

Abstract
Rett syndrome is an X-linked neurodevelopmental disorder caused by mutation in the methyl-CpG-binding protein 2 gene (MECP2) in the majority of cases. We describe an RNA sequencing dataset of postmortem brain tissue samples from four females clinically diagnosed with Rett syndrome and four age-matched female donors. The dataset contains 16 transcriptomes, including two brain regions, temporal and cingulate cortex, for each individual. We compared our dataset with published transcriptomic analyses of postmortem brain tissue from Rett syndrome and found consistent gene expression alterations among regions of the cerebral cortex. Our data provide a valuable resource to explore the biology of the human brain in Rett syndrome.
Subject terms: Genetics research, Brain, Paediatric neurological disorders, Autism spectrum disorders, Gene expression
| Measurement(s) | transcriptome • RNA |
| Technology Type(s) | RNA sequencing |
| Factor Type(s) | Rett syndrome brain versus control brain • brain regions (temporal cortex and cingulate cortex) |
| Sample Characteristic - Organism | Homo sapiens |
Machine-accessible metadata file describing the reported data: 10.6084/m9.figshare.12383291
Background & Summary
Rett syndrome (RTT) is an X-linked neurodevelopmental disorder mostly caused by heterozygous de novo mutation in the methyl-CpG-binding protein 2 gene (MECP2) and predominantly affecting females1. MECP2 duplications have been identified in males with developmental encephalopathy, seizures, autistic features, and recurrent infection2. These clinical disorders illustrate the critical requirement for proper MECP2 expression in human brain development, though how MeCP2 dysfunction leads to the RTT phenotype is unclear.
MeCP2 acts as a global transcriptional regulator by recruiting chromatin-remodeling complexes or regulating higher-order chromatin structures3–8. Thus, MeCP2 may be required for fine-tuning the gene expression for a network of protein-coding genes through both direct and indirect mechanisms. Consistent with this hypothesis, small magnitude changes in gene expression have been detected in brain tissue from either human postmortem RTT samples or mouse Mecp2-mutants9–12. However, most transcriptional studies of postmortem RTT brain have used microarray platforms with small numbers and a lack of age-matched control samples, which impact the sensitivity for detecting transcriptional changes. One study used both microarrays and RNA sequencing (RNA-seq) to examine frontal and temporal cortex from individuals with RTT compared to controls and identified over 200 differentially expressed genes after normalizing data for neuron versus glia composition of samples13. Another larger study used RNA-seq to examine motor cortex and cerebellum and identified over 2,000 differentially expressed genes with a global increase in expression14.
We generated RNA-seq data using brain samples for two distinct brain regions, temporal cortex and cingulate cortex, from four female RTT and four age-matched female donors. Reduced volume and dendritic branching of neurons in the temporal cortex and reduced connectivity of the cingulate cortex have been reported in RTT, indicating the importance of these brain regions in the disorder15–18. We also compared our data with the transcriptomic profiles of RTT brain samples from published RNA-seq datasets13,14. The composite analysis will be useful to facilitate interpretation and further understanding of MECP2-mediated changes in human brain.
Methods
Brain samples
Postmortem brain tissue samples were obtained from the Harvard Brain Bank (http://hbtrc.mclean.harvard.edu/) and the National Institutes of Health (NIH) NeuroBioBank (https://neurobiobank.nih.gov), with approval from the coordinating foundation (https://www.rettsyndrome.org). Consent was obtained from next of kin and tissue was collected with approval from the Partners Human Research Committee for the Harvard Brain Bank and from The University of Maryland Institutional Review Board (IRB) and The Maryland Department of Health and Mental Hygiene IRB for the NeuroBioBank. Work was approved by the University of Southern California and is compliant with all ethical regulations. Frozen temporal (BA36/38) and cingulate cortex samples were obtained from four RTT and four control (CTL) brain donors that were matched in age (Fig. 1). The Harvard Brain Bank sequenced MECP2 coding exons and reported intragenic mutations in two of the four brains. Brain donor characteristics are described in Table 1.
Fig. 1.
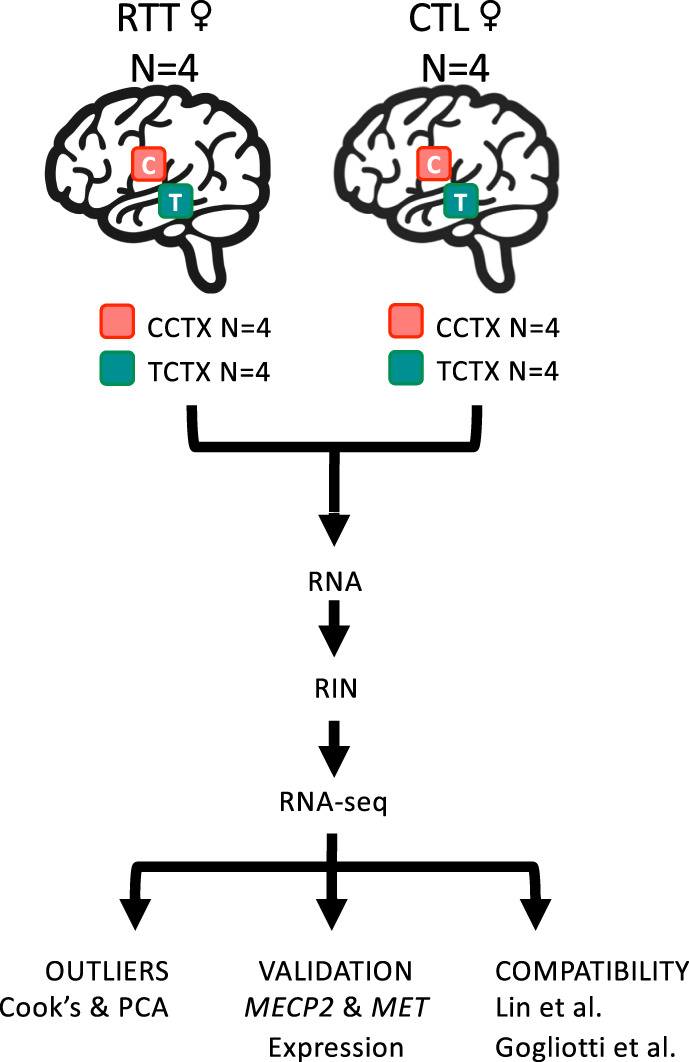
Overview of the experimental workflow.
Table 1.
Brain Donor Characteristics.
| Brain | Group | Gender | Age | PMI | Source | MECP2 | |
|---|---|---|---|---|---|---|---|
| NM_004992.3 | |||||||
| cDNA | Protein | ||||||
| 1038 | CTL | F | 24 | 7 | NBB | NA | NA |
| 1614 | CTL | F | 27 | 18 | NBB | NA | NA |
| 4724 | CTL | F | 16 | 15 | NBB | NA | NA |
| 4725 | CTL | F | 32 | 17 | NBB | NA | NA |
| 6355 | RTT | F | 16 | 28 | HBTRC | NR | NR |
| 7773 | RTT | F | 24 | 25 | HBTRC | c.473 C > T | p.Thr158Met |
| 7783 | RTT | F | 26 | 39 | HBTRC | Exon Del | |
| B7992 | RTT | F | 31 | 30 | HBTRC | NR | NR |
Abbreviations: CTL, control; F, female; HBTRC, Harvard Brain Tissue Resource Center; NA, not applicable; NBB, NIH NeuroBioBank; NR, no mutation in MECP2 reported; PMI, postmortem interval in hours; RTT, Rett syndrome.
MECP2 variant confirmation
Genomic DNA was isolated from brain samples for 7773 and 7783 using the PureLink Genomic DNA Kit (LifeTechnologies) according to the manufacturer’s protocol. We performed Sanger sequencing of MECP2 to verify the reported variants (Table 1). Chromatograms were aligned to MECP2 (ENSG00000169057) using MAFFT v719. No additional genes were screened.
RNA sample and library preparation
Total RNA was previously isolated using the Qiagen RNeasy Kit according to the manufacturer’s instructions20. Double stranded cDNA fragments were synthesized from mRNA, ligated with adapters, and size-selected for library construction according to the TruSeq Sample Preparation v2 protocol using 0.5–1.5 μg of total RNA (Table 2). ERCC RNA spike-in controls were not included in this experiment. Library quality was measured using an Agilent 2100 Bioanalyzer and concentration was assessed by PicoGreen incorporation. Barcoded libraries were pooled and sequenced in two lanes using an Illumina HiSeq 2000 sequencer.
Table 2.
RNA Sample Characteristics.
| Brain | Group | Region | RIN | RNA (μg) | GEO | SRA |
|---|---|---|---|---|---|---|
| 1038 | CTL | CCTX | 9.8 | 1.50 | GSM3673208 | SRX5527579 |
| 1614 | CTL | CCTX | 9.2 | 0.70 | GSM3673209 | SRX5527580 |
| 4724 | CTL | CCTX | 9.1 | 0.60 | GSM3673210 | SRX5527581 |
| 4725 | CTL | CCTX | 9.5 | 1.50 | GSM3673211 | SRX5527582 |
| 6355 | RTT | CCTX | 8.6 | 0.60 | GSM3673212 | SRX5527583 |
| 7773 | RTT | CCTX | 9.8 | 1.50 | GSM3673213 | SRX5527584 |
| 7783 | RTT | CCTX | 8.6 | 0.70 | GSM3673214 | SRX5527585 |
| B7992 | RTT | CCTX | 10 | 1.50 | GSM3673215 | SRX5527586 |
| 1038 | CTL | TCTX (BA38) | 10 | 1.50 | GSM3673216 | SRX5527587 |
| 1614 | CTL | TCTX (BA36) | 8.3 | 0.51 | GSM3673217 | SRX5527588 |
| 4724 | CTL | TCTX (BA38) | 9.7 | 1.28 | GSM3673218 | SRX5527589 |
| 4725 | CTL | TCTX (BA38) | 9.3 | 1.00 | GSM3673219 | SRX5527590 |
| 6355 | RTT | TCTX | 8.6 | 1.28 | GSM3673220 | SRX5527591 |
| 7773 | RTT | TCTX | 9.7 | 1.50 | GSM3673221 | SRX5527592 |
| 7783 | RTT | TCTX | 8.7 | 0.51 | GSM3673222 | SRX5527593 |
| B7992 | RTT | TCTX | 9.6 | 1.00 | GSM3673223 | SRX5527594 |
Abbreviations: BA, Brodmann area; CCTX, cingulate cortex; CTL, control; RIN, RNA integrity number; RNA, quantity of total RNA used as input for library preparation; RTT, Rett syndrome; TCTX, temporal cortex.
RNA-Seq data analysis
Single-end reads (100 bp) were aligned to the Human reference genome (NCBI build 37/hg19) using STAR v2.5.3a21 (see Code Availability 1). Aligned reads mapping to the exons of a gene were summarized into gene counts using featureCounts v1.622 (see Code Availability 2). Picard CollecteRnaSeqMetrics was used to measure the 3′ bias of genes in the RNA-seq data (see Code Availability 3). Gene-level differential expression was analyzed using DESeq223 specifying ~ region + group + bias as the experimental design (see Code Availability 4). Aligned reads mapping to MECP2 isoforms were also summarized using featureCounts v1.622 (see Code availability 2) by substituting isoforms for gene name.
Data Records
Count matrix and normalized count matrix were submitted to the NCBI Gene Expression Omnibus (GEO) under accession number GSE12838024. The raw FASTQ files can be downloaded from the Sequence Read Archive (SRA) under accession number SRP18855525.
Technical Validation
MECP2 variant confirmation
We verified the presence of the MECP2 c.473 C > T (p.Thr158Met) intragenic variant using DNA isolated from brain 7773 (Supplemental Fig. 1). No MECP2 variants were detected in exons 2–4 of brain 7783. Since we were unable to amplify exon 1 in 7783, we infer exon 1 is likely to be the deleted exon. We also examined RNA-seq data for presence of MECP2 variants (Supplemental Fig. 2). The MECP2 c.473 C > T (p.Thr158Met) intragenic variant was also detected in RNA-seq data from CCTX and TCTX for brain 7773. MECP2 variants were not detected in RNA-seq data for other RTT brain samples, possibly due to low sequencing read depth of MECP2 (Supplemental Fig. 3), or because causal variants are present in another gene26,27.
RNA and data quality
RNA quality was determined using the Agilent 2100 Bioanalyzer and the RNA 6000 Pico Kit and high-quality RNA was obtained from all samples (RNA integrity number [RIN] > 8.0; median RIN = 9.4 [Table 2]). At the time the experiment was performed, the TruSeq RNA Sample Prep v2 protocol (Part # 15026495 Rev.C, May 2012) was optimized for 0.1–4 μg of total RNA. Although the quantity of RNA input varied among the samples in our experiment, it was equivalent within each age- and tissue- matched case-control sample pair, and all samples were within the optimized range. On average, RNA-seq generated 21.9 million high-quality reads per sample, 70.3% of which mapped uniquely to the Human reference genome (NCBI build 37/hg19) (Table 3). RIN and RNA quantity were each correlated with the number of uniquely mapped reads (Fig. 2). Cook’s distance was calculated to test for outliers, with none detected (Fig. 3a). The first principal component explained over 50% of the variance (Fig. 3b). A correlation matrix based on the gene expression data indicated that samples mostly cluster by individual and diagnostic group, but also by 3′ bias (Fig. 3c).
Table 3.
RNA-seq Data Mapping Statistics.
| Brain | Group | Region | Total Reads | % Bases ≥ Q30 | Mean Base Quality | Uniquely Mapped Reads | Mapping Rate | Median 5′ to 3′ Bias |
|---|---|---|---|---|---|---|---|---|
| 1038 | CTL | CCTX | 22,666,185 | 77.09 | 31.71 | 18,508,346 | 81.66 | 2.16 |
| 1614 | CTL | CCTX | 20,406,028 | 76.18 | 31.42 | 13,966,751 | 68.44 | 0.76 |
| 4724 | CTL | CCTX | 17,775,778 | 76.21 | 31.45 | 12,903,748 | 72.59 | 0.51 |
| 4725 | CTL | CCTX | 17,152,400 | 76.58 | 31.57 | 13,787,313 | 80.38 | 1.36 |
| 6355 | RTT | CCTX | 16,799,774 | 76.38 | 31.49 | 11,754,027 | 69.96 | 0.71 |
| 7773 | RTT | CCTX | 22,119,416 | 77.01 | 31.70 | 17,627,391 | 79.69 | 1.36 |
| 7783 | RTT | CCTX | 32,207,239 | 77.16 | 31.72 | 22,511,241 | 69.89 | 0.43 |
| B7992 | RTT | CCTX | 21,882,359 | 76.58 | 31.56 | 17,048,617 | 77.91 | 1.09 |
| 1038 | CTL | TCTX (BA38) | 23,689,678 | 77.72 | 31.88 | 16,671,042 | 70.37 | 1.34 |
| 1614 | CTL | TCTX (BA36) | 17,569,738 | 78.01 | 31.97 | 11,367,085 | 64.70 | 0.44 |
| 4724 | CTL | TCTX (BA38) | 22,429,527 | 77.84 | 31.93 | 15,519,596 | 69.19 | 0.98 |
| 4725 | CTL | TCTX (BA38) | 24,262,545 | 78.03 | 31.97 | 16,322,635 | 67.28 | 1.04 |
| 6355 | RTT | TCTX | 25,330,544 | 76.95 | 31.64 | 14,294,799 | 56.43 | 0.36 |
| 7773 | RTT | TCTX | 22,557,491 | 77.85 | 31.92 | 15,330,591 | 67.96 | 0.97 |
| 7783 | RTT | TCTX | 17,060,381 | 77.82 | 31.92 | 11,301,790 | 66.25 | 0.82 |
| B7992 | RTT | TCTX | 26,211,057 | 77.11 | 31.70 | 16,434,103 | 62.70 | 1.01 |
Fig. 2.

RNA quality or RNA quantity versus number of uniquely mapped reads.
Fig. 3.

RNA-seq data quality assessment. (a) Boxplots showing Cook’s distance calculated for each sample. (b) Principal component analysis with samples colored by diagnostic group (CTL, RTT), brain region (CCTX, TCTX), or brain donor. (c) Heatmap of the sample distance matrix. Presence (black) or absence (grey) of 3′ bias in RNA-seq data is indicated for each sample.
MECP2 and MET differential expression
We previously used quantitative reverse transcription PCR to compare expression of MECP2_e1 (NM_004992.3), MECP2_e2 (NM_001110792.1), and MET (NM_000245.3) in the temporal cortex between RTT and CTL brains28. Consistent with our previous results, the RNA-seq data showed no significant difference in MECP2 expression between RTT and CTL brains (FDR adjusted p-value = 0.16 and 0.59, respectively), while MET expression was significantly reduced in RTT brains (FDR = 1.07 × 10−05; Fig. 4).
Fig. 4.

Boxplots showing the expression of MECP2_e1, MECP2_e2, and MET in RTT and CTL brain. Expression values are shown as normalized counts.
Compatibility with published transcriptional profiles
Two RNA-seq datasets of postmortem brain from females with RTT compared to controls have been published13,14 (Table 4). The first dataset examined pooled frontal and temporal cortex (FTTX) for each of three individuals with RTT compared to three CTL and is available from the Sequence Read Archive under accession number PRJNA30268529. The second larger dataset examined motor cortex (Motor) and cerebellum (Cblm) for nine females and six females with RTT, respectively, compared to eight CTL of each tissue, but the primary data were not accessible14. We downloaded the FASTQ files for the available dataset, aligned reads using salmon30 (see Code Availability 6), summarized the aligned reads into gene counts using tximport v1.12.131 (see Code Availability 7), and retained genes with ≥10 counts in ≥3 samples. Count data were converted to logCPM to adjust for the total counts per sample using limma v3.40.232 (see Code Availability 8), then observation-level and sample-level weights were estimated using voom32,33. We also reanalyzed our data using this workflow. Analysis of variance models were fit separately for each of three brain regions (CCTX, FCTX, FTTX), then combined in a random effects meta-analysis using GeneMeta v1.56.034 (see Code Availability 9). Our meta-analysis identified 1,455 genes that were significantly differentially expressed (FDR < 0.05) between brain samples from control individuals and those with RTT.
Table 4.
Published RTT Brain RNA-seq datasets.
| Dataset | Brain Region | # Cases | # Controls | SRA | PMID |
|---|---|---|---|---|---|
| Lin | FTTX | 2 | 3 | PRJNA302685 | 27267200 |
| TCTX | 1 | – | |||
| Gogliotti | Motor | 9 | 8 | – | 29523700 |
| CBLM | 6 | 8 |
Abbreviations: CBLM, cerebellum; FTTX, pooled frontal cortex and temporal cortex from the same individual; Motor, motor cortex; TCTX, temporal cortex; SRA, sequence read archive; PMID, PubMed unique identifier for published reference article.
To verify these results, we compared the results from our meta-analysis with differential gene expression results from previous RTT RNA-seq analyses13,14 (Fig. 5). We compared the Z-score for each of the significantly differentially expressed genes from our meta-analysis with the log2 fold change from our previous analysis (GEO DESeq2) and from each of the three published RNA-seq datasets (Lin et al., Gogliotti et al. Motor, and Gogliotti et al. Cblm; Fig. 5a). We found strong concordance among RTT transcriptional profiles from regions within the cerebral cortex, while RTT transcriptional profiles from the cerebellum were least correlated with the regions from the cerebral cortex (Fig. 5b). We aggregated the gene-wise correlation coefficients among datasets and found an overall positive correlation for 63% of the comparisons among datasets, indicating an overall agreement among the differential gene expression per dataset (Fig. 5c). Not only do our data represent an independent technical and biological replication of molecular alterations in RTT brain, but our meta-analysis demonstrates the power of combining datasets to maximize detectable results among several smaller studies.
Fig. 5.

Replication of differential gene expression between RTT and CTL brain. (a) Meta-analysis Z-Score compared to log2 fold change (FC) between RTT and CTL from our initial analysis (GEO), Lin et al. combined frontal and temporal cortex (from Table S5)13, Gogliotti et al. motor cortex (from Table S2)14, and Gogliotti et al. cerebellum (from Table S3)14. Genes with significant differential expression (False Discovery Rate [FDR] < 0.05) in the dataset represented on the X-axis are in red. (b) Spearman’s correlation between meta-analysis Z-score and logFC for each of the other datasets. Color intensity and circle size are proportional to the correlation coefficients with values displayed below the diagonal. (c) Density of gene-wise correlation coefficients among datasets in (b).
Supplementary information
Acknowledgements
This work was supported by NIH grant MH067842 to P.L. and an Epilepsy Foundation of Greater Los Angeles fellowship to K.A.A.
Author contributions
K.A.A. conceived the project, performed experiments with guidance from O.V.E., analyzed data, and wrote the manuscript. A.E.T., J.W.M. and J.S.H. analyzed data. H.M. performed experiments. T.K.B., J.A.K. and P.L. supervised the work. All authors reviewed and accepted the final version of the manuscript.
Code availability
We used the following software and versions to process our dataset as described in the text:
1. STAR v2.5.3a was used for mapping reads to the Human reference genome NCBI build 37/hg19: https://github.com/alexdobin/STAR
2. featureCounts v1.6 was used to summarize gene counts: http://bioinf.wehi.edu.au/featureCounts/
3. Picard v2.15.0 was used to measure 5′ to 3′ bias: http://broadinstitute.github.io/picard
4. DESeq2 v1.20.0 was used for differential expression analysis: https://bioconductor.org/packages/release/bioc/html/DESeq2.html.
5. IGV v2.8.2 was used to visualize MECP2 coding regions for sequence variation: http://software.broadinstitute.org/software/igv/
6. Salmon was used to align reads to the Human GRCh38 reference transcriptome and estimate counts for each transcript: https://combine-lab.github.io/salmon/
7. tximport v1.12.1 was used to summarize gene counts: https://bioconductor.org/packages/release/bioc/html/tximport.html
8. limma v3.40.2 was used to convert count data to log counts per million (logCPM) and to estimate weights: https://bioconductor.org/packages/release/bioc/html/limma.html
9. GeneMeta v1.56.0 was used to perform a random effects meta-analysis: https://www.bioconductor.org/packages/release/bioc/html/GeneMeta.html
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41597-020-0527-2.
References
- 1.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 2.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker SA, et al. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152:984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho KL, et al. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 5.Lewis JD, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-O. [DOI] [PubMed] [Google Scholar]
- 6.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 7.Nan X, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 8.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colantuoni C, et al. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- 11.Deng V, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- 12.Gibson JH, et al. Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci. 2010;11:53. doi: 10.1186/1471-2202-11-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin P, et al. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q Genes in Rett syndrome. BMC Genomics. 2016;17:427. doi: 10.1186/s12864-016-2746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gogliotti RG, et al. Total RNA Sequencing of Rett Syndrome Autopsy Samples Identifies the M4 Muscarinic Receptor as a Novel Therapeutic Target. J Pharmacol Exp Ther. 2018;365:291–300. doi: 10.1124/jpet.117.246991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armstrong DD, Dunn K, Antalffy B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol. 1998;57:1013–1017. doi: 10.1097/00005072-199811000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Mahmood A, et al. White matter impairment in Rett syndrome: diffusion tensor imaging study with clinical correlations. AJNR Am J Neuroradiol. 2010;31:295–299. doi: 10.3174/ajnr.A1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khong PL, Lam CW, Ooi CG, Ko CH, Wong VC. Magnetic resonance spectroscopy and analysis of MECP2 in Rett syndrome. Pediatr Neurol. 2002;26:205–209. doi: 10.1016/s0887-8994(01)00385-x. [DOI] [PubMed] [Google Scholar]
- 18.Subramaniam B, Naidu S, Reiss AL. Neuroanatomy in Rett syndrome: cerebral cortex and posterior fossa. Neurology. 1997;48:399–407. doi: 10.1212/wnl.48.2.399. [DOI] [PubMed] [Google Scholar]
- 19.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldinger KA, Plummer JT, Levitt P. Comparative DNA methylation among females with neurodevelopmental disorders and seizures identifies TAC1 as a MeCP2 target gene. J Neurodev Disord. 2013;5:15. doi: 10.1186/1866-1955-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aldinger KA. 2019. Transcriptome data of temporal and cingulate cortex in the Rett syndrome brain. Gene Expression Omnibus. GSE128380 [DOI] [PMC free article] [PubMed]
- 25.2019. NCBI Sequence Read Archive. SRP188555
- 26.Sajan SA, et al. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet Med. 2017;19:13–19. doi: 10.1038/gim.2016.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, et al. Rett and Rett-like syndrome: Expanding the genetic spectrum to KIF1A and GRIN1 gene. Mol Genet Genomic Med. 2019;7:e968. doi: 10.1002/mgg3.968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plummer JT, et al. Transcriptional regulation of the MET receptor tyrosine kinase gene by MeCP2 and sex-specific expression in autism and Rett syndrome. Transl Psychiatry. 2013;3:e316. doi: 10.1038/tp.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.2015. NCBI Sequence Read Archive. SRP066394
- 30.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol3, Article3, 10.2202/1544-6115.1027 (2004). [DOI] [PubMed]
- 33.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi JK, Yu U, Kim S, Yoo OJ. Combining multiple microarray studies and modeling interstudy variation. Bioinformatics. 2003;19(Suppl 1):i84–90. doi: 10.1093/bioinformatics/btg1010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Aldinger KA. 2019. Transcriptome data of temporal and cingulate cortex in the Rett syndrome brain. Gene Expression Omnibus. GSE128380 [DOI] [PMC free article] [PubMed]
- 2019. NCBI Sequence Read Archive. SRP188555
- 2015. NCBI Sequence Read Archive. SRP066394
Supplementary Materials
Data Availability Statement
We used the following software and versions to process our dataset as described in the text:
1. STAR v2.5.3a was used for mapping reads to the Human reference genome NCBI build 37/hg19: https://github.com/alexdobin/STAR
2. featureCounts v1.6 was used to summarize gene counts: http://bioinf.wehi.edu.au/featureCounts/
3. Picard v2.15.0 was used to measure 5′ to 3′ bias: http://broadinstitute.github.io/picard
4. DESeq2 v1.20.0 was used for differential expression analysis: https://bioconductor.org/packages/release/bioc/html/DESeq2.html.
5. IGV v2.8.2 was used to visualize MECP2 coding regions for sequence variation: http://software.broadinstitute.org/software/igv/
6. Salmon was used to align reads to the Human GRCh38 reference transcriptome and estimate counts for each transcript: https://combine-lab.github.io/salmon/
7. tximport v1.12.1 was used to summarize gene counts: https://bioconductor.org/packages/release/bioc/html/tximport.html
8. limma v3.40.2 was used to convert count data to log counts per million (logCPM) and to estimate weights: https://bioconductor.org/packages/release/bioc/html/limma.html
9. GeneMeta v1.56.0 was used to perform a random effects meta-analysis: https://www.bioconductor.org/packages/release/bioc/html/GeneMeta.html