVIDIA Pascal graphics processing units (GPUs) per node. These runs were executed for 400 ns at constant temperature and pressure, saving structures every 100,000 time steps (i.e., 100.0 ps) for subsequent analysis, resulting in a total simulation time of 4.0 μs.
Analysis of PKA structural dynamics
As an assessment of R-subunit structural propensities, RMSDs from the 1NE4 and 2QCS x-ray structures were computed for the CBD-A/CBD-B region of the R-subunit (i.e., residues 119 to 357 of PKA R1a), and the CBD-A and CBD-B domains (i.e., residues 119 to 231 and 243 to 357, respectively), over the course of all simulations. While the simulated PKA structures contain additional structural regions at the N and C termini of the R-subunit, these more flexible regions were excluded from the RMSD calculations to avoid the possibility of dynamics in these regions obscuring the structural dynamics of interest. These flexible regions consist of R-subunit residues N-terminal to residue 119 (i.e., the N-terminal linker) and C-terminal to residue 357 (i.e., the CBD-B domain C-helix and C-terminal tail, which adopt a more disordered structure in 2QCS). In addition, RMSDs from the 2QCS x-ray structure were computed for the CBD-A/CBD-B region of the R-subunit, and the CBD-A and CBD-B domains, together with the bound C-subunit over the course of all C-subunit–bound structure simulations to assess dynamics of the R- and C-subunits relative to one another, and the corresponding RMSDs of the C-subunit alone from the 2QCS x-ray structure (considering all C-subunit residues present in the structure) were computed to check for structural dynamics (if any) contributed by the C-subunit itself. The first 50 ns in each trajectory were not included in the computations to account for equilibration. Inter-CM distances between domains were computed as previously described (42). As in the RMSD calculations, residues 119 to 231 and 243 to 357 of the R-subunit were considered in the calculation of the CMs for the CBD-A and CBD-B domains, respectively, while all C-subunit residues present in the structure were considered in the calculation of the CM for the C-subunit. As for the RMSD computations, the first 50 ns in each trajectory were excluded.
Summary of experimental error analyses
The errors on the populations of each conformational state were estimated from the errors on the slopes of the linear regressions in Fig. 2 (C and D, G and H, and M and N). The errors on the Kd were estimated from the errors on the slopes of the Scatchard regressions (fig. S3). The errors associated with the PFs in fig. S1 were propagated from the errors on the H/D exchange rates obtained in the exponential fits. The errors on the minor versus major peak intensity ratio of R:Rp2 and the errors on the WT versus W260A RAB:cAMP2 TROSY intensity ratio were calculated through the SDs of the intensity ratio distributions (fig. S1A). Similarly, the errors for the average fractional inhibition of C:RAB:Rp2 were calculated through the SD of the fractional inhibition distribution from multiple residues (fig. S2A). The errors for the PRE intensity ratios (Iox/Ired) were obtained from the errors on the intensities estimated on the basis of the signal-to-noise ratio of each peak and error propagation. The errors for the kinase activities were obtained from the SD of triplicate measurements. Other reported errors were obtained through error propagation. As to the boxplots used for the MD simulation data, the middle, bottom, and top lines of the central box correspond to the median, 25th percentile, and 75th percentile of the dataset, respectively, while the whiskers denote points falling within 1.5*interquartile range (IQR) above the 75th percentile or below the 25th percentile. The IQR is the difference between the 75th and 25th percentiles. The square symbol within the box and the two “x” symbols represent the mean and the 1st and 99th percentiles of the dataset, respectively.
Supplementary Material
Acknowledgments
We thank L. Konermann, K. Green, C. Kalodimos, R. Ahmed, N. Jafari, S. Boulton, and K. Van for helpful discussions. Funding: This work was supported by the Canadian Institutes of Health Research grant 389522 (to G.M.), the Natural Sciences and Engineering Research Council of Canada grant RGPIN-2019-05990 (to G.M.), and NIH grant R35-GM130389 (to S.S.T.). Author contributions: J.A.B. and M.A. prepared samples; designed, executed, and analyzed all NMR experiments; and contributed to the development of the EAM and to the writing of the manuscript. B.V. carried out all MD simulations. T.S.L. contributed to the PRE measurements and assisted in sample preparation. S.S.T. contributed to the writing of the manuscript. G.M. designed the experiments and contributed to the development of the ensemble allosteric model, the analysis of the data, and the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eabb1250/DC1
REFERENCES AND NOTES
- 1.De Smet F., Christopoulos A., Carmeliet P., Allosteric targeting of receptor tyrosine kinases. Nat. Biotechnol. 32, 1113–1120 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Wodak S. J., Paci E., Dokholyan N. V., Berezovsky I. N., Horovitz A., Li J., Hilser V. J., Bahar I., Karanicolas J., Stock G., Hamm P., Stote R. H., Eberhardt J., Chebaro Y., Dejaegere A., Cecchini M., Changeux J.-P., Bolhuis P. G., Vreede J., Faccioli P., Orioli S., Ravasio R., Yan L., Brito C., Wyart M., Gkeka P., Rivalta I., Palermo G., McCammon J. A., Panecka-Hofman J., Wade R. C., Di Pizio A., Niv M. Y., Nussinov R., Tsai C. J., Jang H., Padhorny D., Kozakov D., McLeish T., Allostery in its many disguises: From theory to applications. Structure 27, 566–578 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen P., Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Whittier S. K., Hengge A. C., Loria J. P., Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 341, 899–903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saleh T., Rossi P., Kalodimos C. G., Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat. Struct. Mol. Biol. 24, 893–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang R., Ripstein Z. A., Rubinstein J. L., Kay L. E., Cooperative subunit dynamics modulate p97 function. Proc. Natl. Acad. Sci. U.S.A. 116, 158–167 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motlagh H. N., Hilser V. J., Agonism/antagonism switching in allosteric ensembles. Proc. Natl. Acad. Sci. U.S.A. 109, 4134–4139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu N. Z., Cidlowski J. A., Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol. Cell 18, 331–342 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Nguyen T. T., Ghirlando R., Venditti V., The oligomerization state of bacterial enzyme I (EI) determines EI’s allosteric stimulation or competitive inhibition by α-ketoglutarate. J. Biol. Chem. 293, 2631–2639 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulton S., Selvaratnam R., Blondeau J.-P., Lezoualc’h F., Melacini G., Mechanism of selective enzyme inhibition through uncompetitive regulation of an allosteric agonist. J. Am. Chem. Soc. 140, 9624–9637 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Dostmann W. R. G., Taylor S. S., Identifying the molecular switches that determine whether (Rp)-cAMPS functions as an antagonist or an agonist in the activation of cAMP-dependent protein kinase I. Biochemistry 30, 8710–8716 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Saloustros E., Salpea P., Starost M., Liu S., Faucz F. R., London E., Szarek E., Song W.-J., Hussain M., Stratakis C. A., Prkar1a gene knockout in the pancreas leads to neuroendocrine tumorigenesis. Endocr. Relat. Cancer 24, 31–40 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veugelers M., Wilkes D., Burton K., McDermott D. A., Song Y., Goldstein M. M., La Perle K., Vaughan C. J., O’Hagan A., Bennett K. R., Meyer B. J., Legius E., Karttunen M., Norio R., Kaariainen H., Lavyne M., Neau J.-P., Richter G., Kirali K., Farnsworth A., Stapleton K., Morelli P., Takanashi Y., Bamforth J.-S., Eitelberger F., Noszian I., Manfroi W., Powers J., Mochizuki Y., Imai T., Ko G. T. C., Driscoll D. A., Goldmuntz E., Edelberg J. M., Collins A., Eccles D., Irvine A. D., McKnight G. S., Basson C. T., Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and prkar1a haploinsufficient mice. Proc. Natl. Acad. Sci. U.S.A. 101, 14222–14227 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boikos S. A., Stratakis C. A., Carney complex: The first 20 years. Curr. Opin. Oncol. 19, 24–29 (2007). [DOI] [PubMed] [Google Scholar]
- 15.de Wit R. J. W., Hoppe J., Stec W. J., Baraniak J., Jastorff B., Interaction of cAMP derivatives with the “Stable” cAMP-binding site in the cAMP-dependent protein kinase type I. Eur. J. Biochem. 122, 95–99 (1982). [DOI] [PubMed] [Google Scholar]
- 16.Poppe H., Rybalkin S. D., Rehmann H., Hinds T. R., Tang X.-B., Christensen A. E., Schwede F., Genieser H.-G., Bos J. L., Doskeland S. O., Beavo J. A., Butt E., Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Taylor S. S., Ilouz R., Zhang P., Kornev A. P., Assembly of allosteric macromolecular switches: Lessons from PKA. Nat. Rev. Mol. Cell Biol. 13, 646–658 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim C., Cheng C. Y., Saldanha S. A., Taylor S. S., PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Huang L. J., Taylor S. S., Dissecting cAMP binding domain A in the RIα subunit of cAMP-dependent protein kinase. Distinct subsites for recognition of cAMP and the catalytic subunit. J. Biol. Chem. 273, 26739–26746 (1998). [DOI] [PubMed] [Google Scholar]
- 20.Akimoto M., McNicholl E. T., Ramkissoon A., Moleschi K., Taylor S. S., Melacini G., Mapping the free energy landscape of PKA inhibition and activation: A double-conformational selection model for the tandem cAMP-binding domains of PKA RIα. PLOS Biol. 13, e1002305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hao Y., England J. P., Bellucci L., Paci E., Hodges H. C., Taylor S. S., Maillard R. A., Activation of PKA via asymmetric allosteric coupling of structurally conserved cyclic nucleotide binding domains. Nat. Commun. 10, 3984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anand G. S., Krishnamurthy S., Bishnoi T., Kornev A., Taylor S. S., Johnson D. A., Cyclic AMP- and (Rp)-cAMPS-induced conformational changes in a complex of the catalytic and regulatory (RIα) subunits of cyclic AMP-dependent protein kinase. Mol. Cell. Proteomics 9, 2225–2237 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J., Jones J. M., Nguyen-Huu X., Ten Eyck L. F., Taylor S. S., Crystal structures of RIalpha subunit of cyclic adenosine 5′-monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′-cyclic monophosphothioate, the phosphothioate analogues of cA. Biochemistry 43, 6620–6629 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Marsiglia W. M., Katigbak J., Zheng S., Mohammadi M., Zhang Y., Traaseth N. J., A conserved allosteric pathway in tyrosine kinase regulation. Structure 27, 1308–1315.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonin J. P., Sapienza P. J., Wilkerson E., Goldfarb D., Wang L., Herring L., Chen X., Major M. B., Lee A. L., Positive cooperativity in substrate binding by human thymidylate synthase. Biophys. J. 117, 1074–1084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Simone A., Richter B., Salvatella X., Vendruscolo M., Toward an accurate determination of free energy landscapes in solution states of proteins. J. Am. Chem. Soc. 131, 3810–3811 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Narayanan C., Bernard D. N., Bafna K., Gagné D., Chennubhotla C. S., Doucet N., Agarwal P. K., Conservation of dynamics associated with biological function in an enzyme superfamily. Structure 26, 426–436.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herberg F. W., Dostmann W. R. G., Zorn M., Davis S. J., Taylor S. S., Crosstalk between domains in the regulatory subunit of cAMP-dependent protein kinase: Influence of amino terminus on cAMP binding and holoenzyme formation. Biochemistry 33, 7485–7494 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Lu T.-W., Wu J., Aoto P. C., Weng J.-H., Ahuja L. G., Sun N., Cheng C. Y., Zhang P., Taylor S. S., Two PKA RIα holoenzyme states define ATP as an isoform-specific orthosteric inhibitor that competes with the allosteric activator, cAMP. Proc. Natl. Acad. Sci. U.S.A. 116, 16347–16356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herberg F. W., Doyle M. L., Cox S., Taylor S. S., Dissection of the nucleotide and metal−phosphate binding sites in cAMP-dependent protein kinase. Biochemistry 38, 6352–6360 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., Varughese K. I., Regulatory subunit of protein kinase A: Structure of deletion mutant with cAMP binding domains. Science 269, 807–813 (1995). [DOI] [PubMed] [Google Scholar]
- 32.Motlagh H. N., Wrabl J. O., Li J., Hilser V. J., The ensemble nature of allostery. Nature 508, 331–339 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilser V. J., Wrabl J. O., Motlagh H. N., Structural and energetic basis of allostery. Annu. Rev. Biophys. 41, 585–609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badireddy S., Yunfeng G., Ritchie M., Akamine P., Wu J., Kim C. W., Taylor S. S., Qingsong L., Swaminathan K., Anand G. S., Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: Conformational selection highlights a new concept in allosteric inhibitor design. Mol. Cell. Proteomics 10, M110.004390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker C., Wang Y., Olivieri C., Karamafrooz A., Casby J., Bathon K., Calebiro D., Gao J., Bernlohr D. A., Taylor S. S., Veglia G., Cushing’s syndrome driver mutation disrupts protein kinase A allosteric network, altering both regulation and substrate specificity. Sci. Adv. 5, eaaw9298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musheshe N., Schmidt M., Zaccolo M., cAMP: From long-range second messenger to nanodomain signalling. Trends Pharmacol. Sci. 39, 209–222 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Aggarwal-Howarth S., Scott J. D., Pseudoscaffolds and anchoring proteins: The difference is in the details. Biochem. Soc. Trans. 45, 371–379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akimoto M., Selvaratnam R., McNicholl E. T., Verma G., Taylor S. S., Melacini G., Signaling through dynamic linkers as revealed by PKA. Proc. Natl. Acad. Sci. U.S.A. 110, 14231–14236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McNicholl E. T., Das R., SilDas S., Taylor S. S., Melacini G., Communication between tandem cAMP binding domains in the regulatory subunit of protein kinase A-Iα as revealed by domain-silencing mutations. J. Biol. Chem. 285, 15523–15537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J., Brown S., Xuong N.-H., Taylor S. S., RIα subunit of PKA: A cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure 12, 1057–1065 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Guex N., Peitsch M. C., SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–2723 (1997). [DOI] [PubMed] [Google Scholar]
- 42.VanSchouwen B., Melacini G., Role of dimers in the cAMP-dependent activation of hyperpolarization-activated cyclic-nucleotide-modulated (HCN) ion channels. J. Phys. Chem. B 122, 2177–2190 (2018). [DOI] [PubMed] [Google Scholar]
- 43.K. A. Dill, S. Bromberg, Molecular Driving Forces. Statistical Thermodynamics in Biology, Chemistry, Physics, and Nanoscience (Garland Science, 2010). [Google Scholar]
- 44.Moleschi K. J., Akimoto M., Melacini G., Measurement of state-specific association constants in allosteric sensors through molecular stapling and NMR. J. Am. Chem. Soc. 137, 10777–10785 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Selvaratnam R., Chowdhury S., VanSchouwen B., Melacini G., Mapping allostery through the covariance analysis of NMR chemical shifts. Proc. Natl. Acad. Sci. U.S.A. 108, 6133–6138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Knape M. J., Ahuja L. G., Bertinetti D., Burghardt N. C. G., Zimmermann B., Taylor S. S., Herberg F. W., Divalent metal ions Mg2+ and Ca2+ have distinct effects on protein kinase a activity and regulation. ACS Chem. Biol. 10, 2303–2315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gjertsen B. T., Mellgren G., Otten A., Maronde E., Genieser H.-G., Jastorff B., Vintermyr O. K., McKnight G. S., Døskeland S. O., Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1β action. J. Biol. Chem. 270, 20599–20607 (1995). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eabb1250/DC1