

A feasible and transformable cancer vaccine was designed for personalized immunotherapy of postoperative tumors.
Abstract
Autologous tumor cell–based vaccines (ATVs) are emerging as a transformable approach for personalized immunotherapy, but their therapeutic efficacy remains unsatisfying in patients with cancer. Here, we design a photodynamic therapy (PDT)–motivated ATV (P-ATV) in Fmoc-KCRGDK–phenylboronic acid (FK-PBA) hydrogel, which mobilizes local immune activation to inhibit relapse of postoperative tumors. The FK-PBA targeting overexpressed sialic acid on tumor cells can enable on-demand gelation in residue tumor areas and maintain continuous vaccination in surgical bed. Unlike neoantigen-based vaccine or adoptive cell therapy that takes several months to prepare, P-ATV can be easily manufactured within a few days and efficiently boost neoepitope-specific CD8+ T cells to activate personalized immunotherapy. This simple and powerful approach of engineered ATVs provides an alternative strategy for personalized immunotherapy and is readily transformable to various kinds of cell-based antigens to inhibit the relapse of postoperative tumors.
INTRODUCTION
Cancer vaccines have gained much attention for cancer treatment by harnessing the host immunity to fight against tumor cells (1), which elicits durable clinical benefits in preventing tumor growth, recurrence, and metastasis (2–5). Among the various strategies, autologous tumor cell–based vaccines (ATVs) show great potential in eliciting robust antitumor immune responses for personalized cancer immunotherapy (6, 7). However, only a small fraction of patients with cancer benefit from ATVs in clinic (8, 9), which could be resulting from the following disadvantages: (i) most of ATVs depend on ex vivo induction and engineering of autologous dendritic cells (DCs), which requires highly trained personnel and definitely increases treatment costs, delays dosing schedule, and causes unpredicted low effectiveness (6, 10, 11); and (ii) ATVs alone fail to elicit an adequate number of cytotoxic T lymphocytes (CTLs) with avid recognition of tumor antigens and barely promote the infiltration and activation of CTLs in tumor stroma (6, 8, 12). Therefore, feasible approaches that can amplify the population and activate the function of tumor-specific CTLs are highly desirable to enhance ATVs-based cancer immunotherapy.
Several strategies have been reported to improve ATVs-based immunotherapy (6, 13–16). The objective responses in patients with ovarian cancer are effectively improved by combining ATVs with chemotherapies (6). Microneedle patch (13) and hydrogels (14–16) have been used to codeliver whole tumor cells or lysates with other adjuvants such as granulocyte-macrophage colony-stimulating factor, cytosine-phosphodiester-guanine oligodeoxynucleotide, or even exogenous DCs, which evoke effective T effector response for tumor rejection in prophylactic and established mouse models. However, when dealing with postoperative tumors by ATVs-based therapy, it is still far from satisfactory by just evoking T cells (17). In practice, the manufacture of engineered ATVs should be simplified to meet the short-time interval between acquisition of autologous cell sources and postoperative vaccination (18). The ATVs-induced immune responses should be precisely controlled in surgical bed to reduce immune-related adverse effects (19). It is of great necessity to combine ATVs with other noninvasive therapeutic strategies that can scavenge residue tumors to consolidate surgery treatment (17, 20). It has been reported that photodynamic therapy (PDT) is a noninvasive strategy for treating tumors in the presence of near-infrared (NIR) light and photosensitizer (21). PDT can not only directly kill tumor cells through generating reactive oxygen species (ROS) but also stimulate an inflammatory environment to recruit DCs and other immune cells (22, 23). PDT enables up-regulation of heat-shock proteins to promote the efficacy of cross-presentation for antigens. PDT-induced proinflammatory effects also facilitate DCs’ migration, maturation, and antigen presentation (22). The photosensitizer-based PDT could perform adjuvant-like effects to mobilize strong antitumor immunity for tumor inhibition (24). Thus, we propose to combine ATVs with PDT for synergistically mobilizing neoepitope-specific CTLs and preventing relapse of postoperative tumors.
Here, we design a PDT-motivated autologous tumor cell vaccine (P-ATV) to locally mobilize antitumor immunity in surgical bed to inhibit tumor relapse. The vaccine is engineered by coating oxidized autologous tumor cells with polyethylenimine-conjugated chlorin e6 (PEI-Ce6) in 9-fluorenyl methoxycarbonyl (Fmoc)–KCRGDK–phenylboronic acid (FK-PBA) hydrogels (namely, PC-Cell@gel). PEI-Ce6 is efficiently coated on cell surface via electrostatic interaction and conducts PDT upon NIR laser irradiation. PBA, a sialic acid–targeting moiety, is conjugated to FK peptide to get FK-PBA. The FK-PBA can target residue tumors via the overexpressed sialic acid on tumor cells (25) (Fig. 1A), while on-demand gelation of FK-PBA can be activated by sequential injection of PEI-Ce6–coated autologous tumor cells (PC-Cell) that dispersed in 1 mM Na2CO3 solution (Fig. 1B). This tumor-specific gelation property will restrict antigens within residue tumor areas and maintain continuous immune activation in surgical bed. We find that PC-Cell@gel coupled with PDT can effectively mature DCs, suppress regulatory T cells (Tregs), boost neoepitope-specific CD8+ T cells, and inhibit tumor relapse in melanoma and colorectal mouse models with no evident systemic toxicity (Fig. 1C). The designed P-ATV is readily transformable to various kinds of cell-based antigens for personalized immunotherapy, which provides innovation for the next generation of ATVs.
Fig. 1. Scheme illustrating the formation and function of PC-Cell@gel.
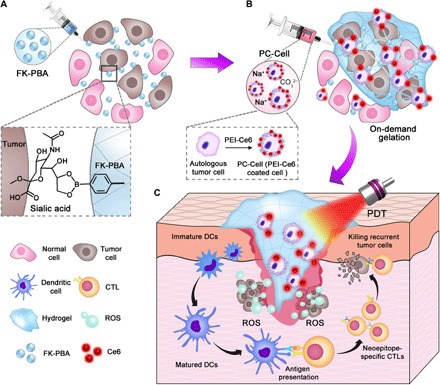
(A) On-demand gelation in residual tumor areas was activated via two steps. First, FK-PBA was locally injected into tumor surgical bed, enabling sialic acid–targeted accumulation of FK-PBA in residual tumors. (B) Then, PC-Cell was prepared by coating oxidized autologous tumor cells with PEI-Ce6. PC-Cell dispersed in 1 mM Na2CO3 solution was sequentially injected to activate the gelation of FK-PBA. (C) Upon NIR laser irradiation, engineered hydrogel vaccine matured DCs and mobilized neoepitope-specific CTLs in surgical bed. The synergetic effects of ATVs and PDT highly efficiently inhibit relapse of postoperative tumors.
RESULTS
Preparation and characterization of engineered ATVs
To prepare photosensitizer loaded autologous tumor cells, first, we conjugated Ce6 with hydrophilic PEI by amide condensation reaction (fig. S1A). Ce6 is a well-studied molecule for PDT (26). The successful synthesis of PEI-Ce6 was confirmed (fig. S1B). The molecular weight of PEI-Ce6 was determined as 5.98 × 103 Da (Mw) and 5.13 × 103 Da (Mn) and its polydispersity index (PDI) was 1.17 (fig. S1C), suggesting a cross-link conjugation between Ce6 and PEI molecules. The PEI-Ce6 conjugates performed similar ultraviolet-visible absorption spectrum as free Ce6 (fig. S1D). The hydrophilic cationic polymer, PEI-Ce6, can be easily coated on tumor cells via electrostatic interaction. Next, we prepared autologous tumor cells by dissociating resected tumor tissues into single cells. The collected cells were treated with hypochlorous acid (HClO) to induce apoptosis/necrosis. HClO is an effective bactericidal agent produced by activated neutrophils in inflammatory microenvironment. HClO can potentiate the immunogenicity of protein antigens by modifying them with an N-linked carbohydrate side chain and unfolding antigens to expose immunogenic peptides, thus promoting the recognition and presentation of antigens by DCs (27). It was reported that treating SKOV-3 ovarian tumor cells with HClO induced rapid primary necrosis, improved the uptake of protein antigens by DCs, and mobilized ovarian tumor-specific T cells (28). Moreover, HClO-treated tumor cells have been used to prepare DC-based vaccines for clinical use in previous studies (6). One hundred micromolar HClO was screened to prepare the oxidized autologous tumor cells, which induced almost total apoptosis/necrosis of the cells (fig. S2A). After that, the oxidized tumor cells were killed by freeze-thaw cycles and dispersed in PEI-Ce6 solution, allowing electrostatic adsorption between the cationic PEI-Ce6 and negatively charged cell membrane. Excess PEI-Ce6 was removed by centrifugation, and the separated PC-Cell was washed twice for further use. Tumor cells were killed by an optimized method without inducing thorough disruption of cell structure. The cells dispersed well in phosphate-buffered saline (PBS) after HClO incubation and two rounds of freeze-thaw cycle (fig. S2B). HClO and the freeze-thaw process triggered damages in cells as many holes were observed on the cell membrane. Compared to the oxidized autologous tumor cells, no obvious morphological change was observed after the coating of PEI-Ce6 on tumor cells (Fig. 2A and fig. S2C). The loading efficacy of PEI-Ce6 on tumor cells was concentration dependent, with a maximum of 44.6 ± 0.4 μg of PEI-Ce6 (equal to 14.7 μg of Ce6) per million cells (Fig. 2B). Note that the preparation of PC-Cell is ultrafast. The surface charge of tumor cells was reversed from −20.3 ± 1.0 mV to 4.3 ± 0.4 mV after incubating with PEI-Ce6 for 2 min, indicating the successful coating of PEI-Ce6 on tumor cells (Fig. 2C). Given that intracellular contents including antigens could be washed away during the preparation of PC-Cell, we investigated the variation of intracellular contents before and after the manufacture. We performed SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis for intracellular proteins. Protein samples were gained from an equal number of B16-F10 tumor cells, oxidized tumor cells, and B16-F10–derived PC-Cell. It was confirmed that consistent protein shift bands were observed in the three groups (Fig. 2D). We further examined four kinds of intracellular proteins including Toll-like receptor 3, nuclear factor κB p65, β-actin, and high-mobility group box 1. PC-Cell still retained these four proteins while showing a decrease in amount compared to the cell group (Fig. 2E). Above results indicate that our manufacturing process causes the decrease of intracellular proteins but does not result in the complete absence of intracellular contents in the product PC-Cell. Moreover, similar results of protein shift bands were observed in B16-ovalbumin (B16-OVA) and CT26-derived PC-Cell complexes (fig. S2D). Above results suggested the PC-Cell was potentially able to donate shared and private mutated protein antigens to prime antitumor immune responses.
Fig. 2. Preparation and characterization of PC-Cell@gel.
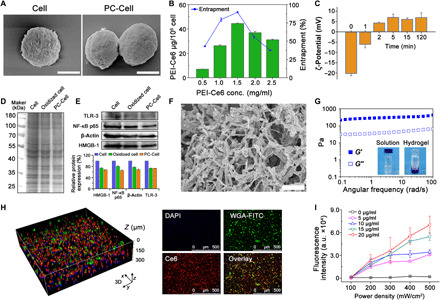
(A) Scanning electron microscopy (SEM) images of oxidized tumor cells and PC-Cell. Scale bars, 5 μm. (B) Loading and encapsulation efficacy of PEI-Ce6 on tumor cells. (C) ζ-Potential of PC-Cell as a function of PEI-Ce6 incubation time. (D) SDS-PAGE electrophoresis of proteins in B16-F10–derived PC-Cell complexes. (E) Western blot assay and semiquantification of intracellular Toll-like receptor 3 (TLR-3), nuclear factor κB p65 (NF-κB p65), β-actin, and high-mobility group box 1 (HMGB-1) proteins. (F) SEM image of FK-PBA hydrogel. Scale bar, 10 μm. (G) Frequency-dependent rheological properties of FK-PBA hydrogel at 37°C (peptide concentration, 0.5 weight %; strain, 1%). Inner photographs are FK-PBA solution and formed hydrogel. (H) Three-dimensional (3D) construction image of FK-PBA hydrogel encapsulating PC-Cell (left) and 2D images of PC-Cell@gel (right). The cell membrane was prelabeled with wheat germ agglutinin (WGA)–fluorescein isothiocyanate (FITC). Scale bars, 500 μm. DAPI, 4′,6-diamidino-2-phenylindole. (I) ROS generation following increased Ce6 concentration (5, 10, 15, and 20 μg/ml) and laser power density (100, 200, 300, 400, and 500 mW/cm2). Data are shown as means ± SD (n = 3). a.u., arbitrary units.
Self-assembling hydrogels show enormous advantages in local delivery of bioactive substances, allowing sustained and controllable drug release (29–33). Peptide-based scaffolds are used to deliver immune modulating molecules or antigens for enhanced cancer immunotherapy (14, 16). In addition, versatile peptides were developed to trigger self-assembly in situ or in living cells for biomedical applications (34). To prepare our tumor-targeting FK-PBA peptide, 4-mercaptophenylboronic acid was first reacted with 2,2′-dipyridyl disulfide (fig. S3) and sequentially reacted with the cysteine residue of FK (fig. S4). The FK-PBA was dissolved in water with a heating process and formed hydrogel with addition of 1 mM Na2CO3 solution to neutralize the residue carboxylic groups. When examined by transmission electron microscopy, it was found that the peptide self-assembled into ~200-nm fibers or ~ 60-nm spherical nanoparticles when dissolved in H2O (fig. S5A). However, the morphology of these nanostructures transformed to continuous network fibers after the addition of Na2CO3 solution to activate gelation (fig. S5B). The diameter and PDI of FK-PBA in H2O were 112.4 ± 0.4 nm and 0.18 while enlarged to 235.0 ± 6.1 nm and 0.55 in Na2CO3 solution by dynamic light scattering (fig. S5C), respectively. The three-dimensional structure morphology of FK-PBA hydrogel was confirmed by scanning electron microscopy (SEM) (Fig. 2F). The rheological behavior of hydrogel showed that angular frequency negligibly affected the stability of hydrogels. The storage modulus (G′) and loss modulus (G′′) were barely influenced by the frequency (0.1 to 100 rad/s) (Fig. 2G). Uniform and ordered dispersion of PC-Cell in FK-PBA hydrogel was observed by confocal laser scanning microscopy (CLSM) examination (Fig. 2H and fig. S6A), suggesting that the peptide hydrogel can effectively fix and maintain adequate antigen sources. We evaluated the impacts of PDT and hydrogel formation on the control-release profile of PEI-Ce6 and melanin from B16-F10–derived PC-Cell@gel (fig. S6, B and C). A low rate of PEI-Ce6 and melanin was released from PC-Cell@gel, while an increased level of PEI-Ce6 and melanin was released from the hydrogel after PDT at 24 hours. The cumulative release of PEI-Ce6 and melanin was improved along with time. PEI-Ce6 (11.7 ± 1.0%) and melanin (24.0 ± 2.0%) were released from PDT-treated PC-Cell@gel at 96 hours, indicating that photodynamic effects could facilitate the release of PC-Cell contents from hydrogel. Next, the ROS generation capability of PC-Cell@gel was determined by using singlet oxygen sensor green (SOSG). Increased production of ROS was detected along with the elevation of laser power density (Fig. 2I). Moreover, sufficient ROS production was achieved with an irradiated duration over 5 min (fig. S6D).
PDT effects and DC maturation in vitro
PBA moiety is able to form fast and stable boronate esters with diols of sialic acid overexpressed on various tumor cells (25), potentially leading to tumor-specific distribution of FK-PBA. The gelation of peptide can be activated with the addition of PC-Cell in 1 mM Na2CO3 solution. The Na2CO3 solution can not only regulate reassembly of FK-PBA but also potentially scavenge H+ in surgical bed to induce M1-like polarization of tumor-associated macrophages and decrease immunosuppressive cells (4, 35). To confirm the on-demand gelation of peptide in vitro, B16-OVA cells were seeded in cell culture dishes and incubated with FK-PBA or FK at 4°C for 4 hours. Then, unbound peptide was removed and PC-Cell in Na2CO3 solution was added to trigger the gelation (Fig. 3A). CLSM images showed that a thin layer of PC-Cell was formed over FK-PBA–treated B16-OVA cells (Fig. 3B), which could attribute to the targeting of FK-PBA with sialic acid on B16-OVA cells. Next, we tested the photodynamic and phototoxicity effects of PC-Cell@gel in vitro. After cocultured with PC-Cell@gel and irradiated by an NIR laser, a highly fluorescent signal of ROS probe 2,7-dichlorofluorescein diacetate (DCF) was observed in the cytoplasm of B16-OVA cells (Fig. 3C). It has been proved that sufficient ROS could lead to apoptosis and necrosis of tumor cells. Phototoxicity results showed that PC-Cell@gel + Laser significantly decreased viability of B16-OVA cells along with increased Ce6 concentration and laser power density (Fig. 3D). To investigate the capability of hydrogel vaccine to mature DCs, we treated bone marrow–derived DCs (BMDCs) with increased concentration of Cell@gel. The vaccine significantly enhanced the population of matured DCs (CD11c+CD80+CD86+), which was ~13.4-fold higher than that of the PBS group at a vaccine cell concentration of 2.0 × 105/ml (Fig. 3E). Similarly, a correlation was found between the DC maturation and the concentration of PC-Cell@gel. PC-Cell@gel–induced photodynamic effect further facilitated the maturation of BMDCs, which could attribute to the mobilized antigen release from oxidized tumor cells after laser irradiation (Fig. 3, F and G). As a result, upon NIR laser irradiation, PC-Cell@gel would generate plentiful ROS for PDT, promote DC maturation, and sequentially expand CTLs in vivo. As the in vitro gelation of FK-PBA has been confirmed, next we determined whether the gelation of FK-PBA could be activated in vivo. Mice with partially resected green fluorescent protein expressed CT26 (CT26-GFP) tumors were locally injected with cyanine 5.5 (Cy5.5; 5 mg/ml) labeled FK-PBA (FK-PBA-Cy5.5) or FK (FK-Cy5.5). One hundred microliter of 1 mM Na2CO3 solution was sequentially injected 4 hours later. Tissues in tumor surgical bed were harvested and sliced for CLSM imaging at 24 hours (Fig. 3H). Fluorescence signals of FK-Cy5.5 were detected in both GFP presence and GFP absence areas, which could be due to the lack of tumor cell targeting capacity of FK. In contrast, obvious colocalization of Cy5.5 and GFP was found in FK-PBA–treated mice, suggesting that FK-PBA enabled specific targeting to tumor cells and achieved on-demand gelation in residual tumor regions. Besides, we also determined the gelation and retention of FK-PBA by using a hydrophilic dye, rhodamine B. Rhodamine B dispersed in 1 mM Na2CO3 was injected 4 hours after the injection of FK-PBA, and tumor slices were prepared at 72 hours. With FK-PBA (1.0 mg/ml), little retention of rhodamine B was found within the tumor. However, bright fluorescence of rhodamine B was observed at the injection site with FK-PBA (5.0 mg/ml) (fig. S6E). The prolonged retention of rhodamine B indicated the formation of peptide hydrogel in surgical bed. Rhodamine B signals were detected in residue tumor areas while barely found in subcutaneous tissues (fig. S6F), which could attribute to the tumor-targeted distribution of FK-PBA.
Fig. 3. PC-Cell@gel induced PDT and DC maturation in vitro.
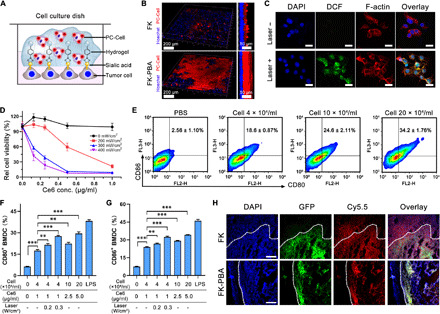
(A) Scheme illustrating the gelation of FK-PBA in vitro. Seeded B16-OVA cells were stained by Hoechst, fixed, and sequentially treated with FK-PBA and PC-Cell in 1 mM Na2CO3 solution. (B) 3D construction of PC-Cell@gel on the surface of B16-OVA cells (right). Scale bars, 200 μm. Side view of the 3D structures (left). Scale bars, 50 μm. (C) PC-Cell@gel induced ROS production in B16-OVA cells. Scale bars, 20 μm. (D) Laser power density–dependent phototoxicity of PC-Cell@gel. (E) Cell@gel promoted BMDC maturation following increased vaccine cell concentrations. (F) Percentage of CD11c+CD80+ BMDCs and (G) CD11c+CD86+ BMDCs after incubated with PC-Cell@gel or laser-pretreated PC-Cell@gel. LPS, lipopolysaccharide. (H) Mice with partially resected CT26-GFP tumors were first locally treated with FK-PBA-Cy5.5 or FK-Cy5.5 at 5 mg/ml, respectively. Four hours later, 1 mM Na2CO3 solution was sequentially injected. Tissues in surgical bed were collected and sliced for CLSM imaging at 24 hours. Scale bars, 200 μm. Data are means ± SD (n = 3). **P < 0.01, ***P < 0.001.
P-ATV inhibited relapse of B16-OVA tumor and elicited neoepitope-specific CTLs
To investigate whether PC-Cell@gel could elicit neoepitope-specific CTLs in vivo, we tested the vaccination capability on a postoperative B16-OVA tumor model (Fig. 4A). PDT alone barely suppressed the recurrence of this advanced malignancy as significant distinction was only found in early stages. However, PC-Cell@gel + Laser showed improved antirelapse performance in contrast to other groups (Fig. 4B). No obvious body weight drop was found in any group during the monitoring period (Fig. 4C). The infiltration of CTLs into tumor stroma was also examined by fluorescent staining of tumor slices. It was found that PDT promoted the infiltration of CTLs in tumors. Moreover, PDT-motivated PC-Cell@gel further effectively expanded the intratumoral infiltration of CTLs. Numerous brightly stained CD8+ T cells were observed in PC-Cell@gel + Laser group (Fig. 4D). Given that the improved therapeutic effects could attribute to activated neoepitope-specific CTLs, we examined the frequency of matured DCs and OVA-specific CTLs in prevaccinated C57BL/6 mice. PC-Cell@gel with PDT effectively promoted the maturation of DCs in vivo (Fig. 4E). It was found that all groups containing oxidized autologous tumor cells generated higher frequency of OVA-specific CD8+ T cells than the PBS or PC@gel + Laser group. The percentage of neoepitope-specific CTLs after PC-Cell@gel + Laser–treated was 13.6-fold higher than that of the PBS group (Fig. 4F). In terms of relapse inhibition in mice, the unsatisfied outcomes of Cell@gel and PC-Cell@gel may be due to the insufficient infiltration of these CTLs in tumors. Fortunately, PDT could not only systemically synergize the priming of neoepitope-specific CTLs but also promote the infiltration of CTLs into tumors by reducing the dense extracellular matrix, which has been reported in previous studies including ours (22, 23). Moreover, the population of central memory CD8+ T cells was more likely to be affected by PDT when examined on day 30 of the antirelapse study, showing an average of 4.2-fold increase in PC-Cell@gel + Laser group in contrast to the PBS group (fig. S7A). The results suggested that PC-Cell@gel coupled with PDT generated long-lasting immune response in vivo, potentially supporting the antirelapse efficacy. As evaluated by hematoxylin and eosin (H&E) analysis, no appreciable injury or inflammation was found in five major organs of C57BL/6 mice in control and treated groups (fig. S7B).
Fig. 4. P-ATV elicited neoepitope-specific CTLs to inhibit relapse of postoperative B16-OVA and B16-F10-Luc tumors.
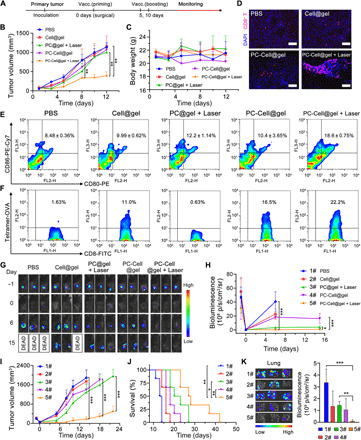
(A) Treatment schedule of antirelapse study in B16-OVA and B16-F10-Luc mouse tumor model. (B) Growth kinetics of recurrent B16-OVA tumors in C57BL/6 mice model (n = 4). (C) Body weight of mice during the antirelapse study (n = 4). (D) Fluorescent staining of CD8+ T cells in tumor slices collected from control and treated groups. (E) Lymphocytes isolated from lymph nodes of vaccinated mice were determined for the presence of matured DC (CD11c+CD80+CD86+). (F) Frequency of OVA-specific CD8+ T cells in peripheral blood 3 days after the second vaccination. (G) Bioluminescence images of B16-F10-Luc tumor–bearing mice collected on days −1, 0, 6, and 15 of the antirelapse study (n = 3). (H) Quantification of BLI in tumor surgical bed at desired time points after different treatments (n = 3). (I) Growth curves of relapsed B16-F10-Luc tumors in C57BL/6 mice (n = 6). (J) Survival kinetics of postoperative B16-F10-Luc tumor–bearing mice in all groups (n = 6). (K) Bioluminescence images (left) and BLI quantification (right) of lungs collected from metastatic B16-F10-Luc mouse model after various treatments (n = 3). Data are means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, we performed an antirelapse study on B16-F10-Luc tumor–bearing mouse model to evaluate the therapeutic effects of P-ATV. Tumor relapse was evaluated via the bioluminescence signals of B16-F10-Luc cells (Fig. 4, G and H). Similar to the results on the B16-OVA tumor model, PBS and Cell@gel treatments showed negligible suppression on the relapse of B16-F10-Luc tumors as strong bioluminescence intensity (BLI) was detected in surgical bed on day 6. PDT moderately inhibited the relapse of tumors as lower BLI and delayed tumor growth kinetics were found in the PC@gel + Laser group compared to PBS-treated mice (Fig. 4, H and I). Obviously decreased BLI and significantly suppressed tumor recurrence were observed in mice treated with PC-Cell@gel + Laser (Fig. 4I). Prolonged survival duration was achieved in mice receiving PC-Cell@gel + Laser in contrast to other groups (Fig. 4J), which could attribute to P-ATV–induced antitumor immunity and PDT-based scavenge of residual tumor cells. Considering the potential risk of tumor metastasis after surgery and PDT, we also evaluated the antimetastasis capacity of P-ATVs in a metastatic B16-F10-Luc mouse tumor model. Strong bioluminescence signals of metastatic lesions were detected in lungs of mice treated by PBS. In stark contrast, extremely low intensity in lungs was found in mice with PC-Cell@gel + Laser (Fig. 4K). These results indicated that P-ATVs could inhibit the metastasis of B16-F10-Luc to lungs by eliciting strong antitumor immunity.
P-ATV promoted antitumor immunity in postoperative CT26 mouse model
P-ATV is readily transformable to other types of tumors for personalized immunotherapy. To prove this, we applied PC-Cell@gel on a postoperative CT26 mouse model. First, we investigated the retention of CT26-derived PC-Cell@gel at injection site in vivo. The mice were examined by an IVIS (In Vivo Imaging System) imaging system on days 0, 7, 14, 21, and 28 after treated by PC-Cell or PC-Cell@gel, respectively. Fluorescent signals were detected in both groups, showing a modest decrease of Ce6 intensity in 28 days (Fig. 5A). Effectively, a stronger retention signal was observed in PC-Cell@gel–treated mice than that of the PC-Cell group from day 14. In addition, signal of PC-Cell@gel could still be detected on day 28 even though its strength decreased by 72% compared with day 0 (Fig. 5B), indicating that the hydrogel enabled prolonged retention of PC-Cells in situ. The long-lasting retention feature of PC-Cell@gel is favorable for continuous stimulation of DCs in vivo. Intrigued by prolonged retention of the hydrogel vaccine, we investigated the influence of PC-Cell@gel on antitumor immune responses in postoperative CT26 mouse models (Fig. 5C). Tumor necrosis factor–α (TNF-α) and interferon-γ (IFN-γ) in mice serum were examined by enzyme-linked immunosorbent assay (ELISA) kits on days 3, 6, and 9 to determine inflammatory status at different stages (Fig. 5, D and E). Compared to PBS-treated mice, the secretion of TNF-α and IFN-γ was significantly elevated in vaccinated groups after the first vaccination. The TNF-α and IFN-γ levels in PC-Cell@gel + Laser–treated mice were further enhanced on day 6 after secondary vaccination. The cytokine levels in all vaccinated groups were decreased on day 9, while mice treated with PC-Cell@gel + Laser maintain higher cytokine levels than the other groups. Following the promotion of inflammatory status in vivo, the frequency of tumor-infiltrated CD8+ T cells performed a similar trend. Significant elevation of CD8+ T cells was found in PC-Cell@gel + Laser group on day 4 and kept >14% over 12 days (Fig. 5F). The population of CD8+ T cells in the PC@gel + Laser group showed a slight decrease on day 12, which was possibly due to the lack of sufficient tumor antigens in situ. After immunized with Cell@gel or PC@gel + Laser on day 8, the frequency of tumor-infiltrated CD8+ T cells was modestly increased to 8.22 ± 2.67% and 11.8 ± 1.16%, respectively. In contrast, PC-Cell@gel + Laser treatment induced significant elevation of CD8+ T cells to 14.5 ± 2.09% (Fig. 5G), suggesting that PDT could synergistically amplify the activation of CTLs in tumor surgical bed. In addition, vaccination with PC-Cell@gel + Laser reduced the population of Tregs in residue tumors. The frequency of Tregs decreased from 32.7 ± 3.41% in the PBS group to 10.6 ± 2.32% in the final group (Fig. 5H), relieving the suppressive effect of Tregs on immunity priming. We further normalized the number of tumor-infiltrated lymphocytes in control and treated groups. Cell@gel and PC-Cell@gel induced a modest increase of CTLs in tumor, while PC@gel-based PDT performed enhanced immune priming effects. The normalized number of infiltrated CD8+ T cells in PC-Cell@gel + Laser–treated mice was 14.2-fold higher than that of the PBS group (Fig. 5I). Moreover, the average number of functional IFN-γ+ CD8+ T cells was also significantly elevated when immunized by PC-Cell@gel + Laser (Fig. 5J). Although the population of Tregs was reduced after the treatment of vaccines, the normalized number of Tregs in surgical bed of the PC@gel + Laser and PC-Cell@gel + Laser groups was still higher than that of the PBS group (fig. S8A), which could be explained by the significantly increased number of CD4+ T cells after PDT and vaccination (fig. S8B). Fortunately, the elicitation of CD8+ T cells in the PC-Cell@gel + Laser group was much stronger than that of Tregs, leading to an enhanced CD8+ T cell–to–Treg ratio compared to the other groups (Fig. 5K). To investigate the potential cytotoxicity of PC-Cell, we examined the effects of PEI and PC-Cell on the viability of tumor-infiltrated lymphocytes. To avoid the influence of various factors in a tumor microenvironment, tumor-infiltrating lymphocytes were separated from mice bearing CT26 tumors and cocultured with gradient concentrations of PEI, PEI-Ce6, and PC-Cell at equal content of PEI. The potential cytotoxicity of the above suspensions was evaluated by an annexin V–fluorescein isothiocyanate (FITC) apoptosis detection kit in early stages. The results showed that PEI (5.0 μg/ml) induced apoptosis in tumor-infiltrated lymphocytes as early as 4 hours, while no significant apoptosis of lymphocytes was observed in PEI-Ce6– and PC-Cell–treated groups at the same time (fig. S9A). The viability of lymphocytes was evaluated by a Cell Counting Kit-8 assay kit. After incubating for 48 hours, free PEI resulted in a sharply decrease in cell viability following increased concentrations. However, much lower cytotoxicity on lymphocytes was found in PC-Cell groups (fig. S9B). As PEI-Ce6 closely coated on ATVs via electrostatic interaction, it could weaken the contact of PEI with lymphocytes, thus potentially leading to the decreased cytotoxicity of PC-Cell. Together, these results indicated that the designed P-ATV elicited robust antitumor immune responses in the postoperative CT26 tumor model.
Fig. 5. P-ATV promoted antitumor immunity in postoperative CT26 tumor model.
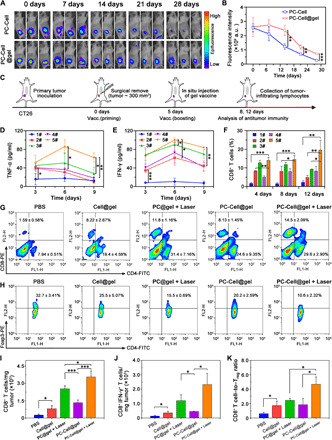
(A) Fluorescent imaging of PC-Cell– and PC-Cell@gel–treated mice by subcutaneous injection within 28 days. (B) Time course quantification of average fluorescence intensity at injection sites (n = 3). (C) Vaccination schedule for immunotherapy. (D) Secretion levels of TNF-α and (E) IFN-γ in serum on days 3, 6, and 9 after treatments, respectively. (F) Frequency of CD8+ T cells in tumor surgical bed on days 4, 8, and 12 in control and treated groups. (G) Frequency of CD4+ and CD8+ T cells in lymphocytes harvested from tumor surgical bed in control and treated groups on day 8. (H) Frequency of Tregs in tumor surgical bed with different treatments on day 8. (I) Weight-normalized number of CD8+ T cells and (J) IFN-γ+ CD8+ T cells in residue tumors of control and treated groups. (K) CD8+ T cell–to–Treg ratio in tumor surgical bed. Data are means ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
PDT-motivated vaccination prevented relapse of postoperative CT26 tumors
Inspired by the amplified antitumor immunity, we investigated therapeutic effects of P-ATV to prevent tumor relapse in partially resected CT26 murine models. As primary tumor maintains an immunosuppressive microenvironment, the immune responses generated by directly PDT on primary tumors could be suppressed. However, in the treatment of postoperative objects with lifted tumor burden, P-ATV could manifestly enhance the therapeutic benefits. The vaccine products were injected into the tumor surgical bed and irradiated by a 655-nm laser 2 hours later for auxiliary elimination of residual tumors (Fig. 6A). Mice immunized with PC-Cell@gel + Laser performed delayed tumor relapse as compared to the other groups, with 40% of mice free of tumor relapse during the monitoring period (Fig. 6B). The inhibited tumor relapse correlated with the survival of mice. None of the mice survived over 25 days in the other four control groups, while significantly prolonged survival of mice was found after PC-Cell@gel + Laser treatment (Fig. 6C). Moreover, no significant drop in mouse body weight was observed after treatment with hydrogel vaccine and PDT (Fig. 6D), indicating that the designed P-ATV is well tolerated by the animal. To further evaluate the biosafety of hydrogel vaccine, we performed serological and physiological-biochemical analysis in mice (Fig. 6E). Vaccination with PC-Cell@gel + Laser showed a negative impact on the number of leukocytes, red blood cells, platelets, and hemoglobin in contrast to the PBS group. However, the percentage of lymphocytes was properly elevated by PC-Cell@gel + Laser, which consisted with the activation of antitumor immune responses in tumor surgical bed. High aspartate aminotransferase–to–alanine aminotransferase (AST/ALT) ratio and total bilirubin (TBL) were detected in PBS-treated mice, while much lower AST/ALT ratio and TBL were found in the PC-Cell@gel + Laser group, indicating that P-ATV–based therapy could not only lift the tumor burden but also potentially reduce liver impairment. Decreased level of creatinine was found in the vaccinated groups, which may correlate with improved kidney function in mice. No significant difference was found in serum lactic dehydrogenase, and improved serum glucose was detected in PC-Cell@gel + Laser–treated mice. The above results provide preliminary investigation of the biosafety of P-ATV. H&E staining of recurrent tumors demonstrated that the hydrogel vaccine cooperating with PDT induced obvious apoptosis/necrosis in tumor cells as a presence of nuclear collapse and cytoplasmic rupture (Fig. 6F). Besides, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) also exhibited that most tumor cells were apoptotic after treated by PC-Cell@gel + Laser, while few events were found in the PBS group (Fig. 6G). These observations indicated that in situ implantation of our hydrogel vaccine was feasible and effective to inhibit relapse of postoperative tumors.
Fig. 6. Inhibiting the relapse of postoperative tumor by PC-Cell@gel.
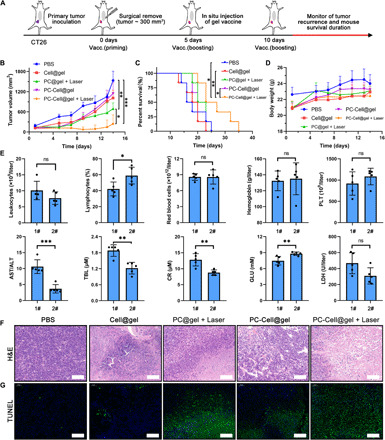
(A) Treatment schedule in CT26 recurrent mouse tumor model. (B) Average growth kinetics of recurrent tumors after treated by different suspensions (n = 5). (C) Survival percentage of CT26 recurrent tumor-bearing mice in control and treated groups (n = 6). (D) Body weight of mice in all groups (n = 5). (E) Serological and physiological-biochemical analysis of vaccinated mice was performed at the end of study (n = 5). 1#, PBS; 2#, PC-Cell@gel + Laser. (F) Histological assessment of recurrent tumors. Scale bars, 100 μm. (G) TUNEL staining of tumor sections in all groups. Scale bars, 100 μm. Data are means ± SD. Statistical significance was calculated by one-way ANOVA with Tukey’s post hoc test and log-rank (Mantel-Cox) test (ns, no significance; *P < 0.05, **P < 0.01, ***P < 0.001).
DISCUSSION
In this study, we have demonstrated that an engineered ATV, with a feasible producing procedure and tumor-specific gelation properties for in situ vaccination, can efficiently generate ROS with an NIR laser, mature BMDCs to present multiple tumor antigens, mobilize neoepitope-specific CTLs, significantly suppress the relapse of residual tumors, and maintain long-lasting immune responses. As previously reported, neoepitope-specific T cells can be boosted following vaccination with synthetic peptides or mRNA, realizing durable therapeutic benefits in partial patients (3, 36). Versatile nanoplatforms and scaffolds have been developed to deliver multiple tumor-specific antigens (37–40), such as a cocktail of neoantigens for cancer immunotherapy (41). Unfortunately, the sequencing and scale-up manufacturing of neoantigens are time consuming and high priced. It takes several months to identify, confirm, and produce those vaccines, which is unfavorable for patients with advanced tumors. In stark contrast, the production of our P-ATV is convenient and practicable to meet the critical time window for malignancy control. The autologous tumor cells can not only provide numerous antigens to expand multiple T cells but also elicit neoepitope-specific T cells for personalized immunotherapy. To overcome the deficiency of ATVs alone, ATVs are engineered with PDT to induce abundant ROS and timely scavenge residual tumors after surgery. PDT is suitable for the treatment of most superficial tumors, while the preferred clinical strategy for these tumors is surgery in general. The amount of residual tumor cells in primary site varies with the operation and tumor’s invasion to adjacent tissues. In practice, most tumor tissues are removed by surgery, leading to insufficient antigen sources left to prime strong antitumor immunity by PDT. Therefore, it is well matched to combine ATVs with PDT to maintain adequate antigens in situ. Our designed P-ATVs provide a noninvasive strategy for both scavenging residual tumor cells and eliciting effective antitumor immunity. In addition, in situ administration of the hydrogel vaccine is readily accessible during the surgical operation for solid tumors, releasing the inaccessibility limitation of most therapeutics in patients with cancer. Moreover, the tumor-specific gelation of FK-PBA fixes antigens and further improves its retention in residue tumor regions, which enables sustained immune stimulation and may reduce the risk of immune-related adverse effects.
Overall, we demonstrate a promising and powerful strategy to design personalized cancer vaccine for postoperative tumor treatment. In the case of future clinical translation, acquiring sufficient individual tumor sources to produce vaccine is vital for ensuring sustainable therapeutic regimen, and thoroughly investigating the biosafety of the hydrogel vaccine is also crucial. Moreover, combining P-ATV with immune checkpoint inhibitors may further amplify the therapeutic benefits by unleashing the function of CTLs. As the urgent demand of personalized approaches for treating individual patients, our vaccination strategy mobilizing neoepitope-specific CTLs at the site of interest may be generally applicable for postoperative treatment of various solid tumors.
MATERIALS AND METHODS
Materials
FK was obtained from Bankpeptide Biological Technology co., Ltd. Mercaptophenylboronic acid, chlorin e6, 2,2′-dipyridyl disulfide, and lipopolysaccharide (LPS) were purchased from J&K Scientific. PEI (Mw = 800 Da) and 2′,7′-dichloro-dihydro-fluorescein diacetate (DCFH-DA) were purchased from Sigma-Aldrich. Annexin V–FITC/propidium iodide (PI) apoptosis detection kit was gained from Meilunbio. Co. Ltd. Rhodamine B–labeled phalloidin was purchased from Yeasen Biotechnology Co., Ltd. Mice peripheral lymphocyte separation buffer and red blood cell lysis buffer were purchased from Dakewe Biotech Co., Ltd. Anti–TNF-α and anti–IFN-γ ELISA kits were obtained from Neobioscience Technology Co., Ltd. Flow cytometry antibodies were purchased from eBioscience, BD Pharmingen, and BioLegend. Other chemicals and reagents were analytical graded and purchased from Sinopharm Group Chemical Reagent Co., Ltd.
Cell lines and animals
Murine CT26 colorectal cancer cells and B16-OVA melanoma cells were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China). B16-F10-Luc melanoma cells were obtained from Shanghai Sciencelight Biology Co., Ltd. CT26 cells were cultured in complete RPMI 1640 cell culture medium containing 10% fetal bovine serum (FBS) and penicillin G sodium and streptomycin sulfate (100 U/ml). B16-OVA and B16-F10-Luc cells were cultured in Dulbecco’s minimum essential medium cell culture medium containing 10% FBS and penicillin G sodium and streptomycin sulfate (100 U/ml). Cell cultures were maintained at 37°C in 5.0% CO2 atmosphere. Four-week-old female BALB/c mice and C57BL/6 mice were obtained from the Shanghai Experimental Animal Center (Shanghai, China). Animal procedures were carried out under the guidelines approved by the Institutional Animal Care and Use Committee of the Shanghai Institute of Material Medica, Chinese Academy of Sciences.
Synthesis of PEI-Ce6 and FK-PBA
To synthesize PEI-Ce6, 100 mg of Ce6, 143.25 mg of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, and 101.25 mg of hydroxybenzotriazole were dissolved in 10 ml of anhydrous dimethyl sulfoxide (DMSO). The solution was stirred for 1.5 hours in the dark and then dropwise added into 800 mg of PEI dissolved in 2 ml of DMSO under stirring. The reaction was protected from light for 24 hours. The crude product was first dialyzed against DMSO, followed by purified water, and then lyophilized. The final product was characterized using proton nuclear magnetic resonance spectrum examination, and the molecule weight was confirmed by gel permeation chromatography. For FK-PBA synthesis, 100 mg of mercaptophenylboronic acid and 171 mg of 2,2′-dipyridyl disulfide were dissolved in methanol and reacted for 2 hours. The product was vacuum-dried and precipitated in n-hexane to get 4-(dipyridyl disulfide)-PBA. Then, 50 mg of 4-(dipyridyl disulfide)-PBA and 200 mg of FK were mixed in 10 ml of methanol and reacted overnight at 50°C. The final product, FK-PBA, was purified by precipitation in diethyl ether and vacuum-dried for further use. To synthesize Cy5.5 labeled FK-PBA and FK, the peptide solutions were reacted with N-hydroxysuccinimide ester–modified Cy5.5 for 6 hours. Then, the products were dialyzed against ethanol and purified water and lyophilized for further use.
Preparation and characterization of PC-Cell
Murine B16-OVA, B16-F10-Luc, and CT26 cells were collected from tumor xenografts. Tumors in mice were surgically removed, collected, and cut into small pieces with a diameter <1.0 mm in biological safety cabinet. Then, cell sources were immersed in RPMI 1640 cell culture medium containing hyaluronidase (100 U/ml), collagenase IV (175 U/ml), and deoxyribonuclease (DNase) (30 U/ml) for 30 min at 37°C. The cell suspensions were filtered with a 75-μm filter. After that, the tumor cells were precipitated and redispersed in 100 μM HClO solutions for 1 hour at 37°C and then transferred to 4°C for 1 hour. The HClO-treated tumor cells were gently washed by cold PBS and redispersed in PBS at a concentration of 1 × 107/ml. The suspensions were completely frozen at −30°C for 30 min and thawed at 4°C. The freeze-thaw cycle was repeated twice, and cell viability was examined by live/dead staining assay. Then, PEI-Ce6 (1.5 mg/ml) was added and the mixture was gently shaken at 37°C, 120 rpm for 20 min to get the final product PC-Cell. To detect the loading efficiency of Ce6 in PC-Cell, the standard curve of Ce6 and PEI-Ce6 was first constructed by a fluorescence spectrophotometer [Ex (excitation)/Em (emission): 405 nm/676 nm]. On the basis of the standard curve, the cell number of PC-Cell was counted and the loading efficacy of Ce6 was detected after the ultrasonic decomposition of PEI-Ce6.
For morphology investigation of oxidized autologous tumor cell and PC-Cell, SEM samples were prepared as following steps. The samples were fixed in 4% paraformaldehyde, washed twice with purified water, and dehydrated with gradient increased ethanol. The products were dropped on the silicon wafer, vacuum-dried, and examined by SEM. To verify the protein contents in PC-Cell complexes, SDS-PAGE electrophoresis of extractive proteins was performed. PC-Cell complexes manufactured from CT26 and B16-OVA cells were first treated with or without a 655-nm laser at a power density of 300 mW/cm2 for 5 min. Then, proteins in all samples were extracted, and electrophoresis was carried out at a voltage of 80 V. After that, the gels were treated by Coomassie blue staining and decolorized until the protein strips were clearly displayed. Last, the gel samples were examined by the Bio-Rad MP Imaging System. For Western blot analysis, protein samples were obtained from an equal number of tumor cells, oxidized tumor cells, and PC-Cell. The proteins were separated by electrophoresis, transferred to a nitrocellulose membrane, and incubated against primary antibodies. Then, the protein shift bands were stained by horseradish peroxidase–conjugated secondary antibodies and visualized by the Bio-Rad MP Imaging System. To investigate the dynamic formation of PC-Cell, cells were cultured with PEI-Ce6 solution for different time durations. Then, the PC-Cell complexes were separated at desired time points and dispersed in 20 mM Hepes for ζ-potential measurement. Blank cells without PEI-Ce6 were set as start point.
Generation of ROS and phototoxicity
The generation of ROS after laser irradiation was determined by using the ROS fluorescent probe SOSG. To evaluate the influence of Ce6 concentration and laser power density on ROS production, 100 μM SOSG was dissolved in 100 μl of PC-Cell@gel. The Ce6 concentration was set at 0, 5, 10, 15, and 20 μg/ml, respectively. The samples were irradiated by a 655-nm NIR laser with gradient power density for 1 min. The fluorescent intensity of SOSG after treatments was examined by a microplate reader (Ex/Em: 504 nm/525 nm) (PerkinElmer, USA). To determine the effect of irradiation duration on ROS production, the laser power density was fixed at 300 mW/cm2, and Ce6 concentration was 10 μg/ml. The samples were exposed to laser irradiation for 1, 3, 5, 7, and 10 min, respectively.
To examine the PC-Cell@gel–based ROS generation in vitro. B16-OVA cells were seeded on glass dishes at a density of 3 × 104 cells per well overnight. The cells were treated by 10 μM DCFH-DA (Ex/Em: 488 nm/520 nm) and PC-Cell@gel at an identical Ce6 concentration of 10 μg/ml for 30 min. Then, selected groups were irradiated by an NIR laser at a power density of 300 mW/cm2 for 5 min. After that, nucleus and F-actin of the cells were stained by 4′,6-diamidino-2-phenylindole (Ex/Em: 405 nm/460 nm) and phalloidin, respectively, and examined by CLSM (Leica TCS-SP8 STED, Germany).
For phototoxicity examination, the B16-OVA cells were seeded at 5000 cells per well in 96-well plates for 24 hours. The cells were treated with increased gradient concentration of PC-Cell@gel and cultured for 6 hours. After that, the cells were irradiated by a 655-nm laser for 1 min at a series of power densities. With another continuous incubation for 24 hours, the viability of B16-OVA cells was evaluated by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay.
DC maturation in vitro
To investigate the effects of PC-Cell@gel on DC maturation in vitro, BMDCs were generated from the bone marrow of 6-week-old C57BL/6 mice with particular cytokines. On the fifth day, immature BMDCs were treated with Cell@gel, PC-Cell@gel, laser-pretreated PC-Cell@gel, and LPS and cultured for another 24 hours. LPS-treated BMDCs (1.0 μg/ml) were set as positive control. Anti–CD80-PE and anti–CD86-PE antibodies were used in individual groups to avoid the interference of Ce6. The maturation of BMDCs was examined by using a FACSCalibur flow cytometer (BD Biosciences, USA).
Postoperative tumor models and treatments
The antitumor effect of the PC-Cell@gel coupled with PDT was performed in postoperative CT26, B16-OVA, and B16-F10-Luc mouse tumor models. To establish the tumor-bearing mice, 4-week-old BALB/c mice or C57BL/6 mice were subcutaneously injected with 1 × 106 of CT26 cells, B16-OVA cells, or B16-F10-Luc cells in the right flank, respectively. The tumors of mice were partially removed when the tumor volume reached 300 mm3. All mice were randomly divided into five groups: PBS, Cell@gel, PC@gel + Laser, PC-Cell@gel, and PC-Cell@gel + Laser group. The mice were treated with FK-PBA (10 mg/ml) in surgical bed for 4 hours and sequentially treated by PC-Cell at an equal Ce6 dose of 1.0 mg/kg. The dosage of Ce6 for per mouse is ~20 μg. According to the loading efficacy of PEI-Ce6, 1.0 × 106 of tumor cells were able to load ~14.7 μg of Ce6. Therefore, it can be calculated that the number of cells used for one mouse in a single treatment is ~1.36 × 106. More than 1.0 × 107 of tumor cell sources could be collected from a resected tumor of 300 mm3, which enabled a dosing regimen more than seven times. The mice in desired groups were irradiated by a 655-nm laser at 300 mW/cm2 for 5 min at injection site. The treatment procedure was repeated three times at a time interval of 4 days. Tumor growth and body weight change of mice were carefully monitored every 2 days. Tumor volume was calculated by using the formula: V = L × W × W/2 (L, the longest dimension; W, the shortest dimension). Bioluminescence images of B16-F10-Luc tumor relapse mice were obtained by using an IVIS imaging system at desired time points (PerkinElmer, USA). At the end of the antitumor study, the tumors and major organs (e.g., heart, liver, lung, spleen, and kidney) were harvested, fixed, dehydrated, and subjected to H&E and TUNEL staining. For long-term survival study, the mice were treated as above, and an event was recorded in the case that the tumor exceeded 2000 mm3 or if the mice performed either death or pathologic behaviors.
Immunoassay
Surgeries were performed on CT26-bearing BALB/c mice or B16-OVA-bearing C57BL/6 mice to construct postoperative tumor models. The mice were randomly grouped, treated with PBS or hydrogel vaccine, and irradiated by a 655-nm laser at 300 mW/cm2 for 5 min in desired groups. At desired time points, the tumor surgical regions were harvested, cut into small pieces, and immersed in the solution of hyaluronidase (100 U/ml), collagenase IV (175 U/ml), and DNase (30 U/ml) for 30 min at 37°C. The tumor suspensions were filtered through 75-μm filters and enriched using lymphocyte separation medium. Last, the separated lymphocytes were stained with antibodies according to the manufacturer’s protocols.
To evaluate the activation of neoepitope-specific CD8+ T cells after vaccination, hydrogel vaccines were derived from oxidized B16-OVA cells, and C57BL/6 mice were subcutaneously treated with PBS, Cell@gel, PC@gel + Laser, PC-Cell@gel, and PC-Cell@gel + Laser. The mice were vaccinated twice on days 0 and 5. Blood and lymph node samples were harvested on day 8. Lymphocytes in lymph nodes were separated and stained with anti–CD11c-FITC, anti–CD80-PE, and anti–CD86-PE-Cy7. Lymphocytes derived from mice blood were stained with anti–CD3-PerCP-Cy5, anti–CD8-FITC, and H-2Kb OVA-tetramer-PE. After incubation in the dark for 40 min, the samples were analyzed by flow cytometry. For long-term memory effect analysis, the lymph nodes of vaccinated mice were collected on day 30. Lymphocytes were enriched, washed, and stained to determine the percentages of central memory CD8+ T cells by flow cytometry.
Statistical analysis
Data are given as the mean ± SD. The statistical significance was displayed by one-way analysis of variance (ANOVA) with Tukey’s post hoc test and two-sided unpaired Student’s t test. Comparisons of survival rates were calculated by the log-rank (Mantel-Cox) test. Statistical significance was set as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Acknowledgments
We thank the National Center for Protein Science Shanghai for the cell sorter BD Influx, and we are grateful to Y. Wang for FACS sorting. Funding: Financial supports from the National Natural Science Foundation of China (81690265, 81803444, 81521005, and 81903548), the Strategic Priority Research Program of CAS (XDA12050307), the Youth Innovation Promotion Association of CAS (2019283), and the Shanghai Sailing Program (19YF1457300) are gratefully acknowledged. Author contributions: L.F. and Z.Z. contributed equally to this work. L.F. and D.W. conceived and designed the project. L.F. and Z.Z. carried out the characterization of the vaccine in vitro and performed animal experiments. J.W. synthesized the FK-PBA peptide. D.W. and Y.L. interpreted the data and wrote the manuscript. All authors discussed the results and reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaba4024/DC1
REFERENCES AND NOTES
- 1.Banchereau J., Palucka K., Immunotherapy: Cancer vaccines on the move. Nat. Rev. Clin. Oncol. 15, 9–10 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Finn O. J., The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 18, 183–194 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Sahin U., Derhovanessian E., Miller M., Kloke B. P., Simon P., Löwer M., Bukur V., Tadmor A. D., Luxemburger U., Schrors B., Omokoko T., Vormehr M., Albrecht C., Paruzynski A., Kuhn A. N., Buck J., Heesch S., Schreeb K. H., Müller F., Ortseifer I., Vogler I., Godehardt E., Attig S., Rae R., Breitkreuz A., Tolliver C., Suchan M., Martic G., Hohberger A., Sorn P., Diekmann J., Ciesla J., Waksmann O., Brück A. K., Witt M., Zillgen M., Rothermel A., Kasemann B., Langer D., Bolte S., Diken M., Kreiter S., Nemecek R., Gebhardt C., Grabbe S., Holler C., Utikal J., Huber C., Loquai C., Türeci O., Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Chen Q., Wang C., Zhang X., Chen G., Hu Q., Li H., Wang J., Wen D., Zhang Y., Lu Y., Yang G., Jiang C., Wang J., Dotti G., Gu Z., In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 14, 89–97 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Han X., Shen S., Fan Q., Chen G., Archibong E., Dotti G., Liu Z., Gu Z., Wang C., Red blood cell-derived nanoerythrosome for antigen delivery with enhanced cancer immunotherapy. Sci. Adv. 5, eaaw6870 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanyi J. L., Bobisse S., Ophir E., Tuyaerts S., Roberti A., Genolet R., Baumgartner P., Stevenson B. J., Iseli C., Dangaj D., Czerniecki B., Semilietof A., Racle J., Michel A., Xenarios I., Chiang C., Monos D. S., Torigian D. A., Nisenbaum H. L., Michielin O., June C. H., Levine B. L., Powell D. J. Jr., Gfeller D., Mick R., Dafni U., Zoete V., Harari A., Coukos G., Kandalaft L. E., Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 10, eaao5931 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Carreno B. M., Magrini V., Becker-Hapak M., Kaabinejadian S., Hundal J., Petti A. A., Ly A., Lie W. R., Hildebrand W. H., Mardis E. R., Linette G. P., Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dillman R. O., Cornforth A. N., Nistor G. I., McClay E. F., Amatruda T. T., Depriest C., Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in metastatic melanoma: 5-year follow up and additional analyses. J. Immunother. Cancer 6, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho D. Y., Yang W. K., Lee H. C., Hsu D. M., Lin H. L., Lin S. Z., Chen C. C., Harn H. J., Liu C. L., Lee W. Y., Ho L. H., Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: A phase II clinical trial. World Neurosurg. 77, 736–744 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Garg A. D., Coulie P. G., Van den Eynde B. J., Agostinis P., Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 38, 577–593 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Aerts J. G. J. V., de Goeje P. L., Cornelissen R., Kaijen-Lambers M. E. H., Bezemer K., van der Leest C. H., Mahaweni N. M., Kunert A., Eskens F. A. L. M., Waasdorp C., Braakman E., van der Holt B., Vulto A. G., Hendriks R. W., Hegmans J. P. J. J., Hoogsteden H. C., Autologous dendritic cells pulsed with allogeneic tumor cell lysate in mesothelioma: From mouse to human. Clin. Cancer Res. 24, 766–776 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg S. A., Yang J. C., Restifo N. P., Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 10, 909–915 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye Y., Wang C., Zhang X., Hu Q., Zhang Y., Liu Q., Wen D., Milligan J., Bellotti A., Huang L., Dotti G., Gu Z., A melanin-mediated cancer immunotherapy patch. Sci. Immunol. 2, eaan5692 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Yang P., Song H., Qin Y., Huang P., Zhang C., Kong D., Wang W., Engineering dendritic-cell-based vaccines and PD-1 blockade in self-assembled peptide nanofibrous hydrogel to amplify antitumor T-cell immunity. Nano Lett. 18, 4377–4385 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Bencherif S. A., Warren Sands R., Ali O. A., Li W. A., Lewin S. A., Braschler T. M., Shih T.-Y., Verbeke C. S., Bhatta D., Dranoff G., Mooney D. J., Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun. 6, 7556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T., Wang D., Yu H., Feng B., Zhou F., Zhang H., Zhou L., Jiao S., Li Y., A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat. Commun. 9, 1532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasikala A. R. K., Unnithan A. R., Thomas R. G., Ko S. W., Jeong Y. Y., Park C. H., Kim C. S., Multifaceted implantable anticancer device for potential postsurgical breast cancer treatment: A single platform for synergistic inhibition of local regional breast cancer recurrence, surveillance, and healthy breast reconstruction. Adv. Funct. Mater. 28, 1704793 (2018). [Google Scholar]
- 18.Barve M., Maples P. B., Orr D., Kuhn J., Magee M., Lamont J., Bedell C., Wallraven G., Pappen B. O., Roth A., Horvath S., Nemunaitis D., Kumar P., Senzer N., Nemunaitis J., Summary of bi-shRNAfurin/GM-CSF augmented autologous tumor cell vaccine (FANGTM) in advanced cancer of the liver. Mol. Ther. 22, S240–S240 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Boudewijns S., Westdorp H., Koornstra R. H. T., Aarntzen E. H. J. G., Schreibelt G., Creemers J. H. A., Punt C. J. A., Figdor C. G., de Vries I. J. M., Gerritsen W. R., Bol K. F., Immune-related adverse events of dendritic cell vaccination correlate with immunologic and clinical outcome in stage III and IV melanoma patients. J. Immunother. 39, 241–248 (2016).27227325 [Google Scholar]
- 20.Conde J., Oliva N., Zhang Y., Artzi N., Local triple-combination therapy results in tumour regression and prevents recurrence in a colon cancer model. Nat. Mater. 15, 1128–1138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan W., Huang P., Chen X., Overcoming the Achilles’ heel of photodynamic therapy. Chem. Soc. Rev. 45, 6488–6519 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Castano A. P., Mroz P., Hamblin M. R., Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 6, 535–545 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D., Wang T., Liu J., Yu H., Jiao S., Feng B., Zhou F., Fu Y., Yin Q., Zhang P., Zhang Z., Zhou Z., Li Y., Acid-activatable versatile micelleplexes for PD-L1 blockade-enhanced cancer photodynamic immunotherapy. Nano Lett. 16, 5503–5513 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Moyer T. J., Zmolek A. C., Irvine D. J., Beyond antigens and adjuvants: Formulating future vaccines. J. Clin. Invest. 126, 799–808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deshayes S., Cabral H., Ishii T., Miura Y., Kobayashi S., Yamashita T., Matsumoto A., Miyahara Y., Nishiyama N., Kataoka K., Phenylboronic acid-installed polymeric micelles for targeting sialylated epitopes in solid tumors. J. Am. Chem. Soc. 135, 15501–15507 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Wang T., Wang D., Yu H., Wang M., Liu J., Feng B., Zhou F., Yin Q., Zhang Z., Huang Y., Li Y., Intracellularly acid-switchable multifunctional micelles for combinational photo/chemotherapy of the drug-resistant tumor. ACS Nano 10, 3496–3508 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Prokopowicz Z. M., Arce F., Biedron R., Chiang C. L.-L., Ciszek M., Katz D. R., Nowakowska M., Zapotoczny S., Marcinkiewicz J., Chain B. M., Hypochlorous acid: A natural adjuvant that facilitates antigen processing, cross-priming, and the induction of adaptive immunity. J. Immunol. 184, 824–835 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Chiang C. L.-L., Kandalaft L. E., Tanyi J., Hagemann A. R., Motz G. T., Svoronos N., Montone K., Mantia-Smaldone G. M., Smith L., Nisenbaum H. L., Levine B. L., Kalos M., Czerniecki B. J., Torigian D. A., Powell D. J. Jr., Mick R., Coukos G., A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 19, 4801–4815 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang C., Wang J., Zhang X., Yu S., Wen D., Hu Q., Ye Y., Bomba H., Hu X., Liu Z., Dotti G., Gu Z., In situ formed reactive oxygen species-responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 10, eaan3682 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Chao Y., Chen Q., Liu Z., Smart injectable hydrogels for cancer immunotherapy. Adv. Funct. Mater. 30, 1902785 (2019). [Google Scholar]
- 31.Culver H. R., Clegg J. R., Peppas N. A., Analyte-responsive hydrogels: Intelligent materials for biosensing and drug delivery. Acc. Chem. Res. 50, 170–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu C., Liu X., Ran W., Meng J., Zhai Y., Zhang P., Yin Q., Yu H., Zhang Z., Li Y., Regulating cancer associated fibroblasts with losartan-loaded injectable peptide hydrogel to potentiate chemotherapy in inhibiting growth and lung metastasis of triple negative breast cancer. Biomaterials 144, 60–72 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Li Y., Kumacheva E., Hydrogel microenvironments for cancer spheroid growth and drug screening. Sci. Adv. 4, eaas8998 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J., Zhan J., Yang Z., Enzyme-instructed self-assembly (EISA) and hydrogelation of peptides. Adv. Mater. , 1805798 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y.-X., Zhao Y.-Y., Shen J., Sun X., Liu Y., Liu H., Wang Y., Wang J., Nanoenabled modulation of acidic tumor microenvironment reverses anergy of infiltrating T cells and potentiates anti-PD-1 therapy. Nano Lett. 19, 2774–2783 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Keskin D. B., Anandappa A. J., Sun J., Tirosh I., Mathewson N. D., Li S., Oliveira G., Giobbie-Hurder A., Felt K., Gjini E., Shukla S. A., Hu Z., Li L., Le P. M., Allesoe R. L., Richman A. R., Kowalczyk M. S., Abdelrahman S., Geduldig J. E., Charbonneau S., Pelton K., Iorgulescu J. B., Elagina L., Zhang W., Olive O., McCluskey C., Olsen L. R., Stevens J., Lane W. J., Salazar A. M., Daley H., Wen P. Y., Chiocca E. A., Harden M., Lennon N. J., Gabriel S., Getz G., Lander E. S., Regev A., Ritz J., Neuberg D., Rodig S. J., Ligon K. L., Suva M. L., Wucherpfennig K. W., Hacohen N., Fritsch E. F., Livak K. J., Ott P. A., Wu C. J., Reardon D. A., Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu W. L., Zou M. Z., Liu T., Zeng J. Y., Li X., Yu W. Y., Li C. X., Ye J. J., Song W., Feng J., Zhang X. Z., Expandable immunotherapeutic nanoplatforms engineered from cytomembranes of hybrid cells derived from cancer and dendritic cells. Adv. Mater. 31, e1900499 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Liu H., Moynihan K. D., Zheng Y., Szeto G. L., Li A. V., Huang B., Van Egeren D. S., Park C., Irvine D. J., Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J., Li W. A., Choi Y., Lewin S. A., Verbeke C. S., Dranoff G., Mooney D. J., Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol. 33, 64–72 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang R., Xu J., Xu L., Sun X., Chen Q., Zhao Y., Peng R., Liu Z., Cancer cell membrane-coated adjuvant nanoparticles with mannose modification for effective anticancer vaccination. ACS Nano 12, 5121–5129 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaba4024/DC1