

Abstract
Nitric oxide (NO)-cyclic GMP (cGMP) signaling is a vasoprotective pathway therapeutically targeted, for example, in pulmonary hypertension. Its dysregulation in disease is incompletely understood. Here we show in pulmonary artery endothelial cells that feedback inhibition by NO of the NO receptor, the cGMP forming soluble guanylate cyclase (sGC), may contribute to this. Both endogenous NO from endothelial NO synthase and exogenous NO from NO donor compounds decreased sGC protein and activity. This effect was not mediated by cGMP as the NO-independent sGC stimulator, or direct activation of cGMP-dependent protein kinase did not mimic it. Thiol-sensitive mechanisms were also not involved as the thiol-reducing agent N-acetyl-L-cysteine did not prevent this feedback. Instead, both in-vitro and in-vivo and in health and acute respiratory lung disease, chronically elevated NO led to the inactivation and degradation of sGC while leaving the heme-free isoform, apo-sGC, intact or even increasing its levels. Thus, NO regulates sGC in a bimodal manner, acutely stimulating and chronically inhibiting, as part of self-limiting direct feedback that is cGMP independent. In high NO disease conditions, this is aggravated but can be functionally recovered in a mechanism-based manner by apo-sGC activators that re-establish cGMP formation.
Subject terms: Pharmacology, Diseases
Introduction
The nitric oxide (NO)-cGMP signaling pathway plays several essential roles in physiology, including cardiopulmonary homeostasis1,2. The main receptor and mediator of NO’s actions is soluble guanylate cyclase (sGC), a heterodimeric heme protein. In its Fe(II)heme-containing state, sGC binds NO and is thereby activated to convert GTP to the second messenger, cGMP, whose steady-state levels are counter-regulated by different phosphodiesterases (PDEs)3. cGMP exerts its cardiopulmonary effects via activating cGMP-dependent protein kinase I (PKG)4. The latter results in protective vasodilation, anti-proliferation, and anti-thrombosis5. In disease, heme loss and the appearance of NO-insensitive apo-sGC impair NO-cGMP signaling6,7.
In addition to acutely activating sGC, NO appears to have further roles in sGC regulation. During enzyme maturation, NO facilitates heme incorporation into apo-sGC8,9, and activation of sGC by NO is followed by acute and rapid desensitization involving protein S-nitrosylation10,11. In addition, chronic exposure to NO donor drugs has been suggested to negatively affect sGC activity in a not fully reversible manner12–14. It is unclear, however, whether this pharmacological effect also pertains to endogenously formed NO and has pathophysiological relevance.
Here, we examine this important knowledge gap in the (patho)biology of NO. As model systems, we chose porcine pulmonary artery endothelial cells (PPAECs) as they relate to the clinical application of NO and cGMP-modulating drugs in pulmonary hypertension15,16. We investigate the effects of chronic exposure to exogenous (from NO donor drugs) and endogenous NO on sGC protein and activity in these cells. In addition, we investigate in health and disease, whether chronic effects of NO on sGC involve canonical cGMP signaling, thiol modulation, or formation of heme-free sGC (apo-sGC). As disease model, we use again a condition related to pulmonary hypertension and chronically elevated levels of NO, i.e., porcine acute respiratory disease syndrome (ARDS)17–19.
Results
NO chronically decreases vascular sGC protein and activity in-vivo and in-vitro
For analyzing the chronic effects of NO at a mechanistic level, PPAECs were incubated for up to 72 h in the presence of the NO synthase (NOS) inhibitor, NG-nitro L-arginine methyl ester (L-NAME), and sGC expression and activity were measured. In the presence of L-NAME to eliminate endogenous NO formation, protein levels of the heme-binding sGCβ1 subunit were increased (Fig. 1A), and this was also associated with increased sGC activity (Fig. 1B). Next, we tested the reverse, i.e., whether an increase of NO to supra-physiological concentrations20–22 by chronic exposure to the long-acting NO donor compound, DETA/NO, downregulates sGC. When establishing the concentration-dependence of DETA/NO on sGC expression, we found 100 µM to exert a maximal downregulation (Supplementary Fig. S1) without affecting cell viability. In line with this, DETA/NO was also used in previous studies at a concentration of 100 µM to mimic chronically high-NO disease conditions20–22. Therefore, in all subsequent experiments, cells were exposed to 100 µM DETA/NO, unless otherwise indicated. Pre-incubating PPAECs with DETA/NO (100 µM) decreased both sGCα1 and sGCβ1 protein (Fig. 1C) and sGC activity (Fig. 1D). Thus, in-vitro in PPAECs, endogenous NO chronically downregulates sGC protein and activity in an L-NAME-reversible manner, and this is further aggravated by exogenous, pharmacologically applied NO in supra-physiological concentrations.
Figure 1.
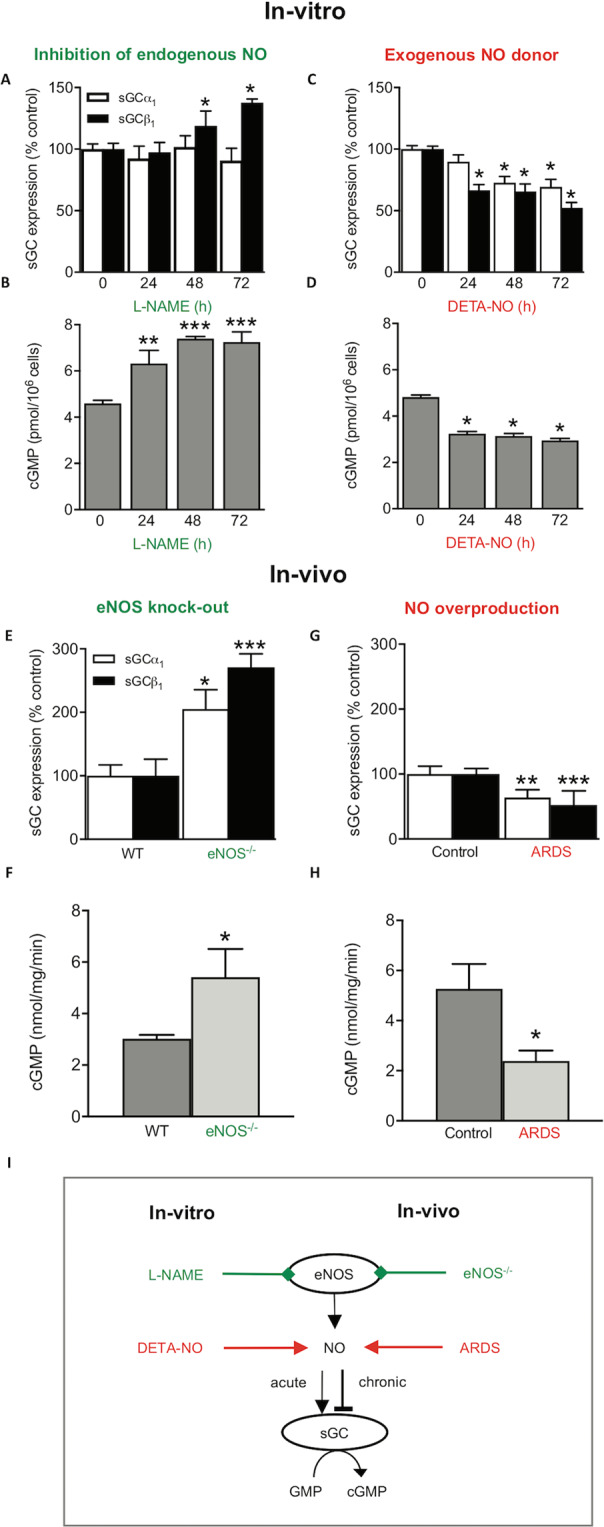
Chronic NO decreases vascular sGC protein and activity in-vivo and in-vitro. (A) Inhibiting basal NO formation in PPAECs by L-NAME (100 μM) for up to 72 h increased sGCβ1 expression (N = 6). (B) This upregulation was associated with increased sGC activity (N = 3). Exposing cells to supra-physiological levels of NO by chronic exposure to the NO donor compound, DETA/NO (100 µM), for up to 72 h decreased both sGCα1 and sGCβ1 protein (C) (N = 6) and sGC activity (D) (N = 5). In-vivo validation of the in-vitro observations showed in eNOS knockout mice (eNOS−/−) mice increased sGC protein (E) and activity levels (F) (N = 9), and in a porcine lung disease model (ARDS) characterized by NO overproduction, decreased sGCα1 and sGCβ1 protein (G) (N = 5) and sGC activity levels (H) (N = 3). Data are expressed as mean ± SEM. *,**,***p < 0.05, 0.01 or 0.001 vs. control, respectively. (I) A schematic summary showing that both in-vitro (porcine lung endothelial cells) and in-vivo (the porcine lung disease model, ARDS) both endogenous and exogenous NO downregulate sGC protein and activity. Representative full-length blots are presented in Supplementary Figure S4.
Next, we wanted to validate these in-vitro observations at an in-vivo level. To eliminate endogenous NO formation similar to the in-vitro L-NAME experiment, we chose eNOS knockout mice (eNOS−/−); as a high-NO condition, the previously extensively validated porcine ARDS model17,19,23. In line with our observations in PPAECs, eNOS−/− mice showed increased protein levels of sGCα1 and sGCβ1 (Fig. 1E) and increased sGC-activity (Fig. 1F). In the high-NO porcine ARDS model, sGCα1 and sGCβ1 protein levels (Fig. 1G) and sGC activity were decreased (Fig. 1H). These data collectively suggest in both in-vitro and in-vivo that decreasing endogenous NO elevates, and increasing it lowers sGC protein subunit levels and sGC activity (Fig. 1I), respectively.
cGMP/PKG does not mediate the downregulation of sGC protein and activity by chronic NO
Next, we aimed to clarify the mechanisms underlying the downregulation of sGC protein and activity by chronic NO. First, we tested whether cGMP/PKG signaling is involved, as it had been shown previously to decrease both sGC activity24 and expression25. Of experimental importance, cell passaging can cause downregulation of PKG and prevent the detection of its-dependent signaling26–29. Hence, we, therefore, restricted our studies to low passage number cells and ensured fully functional PKG signaling by validating the known autoregulation of PKG expression30,31. Indeed, in our PPAEC system, both the PKG activator, 8-Br-cGMP, and the NO-independent sGC stimulator and PDE inhibitor, YC-132, were able to reproduce the reduction of PKG expression (Supplementary Fig. S2) confirming the presence of a fully functional PKG. We then studied whether the observed downregulation of sGC protein and activity by NO can be mimicked by cGMP or is prevented by inhibiting PKG. When we exposed PPAECs, however, for 72 h to different concentrations of the sGC stimulator and PDE inhibitor, YC-1, to raise cGMP in a NO-independent manner, or to the direct PKG activator, 8-Br-cGMP, neither sGC protein nor activity were lowered (cf. to Fig. 1). Unexpectedly, we observed even a slight upregulation of sGC protein (Fig. 2A,B). Consistent with this, the NO-induced downregulation of sGC could not be prevented by co-incubation with the PKG inhibitor, Rp-8-Br-PET-cGMPS (Supplementary Fig. S3). To extend these in-vitro findings to the in-vivo level, we studied sGC expression and activity in PKG knockout mice (PKG−/−)33. Consistent with our in-vitro data, sGC protein levels (Fig. 2D) and sGC activity (Fig. 2E) were unchanged in PKG−/− compared to wildtype mice.
Figure 2.

PKG does not mediate the downregulation of sGC protein and activity by chronic NO. When PPAECs were incubated for 72 h in the absence or presence of increasing concentrations of (A) the NO-independent sGC stimulator, YC-1 (N = 6), this did not cause downregulation of sGCα1 and sGCβ1 expression but rather a small upregulation. Consistent with this, in (B) the direct PKG activator 8-Br-cGMP (N = 6) led to increased sGCα1 protein expression. (C) The scheme summarizes the in-vivo and in-vitro data suggesting that the downregulation of sGC protein and activity by chronic NO is cGMP- and PKG-independent and thus appeared to be due to a non-canonical mechanism. (D) sGC protein expression (N = 4) and (E) activity (N = 4) are not altered in PKG−/− as compared to wildtype mice. Data are expressed as mean ± SEM. *,**,***p < 0.05, 0.01 or 0.001 vs. control, respectively. Representative full-length blots are presented in Supplementary Figure S4.
In conclusion, both our in-vivo and in-vitro data suggested that the downregulation of sGC protein and activity by chronic NO is cGMP- and PKG-independent and thus appeared to be due to a non-canonical mechanism (Fig. 2C). At least two cGMP-independent effects on sGC have been reported, rapid desensitization10,20,34, which is reversible in a thiol-dependent manner35,36, and oxidative heme-loss yielding the NO-insensitive apo-form of sGC (apo-sGC)7,37. These possibilities were tested in our two next sets of experiments.
N-acetyl-L-cysteine does not prevent NO-induced sGC downregulation
Thiol-sensitive mechanisms are involved in both sGC regulation, such as sGC maturation, and airway pathologies such as asthma20. Therefore, we assessed whether NO-posttranslational modification of free-thiol cysteines i.e., S-nitrosylation, contributes to the downregulation of sGC by high chronic NO incubation. For this approach, PPAECs were again exposed for 72 h to DETA-NO (100 µM) in absence or presence, over the full-time frame, of the membrane-permeable thiol-reducing agent, N-acetyl-L-cysteine (NAC; 1 mM). NAC is a membrane-permeable de-nitrosylating agent and glutathione precursor that has been extensively validated to protect sGC from nitrosylation38,39 down to concentrations as low as 1 mM40,41, which we, however, did not re-validate. NAC, however, neither affected sGC protein levels (Fig. 3A) nor sGC activity (Fig. 3B), suggesting that it is unlikely that a thiol-reversible mechanism similar to the acute desensitization is involved in the chronic NO-induced downregulation of sGC. These findings left oxidative heme-loss yielding apo-sGC7,37 as the most likely cGMP-independent effect on sGC.
Figure 3.

NO-induced sGC downregulation is thiol-independent but involves sGC loss and a shift towards apo-sGC. When PPAECs were exposed for 72 h to DETA-NO (100 µM) in the absence and presence of N-acetyl-L-cysteine (NAC; 1 mM), NAC neither affected sGC protein levels (N = 5) (A) nor activity (N = 4) (B). Exposure of PPAECs for 72 h to DETA-NO (100 µM) increased apo-sGC activity, measured as BAY 58-2667-induced cGMP formation (BAY 58-2667, 10 µM) (N = 3) (C). Validation of the above in-vitro mechanistic findings in-vivo in the porcine high-NO ARDS model showing also increased apo-sGC activity (N = 3) (D). (E) A scheme summarizing both our in-vitro and in-vivo data that both endogenous NO or pharmacological NO donor compounds that acutely stimulate sGC, chronically decreased both sGC protein and activity leading to inactivation of sGC and an apparent net shift towards NO-insensitive apo-sGC. Data are expressed as mean ± SEM. *,**p < 0.05 or 0.01 vs. control, respectively. Representative full-length blots are presented in Supplementary Figure S4.
NO-induced sGC downregulation generates NO-insensitive sGC
We, therefore, examined whether chronic NO converts sGC to apo-sGC. To assay for the presence of apo-sGC, we took advantage of the apo-sGC activator, BAY 58-2667 (cinaciguat), which specifically binds to the empty heme-binding pocket of apo-sGC and re-activates cGMP formation in a NO-independent manner42. Indeed, up to 72 h exposure of PPAECs to DETA-NO (100 µM) increased apo-sGC activity, measured as BAY 58-2667-induced cGMP formation (Fig. 3C), and reduced sGC activity (Fig. 1D). To validate this mechanistic finding in-vivo, we re-examined the high-NO porcine ARDS model and found indeed apo-sGC activity to be increased (Fig. 3D). Collectively, this established apo-sGC formation as one possible mechanism of NO-induced reduction in sGC activity in addition to the loss in sGC protein (Fig. 3E).
Discussion
Our findings close important gaps in our understanding of NO-cGMP signaling, in particular on the long-term effects of endogenously formed NO versus NO donors on sGC and the pathophysiological relevance of chronic NO for sGC regulation. Thus, we expand the previously observed notion that NO donor drugs can reduce sGC mRNA43 to the protein level and, importantly, from pharmacology to endogenous NO. Previously, sGC protein levels were not consistently investigated or with antibodies of unclear specificity12,44. Moreover, only in some cases were the effects of PKG on cGMP levels investigated43,45 or in relation to cGMP metabolism rather than cGMP formation43,46,47.
Surprisingly, not only pathological/high levels of NO, as in our porcine ARDS model but already low chronic endogenous NOS activity, suppressed sGC protein and activity in an L-NAME reversible manner. These findings establish a previously not recognized delicate steady state in the interactions between NO and sGC, acutely stimulating and chronically limiting its expression and activity. On a positive note, under conditions of diminished NO synthesis, this may, in turn, rapidly upregulate sGC protein and activity, as we have observed in the presence of the NOS inhibitor, L-NAME, and in-vivo in eNOS−/− mice. In this regard, previous data are controversial. For example, sGC activity was increased in eNOS−/− mice13,48, which agrees with our findings, while others found neither sGC expression nor activity to be changed, neither in eNOS−/− mice49,50, nor upon treatment with high doses of NO donors51. The reasons for this discrepancy are unclear. In our ARDS model, the pan-NOS inhibitor, L-NAME, could not differentiate between eNOS and other isoforms present, such as iNOS52. Activation of the latter, e.g., by nuclear factor kappa B (NFkB)53,54, increases peroxynitrite leading to sGC oxidation55 and downregulation of sGCβ156. In endothelial cells and under physiological conditions, however, iNOS is not expressed or only at low levels57 and thus unlikely to be involved in the here observed non-canonical feedback.
Therapeutically relevant is the previously not recognized risk of chronic use of NO donor drugs as they will lead to a downregulation of both sGC protein and activity. Together with their problematic pharmacokinetic tolerance58–60, this adds to the clinical limitations of this widely used drug class. With the introduction of NO-independent sGC stimulators and cGMP elevating agents into clinical practice61, there is now an alternative. Indeed, we show that the prototypic sGC stimulator and PDE inhibitor, YC-1, does not lead to sGC downregulation.
Concerning the underlying mechanisms, we initially considered two known mechanisms in NO-cGMP physiology, i.e., cGMP/PKG and thiol modification9,10. Surprisingly, both could be excluded, which was reminiscent of earlier observations where long-term exposure to an exogenous NO donor reduced sGC activity in a manner that could not be reversed or prevented with thiol treatment14,46. Instead, our findings suggest that endogenous and exogenous NO chronically induce a net shift from sGC to apo-sGC and that this is not only a pathophysiological mechanism but pertains to NO-cGMP physiology. Our results explain why apo-sGC activator-induced cGMP formation and functional effects are enhanced in disease but not exclusive to these conditions7. Nevertheless, the availability of sGC activator compounds allows now to overcome such conditions in which sGC protein and activity are diminished in favor of apo-sGC and still induce cGMP formation. As a limitation, other potential underlying mechanisms such as decreased mRNA43, increased degradation of sGC, or high NO-induced NFkB activation that downregulate sGC56,62 have not been addressed by our study63 and cannot be excluded.
Our findings also add to our understanding of apo-sGC as a therapeutic target. Hitherto apo-sGC has been mainly studied by using the heme oxidant, ODQ, or by expressing enzyme where the proximal heme ligating histidine had been deleted64. The mechanisms by which apo-sGC forms in pathophysiology were less clear. Now chronic exposure to (high) levels of NO can be considered as one of these conditions. There are at least three possible non-canonical mechanisms by which high levels of NO can induce the transition from sGC to apo-sGC. First, NO can interact with reactive oxygen species to form the potent oxidant, peroxynitrite65–68. Second, NO can impair heme synthesis69 or activate heme oxygenase-1 (HO-1)70,71, which increases sGC heme degradation72. However, this effect is controversial since high concentrations of NO donors (including DETA-NO) inhibit the heme degradation in endothelial cells73. Third, high NO can increase the association of sGCβ1 with heat shock protein 90 (Hsp90), but not with sGCα1, resulting in the formation of NO-insensitive sGC20.
Of note, the shift from sGC to apo-sGC is not 1-to-1. Some sGC appears to be lost due to inactivation beyond recovery by apo-sGC activators, e.g., by channeling into the ubiquitylation-proteasome pathway74. Nevertheless, an apparent net shift from sGC to apo-sGC as the primary source of cGMP formation is a common denominator and has recently been observed by us in another high NO model of ischemic stroke6 and others in an asthma model20. In contrast to other observations, in our settings, chronic NO incubation for 72 h versus overnight20, did affect sGC β1 expression independent of S-nitrosylation.
In conclusion, our data suggest that both in-vitro and in-vivo, and both under physiological conditions and in disease, NO self-limits its ability to induce cGMP formation via chemical redox feedback, which inactivates sGC and causes an apparent net shift towards apo-sGC. Our findings are of direct therapeutic importance as a pathological sGC/apo-sGC ratio can be treated with sGC activator compounds74, thereby reinstalling cGMP synthesis and PKG signaling7,37. Moreover, concerning the long-established class of NO donor drugs and the use of inhaled NO, a cautionary note needs to be added. Not only do they cause reversible tolerance, but also, as we now find, irreversible downregulation of sGC and apo-sGC formation. Our data explain, thus, also the superiority of the novel NO-independent sGC stimulators, at least in indications such as pulmonary hypertension15.
Materials and Methods
Chemicals
Polyclonal antibodies specific for sGCα1 and sGCβ1 have been described elsewhere (30). Actin monoclonal antibody (Oncogene Research Products, Boston, USA); collagenase type CLS II (Merck, Netherlands); 8-Bromo-cGMP (BIOLOG, Germany); L-NAME, DETA/NO, DEA/NO, IBMX and GTP (Enzo Life Sciences, Netherlands); BAY 58-2667 was synthesized as described75. All other chemicals were of the highest purity grade available and obtained from Sigma or Merck (Netherlands). DETA/NO and DEA/NO were dissolved in 10 mM NaOH, BAY 58-2667 and YC-1 in DMSO.
Tissue isolation
Thoracic aortae from i) 6- to 8-months old male PKG−/− and age-matched control mice were obtained from Prof. Franz Hofmann, Department of Pharmacology and Toxicology at the Technical University Munich (genetic background 129/Sv)33, and ii) 6- to 8-months old male eNOS−/− mice and age-matched control were obtained from the Department of Physiology at Heinrich-Heine-Universität Düsseldorf (genetic background C57BL/6)36. Animals’ care was in accordance with guidelines of Technical University Munich and Heinrich-Heine-Universität Düsseldorf. Experimental protocols were approved by the animal ethics committees of Technical University Munich and Heinrich-Heine-Universität Düsseldorf. Thoracic aortae were grounded in a mortar to a powder that was used for protein determination, Western blots, or sGC activity assays. About 50 mg of tissue powder was suspended in homogenization buffer and shortly homogenized in an Ultra Turrax at 4 °C. Samples were diluted with four volumes of Rotiload buffer (Roth, Germany) and boiled for 10 min at 95 °C, centrifuged for 2–3 min at 14,000 rpm, and the supernatant used for protein determination and protein immunoblot analysis.
Preparation of pulmonary arteries from a porcine ARDS model
Pigs were acclimated for at least 24 h before use in the study and handled carefully to avoid any stress. Body temperature was kept constant using a circulating-water heating pad and cage heating. The porcine ARDS model was induced as previously described76. Briefly, pigs (30–35 kg) were pre-medicated with midazolam (1 mg/kg i.m.) and ketamine (10 mg/kg i.m.); intravenous anesthesia was induced and maintained with midazolam (bolus 0.5–1 mg/kg, followed by 1–2 mg/kg/h) and ketamine (bolus 3–4 mg/kg, followed by 10–18 mg/kg/h). Neuromuscular block was achieved with atracurium (bolus 3–4 mg/kg, followed by 1–2 mg/kg/h). Pigs were mechanically ventilated in a volume-controlled mode with the following settings: Tidal volume (VT) of 8 ml/kg, positive end-expiratory pressure (PEEP) of 5 cm H2O, the fraction of inspired oxygen (FIO2) of 1.0, respiratory rate (RR) adjusted in accordance to an arterial partial pressure of carbon dioxide (PaCO2) between 35–45 mmHg and inspiration to expiration (I:E) of 1:1. Following this preparation, acute lung injury (ALI) was established according to a two-hit model76–79. After established ALI, a lung-protective ventilation strategy was initiated with the following settings: pressure control ventilation (PCV), VT of 6 ml/kg, PEEP of 16 cm H2O, FIO2 of 0.5, RR adjusted in accordance to a PaCO2 between 35-45 mmHg and I:E of 1:1. Mild hypovolemia was induced by means of drainage of 25% of circulating blood volume. After resuscitation with different colloids (HAES 6%, 130/0.4, and gelatin 4%) and crystalloid solutions (Ringer Acetate, Baxter), the lung-protective ventilation was maintained for 4 h. The overall duration of mechanical ventilation was 10-11 h. After that, animals were sacrificed by intravenous boluses of 2 g thiopental and 50 ml KCl 1 M, and organs were snap-frozen in liquid nitrogen. Pulmonary arteries were removed immediately after death, snap-frozen in liquid nitrogen and stored at minus 80 °C or otherwise processed immediately to tissue powder and subsequently suspended in homogenization-buffer and homogenized in an Ultra Turrax at 4 °C. These samples were then used further for protein determination, protein immune blots, and sGC activity assays.
PPAECs
Fresh porcine pulmonary arteries were obtained from a local slaughterhouse and maintained in phosphate-buffered saline (PBS; 10 mM Na2HPO4, 1.8 mM KH2PO4, 140 mM NaCl, 2.7 mM KCl, pH 7.4) at 37 °C. PPAECs were isolated enzymatically by incubation of the aorta inner surface with collagenase type CLS II (0.5 mg/mL for 10 min at room temperature) and then collected in HEPES-buffered medium 199. After centrifugation (250 × g, 10 min) the pellet was re-suspended in growth medium (medium 199 supplemented with 10% fetal calf serum, 100 U/mL penicillin, 100 µg/mL streptomycin) and cells were propagated in coated plastic flasks and incubated (37 °C, 6% CO2). Upon confluence, endothelial cell monolayers were sub-cultured in 35-mm (for Western blot) or 60-mm (for cGMP determination) gelatin-coated dishes. Confluent cell monolayers from the second passage were used for experiments. PPAECs consistently formed a viable confluent monolayer without signs of significant cell loss, e.g., cell detachment or clumping. In all pilot experiments and intermittent controls, no cell death occurred during the 72 h exposure to 100 µM DETA/NO consistent with previous publications80–82. The growth medium was replaced either every 12 or 24 hours if applicable containing the indicated compounds. After incubation time, cells were subsequently used for sGC activity measurements or western blot analysis.
Detection and quantification of sGC protein
Western blotting procedures were described previously83. Briefly, cells were lysed in 250 µL Roti-Load sample buffer (ROTH, Karlsruhe, Germany), preheated to 95 °C and then boiled for an additional 10 min before loading on SDS gel electrophoresis. Primary antibodies were diluted 1:4000 for anti-sGCα1 and 1:2000 for anti-sGCβ1 antibody in 3% dry milk in TBST and incubated with nitrocellulose membranes at 4 °C over-night following challenge of membranes with secondary goat anti-rabbit antibody (1:2000 in 3% milk in TBST) conjugated to horseradish peroxidase (Dako A/S, Denmark). Immuno-complexes were visualized using an enhanced chemiluminescence kit (Amersham Pharmacia Biotech, Freiburg). Samples were quantified with a Kodak Imager Station 440 CF and with the NIH 1.6 software, and all blots were standardized to ß-actin or GAPDH expression that was not affected by the treatments. Representative western blot examples are shown in Supplementary Fig. S4.
Measurement of cGMP levels
For measuring cGMP levels, cells were pretreated for 30 min with the phosphodiesterase inhibitors84, IBMX (1 mM) and zaprinast (100 μM). Then, sGC was consistently stimulated with 250 µM DEA/NO or 10 µM BAY 58-2667 for 3 min at 37 °C. The high concentration of DEA/NO (250 µM) was chosen based on previous studies using endothelial cells, including PPAECs63,67,85. However, other studies showed that cGMP accumulation in endothelial cells in response to DEA/NO peaked at about 1 µM86. Of note, however, there are controversial data regarding the NO peak concentration released from DEA/NO. One study87 showed that 250 µM DEA/NO would give a peak NO concentration of about 30 µM while another study88 showed that 1 mM DEA/NO would release 2.87 μM/min of NO during 3 minutes. Therefore, 250 µM would release 0.71 μM/min of NO. Additional factors, such as medium components may be responsible for this broad range of apparent free concentrations of NO.
After sGC stimulation, cells were immediately lysed in 80% ethanol. Cells were scraped and, after evaporation of ethanol, resuspended in assay buffer and sonicated. Measurement of sGC activity in crude homogenates of porcine tissue was performed as previously described83. Briefly, all samples were measured as the formation of cGMP at 37 °C during 10 min in a total incubation volume of 100 µl containing 50 mM triethanolamine-HCl (pH 7.4), 3 mM MgCl2, 3 mM glutathione, 1 mM IBMX, 100 mM zaprinast, 5 mM creatine phosphate, 0.25 mg/ml creatine kinase and 1 mM or 0.5 mM GTP. The reaction was started by the simultaneous addition of the sample and either DEA/NO or BAY 58-2667, respectively. After incubation of each sample for 10 min, the reaction was stopped by boiling for 10 min at 95 °C. Thereafter the amount of cGMP was subsequently determined by a commercial enzyme immunoassay kit (Enzo Life Sciences, Netherlands).
Statistics
For comparisons, Student’s t-test or multiple comparisons one-way analysis of variance (ANOVA) was followed by Bonferroni’s test. Calculations were performed using GraphPad Prism 6.0 (GraphPad Software, San Diego, USA). All data are expressed as mean ± SEM. P-value <0.05 was considered significant.
Supplementary information
Acknowledgements
We thank Franz Hofmann for providing the PKG−/− mice tissues; Helmut Müller, for expert technical assistance; Kirstin Wingler, Rob Hermans, Sabine Meurer and Merlijn Meens for carefully reading and editing earlier versions of this manuscript. This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 777111 (REPO-TRIAL) and European Research Council Advanced Grant (RadMed) No. 294683. This reflects only the author’s view, and the European Commission is not responsible for any use that may be made of the information it contains.
Author contributions
H.H.H.W.S. designed the research; V.T.D., M.D., P.I.N., A.Gü., C.I.A. performed research; A.Gü. and A.Gö. contributed to new reagents/analytic tools; V.T.D., M.E., M.D., P.I.N., C.I.A. and H.H.H.W.S. analyzed data; and V.T.D., M.E., A.Gö. and H.H.H.W.S. wrote the paper.
Data availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Vu Thao-Vi Dao and Mahmoud H. Elbatreek.
Contributor Information
Mahmoud H. Elbatreek, Email: melbatreek@ppmlab.net
Harald H. H. W. Schmidt, Email: hschmidt@ppmlab.net
Supplementary information
is available for this paper at 10.1038/s41598-020-66639-w.
References
- 1.Bian K, Doursout M-F, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. Journal of clinical hypertension (Greenwich, Conn.) 2008;10:304–310. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. Journal of physiology and pharmacology: an official journal of the Polish Physiological Society. 2002;53:503–514. [PubMed] [Google Scholar]
- 3.Cary SPL, Winger JA, Marletta MA. Tonic and acute nitric oxide signaling through soluble guanylate cyclase is mediated by nonheme nitric oxide, ATP, and GTP. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13064–13069. doi: 10.1073/pnas.0506289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacology & therapeutics. 2009;122:216–238. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Förstermann U, Xia N, Li H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circulation research. 2017;120:713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 6.Langhauser F, et al. A diseasome cluster-based drug repurposing of soluble guanylate cyclase activators from smooth muscle relaxation to direct neuroprotection. NPJ systems biology and applications. 2018;4:8. doi: 10.1038/s41540-017-0039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stasch J-P, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. The Journal of clinical investigation. 2006;116:2552–2561. doi: 10.1172/JCI28371DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh A, Stasch J-P, Papapetropoulos A, Stuehr DJ. Nitric oxide and heat shock protein 90 activate soluble guanylate cyclase by driving rapid change in its subunit interactions and heme content. The Journal of biological chemistry. 2014;289:15259–15271. doi: 10.1074/jbc.M114.559393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh A, Stuehr DJ. Soluble guanylyl cyclase requires heat shock protein 90 for heme insertion during maturation of the NO-active enzyme. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:12998–13003. doi: 10.1073/pnas.1205854109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beuve, A. Thiol-Based Redox Modulation of Soluble Guanylyl Cyclase, the Nitric Oxide Receptor. Antioxidants & redox signaling, ars.2015.6591, 10.1089/ars.2015.6591 (2016). [DOI] [PMC free article] [PubMed]
- 11.Mayer B, et al. Inactivation of soluble guanylate cyclase by stoichiometric S-nitrosation. Mol Pharmacol. 2009;75:886–891. doi: 10.1124/mol.108.052142. [DOI] [PubMed] [Google Scholar]
- 12.Brandes RP, et al. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35:231–236. doi: 10.1161/01.HYP.35.1.231. [DOI] [PubMed] [Google Scholar]
- 13.Faraci FM, Sigmund CD, Shesely EG, Maeda N, Heistad DD. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. The American journal of physiology. 1998;274:H564–570. doi: 10.1152/ajpheart.1998.274.2.H564. [DOI] [PubMed] [Google Scholar]
- 14.Sardón T, Baltrons MA, García A. Nitric oxide-dependent and independent down-regulation of NO-sensitive guanylyl cyclase in neural cells. Toxicology letters. 2004;149:75–83. doi: 10.1016/j.toxlet.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 15.Auger, W. R. & Jamieson, S. W. Pulmonary Thromboendarterectomy Program at University of California, S. D. Riociguat for pulmonary hypertension. The New England journal of medicine369, 2266–2268, 10.1056/NEJMc1312903 (2013). [DOI] [PubMed]
- 16.Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primary pulmonary hypertension. The New England journal of medicine. 2000;343:1342–1342. doi: 10.1056/NEJM200011023431814. [DOI] [PubMed] [Google Scholar]
- 17.Lange M, Enkhbaatar P, Nakano Y, Traber DL. Role of nitric oxide in shock: the large animal perspective. Frontiers in bioscience (Landmark edition) 2009;14:1979–1989. doi: 10.2741/3357. [DOI] [PubMed] [Google Scholar]
- 18.Muzaffar S, Jeremy JY, Angelini GD, Stuart-Smith K, Shukla N. Role of the endothelium and nitric oxide synthases in modulating superoxide formation induced by endotoxin and cytokines in porcine pulmonary arteries. Thorax. 2003;58:598–604. doi: 10.1136/thorax.58.7.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sittipunt C, et al. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:503–510. doi: 10.1164/ajrccm.163.2.2004187. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh A, et al. Soluble guanylate cyclase as an alternative target for bronchodilator therapy in asthma. Proc Natl Acad Sci USA. 2016;113:E2355–2362. doi: 10.1073/pnas.1524398113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridnour LA, et al. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102:13147–13152. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung A, Newland PL, Zaben M, Attard GS, Gray WP. Intracellular nitric oxide mediates neuroproliferative effect of neuropeptide y on postnatal hippocampal precursor cells. J Biol Chem. 2012;287:20187–20196. doi: 10.1074/jbc.M112.346783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rehberg S, et al. The role of nitric oxide and reactive nitrogen species in experimental ARDS. Front Biosci (Schol Ed) 2010;2:18–29. doi: 10.2741/s43. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Z, et al. Protein kinase G phosphorylates soluble guanylyl cyclase on serine 64 and inhibits its activity. Arterioscler Thromb Vasc Biol. 2008;28:1803–1810. doi: 10.1161/ATVBAHA.108.165043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Browner NC, Dey NB, Bloch KD, Lincoln TM. Regulation of cGMP-dependent protein kinase expression by soluble guanylyl cyclase in vascular smooth muscle cells. J Biol Chem. 2004;279:46631–46636. doi: 10.1074/jbc.M408518200. [DOI] [PubMed] [Google Scholar]
- 26.Browner NC, Dey NB, Bloch KD, Lincoln TM. Regulation of cGMP-dependent protein kinase expression by soluble guanylyl cyclase in vascular smooth muscle cells. The Journal of biological chemistry. 2004;279:46631–46636. doi: 10.1074/jbc.M408518200. [DOI] [PubMed] [Google Scholar]
- 27.Cornwell TL, Lincoln TM. Regulation of intracellular Ca2+ levels in cultured vascular smooth muscle cells. Reduction of Ca2+ by atriopeptin and 8-bromo-cyclic GMP is mediated by cyclic GMP-dependent protein kinase. The Journal of biological chemistry. 1989;264:1146–1155. [PubMed] [Google Scholar]
- 28.Draijer R, Atsma DE, van der Laarse A, van Hinsbergh VW. cGMP and nitric oxide modulate thrombin-induced endothelial permeability. Regulation via different pathways in human aortic and umbilical vein endothelial cells. Circulation research. 1995;76:199–208. doi: 10.1161/01.RES.76.2.199. [DOI] [PubMed] [Google Scholar]
- 29.Draijer R, et al. Expression of cGMP-dependent protein kinase I and phosphorylation of its substrate, vasodilator-stimulated phosphoprotein, in human endothelial cells of different origin. Circulation research. 1995;77:897–905. doi: 10.1161/01.RES.77.5.897. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Z, et al. Protein kinase G phosphorylates soluble guanylyl cyclase on serine 64 and inhibits its activity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28:1803–1810. doi: 10.1161/ATVBAHA.108.165043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sellak H, Choi C-s, Dey NB, Lincoln TM. Transcriptional and post-transcriptional regulation of cGMP-dependent protein kinase (PKG-I): pathophysiological significance. Cardiovascular research. 2013;97:200–207. doi: 10.1093/cvr/cvs327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galle J, et al. Effects of the soluble guanylyl cyclase activator, YC-1, on vascular tone, cyclic GMP levels and phosphodiesterase activity. Br J Pharmacol. 1999;127:195–203. doi: 10.1038/sj.bjp.0702495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfeifer A, et al. Defective smooth muscle regulation in cGMP kinase I-deficient mice. The EMBO journal. 1998;17:3045–3051. doi: 10.1093/emboj/17.11.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernhoff NB, Derbyshire ER, Underbakke ES, Marletta MA. Heme-assisted S-nitrosation desensitizes ferric soluble guanylate cyclase to nitric oxide. The Journal of biological chemistry. 2012;287:43053–43062. doi: 10.1074/jbc.M112.393892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sayed N, et al. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circulation research. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evgenov OV, et al. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nature reviews. Drug discovery. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayed N, et al. Nitroglycerin-induced S-nitrosylation and desensitization of soluble guanylyl cyclase contribute to nitrate tolerance. Circ Res. 2008;103:606–614. doi: 10.1161/CIRCRESAHA.108.175133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sayed N, Baskaran P, Ma X, van den Akker F, Beuve A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc Natl Acad Sci U S A. 2007;104:12312–12317. doi: 10.1073/pnas.0703944104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parinandi NL, Scribner WM, Vepa S, Shi S, Natarajan V. Phospholipase D activation in endothelial cells is redox sensitive. Antioxid Redox Signal. 1999;1:193–210. doi: 10.1089/ars.1999.1.2-193. [DOI] [PubMed] [Google Scholar]
- 41.Morrell CN, et al. Regulation of platelet granule exocytosis by S-nitrosylation. Proc Natl Acad Sci U S A. 2005;102:3782–3787. doi: 10.1073/pnas.0408310102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt P, Schramm M, Schröder H, Stasch J-P. Mechanisms of nitric oxide independent activation of soluble guanylyl cyclase. European journal of pharmacology. 2003;468:167–174. doi: 10.1016/S0014-2999(03)01674-1. [DOI] [PubMed] [Google Scholar]
- 43.Ujiie K, et al. Homologous and heterologous desensitization of a guanylyl cyclase-linked nitric oxide receptor in cultured rat medullary interstitial cells. The Journal of pharmacology and experimental therapeutics. 1994;270:761–767. [PubMed] [Google Scholar]
- 44.Yuen PS, Doolittle LK, Garbers DL. Dominant negative mutants of nitric oxide-sensitive guanylyl cyclase. The Journal of biological chemistry. 1994;269:791–793. [PubMed] [Google Scholar]
- 45.Filippov G, Bloch DB, Bloch KD. Nitric oxide decreases stability of mRNAs encoding soluble guanylate cyclase subunits in rat pulmonary artery smooth muscle cells. The Journal of clinical investigation. 1997;100:942–948. doi: 10.1172/JCI119610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrero R, Torres M. Prolonged exposure of chromaffin cells to nitric oxide down-regulates the activity of soluble guanylyl cyclase and corresponding mRNA and protein levels. BMC biochemistry. 2002;3:26. doi: 10.1186/1471-2091-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papapetropoulos A, Go CY, Murad F, Catravas JD. Mechanisms of tolerance to sodium nitroprusside in rat cultured aortic smooth muscle cells. British journal of pharmacology. 1996;117:147–155. doi: 10.1111/(ISSN)1476-5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hussain MB, Hobbs AJ, MacAllister RJ. Autoregulation of nitric oxide-soluble guanylate cyclase-cyclic GMP signalling in mouse thoracic aorta. Br J Pharmacol. 1999;128:1082–1088. doi: 10.1038/sj.bjp.0702874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brandes RP, et al. Increased nitrovasodilator sensitivity in endothelial nitric oxide synthase knockout mice: role of soluble guanylyl cyclase. Hypertension. 2000;35:231–236. doi: 10.1161/01.hyp.35.1.231. [DOI] [PubMed] [Google Scholar]
- 50.Cortese-Krott MM, et al. Identification of a soluble guanylate cyclase in RBCs: preserved activity in patients with coronary artery disease. Redox Biol. 2018;14:328–337. doi: 10.1016/j.redox.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oppermann M, Dao VT, Suvorava T, Bas M, Kojda G. Effect of oral organic nitrates on expression and activity of vascular soluble guanylyl cyclase. Br J Pharmacol. 2008;155:335–342. doi: 10.1038/bjp.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi A, et al. Expression of inducible nitric oxide synthase and inflammatory cytokines in alveolar macrophages of ARDS following sepsis. Chest. 1998;113:1632–1639. doi: 10.1378/chest.113.6.1632. [DOI] [PubMed] [Google Scholar]
- 53.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–L645. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 54.Ding J, Song D, Ye X, Liu SF. A pivotal role of endothelial-specific NF-kappaB signaling in the pathogenesis of septic shock and septic vascular dysfunction. J Immunol. 2009;183:4031–4038. doi: 10.4049/jimmunol.0900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leo MD, Kandasamy K, Subramani J, Tandan SK, Kumar D. Involvement of inducible nitric oxide synthase and dimethyl arginine dimethylaminohydrolase in Nomega-nitro-L-arginine methyl ester (L-NAME)-induced hypertension. Cardiovasc Pathol. 2015;24:49–55. doi: 10.1016/j.carpath.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Park M, et al. NF-kappaB-responsive miR-155 induces functional impairment of vascular smooth muscle cells by downregulating soluble guanylyl cyclase. Exp Mol Med. 2019;51:17. doi: 10.1038/s12276-019-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu F, Tyml K, Wilson JX. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J Cell Physiol. 2008;217:207–214. doi: 10.1002/jcp.21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 59.Mayer B, Beretta M. The enigma of nitroglycerin bioactivation and nitrate tolerance: news, views and troubles. Br J Pharmacol. 2008;155:170–184. doi: 10.1038/bjp.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oelze M, et al. Chronic therapy with isosorbide-5-mononitrate causes endothelial dysfunction, oxidative stress, and a marked increase in vascular endothelin-1 expression. Eur Heart J. 2013;34:3206–3216. doi: 10.1093/eurheartj/ehs100. [DOI] [PubMed] [Google Scholar]
- 61.Ghofrani HA, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–340. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 62.Connelly L, Palacios-Callender M, Ameixa C, Moncada S, Hobbs AJ. Biphasic regulation of NF-kappa B activity underlies the pro- and anti-inflammatory actions of nitric oxide. J Immunol. 2001;166:3873–3881. doi: 10.4049/jimmunol.166.6.3873. [DOI] [PubMed] [Google Scholar]
- 63.Nedvetsky PI, et al. Heat shock protein 90 regulates stabilization rather than activation of soluble guanylate cyclase. FEBS Lett. 2008;582:327–331. doi: 10.1016/j.febslet.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 64.Thoonen R, et al. Cardiovascular and pharmacological implications of haem-deficient NO-unresponsive soluble guanylate cyclase knock-in mice. Nature communications. 2015;6:8482. doi: 10.1038/ncomms9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kagota S, et al. Chronic production of peroxynitrite in the vascular wall impairs vasorelaxation function in SHR/NDmcr-cp rats, an animal model of metabolic syndrome. J Pharmacol Sci. 2009;109:556–564. doi: 10.1254/jphs.08273fp. [DOI] [PubMed] [Google Scholar]
- 66.Hoffmann LS, et al. Distinct molecular requirements for activation or stabilization of soluble guanylyl cyclase upon haem oxidation-induced degradation. Br J Pharmacol. 2009;157:781–795. doi: 10.1111/j.1476-5381.2009.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stasch JP, et al. Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552–2561. doi: 10.1172/JCI28371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Armitage ME, Wingler K, Schmidt HH, La M. Translating the oxidative stress hypothesis into the clinic: NOX versus NOS. Journal of molecular medicine (Berlin, Germany) 2009;87:1071–1076. doi: 10.1007/s00109-009-0544-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mikhael MR, Roshan T, Soe-Lin S, Apte S, Ponka P. Nitrogen monoxide inhibits haem synthesis in mouse reticulocytes. Biochem J. 2013;451:61–67. doi: 10.1042/BJ20121649. [DOI] [PubMed] [Google Scholar]
- 70.Pae HO, Oh GS, Choi BM, Kim YM, Chung HT. A molecular cascade showing nitric oxide-heme oxygenase-1-vascular endothelial growth factor-interleukin-8 sequence in human endothelial cells. Endocrinology. 2005;146:2229–2238. doi: 10.1210/en.2004-1431. [DOI] [PubMed] [Google Scholar]
- 71.Polte T, Abate A, Dennery PA, Schroder H. Heme oxygenase-1 is a cGMP-inducible endothelial protein and mediates the cytoprotective action of nitric oxide. Arterioscler Thromb Vasc Biol. 2000;20:1209–1215. doi: 10.1161/01.atv.20.5.1209. [DOI] [PubMed] [Google Scholar]
- 72.Mingone CJ, Ahmad M, Gupte SA, Chow JL, Wolin MS. Heme oxygenase-1 induction depletes heme and attenuates pulmonary artery relaxation and guanylate cyclase activation by nitric oxide. Am J Physiol Heart Circ Physiol. 2008;294:H1244–1250. doi: 10.1152/ajpheart.00846.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Juckett M, et al. Heme and the endothelium. Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J Biol Chem. 1998;273:23388–23397. doi: 10.1074/jbc.273.36.23388. [DOI] [PubMed] [Google Scholar]
- 74.Meurer S, et al. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circulation research. 2009;105:33–41. doi: 10.1161/CIRCRESAHA.109.198234. [DOI] [PubMed] [Google Scholar]
- 75.Stasch J-P, et al. NO- and haem-independent activation of soluble guanylyl cyclase: molecular basis and cardiovascular implications of a new pharmacological principle. British journal of pharmacology. 2002;136:773–783. doi: 10.1038/sj.bjp.0704778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silva PL, et al. Effects of intravascular volume replacement on lung and kidney function and damage in nonseptic experimental lung injury. Anesthesiology. 2013;118:395–408. doi: 10.1097/ALN.0b013e31827e554c. [DOI] [PubMed] [Google Scholar]
- 77.Lachmann B, Robertson B, Vogel J. In vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta anaesthesiologica Scandinavica. 1980;24:231–236. doi: 10.1111/j.1399-6576.1980.tb01541.x. [DOI] [PubMed] [Google Scholar]
- 78.Spieth PM, et al. Effects of vaporized perfluorohexane and partial liquid ventilation on regional distribution of alveolar damage in experimental lung injury. Intensive Care Med. 2007;33:308–314. doi: 10.1007/s00134-006-0428-7. [DOI] [PubMed] [Google Scholar]
- 79.Wittenstein, J. et al. Effects of variable versus nonvariable controlled mechanical ventilation on pulmonary inflammation in experimental acute respiratory distress syndrome in pigs. Br J Anaesth, 10.1016/j.bja.2019.12.040 (2020). [DOI] [PMC free article] [PubMed]
- 80.Walford GA, Moussignac RL, Scribner AW, Loscalzo J, Leopold JA. Hypoxia potentiates nitric oxide-mediated apoptosis in endothelial cells via peroxynitrite-induced activation of mitochondria-dependent and -independent pathways. J Biol Chem. 2004;279:4425–4432. doi: 10.1074/jbc.M310582200. [DOI] [PubMed] [Google Scholar]
- 81.Kang JJ, et al. alpha-galactosidase A deficiency promotes von Willebrand factor secretion in models of Fabry disease. Kidney Int. 2019;95:149–159. doi: 10.1016/j.kint.2018.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Snyder CM, Shroff EH, Liu J, Chandel NS. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. Plos One. 2009;4:e7059. doi: 10.1371/journal.pone.0007059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nedvetsky PI, Kleinschnitz C, Schmidt HHHW. Regional distribution of protein and activity of the nitric oxide receptor, soluble guanylyl cyclase, in rat brain suggests multiple mechanisms of regulation. Brain research. 2002;950:148–154. doi: 10.1016/S0006-8993(02)03015-9. [DOI] [PubMed] [Google Scholar]
- 84.Schmidt K, Mayer B, Kukovetz WR. Effect of calcium on endothelium-derived relaxing factor formation and cGMP levels in endothelial cells. Eur J Pharmacol. 1989;170:157–166. doi: 10.1016/0014-2999(89)90536-0. [DOI] [PubMed] [Google Scholar]
- 85.Raoch V, et al. Nitric oxide decreases the expression of endothelin-converting enzyme-1 through mRNA destabilization. Arterioscler Thromb Vasc Biol. 2011;31:2577–2585. doi: 10.1161/ATVBAHA.111.232025. [DOI] [PubMed] [Google Scholar]
- 86.Schmidt K, Schrammel A, Koesling D, Mayer B. Molecular mechanisms involved in the synergistic activation of soluble guanylyl cyclase by YC-1 and nitric oxide in endothelial cells. Mol Pharmacol. 2001;59:220–224. doi: 10.1124/mol.59.2.220. [DOI] [PubMed] [Google Scholar]
- 87.Schmidt K, Desch W, Klatt P, Kukovetz WR, Mayer B. Release of nitric oxide from donors with known half-life: a mathematical model for calculating nitric oxide concentrations in aerobic solutions. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:457–462. doi: 10.1007/pl00004969. [DOI] [PubMed] [Google Scholar]
- 88.Zou MH, Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996;382:101–104. doi: 10.1016/0014-5793(96)00160-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.