

Nanofluidic model pores are used to visualize single catalyst particle activity dynamics induced by local reactant conversion.
Abstract
Nanoconfinement in porous catalysts may induce reactant concentration gradients inside the pores due to local conversion. This leads to inefficient active material use since parts of the catalyst may be trapped in an inactive state. Experimentally, these effects remain unstudied due to material complexity and required high spatial resolution. Here, we have nanofabricated quasi–two-dimensional mimics of porous catalysts, which combine the traits of nanofluidics with single particle plasmonics and online mass spectrometry readout. Enabled by single particle resolution at operando conditions during CO oxidation over a Cu model catalyst, we directly visualize reactant concentration gradient formation due to conversion on single Cu nanoparticles inside the “model pore” and how it dynamically controls oxidation state—and, thus, activity—of particles downstream. Our results provide a general framework for single particle catalysis in the gas phase and highlight the importance of single particle approaches for the understanding of complex catalyst materials.
INTRODUCTION
Ensemble averaging effectively denies access to the detailed understanding of how catalyst material structure and composition dictate activity and selectivity, and of how the dynamics of (surface) oxidation and transient structural changes induced locally by the reaction control catalyst function (1–4). This has spurred the development of “single particle catalysis” and corresponding experimental techniques (1–9). However, despite substantial progress in this field, it remains unaddressed that, at the level of the individual nanoparticle, local conversion of reactants can lead to the formation of reactant concentration gradients. As a result, the catalyst can locally attain different oxidation states or experience different reactant compositions or temperatures at different positions, e.g., due to proximity to other active particles. This, in turn, may give rise to widely varying operation regimes in terms of kinetic or mass transport reaction limitations for the individual nanoparticle (10–12) and, therefore, also lead to erroneous conclusions about the active phase. These effects are expected to be particularly pronounced in highly confined environments such as nano- and mesoporous support materials widely used in catalysis, due to the limited reactant supply inside a pore because of mass transport constraints (13). However, the lack of suitable experimental techniques makes it difficult to isolate and study such phenomena, thereby limiting the understanding and optimization of complex catalyst materials.
To address this challenge, we have nanofabricated quasi–two-dimensional (2D) mimics of a porous catalyst that we call model pores. They are composed of a tailored nanofabricated nanofluidic structure that can be simultaneously experimentally addressed by in operando single particle plasmonic nanospectroscopy and online mass spectrometry in the gas phase. As we demonstrate, this approach enables the scrutiny of gas-phase catalytic reactions and reactant conversion effects at the individual nanoparticle level at reaction conditions up to 4 bar, inside a volume that is several orders of magnitude smaller than for microreactors traditionally used to study small amounts of catalyst material (14–17). Furthermore, it enables the direct comparison between optical single particle plasmonic nanospectroscopy readout from the reaction zone, where individual nanoparticles are located, and catalytic activity simultaneously measured by an online quadrupole mass spectrometer (QMS) that samples an ensemble with a well-controlled population of 3 × 104 nominally identical nanoparticles located on the same chip. This ensures that the obtained single particle results can provide the anticipated guidance for improving the performance of real catalysts, which naturally are composed of a large ensemble. To this end, the demonstrated use of nanofluidic structures in single particle catalysis in the gas phase also constitutes the first application of the nanofluidics paradigm to a gaseous fluid in general and therefore goes substantially beyond its recently introduced liquid-phase counterpart (18).
RESULTS
Experimental setup and nanofluidic model pore characterization
We have designed a platform in which a nanofluidic chip is connected via a sample holder to a stainless steel gas handling system compatible with up to 4 bar gas pressure, a power controller for the on-chip heater enabling operation at up to 723 K, and a QMS that enables continuous quantitative reactant and product analysis in continuous flow mode operation. This platform is then mounted on an upright optical microscope connected to a spectrometer equipped with an electron multiplying charge-coupled device (EMCCD) camera that facilitates simultaneous single particle plasmonic nanospectroscopy (19) from multiple single catalyst particles located inside a nanofluidic channel that serves as the model pore (Fig. 1A and fig. S1, and further details in Materials and Methods). In brief, plasmonic nanospectroscopy relies on measuring visible light scattered from single metal nanoparticles, enhanced by localized surface plasmon resonance (LSPR) excitation, and correlating changes in the measured scattered light with structural or chemical changes that occur within/on or in close vicinity of the nanoparticle (19). The nanofluidic chip itself is nanofabricated in a thermally oxidized silicon wafer, as described in detail in Materials and Methods and fig. S2. It is composed of a microfluidic inlet and outlet system that connects to the sample holder toward the high-pressure gas supply side and the low-pressure QMS side (Fig. 1B). The U design of the microfluidic system at the inlet serves the purpose of enabling fast gas exchange using conventional mass flow controllers. On the other end, the microfluidic system connects to the nanofluidic system comprising six identical model pores (Fig. 1C and fig. S3). They are 600 μm long and designed as symmetric funnels that narrow down to a center region that is 100 μm long, has a width of 400 nm, and a height of 100 nm. The individual model catalyst nanoparticles, in the 100-nm size range, are placed 20 μm apart (Fig. 1D) by means of electron beam lithography (EBL) (20). Furthermore, in the wider funnel areas, patches of high-density arrays of catalyst particles are fabricated (Fig. 1D), such that a total of 3 × 104 nominally identical nanoparticles are present on the chip. They are distributed within the six model pores in different amounts to mimic potentially different loading as in individual nano- or mesopores of a real catalyst support (Fig. 1C and fig. S3). In this way, the QMS response is obtained from a total particle number that is large enough to constitute a statistically relevant ensemble, which allows the critical direct comparison between single particle response obtained by plasmonic nanospectroscopy from a single model pore and the corresponding ensemble response in one and the same experiment (5).
Fig. 1. Nanofluidic chip design, implementation, and gas flow characterization.
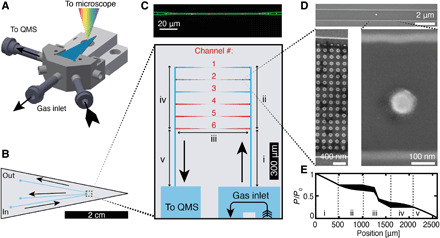
(A) Schematic of the nanofluidic chip (blue) mounted in the sample holder containing gas and electrical connections. (B) Schematic of the nanofluidic chip and the microfluidic channels that connect the nanofluidic model pores to the inlet and outlet holes. (C) Top: Dark-field optical micrograph of a single model pore (channel 3 in the bottom) decorated with individual catalyst nanoparticles (red). Bottom: Nanofluidic system consisting of six parallel channels acting as the model pores. Each channel contains nanofabricated model catalyst particles in the 100-nm size range (details in fig. S3). The roman numbers highlight different regions relevant for the flow model calculations shown in (E). (D) Representative scanning electron microscopy (SEM) micrographs of a model pore decorated with a single Cu nanoparticle in its center region and an array of nanoparticles placed in its tapered parts connecting to the microfluidic system. (E) Simulated pressure profile of the model pores. The widened black line corresponds to the pressure difference between the six channels that stem from the fact that they have a different total length (more details in fig. S6). The roman numbers denote the different regions depicted in (C). Black arrows denote the gas flow direction in all panels.
As a first characterization step of this setup as a whole, we used the QMS to determine the mass flow (fig. S4) and temporal response (fig. S5). It revealed a mass flow rate of approximately 1 × 1011 molecules/s through each model pore, resulting in a residence time of 0.07 s, a gas exchange time of 10 min due to mixing in the external tubing, and an instantaneous response of the QMS to changes in reaction temperature (details in section S2). Furthermore, we performed calculations based on a unified flow model (21) to obtain pressure profiles of the system (details in Materials and Methods). As the main result, we find a moderate symmetric pressure drop across the chip, with very small variation between the six nanofluidic model pores (Fig. 1E and fig. S6).
To experimentally benchmark single particle plasmonic nanospectroscopy inside a model pore and to further corroborate the pressure modeling results and verify the pressure profile, we monitored 10 hybrid Au-Pd nanostructures placed inside a nanochannel (channel 3 in Fig. 1C) using single particle scattering spectroscopy (Fig. 2, A and B). The hybrid Au-Pd nanostructure comprises a bottom Au nanodisk (120-nm diameter, 40 nm thick) and a top Pd disk (80-nm diameter, 15 nm thick), separated by a 7-nm SiO2 layer (Fig. 2C, inset). In this arrangement, the Au disk is sensitive to the changes in permittivity and volume expansion induced in Pd upon hydrogen sorption and Pd hydride formation (22–26). The Au particle, thus, serves as a plasmonic nanoantenna that itself is inert (27) and probes the adjacent Pd particle (26). We then make use of the first-order phase transition upon hydride formation, which for polycrystalline Pd nanoparticles occurs in a narrow pressure range (19). Specifically, we tracked the single particle dark-field scattering spectra of a row of 10 Au-Pd hybrid nanoparticles located within a nanochannel model pore (Fig. 2D). In this way, we are able to map the pressure drop across the pore by tracking the change in full width at half maximum (ΔFWHM) of the LSPR scattering peak upon exposure to H2 pulses in Ar carrier gas (Fig. 2E, figs. S7 and S8, and section S2). Using the theoretical calculations as a guide, we applied H2 pulses of different concentration at 303 K and 2 bar inlet pressure, predicted to result in H2 partial pressures of 100, 70, 50, 20, and 10 mbar, respectively, at the position of the particles in the center of the nanofluidic model pore. The corresponding single particle response reveals that all particles exhibit a distinct ΔFWHM signal down to 50 mbar H2 (Fig. 2E and fig. S7). In contrast, at 25 mbar, only two particles respond, whereof one with a notable time delay, and at 10 mbar, no response is observed for any of the particles. We also note a trend, where particles placed further downstream require a higher nominal H2 concentration to respond (fig. S7), again in good agreement with the predicted pressure drop in the channel. Furthermore, the apparent threshold of ca. 25 mbar H2 for hydride formation to take place in the center of the channel is in excellent agreement with our previous single particle results on similar Pd nanoparticles measured on an open surface (19). Last, we observe slight differences in the magnitude of the ΔFWHM shifts and that not all particles follow the pressure trend perfectly. These effects can also be explained by our previous work, which identified that the exact particle morphology in terms of grain structure dictates both the optical response and the Pd hydride formation pressure (19). Hence, this experiment demonstrates the single particle readout capabilities of our platform, and it corroborates the validity of the pressures predicted by our model, since they were derived from the calculations without any fitting to the experimental data. This, in turn, justifies the use of the model for experiment design and data interpretation as we do below.
Fig. 2. Plasmonic nanospectroscopy from multiple single nanoparticles inside a nanofluidic model pore.
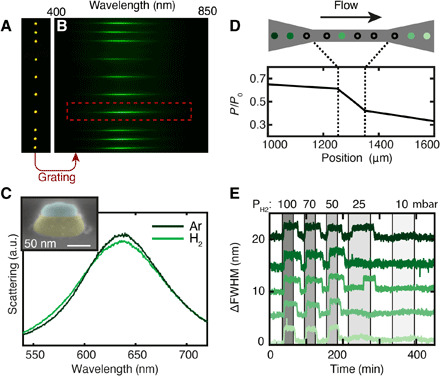
(A) Optical dark-field microscope image of 10 hybrid Au-Pd nanostructures placed inside a nanofluidic model pore (channel 3 in Fig. 1C), schematically depicted in (D). (B) Scattering spectra of the 10 nanoparticles in (A) after insertion of a dispersive grating between the microscope and the CCD camera. (C) Single particle dark-field scattering spectra of an Au-Pd hybrid nanostructure [red outline in (B), side view SEM image in inset] inside the nanofluidic model pore in pure Ar and 100 mbar H2. These structures are composed of an inert Au plasmonic nanoantenna (shaded yellow) that serves as a nanoprobe of the Pd particle (shaded blue) deposited on top of a 7-nm SiO2 support layer sandwiched between the two (43). a.u., arbitrary units. (D) Pressure profile along the model pore containing the nanoparticles, which in the experiment are simultaneously optically probed by plasmonic nanospectroscopy. The black arrow denotes the gas flow direction. (E) Change in scattering peak FWHM (ΔFWHM) of a selection of five nanoparticles along the model pore, induced by H2 pulses with decreasing concentration in Ar carrier gas. Changes in ΔFWHM are due to the formation of Pd hydride at a critical H2 partial pressure (see also fig. S7).
Oxidation state and activity control by CO and O2 conversion over single Cu nanoparticles
Cu catalysts are used in a wide range of important reactions in organic synthesis (28) as well as for the water gas shift process that converts CO and H2O to CO2 and H2 (29–31). Hence, from a catalysis perspective, Cu is a very important but at the same time complex material due to its capability to obtain different oxidation states, depending on the specific conditions. For example, Cu nanoparticles used as CO oxidation catalysts require almost full conversion of O2 to retain a high activity by inhibiting oxidation of Cu (32). This availability of multiple states, in turn, can make it very challenging to identify correctly the active phase for a specific reaction at operando conditions because local variations in the catalyst due to, for instance, mass transport limitations or local conversion effects may induce locally different catalyst states and, thus, activities, which are easily overlooked due to, for example, ensemble averaging.
Here, we apply the model pore concept to visualize and quantify this effect at the single nanoparticle level for the oxidation of CO over a Cu model catalyst. Using the nanofluidic chip design depicted in Fig. 1 and fig. S3, we decorated the model pores with disk-shaped Cu nanoparticles with 100-nm diameter and 40-nm thickness (Fig. 3A). We note that, in this case, the Cu catalyst particles themselves act as the optical probes for the plasmonic nanospectroscopy readout (i.e., no adjacent Au nanoantenna necessary as for the Pd case above) due to their strong intrinsic LSPR (33). The chip was heated to 493 K and flushed at 4 bar inlet pressure with constant 6% CO in Ar carrier gas, while increasing the O2 concentration from 0 to 0.4% in steps of 0.025% (Fig. 3B). Simultaneously using the QMS, we measured the amount of CO and O2 exiting the system, together with the reaction product CO2. Initially, for low O2 concentrations, full O2 conversion was recorded by the QMS, and the CO2 production rate increased proportionally with the increasing O2 concentration in the feed. However, at 0.325% O2 (t ~ 12 hours), a decrease in the QMS CO2 signal becomes apparent, while the amount of O2 exiting the system increases.
Fig. 3. CO oxidation over single Cu nanoparticles in a nanofluidic model pore.
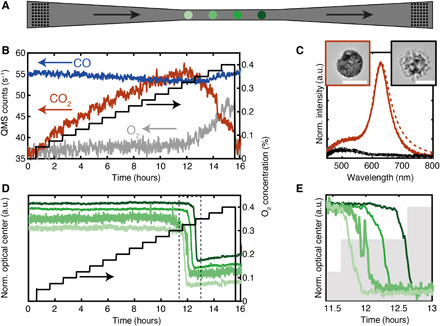
(A) Schematic of the center part of a nanofluidic model pore (channel 2 in Fig. 1C) containing four disk-shaped Cu nanoparticles (100-nm diameter and 40-nm thickness; green circles) in its narrowest region and high-density “patches” of identical particles in the tapered wider regions (black dots; cf. Fig. 1D). The nanoparticles are separated by 20 μm, and the drawing is not to scale. (B) Nominal O2 concentration in the system (black) together with the measured QMS response for CO, O2, and CO2. The CO signal is scaled by a factor of 1/8. (C) Experimental (solid lines) and FDTD simulated (dashed lines) single particle scattering spectra of a single Cu nanoparticle in its metallic (orange) and oxidized state (black). Insets show TEM micrographs of a representative Cu particle in the metallic (left) and oxidized state (right). Scale bars, 100 nm. (D) Optical response of four Cu nanoparticles in the model pore (green lines) upon increasing O2 concentration (black line) during reaction with CO. (E) Scaled zoom-in on the optical response from (D) revealing distinct time delays on the order of 5 min between the oxidation of the four nanoparticles. Shaded regions indicate changes in inlet O2 concentration.
To understand the origin of this observed loss in activity, we take a look at the simultaneously recorded optical response of a single Cu nanoparticle before and after the activity drop and find that the LSPR peak essentially disappears in the latter state (Fig. 3C). This change is explained by oxidation of the Cu nanoparticle to a mix of Cu2O and CuO identified by ex situ x-ray photoelectron spectroscopy measurements (fig. S9) and in agreement with the literature (34). It is further corroborated by corresponding finite-difference time-domain (FDTD) simulations of the expected LSPR response upon Cu nanoparticle oxidation (Fig. 3C) and transmission electron microscopy (TEM) analysis of a particle before and after reaction (insets; Fig. 3C). Correspondingly, now looking at the simultaneously recorded time-resolved optical response of four single Cu nanoparticles in the model pore within the period where the activity loss occurs, we see that a distinct spectral blueshift of the optical center takes place (Fig. 3D; optical center is here defined as the wavelength at which the integrated scattering spectrum equals half the total integrated spectrum (as detailed in section S5 and fig. S10).
Comparing this optical single particle response with the QMS data reveals a notably less sharp transition in global activity (several hours), which cannot be explained by the response time of our setup (fig. S5). Thus, to understand this effect, we zoom in on the oxidation state transition measured optically for the single Cu nanoparticles (Fig. 3E). We observe delays in the onset of the oxidation transition between the individual nanoparticles on the order of 5 min. At the same time, during Cu back-reduction in 6% CO in Ar, all particles respond simultaneously (Fig. 4). Repeating the same experiment results in the same qualitative trend with slight variation in the threshold O2 concentration and time delay between the particles, explained by structural changes to the Cu nanoparticles after each oxidation-reduction cycle (fig. S11). Furthermore, a control experiment with stepwise increase in the O2 concentration in the absence of CO shows that the Cu oxidation, in this case, occurs at much lower O2 concentration (0.02%) and simultaneously on all particles (fig. S12). Hence, the model pore experiment directly visualizes how reactant conversion on a single nanoparticle in a nanoconfined volume controls the oxidation state and, thus, the local activity of the catalyst in its vicinity.
Fig. 4. Cu nanoparticle back-reduction in CO flow through the model pore at 493 K.
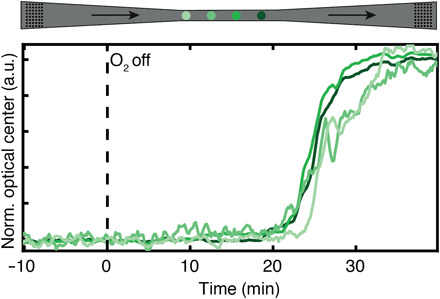
Normalized optical center response of the four single Cu nanoparticles after an experimental sequence identical to the one depicted in Fig. 3. At time, t = 0, indicated by the black dashed line, the O2 concentration in the feed is reduced to zero while keeping the CO flow constant at 6% in Ar carrier gas. This results in the simultaneous back-reduction of all four Cu particles to the metallic state after ca. 20 min, in stark contrast to the reverse oxidation process during the CO oxidation reaction, where delays of ca. 5 min between the phase transformation of the particles from the metallic to the oxidized state were observed due to local reactant conversion (cf. Fig. 3E).
Mechanistically, these single particle observations can be understood as the consequence of the interplay between upstream reactant conversion [which is occurring at high rate as long as the upstream particle(s) is/are in the metallic state] and the limited amount of O2 molecules supplied to the nanoparticles, due to the small volume of the model pore. In other words, when a critical O2 concentration is reached at a specific position/particle in the pore, the surface coverage equilibrium between O and CO changes in favor of O, eventually leading to the nucleation and growth of the now locally stable oxide phase. However, because of the limited supply of O2 in the system due to its small volume, oxidation cannot begin on all particles simultaneously but must start upstream where the required critical O2 concentration is reached first. It is then not until the upstream particle has oxidized, and thus has attained a state of lower activity, that enough O2 reaches the downstream particle and eventually initiates its oxidation. As a consequence, we predict this effect to be further enhanced for smaller pore dimensions. Accordingly, we do not only observe this effect in the narrowest part of the pore where the single particles are located but also for high-particle-density arrays that simulate a nanoparticle ensemble located upstream, as well as in another channel (channel 4 in Fig. 1C) with different particle loading (fig. S13 and movie S1). This confirms that the effect is not an artificial consequence of a single particle experiment. In addition, when there is no catalytic reaction competing for reactants, the oxidation (or reduction) of all Cu particles occurs simultaneously (Fig. 4 and fig. S12), which further confirms that the delay is due to concentration gradients formed by the CO oxidation reaction that consumes a sizable amount of O2 in the model pore.
Finite-volume simulations of reactant conversion over a single particle in a model pore
To further corroborate the mechanism discussed above, we first theoretically analyze the two possible rate-determining factors: (i) surface reaction limitation or (ii) mass transport limitation (section S6). This analysis shows that the turnover frequency required for a sizable reduction in the O2 concentration downstream of a single Cu nanoparticle in a nanofluidic model pore due to conversion is physically reasonable based on reaction rates derived by density functional theory calculations (35). Second, finite-volume simulations reveal that each active particle induces a step in the concentration profile along the channel, with the magnitude determined by the reaction rate used in the simulation (Fig. 5, A and B, and fig. S14). This is a consequence of the high flow rate through the pore-type structure, which effectively limits the influence of diffusion of reactants in the streamwise direction (fig. S14B). In contrast, perpendicular to the flow direction, solely diffusion is important and quickly produces a fully mixed concentration profile only 200 nm after an active particle (Fig. 5C and fig. S15). Consequentially, the concentration of reactants (here, O2) is lowered for particles placed downstream, which explains the delay we observe in the oxidation of the individual particles. By decreasing the activity of the particle (to mimic the effect of oxidation in the simulation), the resulting concentration profile changes such that the next particle in line will experience a higher O2 concentration, inducing oxidation and the corresponding decrease in activity (Fig. 5B).
Fig. 5. Simulated reactant concentration profile in a nanofluidic model pore during CO oxidation on a single Cu nanoparticle.
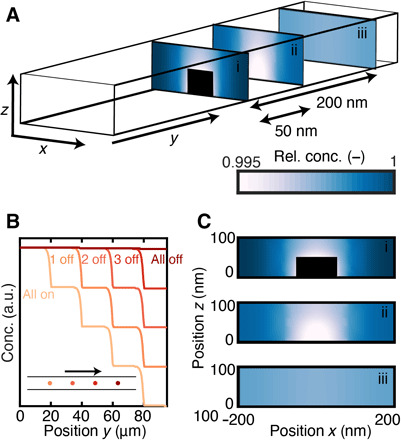
(A) Schematic illustration of the simulation geometry with representative 2D reactant concentration profiles obtained at different positions along a nanofluidic model pore with the same dimensions as in the experiment, that is, width x = 400 nm, length y = 100 μm, height z = 100 nm. The color coding corresponds to the concentration profile presented in the color bar, and the black area corresponds to the particle. (B) Simulated tracer molecule concentration profile along the flow direction due to reactant conversion over four individual nanoparticles placed in sequence. The color code indicates the number of “active” particles (i.e., Cu particles in the metallic state) along the model pore. The inset illustrates the corresponding particle positions, and the arrow indicates the reactant flow direction. (C) 2D contour plots of calculated reactant tracer concentration across the nanofluidic model pore at different positions along it. The three panels are the same as the ones presented in (A) and correspond to the positions: (i) at the nanoparticle center, (ii) 50 nm after, and (iii) 200 nm after the center of the nanoparticle. The color code is the same as in (A). Extended data set in fig. S15.
These findings are of high relevance because they show that nanometer spatial resolution is critical in studies that attempt the identification of catalyst active phase. However, as the key point here, they also show that even single particle spatial resolution may not be enough if not multiple single particles can be probed simultaneously at different spatial locations of a sample because their oxidation state may be different, even if they are direct neighbors. This becomes even more clear when considering the ensemble response tracked by the QMS in our experiments, which shows a continuous slow decline of activity over the course of several hours, during which part of the catalyst is in the oxidized state and part of it in the metallic state. This means that depending on the chosen spatial location of the probe determining the oxidation state, completely different conclusions about the active phase could be made. We, thus, argue that this effect is one important reason for often contradictory experimental reports about catalyst active phase for many reactions, including CO oxidation over Cu studied here (32, 36–39).
DISCUSSION
The performance of complex catalyst materials is strongly affected by the interplay between the porous support and the active material, such as metal nanoparticles. Here, we have shown how a combination of nanofluidics, single particle plasmonic nanospectroscopy, and mass spectrometry can allow the operando detection of local gas-phase reactant conversion-controlled single catalyst nanoparticle activity inside a nanofluidic model pore that mimics a porous catalyst support material. Information obtained from single particle plasmonic nanospectroscopy at operando conditions inside such a model pore indicates that the CO oxidation reaction taking place on a single Cu particle can locally lower the O2 concentration in the feed to transiently prohibit oxide formation on a particle placed further downstream, thus trapping it in a state of higher activity. The ability to visualize these reactant gradients is the direct result of our model pore design that allows for a high reactant flow rate and a reactant residence time of only 0.07 s, which prevents diffusion from equilibrating the local reactant concentration at the position of each individual nanoparticle. These results, thus, directly visualize how local conversion of reactants can lead to the formation of gas concentration gradients inside a nanoconfined volume and, thus, to widely varying and simultaneously existing oxidation states at the level of the single catalyst particle, which are stable for minutes to hours. This is an important finding because it offers a widely applicable explanation for often contradicting experimental results about catalyst active phase in the literature. Specifically, it highlights the critical importance of single particle spatial resolution in studies of complex catalyst materials at realistic application conditions, since otherwise wrong conclusions about the active phase may easily be drawn if catalyst characterization is done in the “wrong” spatial location or averaged over an ensemble in which multiple catalyst phases coexist. More generally, our combination of nanofluidics, single particle plasmonic nanospectroscopy, and mass spectrometry offers a platform to probe and identify the active catalyst phase under nanoconfinement in operando conditions. In this way, it, for example, enables screening for champion particles (31) for a variety of catalyst materials and reactions, as well as investigation of related processes, which may range from the study of metal-support interactions, to spillover effects, to the dynamics of phase separation in alloy catalyst systems. Last, extrapolations of the minimal required active catalyst surface area for online QMS analysis of reaction products from our system reveal that a reduction of one to two orders of magnitude compared to the lower limit proposed for microreactors (17) is within reach.
MATERIALS AND METHODS
Experimental setup
The experiments were performed on a setup consisting of a gas mixing/handling unit, an optical microscope equipped with an optical spectrometer, an EMCCD camera, and the nanofluidic chip holder, which is connected to a QMS (see fig. S1). The gas mixing/handling unit was made from one-fourth inch stainless steel tubing with Vacuum Coupling Radiation (VCR) (Swagelok) fittings as connectors. A connection block manufactured in stainless steel and equipped with welded VCR fittings to provide the gases and mass spectrometry connection hosts the nanofluidic chip. Gas lines were manufactured in the interior of the connection block and connected to flexible stainless steel tubing at the outside. An illustration and technical drawing of the connection block is shown in fig. S1. The gas inlet and outlet of the chip are sealed with perfluoroelastomer (FPM) O-rings to the connection block. To avoid unwanted background signal of potential reaction gases due to diffusion of air through the O-rings into the mass spectrometer, a constant flow of Ar (18 ml/min) is flushed around the O-rings. Therewith, the unavoidable diffusion through the O-rings is limited to Ar. The connector “Gas 1” in fig. S1 is connected to four Bronkhorst Low ΔP mass flow controllers and the connector “P controller” to a pressure controller (maximum pressure of 10 bar). With this system, a mixture of up to four gases can be fed into the system at a defined inlet pressure of up to 10 bar. All gas flows and pressures are controlled and monitored by a customized LabVIEW program.
The system can be heated up to 723 K using a resistive heater on the backside of the chip. Temperature readout is done via a four-wire resistance temperature detector probe at the position of the nanofluidic system. Electrical connection to the chip for heating and temperature readout is enabled by six gold-plated electronic spring spins embedded in a machinable ceramic block and connected to a temperature controller (Lakeshore 335) operated via a LabVIEW program. To compensate for the heating of the chip, the holder is equipped with a water-cooled copper block. In this way, a constant temperature of the chip holder is ensured, helping to keep the leaking of Ar through the O-rings constant, as well as reducing mechanical movements due to heat transfer into the holder assembly.
Gas detection is performed with a triple filter QMS equipped with a pulsed ion counting detector (Hiden HAL/3F PIC) and a gold-plated ion source. The QMS is hosted in an ultrahigh vacuum chamber with a base pressure of 10−10 mbar. During measurements, the pressure rises up to 5∙10−9 mbar depending on the chip design and pressure settings on the gas inlet side.
The optical readout is performed with a spectrometer (Andor Kymera 193i) and EMCCD camera (iXon Ultra 888) connected to a Nikon LV150 microscope with a Nikon LU Plan extra-long working distance (ELWD) 50×/0.55 objective. For spectroscopic measurements, a grating with 150 liters/mm, 630-nm central wavelength, and an integration time of 0.5 s is used. Spectra of several individual nanoparticles are collected by using the multitrack option in the Andor Solis software and integrating the light from a region 5 to 10 pixel rows above and below each particle of interest. Background subtraction is done for each particle individually by taking a spectrum next to each nanoparticle. The final signal is calculated as Isc (λ) = (S − B)/CRS, where S is the signal measured from a particle, B is the background signal, and CRS is the spectrum of the 50-W halogen lamp collected from a certified diffuse white reflectance standard reference sample (Labsphere SRS-99-020).
For the hydride formation pressure gauge experiments, the temperature of the chip was kept at 303 K, and ultrapure H2 (4% in Ar) was introduced at different concentrations to the flow of Ar carrier gas (99.99999% purity). A total flow of 10 ml/min was established through the microchannels, and an inlet pressure of 2 bar was used.
For the CO oxidation experiments over Cu nanoparticles, ultrapure CO (10% in Ar) and O2 (2% in Ar) were used with Ar carrier gas (99.99999% purity) and fed with different concentrations into the chip. The inlet pressure was set to 4 bar, and a total flow of 10 ml/min through the microchannels was applied.
Nanofabrication
Nanofabrication was carried out in cleanroom facilities of Fed. Std.209E Class 10–100, using EBL (JBX-9300FS/JEOL Ltd.), direct laser lithography (Heidelberg Instruments DWL 2000), photolithography (MA 6/SUSS MicroTec), reactive-ion etching (RIE) (Plasmalab 100 ICP180/Oxford Plasma Technology and STS ICP), electron beam evaporation (PVD 225/Lesker), magnetron sputtering (MS150/FHR), deep RIE (STS ICP/STS) and wet oxidation (wet oxidation/Centrotherm), fusion bonding (AWF 12/65/Lenton), and dicing (DAD3350/Disco). To achieve precise alignment for consecutive lithography steps, alignment marks, fabricated using EBL, were used. These marks allowed us to achieve a positioning error of approximately 10 nm using EBL and 1 μm using photolithography. In particular, the fabrication steps (illustrated in fig. S2) comprised the following processing steps of a 4″ silicon (p-type) wafer.
Fabrication of alignment marks
(i) Spin coating hexamethyldisilizane (HMDS) adhesion promoter (MicroChem Corporation) at 3000 revolutions per minute (rpm) for 30 s and soft baking on a hot plate (HP) at 115°C for 120 s. Spin coating UV5 (MicroChem Corporation) at 3000 rpm for 60 s and soft baking (HP) at 130°C for 120 s. (ii) Electron beam exposure of alignment marks for both optical and EBL at 10 nA with a shot pitch of 24 nm and 25 μC/cm2 exposure dose. (iii) Postexposure bake (HP) at 130°C for 90 s. (iv) Development in MF-24A (Microposit) for 90 s, rinsing in water, and drying under N2 stream. (v) RIE for 15 s at 60 mtorr chamber pressure, 60 W radio frequency (RF) power, 60 standard cubic centimeter per minute (sccm) O2 flow (descum). RIE for 30 min at 40 mtorr chamber pressure, 50 W RF power, 100 W inductively coupled plasma power (ICP) power, 50 sccm Cl2 flow (1200-nm etch depth in silicon).
Thermal oxidation
(i) Cleaning in 50 ml H2O2 + 100 ml H2SO4 at 130°C for 10 min, rinsing in water, and drying under N2 stream. (ii) Wet oxidation in water atmosphere for 45 min at 950°C (200-nm thermal oxide).
Nanofluidic model pores
(i) Electron beam evaporation of 20-nm Cr (hard mask). (ii) Spin coating ZEP520A:anisole (2:1) (ZEONEX Electronic Chemicals) at 2000 rpm for 60 s and soft baking (HP) at 180°C for 10 min. (iii) Electron beam exposure of lines at 2 nA with a shot pitch of 4 nm and 280 μC/cm2 exposure dose. (iv) Development in n-amyl acetate for 120 s, rinsing in isopropanol, and drying under N2 stream. Covering alignment marks with Kapton tape. (v) RIE for 10 s at 40 mtorr chamber pressure, 40 W RF power, and 40 sccm O2 flow (descum). RIE for 90 s at 20 mtorr chamber pressure, 50 W RF power, 200 W ICP power, 20 sccm O2 flow, and 50 sccm Cl2 flow (selective Cr hard-mask etch). RIE for 30 s at 8 mtorr chamber pressure, 50 W RF power, and 50 sccm NF3 flow (30-nm etch depth in thermal oxide). The resulting channels have a depth of 100 nm measured with a Dektak 150 surface profiler.
Microchannels
(i) Spin coating HMDS at 3000 rpm for 30 s and soft baking (HP) at 115°C for 2 min. Spin coating S1813 (Shipley) at 3000 rpm for 30 s and soft baking (HP) at 115°C for 2 min. (ii) Expose microchannels for 8 s in contact aligner at 6 mW/cm2 intensity. (iii) Development in MF-319 (Microposit) for 60 s, rinsing in water, and drying under N2 stream. (iv) Buffered oxide etch for 2 min to remove thermal oxide, rinsing in water, and drying under N2 stream. (v) Deep RIE for 100 cycles of 7 s at 6 mtorr chamber pressure, 800 W RF power, 8 W platen power, 130 sccm SF6 flow (Si etch), and of 5 s at 6 mtorr chamber pressure, 800 W RF power, 8 W platen power, and 85 sccm C4F8 flow (passivation) at a rate of 600 nm/cycle. (vi) Removal of resist in 50 ml H2O2 + 100 ml H2SO4 at 130°C for 10 min, rinsing in water, and drying under N2 stream. The resulting channels have a depth of 60 μm measured using a Dektak 150 surface profiler.
Inlets (from backside)
(i) Magnetron sputtering of 200-nm Al (hard mask). (ii) Spin coating S1813 at 3000 rpm for 30 s and soft baking (HP) at 115°C for 2 min. (iii) Expose inlets for 10 s in contact aligner at 6 mW/cm2 intensity. (iv) Development in MF-319 for 60 s, rinsing in water, and drying under N2 stream. (v) Aluminum wet etch [H3PO4:CH3COOH:HNO3:H2O (4:4:1:1)] for 10 min to clear the hard mask at inlet positions. (vi) Deep RIE for 300 cycles of 12 s at 5 mtorr chamber pressure, 600 W RF power, 10 W platen power, 130 sccm SF6 flow (Si etch), and of 7 s at 5 mtorr chamber pressure, 600 W RF power, 10 W platen power, and 85 sccm C4F8 flow (passivation) at a rate of 2 μm/cycle. (vii) Removal of Al hard mask in 50 ml H2O2 + 100 ml H2SO4 at 130°C for 10 min, rinsing in water, and drying under N2 stream.
Heater elements on the backside
(i) Spin coating HMDS at 3000 rpm for 30 s and soft baking (HP) at 115°C for 2 min. Spin coating LOR3A (MicroChem Corporation) at 3000 rpm for 30 s and soft baking (HP) at 180°C for 5 min. Spin coating S1813 (Shipley) at 3000 rpm for 30 s and soft baking (HP) at 115°C for 2 min. (ii) Expose heater elements with direct laser lithography at 6 mW/cm2 intensity. (iii) Development in MF-319 (Microposit) for 60 s, rinsing in water, and drying under N2 stream. (iv) Electron beam evaporation of 10 nm Cr/100 nm Pt. (v) Liftoff in remover Rem400 (MicroChem Corporation), rinsing in isopropanol, and drying under N2 stream.
Nanoparticles inside nanofluidic model pores
(i) Spin coating copolymer methyl methacrylate (8.5) methacrylic acid [MMA(8.5)MAA] (10 weight % diluted in anisole; MicroChem Corporation) at 6000 rpm for 60 s and soft baking (HP) at 180°C for 10 min. Spin coating ZEP520A:anisole (1:2) at 3000 rpm for 60 s and soft baking (HP) at 180°C for 10 min. (ii) Electron beam exposure at 1 nA with a shot pitch of 2 nm and 280 μC/cm2 exposure dose. (iii) Development in n-amyl acetate for 120 s, rinsing in isopropanol, and drying under N2 stream. Development in methyl isobutyl ketone:isopropanol (1:1) for 120 s, rinsing in isopropanol, and drying under N2 stream. (iv) Electron beam evaporation of respective materials. (v) Liftoff in acetone, rinsing in isopropanol, and drying under N2 stream.
Fusion bonding
(i) Cleaning of the substrate together with a lid (175-μm-thick 4″ pyrex; UniversityWafer) in H2O:H2O2:NH3OH (5:1:1) for 10 min at 80°C. (ii) Prebonding the lid to the substrate by bringing surfaces together and manually applying pressure. (iii) Fusion bonding of the lid to the substrate for 5 hours in N2 atmosphere at 550°C (5°C/min ramp rate).
Dicing of bonded wafers
Cutting nanofluidic chips from the bonded wafer using a resin-bonded diamond blade of 250-μm thickness (Dicing Blade Technology) at 35 krpm and 1 mm/s feed rate.
Flow and pressure calculations
The gas flow through the nanofluidic chip covers all the rarefied flow regimes, from continuum flow in the upstream model pore (Kn→0) to free molecular flow at the exit to the mass spectrometer (Kn→∞), with the slip and transitional flow regimes occurring inside the main channel system. The mass flow rate and the pressure distribution for the model pore are obtained using the unified channel flow model of Beskok and Karniadakis (21). The nanofluidic system is described as a number of parallel and sequential segments for which the major losses are obtained by using a constant value = 2.2 in the expression for the rarefaction coefficient, and a slip coefficient b = −1, which have been shown to yield predictions similar to those of comprehensive direct simulation Monte Carlo (DSMC) simulations for nitrogen and helium (21). The tangential momentum accommodation coefficient is set equal to unity, and minor losses are neglected. The law of continuity requires that the mass flow rate through the system is constant at steady-state conditions, and so the intermediate pressures at the junction points between the segments can be determined from the pressures at the upstream microchannel duct and at the exit point after solving a nonlinear equation system equating the mass flow rates at the junction points. When the pressures at the junction points are known, the mass flow rate can be calculated from the pressure drop correlation for an arbitrary segment. Last, the pressure distribution along each segment can be retrieved at arbitrary resolution using the implicit relation for pressure provided by Beskok and Karniadakis (21). The calculated pressure profile is presented in Fig. 1E and fig. S6.
Simulating spatial distribution of reactants
To simulate the spatial distribution of O2 inside a nanofluidic model pore, finite-volume simulations were performed. In these simulations, the gas mixture passes through a 3D computational domain representing a section of the pore with cylindrical Cu particles (Ø = 100 nm, H = 40 nm) at a given spacing. The coupled system of partial differential equations used to describe the gas flow, and the tracer transport and reaction, is discretized in a finite-volume framework and solved using the computational fluid dynamics code Ansys Fluent 19.1.0. The flow is assumed to slip freely over the surfaces, and a convection-diffusion-reaction equation is solved for a tracer species, the diffusivity of which is set equal to the Knudsen diffusivity of the nanofluidic model pore
| (1) |
Here, ρ is the gas phase density (kg/m3), u is the velocity vector (m/s), Y is the mass fraction of the tracer species, DKn is the Knudsen diffusivity (m2/s), and R is the reaction rate (kg/m3,s). The boundary condition for the tracer species is a mass fraction of 0.001 at the inlet and zero flux at all surfaces. The source term R is zero everywhere except below the surface of each Cu particle, where a first-order reaction is activated: R = −AρY. The parameter A (s−1) is to be interpreted as an effective global reaction rate constant for the situation where the intrinsic reaction kinetics are approximated by a single-step first-order reaction. The single-step reaction can then be thought of as representing the rate-limiting step of a more complex mechanism while assuming a first-order dependence on the reactant (tracer) concentration. In the specific situation of a distinct rate-limiting step that is of first order, A would be defined as
| (2) |
where A0 is the Arrhenius frequency factor, f is a function describing the dependence of the reaction rate on the surface coverages, S is a parameter relating the finite-volume cell volume to the availability of active sites, and exp(−Ea/RT) is the Arrhenius reaction probability term. In situations where a single rate-limiting step cannot be unambiguously identified, A will be a lumped parameter characterizing the global rate of conversion for the complete mechanism cast in the form of a single-step first-order reaction. Hence, A becomes a model parameter that represents the overall rate of reaction. If a value of A is chosen so that the global conversion matches that of an experiment, then the detailed finite-volume simulation results can be used to obtain spatially resolved insight into the combined effects of reaction and transport inside the model pore on the species concentration fields.
FDTD simulations
FDTD simulations, performed using FDTD Solutions (Lumerical), were used to evaluate the optical response of the plasmonic nanostructures. The nanofluidic model pore was simulated as a gas-filled channel with cross-sectional dimensions of 400 × 100 nm surrounded by SiO2 with an Si layer placed 98 nm below the channel. In the center of the channel, a Cu particle was modeled as a single truncated cone with rounded corners (bottom rounding, 14 nm; and top rounding, 8 nm), and the change from metal to oxide was simulated by changing the refractive index of the structure. SiO2 was simulated as a material with a dielectric function taken from Palik (40) and the gas in the channel as vacuum with refractive index n = 1. The dielectric function for Cu was taken from Hagemann et al. (41) and the one for Cu2O from Tahir et al. (42). To correctly resolve the field close to the nanoparticle, a mesh overlay with a step size of 0.3 nm was used around it. Light was introduced as a linearly polarized plane wave via a total-field/scattered-field source, and the backward scattering was collected by integrating the Poynting vector of the field in the backward direction with respect to the incident light, using an area corresponding to the numerical aperture (NA) of the microscope objective used in the experiment (NA = 0.55).
Supplementary Material
Acknowledgments
We acknowledge fruitful discussions with D. B. Trimarco, P. Vesborg, I. Chorkendorff, M. Jørgensen, and H. Grönbeck. Funding: This research has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (678941/SINCAT) and from the Knut and Alice Wallenberg Foundation project 2015.0055. Part of this work was carried out at the MC2 cleanroom facility and at the Chalmers Materials Analysis Laboratory. Author contributions: C.L., D.A., and S.B. conceptualized the ideas for the device. D.A. and S.B. developed the experimental platform, performed the experiments, and analyzed the experimental data. D.A. and J.F. fabricated the nanofluidic chips. D.A. performed FDTD simulations and made the figures. H.S. performed the fluid dynamic calculations and simulations. D.A. and H.S. wrote parts of the manuscript. S.N. performed the TEM measurements. C.L. coined the entire concept, wrote and edited the paper, and supervised the project. All authors gave feedback on the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional and raw data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaba7678/DC1
REFERENCES AND NOTES
- 1.Sambur J. B., Chen P., Approaches to single-nanoparticle catalysis. Annu. Rev. Phys. Chem. 65, 395–422 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Zhou X., Andoy N. M., Liu G., Choudhary E., Han K.-S., Shen H., Chen P., Quantitative super-resolution imaging uncovers reactivity patterns on single nanocatalysts. Nat. Nanotechnol. 7, 237–241 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Buurmans I. L. C., Weckhuysen B. M., Heterogeneities of individual catalyst particles in space and time as monitored by spectroscopy. Nat. Chem. 4, 873–886 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Vendelbo S. B., Elkjær C. F., Falsig H., Puspitasari I., Dona P., Mele L., Morana B., Nelissen B. J., van Rijn R., Creemer J. F., Kooyman P. J., Helveg S., Visualization of oscillatory behaviour of Pt nanoparticles catalysing CO oxidation. Nat. Mater. 13, 884–890 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Wang W., Imaging the chemical activity of single nanoparticles with optical microscopy. Chem. Soc. Rev. 47, 2485–2508 (2018). [DOI] [PubMed] [Google Scholar]
- 6.van Schrojenstein Lantman E. M., Deckert-Gaudig T., Mank A. J. G., Deckert V., Weckhuysen B. M., Catalytic processes monitored at the nanoscale with tip-enhanced Raman spectroscopy. Nat. Nanotechnol. 7, 583–586 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Karim W., Spreafico C., Kleibert A., Gobrecht J., VandeVondele J., Ekinci Y., van Bokhoven J., Catalyst support effects on hydrogen spillover. Nature 541, 68–71 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Meirer F., Weckhuysen B. M., Spatial and temporal exploration of heterogeneous catalysts with synchrotron radiation. Nat. Rev. Mater. 3, 324–340 (2018). [Google Scholar]
- 9.Hartman T., Geitenbeek R. G., Whiting G. T., Weckhuysen B. M., Operando monitoring of temperature and active species at the single catalyst particle level. Nat. Catal. 2, 986–996 (2019). [Google Scholar]
- 10.Stötzel J., Frahm R., Kimmerle B., Nachtegaal M., Grunwaldt J.-D., Oscillatory behavior during the catalytic partial oxidation of methane: Following dynamic structural changes of palladium using the QEXAFS technique. J. Phys. Chem. C 116, 599–609 (2012). [Google Scholar]
- 11.Gänzler A. M., Casapu M., Boubnov A., Müller O., Conrad S., Lichtenberg H., Frahm R., Grunwaldt J. D., Operando spatially and time-resolved X-ray absorption spectroscopy and infrared thermography during oscillatory CO oxidation. J. Catal. 328, 216–224 (2015). [Google Scholar]
- 12.Gross E., Uncovering the deactivation mechanism of Au catalyst with operando high spatial resolution IR and X-ray microspectroscopy measurements. Surf. Sci. 648, 136–140 (2016). [Google Scholar]
- 13.Bell A. T., The impact of nanoscience on heterogeneous catalysis. Science 299, 1688–1691 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Tanimu A., Jaenicke S., Alhooshani K., Heterogeneous catalysis in continuous flow microreactors: A review of methods and applications. Chem. Eng. J. 327, 792–821 (2017). [Google Scholar]
- 15.Rizkin B. A., Popovic F. G., Hartman R. L., Review Article: Spectroscopic microreactors for heterogeneous catalysis. J. Vac. Sci. Technol. A 37, 050801 (2019). [Google Scholar]
- 16.Vesborg P. C. K., Olsen J. L., Henriksen T. R., Chorkendorff I., Hansen O., Gas-phase photocatalysis in μ-reactors. Chem. Eng. J. 160, 738–741 (2010). [Google Scholar]
- 17.Henriksen T. R., Olsen J. L., Vesborg P., Chorkendorff I., Hansen O., Highly sensitive silicon microreactor for catalyst testing. Rev. Sci. Instrum. 80, 124101 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Levin S., Fritzsche J., Nilsson S., Runemark A., Dhokale B., Ström H., Sundén H., Langhammer C., Westerlund F., A nanofluidic device for parallel single nanoparticle catalysis in solution. Nat. Commun. 10, 4426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alekseeva S., da Silva Fanta A. B., Iandolo B., Antosiewicz T. J., Nugroho F. A. A., Wagner J. B., Burrows A., Zhdanov V. P., Langhammer C., Grain boundary mediated hydriding phase transformations in individual polycrystalline metal nanoparticles. Nat. Commun. 8, 1084 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fritzsche J., Albinsson D., Fritzsche M., Antosiewicz T. J., Westerlund F., Langhammer C., Single particle nanoplasmonic sensing in individual nanofluidic channels. Nano Lett. 16, 7857–7864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beskok A., Karniadakis G. E., Report: A model for flows in channels, pipes, and ducts at micro and nano scales. Microsc. Thermophys. Eng. 3, 43–77 (1999). [Google Scholar]
- 22.Langhammer C., Kasemo B., Zorić I., Absorption and scattering of light by Pt, Pd, Ag, and Au nanodisks: absolute cross sections and branching ratios. J. Chem. Phys. 126, 194702 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Langhammer C., Larsson E. M., Kasemo B., Zorić I., Indirect nanoplasmonic sensing: Ultrasensitive experimental platform for nanomaterials science and optical nanocalorimetry. Nano Lett. 10, 3529–3538 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Liu N., Tang M. L., Hentschel M., Giessen H., Alivisatos A. P., Nanoantenna-Enhanced Gas Sensing in a Single Tailored Nanofocus. Nat. Mater. 10, 631–636 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Shegai T., Langhammer C., Hydride formation in single palladium and magnesium nanoparticles studied by nanoplasmonic dark-field scattering spectroscopy. Adv. Mater. 23, 4409–4414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syrenova S., Wadell C., Nugroho F. A. A., Gschneidtner T. A., Diaz Fernandez Y. A., Nalin G., Świtlik D., Westerlund F., Antosiewicz T. J., Zhdanov V. P., Moth-Poulsen K., Langhammer C., Hydride formation thermodynamics and hysteresis in individual Pd nanocrystals with different size and shape. Nat. Mater. 14, 1236–1244 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Hammer B., Norskov J. K., Why gold is the noblest of all the metals. Nature 376, 238–240 (1995). [Google Scholar]
- 28.Chemler S. R., Copper catalysis in organic synthesis. Beilstein J. Org. Chem. 11, 2252–2253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeValley T. L., Richard A. R., Fan M., The progress in water gas shift and steam reforming hydrogen production technologies – A review. Int. J. Hydrogen Energy 39, 16983–17000 (2014). [Google Scholar]
- 30.Ratnasamy C., Wagner J. P., Water gas shift catalysis. Catal. Rev. 51, 325–440 (2009). [Google Scholar]
- 31.Zhang Z., Wang S.-S., Song R., Cao T., Luo L., Chen X., Gao Y., Lu J., Li W.-X., Huang W., The most active Cu facet for low-temperature water gas shift reaction. Nat. Commun. 8, 488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bu Y., Niemantsverdriet J. W. H., Fredriksson H. O. A., Cu model catalyst dynamics and CO oxidation kinetics studied by simultaneous in situ UV–Vis and mass spectroscopy. ACS Catal. 6, 2867–2876 (2016). [Google Scholar]
- 33.Chan G. H., Zhao J., Hicks E. M., Schatz G. C., Van Duyne R. P., Plasmonic properties of copper nanoparticles fabricated by nanosphere lithography. Nano Lett. 7, 1947–1952 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Nilsson S., Albinsson D., Antosiewicz T. J., Fritzsche J., Langhammer C., Resolving single Cu nanoparticle oxidation and Kirkendall void formation with in situ plasmonic nanospectroscopy and electrodynamic simulations. Nanoscale 11, 20725–20733 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Falsig H., Hvolbæk B., Kristensen I. S., Jiang T., Bligaard T., Christensen C. H., Nørskov J. K., Trends in the catalytic CO oxidation activity of nanoparticles. Angew. Chem. Int. Ed. 47, 4835–4839 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Eren B., Heine C., Bluhm H., Somorjai G. A., Salmeron M., Catalyst Chemical State during CO Oxidation Reaction on Cu(111) Studied with Ambient-Pressure X-ray Photoelectron Spectroscopy and Near Edge X-ray Adsorption Fine Structure Spectroscopy. J. Am. Chem. Soc. 137, 11186–11190 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Jernigan G. G., Somorjai G. A., Carbon monoxide oxidation over three different oxidation states of copper: Metallic copper, copper (I) oxide, and copper (II) oxide - a surface science and kinetic study. J. Catal. 147, 567–577 (1994). [Google Scholar]
- 38.Xu F., Mudiyanselage K., Baber A. E., Soldemo M., Weissenrieder J., White M. G., Stacchiola D. J., Redox-mediated reconstruction of copper during carbon monoxide oxidation. J. Phys. Chem. C 118, 15902–15909 (2014). [Google Scholar]
- 39.Huang T.-J., Tsai D.-H., CO oxidation behavior of copper and copper oxides. Catal. Lett. 87, 173–178 (2003). [Google Scholar]
- 40.E. D. Palik, Handbook of Optical Constants of Solids (Academic Press, 1998), vol. 2, 1096pp. [Google Scholar]
- 41.Hagemann H. J., Gudat W., Kunz C., Optical constants from the far infrared to the x-ray region: Mg, Al, Cu, Ag, Au, Bi, C, and Al2O3. J. Opt. Soc. Am. 65, 742–744 (1975). [Google Scholar]
- 42.Tahir D., Tougaard S., Electronic and optical properties of Cu, CuO, and Cu2O studied by electron spectroscopy. J. Phys. Condens. Matter 24, 175002 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Alekseeva S., Nedrygailov I. I., Langhammer C., Single particle plasmonics for materials science and single particle catalysis. ACS Photonics 6, 1319–1330 (2019). [Google Scholar]
- 44.Manchester F. D., San-Martin A., Pitre J. M., The H-Pd (hydrogen-palladium) system. J. Phase Equilib. 15, 62–83 (1994). [Google Scholar]
- 45.Behm R. J., Penka V., Cattania M. G., Christmann K., Ertl G., Evidence for “subsurface” hydrogen on Pd(110): An intermediate between chemisorbed and dissolved species. J. Chem. Phys. 78, 7486–7490 (1983). [Google Scholar]
- 46.Fredriksson H., Alaverdyan Y., Dmitriev A., Langhammer C., Sutherland D. S., Zäch M., Kasemo B., Hole-mask colloidal lithography. Adv. Mater. 19, 4297–4302 (2007). [Google Scholar]
- 47.He X., Wang Y., Zhang X., Dong M., Wang G., Zhang B., Niu Y., Yao S., He X., Liu H., Controllable in situ surface restructuring of cu catalysts and remarkable enhancement of their catalytic activity. ACS Catal. 9, 2213–2221 (2019). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaba7678/DC1