

Summary
The progesterone receptor (PR) is an inducible transcription factor that plays critical roles in female reproductive processes and in several aspects of breast cancer tumorigenesis. Our report describes the type I protein arginine methyltransferase 1 (PRMT1) as a cofactor controlling progesterone pathway, through the direct methylation of PR. Mechanistic assays in breast cancer cells indicate that PRMT1 methylates PR at the arginine 637 and reduces the stability of the receptor, thereby accelerating its recycling and finally its transcriptional activity. Depletion of PRMT1 decreases the expression of a subset of progesterone-inducible genes, controlling breast cancer cells proliferation and migration. Consistently, Kaplan-Meier analysis revealed that low expression of PRMT1 predicts a longer survival among the subgroup with high PR. Our study highlights PR methylation as a molecular switch adapting the transcription requirement of breast cells during tumorigenesis.
Subject Areas: Molecular Biology, Molecular Mechanism of Gene Regulation, Molecular Interaction, Cancer
Graphical Abstract
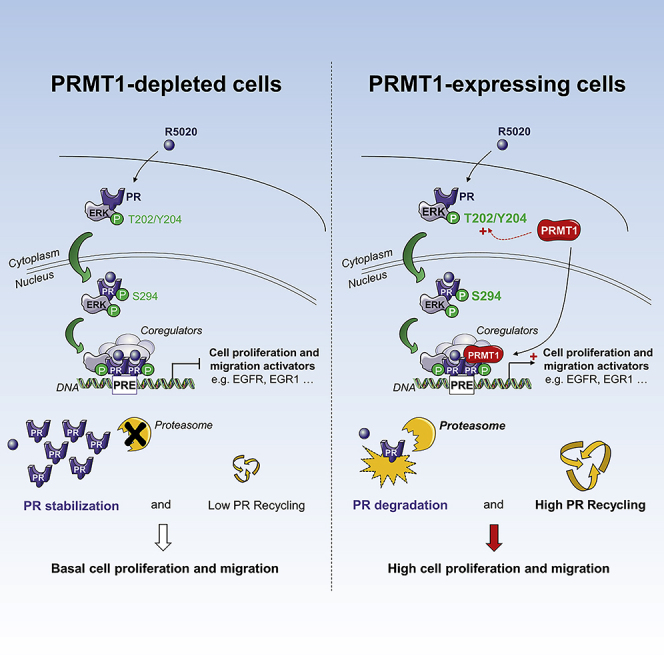
Highlights
-
•
The progesterone receptor (PR) is methylated by PRMT1 upon progestin treatment
-
•
PRMT1-dependent methylation controls the stability of PR
-
•
PRMT1 activates genes involved in the regulation of cell migration and invasion
-
•
PRMT1 expression influences the survival of patients with PR-positive breast cancer
Molecular Biology; Molecular Mechanism of Gene Regulation; Molecular Interaction; Cancer
Introduction
The progesterone receptor (PR) is a member of the nuclear hormone receptor family of ligand-dependent transcription factors (Mangelsdorf et al., 1995). Acting through its cognate steroid hormone progesterone, PR regulates the expression of gene networks to control development, differentiation, and proliferation of female reproductive tissues during the ovarian cycle and pregnancy (Brisken and O'Malley, 2010). Furthermore, numerous studies have established that PR is an important regulator of several aspects of breast cancer tumorigenesis and progression, including cell migration and invasiveness (Grimm et al., 2016, Knutson and Lange, 2014). Two major isoforms of PR exist across species, the longer PR-B and the shorter PR-A, which differ in promoter usage (Kastner et al., 1990). They are differentially expressed and exhibit distinct functions in vivo; PR-A is more responsible for progesterone actions in uterus and ovary, whereas PR-B is required for mammary gland development (Mulac-Jericevic et al., 2003), mediating the proliferative actions of progestins (Boonyaratanakornkit et al., 2001, Faivre and Lange, 2007).
PR activity is regulated by extensive post-translational modifications (PTMs) that include phosphorylation, acetylation, ubiquitination, SUMOylation, and methylation (reviewed in Abdel-Hafiz and Horwitz, 2014, Knutson and Lange, 2014). These modifications affect PR hormone sensitivity, subcellular localization, protein stability, or interactions with cofactors. For example, progesterone stimulation induces MAPK-dependent phosphorylation of its receptor on the serine 294 (Ser-294 or S294), a key modification essential for the activation and the enhancement of PR transcriptional activity. Paradoxically, this event is also a signal for the ligand-dependent degradation of the receptor, essential for its regulated transcriptional functions (Lange et al., 2000, Shen et al., 2001).
The molecular mechanisms underlying PR-dependent transcription have been extensively studied, both in the absence of, and upon exposure to progesterone (Beato and Vicent, 2012, Jacobsen and Horwitz, 2012). These studies converge to a simplified model of gene expression induced by progestins: before hormonal treatment, PR binds genomic sites within a repressive complex maintaining these genes silenced prior to hormone treatment (Vicent et al., 2013). Progestin stimulation leads to the activation of cytoplasmic downstream kinase cascades, including ERK1/2, and finally to PR phosphorylation, especially on Ser-294 (Migliaccio et al., 1998). This active phosphorylated form of PR, associated with kinases, induces the recruitment of histone-modifying enzymes. This is followed by a local chromatin remodeling and the assembly of the transcription initiation complex on progestin-regulated genes (Beato and Vicent, 2012).
Protein arginine methyltransferases (PRMTs) are one such class of histone-modifying enzymes that regulate transcription. Two primary transcriptional coactivators in this family are PRMT1 and CARM1 (Coactivator Associated arginine (R) Methyltransferase 1), recruited as coregulators on promoters of genes targeted by nuclear receptors (Koh et al., 2001, Wang et al., 2001). PRMT1 is the predominant asymmetric arginine methyltransferase in humans and functions as a general transcriptional coactivator, by depositing dimethylarginines on histone 4 (H4R3me2as). However, PRMT1 also dimethylates a large variety of non-histone substrates, regulating many cellular processes required for tissue homeostasis, including RNA processing, transcriptional regulation, signal transduction, DNA repair, and protein-protein interactions (Bedford and Clarke, 2009, Blanc and Richard, 2017). Moreover, aberrant expression of PRMT1 has been reported in several malignancies, including breast cancer, although how PRMT1 contributes to oncogenesis remains largely elusive (Poulard et al., 2016, Yang and Bedford, 2013). We have previously shown that PRMT1 regulates the function of the estrogen receptor ERα through the methylation of the arginine 260, leading to the formation of a cytoplasmic signaling complex activated in aggressive human breast tumors (Le Romancer et al., 2008, Poulard et al., 2012).
We herein highlight the involvement of PRMT1 in breast tumorigenesis through, at least in part, the direct methylation of the progesterone receptor at the arginine 637. PRMT1 interacts with and regulates PR functions, acting as an essential actor of progesterone signaling in breast cancer cells. Our results indicate that PRMT1 and arginine methylation control PR stability, leading to the modulation of its transcriptional activity. PRMT1 allows the activation of a subset of genes controlling proliferation and survival of breast cancer cells and its expression clearly influences the clinical outcome of breast cancer according to PR expression.
Results
PRMT1 Interacts with the Progesterone Receptor in T47D Cancer Cells
To test whether PR might be regulated by arginine methylation, we initially examined a physical association between PR and PRMT1 in vitro and in T47D mammary carcinoma cells, which contain constitutive high levels of PR (Smith et al., 2017). Using a GST-binding assay, we showed the direct binding between PR and PRMT1 (Figure 1A). When T47D cell extracts were immunoprecipitated using an anti-PRMT1 antibody, coimmunoprecipitation (coIP) of both PR isoforms were observed (Figure 1B). Given the functional specificity of PR-B in breast (Boonyaratanakornkit et al., 2001, Faivre and Lange, 2007), we focused our study essentially on this isoform (called PR). As PR is a ligand-regulated nuclear transcription factor, we investigated whether the PR-PRMT1 interaction was hormone dependent and in which cellular compartment it occurred. For that, T47D cells were starved in medium deprived of steroids for 48 h (time 0) prior to the treatment for the indicated times with R5020 (also known as Promegestone), a synthetic agonist of progesterone used in scientific studies because of its stability (Read et al., 1988, Vignon et al., 1983). To localize and quantify these interactions more precisely, we used the in situ proximity ligation assay (PLA) (Poulard et al., 2014, Söderberg et al., 2006). The presence of red dots indicates interactions between endogenous PR and PRMT1 that occurred mainly in the nucleus and varied during the course of R5020 induction (Figure 1C). The graph representing the counting of dots per 100 cells indicated a high number of interactions between the two proteins in the absence of hormonal induction (Figure 1C, lower left panel). Notably, 15 min of R5020 treatment engendered a significant reduction in the signal abundancy, reflecting the dissociation of the PR-PRMT1 complex; then a second interaction peak was detected after 1 h of treatment (Figure 1C, lower left panel). A strong decrease in dot numbers was observed when the expression of PRMT1 or PR was knocked down using a pool of siRNAs, compared with mock T47D cells transfected with scramble siRNA (siCT) (Figure 1C, lower right panel and Figures S1A–S1C), validating the specificity of the PR-PRMT1 interaction, which is nuclear, dynamic, and progesterone-regulated.
Figure 1.

PRMT1 Interacts with PR in R5020-Stimulated T47D Breast Cancer Cells
(A) GST pull-down experiment: 35S-labeled in vitro translated PR-B, and ERα used as a positive control, were incubated with GST and GST-PRMT1 bound to glutathione Sepharose beads. The eluted proteins were analyzed by SDS-PAGE and visualized by autoradiography. Autoradiograph (upper) and Coomassie staining (lower) are shown.
(B) Whole-cell extracts (WCE) of T47D were subjected to immunoprecipitation (IP) using anti-PRMT1 antibody, or control IgG, and immunoblotted (IB) with anti-PR antibody.
(C) Proximity ligation assay (PLA) was used to detect the endogenous interaction between PRMT1 and PR in T47D cells, using anti-PR and anti-PRMT1 antibodies. T47D cells were transfected with control siRNA (siCT) or with anti-PRMT1 siRNAs (siPRMT1) and were cultured in medium deprived of steroids for 48 h, prior to the addition of R5020 (10 nM) for the indicated times. The nuclei were counterstained with DAPI (blue) (Obj: X60). The interactions are represented by red dots. Lower panel (left) shows the quantification of the number of signals per cell, as described in the Transparent Methods section. The mean ± SD of one experiment representative of three experiments is shown. The p value was determined using the Student's t test: ∗∗∗ indicates p ≤ 0.001. The efficacy of PRMT1 siRNA treatment is analyzed by IB and shown in the lower panel (right).
PRMT1 Methylates PR at a Conserved Site In Vitro and in Cells
We then investigated whether PR was methylated in T47D cells using two complementary approaches. First, extracts from V5-tagged PR T47D-transfected cells stimulated with R5020 for 1 h were immunoprecipitated using a pan-methylarginine antibody against asymmetric dimethyl-arginine (adme-R, note pan-meth-R), the type of methylation that marks PRMT1 deposits, and probed with anti-V5 antibody. We found that PR was dimethylated in cells mostly after hormonal treatment (Figure 2A). To confirm, we used PLA analysis with anti-PR and anti-pan-meth-R antibodies to detect asymmetrically dimethylated forms of PR (Poulard et al., 2019). We observed a nuclear signal, increased after R5020 stimulation in T47D cells (Figure 2B). The methylation signal was significantly decreased after PR knockdown and in presence of MS 023, a selective inhibitor of type I PRMT-dependent methylation (Eram et al., 2016), validating the signal specificity (Figure 2B). The effect and the optimal concentration of this inhibitor on PRMT1 activity was validated on H4R3 dimethylation, a known target of PRMT1, by immunofluorescence (IF) and immunoblot (IB), and the concentration of 60 nM was selected for our experiments (Figures S2A and S2B). Collectively, these data identify PR as a potential novel substrate for PRMT1.
Figure 2.

PRMT1 Methylates PR at Arginine 637 under Progesterone Treatment
(A) WCE from T47D, transfected with V5-tagged wild-type PR(B) (PR(B)-WT) plasmid and treated for 1h with R5020 were collected and immunoprecipitated (IP) with anti-pan-meth-R, or anti-immunoglobulin G (IgG), followed by IB using V5 antibody.
(B) PLA used to detect endogenous PR asymmetrically dimethylated on arginine sites in T47D cells, using anti-PR and anti-pan-meth-R antibodies. Cells were transfected with control siRNA (siCT) or with anti-PR siRNAs (siPR) or treated with MS 023 inhibitor (60 nM), then treated with R5020 (1 h) or vehicle ethanol (0). The analysis and the quantification of PLA experiments were performed as in Figure 1C.
(C) Alignment of the carboxy-terminal extensions of steroid receptors with conserved arginine (R) sequence in a similar position.
(D) An in vitro methylation assay using recombinant GST-PR-3 fragments, wild-type (WT) or mutated at R637 (PR-3 R637A) incubated with recombinant GST-PRMT1, in the presence of [methyl-3H] SAM. Reaction products were analyzed by SDS-PAGE followed by fluorography. The migration and the quality of recombinant GST-PR fragments were verified by a Coomassie-stained SDS-PAGE gel, shown in the right panel.
(E) WCE from T47D cells transfected with V5-tagged PR-WT or -R637K plasmids and treated for 1 h with R5020, were used for IP with pan-meth-R antibody and analyzed by IB using V5 and PR antibodies.
(F) Polyclonal anti-met-R637-PR antibody was generated using the annotated peptide encompassing asymmetrically dimethylated-R637 as antigen.
(G) In vitro methylation assay, using GST-PRMT1 and cold SAM, showed that met-R637-PR antibody only detected wild-type GST-PR-3. The membrane was colored by Ponceau S stain and is shown in the lower panel.
(H) Cos7 cells were transfected with V5-tagged PR-WT or -R637K encoding plasmids and treated for 1 h with R5020. WCE were immunoprecipitated with the met-R637-PR antibody followed by IB with anti-PR antibody. Quantification of immunoprecipitated methylated PR was determined relative to the input using ChemiDoc MP (Bio-Rad), for one experiment representative of three independent experiments.
(I) Immunofluorescence assay was performed using T47D cells, transfected with siCT or siPRMT1 and stimulated with R5020 (10 nM, 1 h) using the met-R637-PR and anti-PR antibodies. The nuclei were counterstained with DAPI (blue) (Obj: X40). PRMT1 expression was verified by IB (right).
Then, we performed in vitro methylation assays using the purified GST-tagged PR fragments illustrated in Figure S2C, incubated with recombinant GST-PRMT1 (Figure S2D), in order to identify the region(s) of PR that are methylated by PRMT1. Among the various functional domains of PR, PRMT1 specifically dimethylated only the fragments 3 and 4, spanning in the DNA-binding domain (DBD) and the hinge region, suggesting that arginine methylation by PRMT1 mainly occurs in the 587–687 amino acids region (Figures S2C and S2D). Of interest, this region encompasses the C-terminal extension (CTE) region, previously described as a dynamic region involved in DNA binding, nuclear localization, interaction with coregulators of the receptor (Daniel et al., 2010, Hill et al., 2009), as well as a hot spot for PTMs, including the PRMT1-dependent methylation of ERα (Le Romancer et al., 2008). Sequence alignment of steroid receptor CTEs showed a conserved position for the arginine 637 (Arg-637 or R637) residue, within a GGR motif that PRMT1 preferentially targets, localized in the PR-3 fragment (Figure 2C). To assess whether this motif was a direct target for the enzyme, we mutated the R637 to alanine residue (R637A) into the GST-PR3 fragment, leading to a complete loss of the methylation signal in vitro (Figure 2D).
Further analyses were conducted in the context of full-length PR in T47D cells. For in vivo studies, the R637 was substituted to lysine (R637K) in order to preserve its positive charge. The wild-type (PR(B)-WT) and mutated versions (PR(B)-R637K) of V5-tagged PR were expressed into T47D cells, immunoprecipitated with the pan-meth-R antibody, and revealed with anti-V5 antibody. Figure 2E shows that PR methylation was induced after hormonal stimulation. The R637K mutation strongly impaired PR modification, confirming this residue as the principal site of arginine methylation on PR.
We therefore focused on PR-R637 methylation and analyzed its role in progesterone signaling. We generated an antibody recognizing the asymmetric dimethylation of PR on R637 (R637me2(as), named anti-met-R637-PR). As an epitope, we used a branched peptide that contains the asymmetric dimethylated arginine (Figure 2F). The functionality and specificity of the antibody were established using several approaches in vitro and in cells. By western blot analysis, the anti-met-R637-PR specifically detected only the in vitro methylated WT PR-3 fragment and not the R637A mutant (Figure 2G). Dot blot experiments confirmed this specificity, as the antibody detected only the asymmetric dimethylated R637 peptide (Figure S2E). V5-tagged-wild-type (PR(B)-WT) or -mutated forms of PR (PR(B)-R637K) were expressed in PR-negative Cos7 cells, immunoprecipitated with the anti-met-R637-PR antibody and then probed with anti-PR antibody. Figure 2H shows that wild-type PR was methylated after hormonal treatment, whereas the methylation signal was strongly reduced in PR(B)-R637K protein. Additionally, we used this antibody to explore the subcellular localization of methylated PR at the endogenous level. Immunofluorescence experiments confirmed the methylation of endogenous PR in the nucleus, upon hormonal treatment, as observed with PLA (Figures 2I and 2B). Importantly, PRMT1-knockdown cells, as well as treated with the MS 023 inhibitor, displayed a marked decrease in nuclear methyl-PR signals (Figures 2I and S2F). Taken together, these data indicate that hormonal-activated PR is specifically methylated by PRMT1 on R637 in breast cancer cells.
PRMT1 Influences Progesterone Signaling by Regulating PR Stability
To pinpoint the role of PRMT1-dependant methylation in progesterone signaling, starved T47D cells were treated with 10 nM of R5020, inducing the rapid and transient activation of ERK 1 and 2 kinase activities, by phosphorylation of threonine 202 (T202) and tyrosine 204 (Y204), respectively (Hagan et al., 2012). The activation of these kinases led to the phosphorylation of PR on serine 294 (S294), described as the transcriptionally “primed” form of the receptor (Figure 3A, left panel) (Faivre et al., 2008, Lange et al., 2000). Of note, PRMT1-knockdown cells, as well as cells treated with the MS 023 inhibitor, displayed an impaired ERK activation following progestin treatment and an increase of PR protein level (Figure 3A, left panel). The quantification of the S294 phosphorylation signal, normalized to the signal of total PR, indicated that the inhibition of PRMT1 and its activity decreased the agonist-induced S294 phosphorylation of PR (Figure 3A, right panel). Supporting these data, over-expression of PRMT1 resulted in an increased activation of ERK and a decreased amount of PR following progestin treatment (Figure 3B). In contrast, the silencing of PRMT4 or PRMT6, both members of class I PRMT, did not increase the level of PR or impaired ERK activation after R5020 treatment, confirming a specific role of PRMT1 on PR signaling (Figure 3C). As the increased PR protein level was not associated with an increased synthesis of its mRNA (Figure 3D), we primarily focused on elucidating how PRMT1 controls the stability of the PR protein. Importantly, in chase experiments with the protein synthesis inhibitor cycloheximide, the half-life of endogenous PR protein was extended after depleting PRMT1 (Figure 3E), suggesting that PRMT1 controls PR expression largely through a post-translational mechanism. In agreement with these data, proteasome inhibitor MG132 treatment increased the R5020-activated PR levels, both in control and in PRMT1-depleted cells (Figure 3F), confirming the proteasome machinery involvement in the PR degradation, as described for the majority of nuclear receptors (Helzer et al., 2015).
Figure 3.

PRMT1 Regulates Progesterone Signaling, Inhibiting the Phosphorylation and the Proteasomal Degradation of PR
(A) WB of T47D cells silenced for PRMT1 (siPRMT1) or transfected with siRNA control (siCT), or treated with MS 023 (60 nM), and stimulated with R5020 (10 nM) for the indicated times (left). Quantification of phospho-PR [P-PR-B (S294)] band intensity relative to total PR-B was measured by ChemiDoc MP (Biorad). The ratio was calculated for each time point and is shown graphically (right).
(B) WB of T47D cells transfected with HA-PRMT1 or empty-HA plasmids and treated with R5020 (left). Quantification of PR-B band intensity for each time was measured by ChemiDoc software (Biorad) (right).
(C) WCE from T47D cells, depleted for PRMT1, PRMT4, or PRMT6 by specific siRNA pool or treated with 60 nM of MS 023 inhibitor, then stimulated with R5020, were analyzed by IB.
(D) RT-qPCR of PR mRNA from T47D cells, transfected with siPRMT1 or with siCT and treated with R5020 (10 nM) for 6 h. The mean ± SEM of at least three experiments is shown. Mean values were normalized against the expression of 28S ribosomal mRNA as reference. The p value was calculated using a paired t test: ∗∗∗ indicates p ≤ 0.001.
(E) Half-life study of endogenous PR-B protein. Lysates from T47D cells depleted or not for PRMT1 as in (A) were collected at the indicated time points after addition of cycloheximide and subjected to IB (top). The amount of PR-B was quantified by densitometry using the ChemiDoc software (Biorad).
(F) T47D cells were transfected with siCT or with siPRMT1 and treated with the proteasome inhibitor MG132 (10 μM) for 8 h or vehicle DMSO, before R5020 treatment. WCE were analyzed by IB. IB quantifications were done for one experiment, representative of three independent ones.
Collectively, these results suggest that PRMT1 and its enzymatic activity are required for the progestin-dependent degradation of the receptor. The effects of PRMT1 knockdown are reminiscent of the phenomena reporting that, inhibitors of MAPK or of the 26S proteasome, blocked PR turnover, leading to a decrease of its transcriptional activity (Lange et al., 2000).
PRMT1 Acts as a PR Coregulator in Breast Cancer Cells
Because PRMT1 has been previously described as a coactivator of several nuclear receptors (Koh et al., 2001, Stallcup et al., 2000), we addressed its role in regulating the transcriptional activity of PR. A luciferase-based transcription assay established that PRMT1 enhanced reporter gene activity of PR and that its enzymatic function was required for this effect (Figure 4A). We next asked whether PRMT1 was recruited to some PR-binding sites, identified by genome-wide mapping of, in T47D cells stimulated with R5020 (Ballaré et al., 2013, Kougioumtzi et al., 2014). Chromatin immunoprecipitation experiments followed by qPCR analysis (ChIP-qPCR) were performed in T47D cells stimulated with R5020 (or vehicle ethanol) for 1 h. Figures 4B–4D illustrate the recruitment of PRMT1, as well as PR and the activation mark H3K4me3, to chromatin PR-binding sites of three endogenous well-characterized progesterone target genes, EGFR, STAT5, and FKBP5 (Ballaré et al., 2013, Vicent et al., 2013). To analyze the expression of these genes, starved T47D cells were treated with R5020 and mRNAs were collected several times after the treatment. By quantitative real-time RT-PCR (RT-qPCR), we found a robust mRNA expression at 6 h (Figure S3A), and we chose this time for the remaining experiments. To determine whether PRMT1 was a coregulator of endogenous PR, we knocked down PRMT1 in T47D cells using a pool of three specific siRNAs and examined the expression of these PR target genes. RT-qPCR measurements revealed that, compared with the untreated cells, PRMT1 depletion differentially affected the expression of these PR-target genes: EGFR gene expression was decreased, STAT5 mRNA level was increased, whereas FKBP5 expression was not significantly changed (Figure 4E). As such, the impact of PRMT1 on the PR-dependent transcription appears to be gene specific. We confirmed the effect of PRMT1 depletion on the PR-dependent transcription in a second PR-positive breast cancer cell line, ZR-75 (Figures S3B and S3C).
Figure 4.

PRMT1 Acts as a PR Coregulator in Breast Cancer Cells
(A) HeLa cells were transfected with MMTV-LUC reporter plasmid and expression vectors encoding PR and wild-type or catalytic-mutant PRMT1 (E/Q). After R5020 treatment (10 nM), cell extracts were tested for luciferase activity using the Promega luciferase assay kit. The p value was determined using the Student's t test: ∗ indicates p ≤ 0.05.
(B–D) Starved T47D cells untreated (Eth) or treated with R5020 (10 nM for 1 h) were collected and subjected to chromatin immunoprecipitation (ChIP) with (B) anti-PRMT1, (C) anti-PR, and (D) anti-H3K4met3 antibodies, or control IgG. The precipitated DNA fragments were used for qPCR analysis using specific primers with respect to the input DNA and normalized to a reference locus (human chromosome 1 gene). The p value was calculated using a paired t test: ∗ indicates p ≤ 0.05 and ∗∗p ≤ 0.01.
(E) T47D cells, transfected with siCT or siPRMT1, were treated 6 h with R5020 (10 nM). Total RNA was prepared and complementary DNAs (cDNAs) were analyzed by RT-qPCR with primers for EGFR, STAT5A, and FKBP5. Mean values were normalized against the expression of 28S ribosomal mRNA as reference. The p value was calculated using a paired t test: ∗ indicates p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001. All the graphs show mean ± SEM for, at least, three independent experiments.
PRMT1 Is Required for Progestin-Induced Expression of PR Target Genes Regulating Cell Growth and Migration and Predicts Poor Survival in Patients with Breast Cancer
To address the relevance and the functional significance of the above results, we performed RNA sequencing (RNA-seq) analyses using PRMT1-depleted T47D cells by siRNAs, stimulated 6 h with R5020 treatment (Figure 5A, left panel). The efficacy of hormonal treatment and PRMT1 knockdown were confirmed by IB and RT-qPCR (Figures S4A–S4C). RNA-seq results indicated that, among the 795 genes activated after hormonal exposure (Figure 5A, right panel, pink color), 235 genes were impacted by siPRMT1 (about 30% of the total R5020-regulated genes) (Figure 5A, red color). Among those, 64% of genes were downregulated when PRMT1 expression was decreased, indicating that their R5020-induced activation required PRMT1 (PRMT1-activated genes). Conversely, 36% are characterized as PRMT1-repressed genes (Figure 5B). RT-qPCR analysis of genes randomly selected from the list of PRMT1-dependent genes confirmed that PRMT1 is required for their R5020-induced transcription (Figure S4D). Among the 235 genes regulated by R5020 and PRMT1, we analyzed EGFR, EGR1, SGK1, and CD44, which are functionally connected and described for their roles in the regulation of mammary epithelium differentiation under normal physiology and in cell migration and invasion during breast cancer progression (Figure S4E) (Kovacevic et al., 2016, Menezes et al., 2017). RT-qPCR and IB analysis confirmed that depletion of PRMT1 significantly reduced the R5020-dependent expression of these key targets (Figures 4E and 5C). Of interest, PRMT1 knockdown also inhibited the phosphorylation of PDK1 and p38 MAPK, two enzymes involved in the activation of SGK1 (Liu et al., 2019), leading to the activation of downstream targets, such as NDRG1 (Castel et al., 2016, Godbole et al., 2018, McCaig et al., 2011) (Figures 5D and S4F). To rule out the possibility of off-target effects and further confirm the specificity of the PRMT1 silencing, we expressed a tagged form of rat PRMT1 cDNA (flag-PRMT1, resistant to siRNAs silencing) in PRMT1-silenced cells. In this rescue experiment, we found that the expression of PRMT1 restored the expression of previously tested genes affected by PRMT1 knockdown, almost to the levels of control cells (Figures 5E, S4G, and S4H). Altogether, these data strongly indicate that PRMT1 silencing is specific and reversible.
Figure 5.

PRMT1 Is Required for Progesterone-Dependent Expression of a Subset of PR Target Genes
(A) Genome-wide RNA-seq analysis was performed using T47D cells to identify genes dependent on PRMT1 for R5020-regulated expression. Left panel: Hypothetical results of gene expression profiles for a given gene, illustrating how specific pairwise comparisons were performed between datasets for individual samples. Numbered bars represent hypothetical mRNA levels from RNA-seq data for cells expressing the indicated siRNAs (PRMT1 or CT) and treated for 6 h with ethanol (Eth) or R5020 (10 nM). Colored numbers represent pairwise comparisons performed to determine sets of genes for which mRNA levels were significantly different between the samples. For instance, comparison 1 = set of R5020-regulated genes (fold change ≥2, adjusted p < 0.01); comparison 4 = set of PRMT1-dependent genes (fold change >1.5, adjusted p < 0.01). Right panel: Pink and white Venn diagram represents the R5020-regulated genes in cells expressing siCT (comparisons 1 and 2); blue Venn diagram for PRMT1-dependent genes in R5020-treated cells (comparisons 3 and 4). Overlap area (in red) indicates the number of genes shared among sets. Controls for T47D cell treatments are provided in the Figures S4A–S4C.
(B) Representation of fold changes (log2FC) of all target gene expressions identified by RNA-seq analysis (235 genes). On the left (light blue), genes that are downregulated with siPRMT1 (64%), thus positively regulated by PRMT1. On the right (dark blue), genes that are negatively regulated by PRMT1 (36%).
(C and D) T47D cells were transfected with siRNAs against PRMT1 (or control) and treated for 6 h with ethanol (Eth) or R5020 (10 nM). Total RNA was prepared and cDNAs were analyzed by RT-qPCR with the indicated primers.
(E) cDNAs of T47D cells transfected with anti-PRMT1 siRNAs and co-transfected with a plasmid expressing a rat-PRMT1, then treated for 6 h with R5020 (10 nM) or vehicle ethanol (Eth), were analyzed by RT-qPCR.
All the cDNA mean values were normalized against the expression of 28S ribosomal mRNA as reference. Results shown are mean ± SEM for, at least, three independent experiments. The p value was calculated using a paired t test: ∗ indicates p ≤ 0.05, ∗∗p ≤ 0.01 and ∗∗∗p ≤ 0.001.
Moreover, a pathway analysis of the 235 differentially regulated genes obtained by RNA-seq (list given in Table S1) revealed an enrichment of genes involved in cell movement, morphology, and proliferation (Figure 6A). Consistently, depletion of PRMT1 significantly reduced the proliferation and the cellular migration of R5020-stimulated T47D cells, compared with the vehicle (eth)-treated cells, both analyzed with Incucyte technology (Figures 6B, 6C, and S5A–S5C). To support these phenotypes, we then investigated the correlation between PR and PRMT1 expression and patient survival, in a cohort of 1,764 breast tumors expression, by Kaplan-Meyer survival analysis (Györffy et al., 2010). After confirming that high-PR expression tumors depicted a better relapse-free survival (RFS) in this cohort, compared with low-PR expression ones (Figure 6Da), patients were separated into two groups according to PRMT1 expression (low and high PRMT1 following the median). Interestingly, patients with high expression of PR but low expression of PRMT1 reflected a significantly longer RFS (p = 1 × 10−11) (Figure 6Db). Similar observations were done with overall (OS) (p = 0.00012) and distal metastasis-free survival (DMFS) (p = 1 × 10−4) (Figures S5Db and S5Eb). Collectively, these results underscore that, in PR-positive tumors, low level of PRMT1, causing a decreased expression of some PR target genes including EGFR and EGR1, can inhibit cell proliferation and migration in hormonally responsive breast cancer, leading to a better survival outcome for patients (Figures 6D, S5D, and S5E).
Figure 6.

Low PRMT1 Reduces R5020-Induced Proliferation and Migration of T47D Breast Cancer Cells and Predicts Improved Survival of Patients with Breast Cancer
(A) Ingenuity Pathway Analysis (IPA) of cellular functions for the 235 R5020-regulated genes dependent on PRMT1. The orange vertical line represents the fold of statistical significance.
(B) Analysis of T47D cell proliferation by Incucyte technology. Cells expressing the indicated siRNAs (PRMT1 or CT) were stimulated with R5020 (10 nM) every 48 h for 7 days. Image acquisition was conducted every hour using the Incucyte software, which calculates the percentage of cell confluency according to time over 7 days. Results are represented as a graph showing the proliferation rate every 24 h.
(C) Migration of T47D cells expressing the indicated siRNAs (PRMT1 or CT) and treated with R5020 (10 nM) for 12, 24, and 36 h was analyzed in a wound scratch assay with the Incucyte Live-Cell Imaging System and dedicated software (Essen Bioscience), as reported in the Transparent Methods section. Left panel: The bar plots indicate the cellular migration rate, with a direct comparison between control and PRMT1-depleted cells. Right panel: Images of cells 24 h after the scratch wound. The blue line corresponds to the initial area of the wound. White arrows indicate cell migration areas. Both (B) and (C) graphs show the mean ± SD of one experiment representative of three. The p value was determined using the Student's t test: ∗ indicates p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
(D) Kaplan-Meier estimates relapse-free survival in patients, in GEO, EGA, TCGA datasets, with low (black) or high (red) PR expression, as indicated using KM-plotter in a cohort of 1,764 breast tumors (a), or stratified in 2 groups following low-PRMT1 (b) or high-PRMT1 expression (c).
Loss of PR Methylation at R637 Affects PR Degradation and T47D Cell Proliferation
To directly investigate the function of PRMT1-dependant PR methylation in cells, we used the CRISPR-Cas9 technology in T47D cells. We first knocked out endogenous PR, and the characterization of representative PR KO clones (T47D PRKO) is illustrated in Figures S6A and S6B. Next, we stably re-expressed the wild-type (WT) and mutated (R637K) forms of PR-B in the T47D PRKO cells and named these derived cell lines T47DWT and T47DR637K, respectively. The cellular localization of WT- and R637K-PR proteins was analyzed by immunofluorescence using an anti-PR antibody (Figure 7A). We showed that, in both T47DWT and T47DR637K cell lines, the receptor was correctly localized in the nucleus (Figure 7A). Importantly, we also confirmed that PR was dimethylated after hormonal treatment in T47DWT cells, whereas in T47DR637K cells, the methylation signal was strongly reduced (Figure 7B). Together, these results confirm not only that these cell lines are functionally comparable with the native T47D cells but also that the R637 is the major methylated residue of PR.
Figure 7.

Inhibiting PR Methylation Affects Breast Cancer Cell Proliferation and PR Stability
(A) Immunofluorescence of T47D PRKO, T47DWT, and T47DR637K cells, before and after stimulation with 10 nM of R5020, stained with anti-PR antibody. The nuclei were counterstained with DAPI (blue) (Obj: X40).
(B) WCE of T47DWT or T47DR637K, stimulated with R5020 (10 nM) for 1 h, were used for IP with anti-PR antibody or control IgG and immunoblotted with pan-meth-R and PR antibodies.
(C) WCE from T47DWT and T47DR637K cells stimulated with R5020 for the indicated times were immunoblotted.
(D) Half-life of the endogenous PR in T47DWT and T47DR637K cells. Left panel: WCE from T47DWT and T47DR637K cells treated with cycloheximide before the stimulation with R5020 were immunoblotted with the indicated antibodies. The amount of PR was quantified by densitometry; two different expositions are shown to well quantify the PR band intensity for each time. Right panel: The half-life curves for each cell line is graphically represented. This experiment is representative of three independent experiments.
(E) Analysis of T47DWT and T47DR637K cell proliferation by Incucyte technology, performed as described in Figure 6B.
(F) T47DWT and T47DR637K cells were stimulated with 10 nM of R5020, and colony growth was measured at 10 days after staining with crystal violet. Both (E) and (F) graphs show the mean ± SD of one experiment representative of three. The p value was determined using the Student's t test: ∗∗ indicates p ≤ 0.01 and ∗∗∗p ≤ 0.001.
(G and H) T47DWT and T47DR637K cells were stimulated with R5020 (10 nM). EGFR and EGR1 expression were analyzed (G) by IB and (H) by RT-qPCR (after 6 h of R5020 induction). The mean ± SEM of, at least, three independent experiments is shown. The p value was calculated using a paired t test: ∗ indicates p ≤ 0.05.
Using these two cell lines, we analyzed the impact of the R637K mutation on progesterone signaling. As expected, treatment of T47DWT and T47DR637K cells with R5020 induced a rapid and transient activation of ERK kinases (Figure 7C). However, as observed after PRMT1 depletion, the mutation affected ERK activation, even if to a lesser extent (see Figure 3A). As with PRMT1 inhibition, loss of PR methylation at R637 resulted in a higher basal PR protein level, which appeared stable after R5020 stimulation (Figures 7A–7C). Cycloheximide treatment showed that more than 90% of PR-R637K was still present after 6 h of R5020 exposure, compared with less than 50% in T47DWT cells (Figure 7D). The difference in protein level is caused by the shorter half-life of the wild-type PR compared with the mutant, which corresponds to the increased steady-state level of PR-R637K. Thus, PRMT1-mediated methylation seems to participate in the regulation of PR turnover, required for active hormonal-dependent transcription (Métivier et al., 2003). In line with these results, absence of PR methylation at R637 markedly decreased oncogenic PR functions, leading to reduced cell growth and colony formation of T47DR637K cells, compared with T47DWT cells, when treated with R5020 (Figures 7E, 7F, and S6C). Importantly, expression of some PRMT1-dependent PR-downstream targets identified in Figure 5, as EGFR and EGR1, appeared lower in T47DR637K cells, after progesterone induction (Figures 7G and 7H).
Interestingly, the loss of R637 methylation consequences observed under progesterone treatment are similar to those obtained with PRMT1-knockdown cells, suggesting that PRMT1 effects on the progesterone pathway occur, at least in part, through the methylation of PR.
Discussion
Our study highlights the direct and functional cross talk between PRMT1, arginine methylation, and progesterone signaling, uncovering the molecular mechanisms by which PRMT1 functions as an important modulator of the progesterone response pathway. In the context of hormonal activation, we demonstrated that PRMT1 is recruited on PR-associated chromatin regions of some progestin-regulated genes. RNA-seq data support these results, showing that PRMT1 knockdown in breast cancer cells can affect the expression of thousand progesterone-targeted genes, positively or negatively. Moreover, PRMT1-regulated genes are involved in relevant cell functions including cell movement, morphology, and proliferation. We notably showed that PRMT1 positively regulates the expression of a subset of genes involved in the regulation of cell migration and invasion during breast cancer progression. Consistently, silencing of PRMT1 in T47D cells leads to a significant reduced ability to proliferate and migrate, supporting the involvement of PRMT1 in breast tumorigenesis. Therefore, it is possible that high-level expression of PRMT1 facilitates oncogenesis by providing tumor cells with a survival advantage, in part by enhancing the progestin-dependent receptor degradation and thereby maintaining cells in a proliferative mode. In line with this idea, the analysis of PRMT1 prognostic value in patients with breast cancer showed that high expression of PRMT1 associated with high expression of PR predicts poor relapse-free (RFS), overall (OS), and distal metastasis-free survival (DMFS). This suggests that PRMT1 expression influences the survival of patients with PR-positive breast cancer and that the prognostic value and pathophysiological role of PR depend on PRMT1 expression.
Many studies have already revealed the impact of PRMT1 in breast tumorigenesis (Liu et al., 2019, Morettin et al., 2015). Its expression is often upregulated in tumor samples compared with adjacent normal tissue. Moreover, these studies have highlighted different mechanisms by which PRMT1 regulates the proliferation of tumor cells (regulation of the epithelio-mesenchymal transition EMT, sensitization of cells to a therapy, etc.) (Yang and Bedford, 2013). Here, we report that the direct methylation of the progesterone receptor, a key driver of breast cells proliferation, affects breast cancer progression. Since PRMT1 enhances the transcriptional activity of PR (Figure 4A), we can consider that PRMT1-mediated transactivation is mainly due to direct methylation of PR, leading to increased transcription via the tight control of PR turnover. This is supported by the biological consequences of PR methylation at R637 under physiological conditions, using the T47DWT and T47DR637K cell lines, engineered to stably express the wild-type and mutant forms of PR-B in PRKO T47D cells. We established that the effects of the methylation loss at R637 are consistent with the results obtained in PRMT1 knocked-down cells: T47DR637K cells display decreased oncogenic PR functions, such as a retarded cell growth and a reduced expression of some PRMT1-dependent PR downstream targets identified by RNA-seq (Figures 5 and 7). Moreover, the methylation of R637 directly affects PR stability (Figure 7), suggesting that PRMT1 effects on progesterone pathway occur, at least in part, through the methylation of PR on this conserved residue. Notably, the arginine R637 is located within the hinge region of the steroid receptor, which contains sites for posttranslational modifications, like phosphorylation and acetylation. Precisely, this conserved R637 flanks an acetylation consensus site, in which three lysine residues K638-640-641 are modified following progesterone stimulation (Daniel et al., 2010). It is interesting to note that non-acetylatable mutants (PRK-A) exhibit defective transcriptional activation and are more stable than wild-type receptors, namely, a phenotype similar to the non-methylable mutant PRR637K (Figure 7). Thus, it could exist as a functional communication between these two modifications, as we currently observed with the histone tails on chromatin (Bannister and Kouzarides, 2011).
In response to ligand binding, MAPK activation modulates PR activity by phosphorylating the receptor on serine 294 (Lange et al., 2000, Shen et al., 2001). This modification is crucial for PR nuclear activity, priming the receptor for robust transcriptional activation, also influencing its promoter selectivity (Daniel et al., 2009). Our report demonstrates that progestin treatment induces the recruitment of PRMT1 on several PR-target genes, implying that PRMT1 acts as a coregulator of PR. Furthermore, we also describe that PRMT1 directly dimethylates PR under progestin treatment, primarily at the conserved R637 residue on an RGG methylation consensus motif, in vitro and in vivo (Figures 2D–2I). This methylation occurs in the nucleus and facilitates PR degradation, which in turn speeds up its transcriptional properties. Previous studies have demonstrated the critical role of PR degradation/re-synthesis in the active transcription of the receptor (Dennis et al., 2005). Indeed, the degradation constitutes a stimulatory switch that accelerates the recycling of receptors from pre-initiation complexes, required for active hormonal-dependent transcription (Métivier et al., 2003). Our results show that this mechanism involves the asymmetric dimethylation of PR by PRMT1, which reduces PR stability by affecting the proteasome machinery, thereby accelerating its transcriptional activity. It is tempting to speculate that the effects of PRMT1 on PR degradation/stability can be mediated by a cross talk with PR ubiquitination. Interestingly, recent data have shown that the RNA-binding protein RBM15 is methylated by PRMT1, which triggers its ubiquitination and degradation by the E3 ligase CNOT4 (Zhang et al., 2015). CNOT4 is a subunit of the CCR4-NOT complex (Albert et al., 2002), and we previously showed that PRMT1 physically interacts with the CCR4-NOT complex, regulating its methyltransferase activity (Chapat et al., 2017, Robin-Lespinasse et al., 2007). In future studies, it will be interesting to investigate whether PRMT1-dependent methylation of PR induces its degradation through the recruitment of the E3 ligase CNOT4.
In conclusion, our findings reveal important insights linking PRMT1-dependent arginine methylation to the maintenance of the balance between transcriptional activity and degradation of PR. This result implies that altered methylation of the receptor can induce aberrant cellular response to hormonal stimuli contributing to pathogenesis. However, most of the mechanisms described above are based on in vitro experiments, obtained using a breast cancer cell line with a phenotype particularly useful to dissect the regulatory steps of progesterone signaling, independently of the estrogen receptor (ERα). Future studies will be needed in preclinical models, notably mouse and patient-derived xenografts or directly in patient samples, to determine the clinical relevance of our in vitro mechanistic findings. In addition, improved knowledge of these mechanisms could have a significant impact on long-term outcome for patients with breast cancer, considering the differential effects of progesterone in breast cancers. In fact, recent studies (Finlay-Schultz et al., 2017, Mohammed et al., 2015) indicate that functional relationships between ERα and PR signaling occur in ER+/PR+ breast cancers. Indeed, PR can inhibit the growth-promoting functions of estrogen by directly reprogramming chromatin binding of ERα or indirectly, by reducing the bio-availability of molecules needed for tumor growth. These new findings may provide additional strategies to treat ERα-positive breast cancer, by modulating the interactions between ERα and PR. Given that both receptors are regulated by PRMT1 via their direct methylation, and that PRMT1 clearly influences the clinical outcome of breast cancer patients according to PR expression, we speculate that PRMT1 can be involved in the ERα/PR cross talk, whose mechanistic details are not yet well understood. Therefore, targeting PRMT1-mediated methylation may be a promising strategy for breast cancer treatments; the recent development of pre-clinical small molecules targeting PRMT1 (Fedoriw et al., 2019) may offer a path toward that goal.
Limitations of the Study
Our study depicts a new post-translational modification in the DNA-binding domain of the progesterone receptor, the arginine methylation, deposited by the enzyme PRMT1. Although our data highlight PRMT1 as a transcriptional coregulator of a subset of PR-target genes, involved in breast cancer cell proliferation and migration, we were unable to directly link the arginine methylation to this transcriptional effect. In fact, our home-made antibody specifically directed against the methylated form of PR did not give specific signal in chromatin immunoprecipitation (ChIP) experiment. Thus, we actually do not know if the methylated form of the receptor is bound to DNA and if the methylation affects the association between PR and its coregulators. Most of the molecular mechanisms described in this article come from in vitro studies using the PR-positive cell line T47D, which permits to dissect the PR progesterone signaling in the presence of progestin only and thus independently of ERα. On the contrary, recent studies have shown that estrogen and progestin have different biological consequences when analyzed individually or when both hormones are present, as these hormonal receptors are functionally interconnected. Therefore, these mechanistic studies need to be validated using more physiological preclinical models, such as mouse and patient-derived xenografts, or directly in patient samples, to assess their clinical relevance.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Laura Corbo (laura.corbo@lyon.unicancer.fr).
Materials Availability
All unique reagents generated in this study (CRISPR cell lines, primers, plasmids) are available from the Lead Contact without restriction. There are restrictions for the availability of the methylated-R637-PR antibody, owing to our inability to produce it and our need to maintain the stock.
Data and Code Availability
RNA-seq data have been submitted to Gene Expression Omnibus (GEO) and are available with the GSE134194 submission number.
Original membranes for all the immunoblots presented in the paper have been deposited to Mendeley Data: [https://doi.org/10.17632/rgctdkjyzx.2].
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Helene Joly for starting the project and Julien Jacquemetton, Farida Nasri, Maéva Ruel, and Aurore Souef for helpful technical assistance. We are grateful to Dr. Loredana D'Amato, Dr. Samuele Gherardi, and Dr. Romain Teinturier for their valuable advice and discussions. We also thank Prof. Pierre Chambon for kindly providing the pSG5PR-B and MMTV-Luc plasmids, Dr. Catherine Teyssier for the PRMT1 constructs, and Dr. Stéphane Ansieau for the plasmid pPRUpu. We are also grateful to Benjamin Gillet and Sandrine Hughes for performing the RNA sequencing at the IGFL platform in Lyon. A special thanks to Brigitte Manship and Clément Chapat for reading the manuscript. This study was partially supported by the “Ligue Nationale Contre le Cancer,” the “Association pour la Recherche sur le Cancer,” and the association “Odyssea Chambery.” L.M. is supported by a fellowship from the French Ministry of Research and by the “Fondation ARC pour la Recherche sur le Cancer” and C.P. by the “Fondation de France.”
Author Contributions
L.C., M.L.R., and L.M. conceived the study, designed the experiments, and analyzed data. L.M. carried out most of the experiments, C.L. assisted with the experiments, I.M. performed the CRISPR experiments, C.P. and F.F. helped L.M. with bioinformatics analysis of RNA-seq data. M.L.R. and I.M. contributed to result discussions and provided material. L.C. coordinated the project. The manuscript was written by L.C., with a strong contribution from L.M.
Declaration of Interests
The authors have declared that no competing interest exist.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101236.
Contributor Information
Muriel Le Romancer, Email: muriel.leromancer@lyon.unicancer.fr.
Laura Corbo, Email: laura.corbo@lyon.unicancer.fr.
Supplemental Information
References
- Abdel-Hafiz H.A., Horwitz K.B. Post-translational modifications of the progesterone receptors. J. Steroid Biochem. Mol. Biol. 2014;140:80–89. doi: 10.1016/j.jsbmb.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert T.K., Hanzawa H., Legtenberg Y.I.A., de Ruwe M.J., van den Heuvel F.A.J., Collart M.A., Boelens R., Timmers H.T.M. Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J. 2002;21:355–364. doi: 10.1093/emboj/21.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballaré C., Castellano G., Gaveglia L., Althammer S., González-Vallinas J., Eyras E., Le Dily F., Zaurin R., Soronellas D., Vicent G.P. Nucleosome-driven transcription factor binding and gene regulation. Mol. Cell. 2013;49:67–79. doi: 10.1016/j.molcel.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beato M., Vicent G.P. Impact of chromatin structure and dynamics on PR signaling. The initial steps in hormonal gene regulation. Mol. Cell. Endocrinol. 2012;357:37–42. doi: 10.1016/j.mce.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Bedford M.T., Clarke S.G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc R.S., Richard S. Arginine methylation: the coming of age. Mol. Cell. 2017;65:8–24. doi: 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Boonyaratanakornkit V., Scott M.P., Ribon V., Sherman L., Anderson S.M., Maller J.L., Miller W.T., Edwards D.P. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol. Cell. 2001;8:269–280. doi: 10.1016/s1097-2765(01)00304-5. [DOI] [PubMed] [Google Scholar]
- Brisken C., O’Malley B. Hormone action in the mammary gland. Cold Spring Harb. Perspect. Biol. 2010;2:a003178. doi: 10.1101/cshperspect.a003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel P., Ellis H., Bago R., Toska E., Razavi P., Carmona F.J., Kannan S., Verma C.S., Dickler M., Chandarlapaty S. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kα inhibition. Cancer Cell. 2016;30:229–242. doi: 10.1016/j.ccell.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapat C., Chettab K., Simonet P., Wang P., De La Grange P., Le Romancer M., Corbo L. Alternative splicing of CNOT7 diversifies CCR4-NOT functions. Nucleic Acids Res. 2017;45:8508–8523. doi: 10.1093/nar/gkx506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel A.R., Knutson T.P., Lange C.A. Signaling inputs to progesterone receptor gene regulation and promoter selectivity. Mol. Cell. Endocrinol. 2009;308:47–52. doi: 10.1016/j.mce.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel A.R., Gaviglio A.L., Czaplicki L.M., Hillard C.J., Housa D., Lange C.A. The progesterone receptor hinge region regulates the kinetics of transcriptional responses through acetylation, phosphorylation, and nuclear retention. Mol. Endocrinol. 2010;24:2126–2138. doi: 10.1210/me.2010-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis A.P., Lonard D.M., Nawaz Z., O’Malley B.W. Inhibition of the 26S proteasome blocks progesterone receptor-dependent transcription through failed recruitment of RNA polymerase II. J. Steroid Biochem. Mol. Biol. 2005;94:337–346. doi: 10.1016/j.jsbmb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Eram M.S., Shen Y., Szewczyk M., Wu H., Senisterra G., Li F., Butler K.V., Kaniskan H.Ü., Speed B.A., Dela Seña C. A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 2016;11:772–781. doi: 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre E.J., Lange C.A. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol. Cell. Biol. 2007;27:466–480. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre E.J., Daniel A.R., Hillard C.J., Lange C.A. Progesterone receptor rapid signaling mediates serine 345 phosphorylation and tethering to specificity protein 1 transcription factors. Mol. Endocrinol. 2008;22:823–837. doi: 10.1210/me.2007-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw A., Rajapurkar S.R., O’Brien S., Gerhart S.V., Mitchell L.H., Adams N.D., Rioux N., Lingaraj T., Ribich S.A., Pappalardi M.B. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell. 2019;36:100–114.e25. doi: 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Finlay-Schultz J., Gillen A.E., Brechbuhl H.M., Ivie J.J., Matthews S.B., Jacobsen B.M., Bentley D.L., Kabos P., Sartorius C.A. Breast cancer suppression by progesterone receptors is mediated by their modulation of estrogen receptors and RNA polymerase III. Cancer Res. 2017;77:4934–4946. doi: 10.1158/0008-5472.CAN-16-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbole M., Togar T., Patel K., Dharavath B., Yadav N., Janjuha S., Gardi N., Tiwary K., Terwadkar P., Desai S. Up-regulation of the kinase gene SGK1 by progesterone activates the AP-1-NDRG1 axis in both PR-positive and -negative breast cancer cells. J. Biol. Chem. 2018;293:19263–19276. doi: 10.1074/jbc.RA118.002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S.L., Hartig S.M., Edwards D.P. Progesterone receptor signaling mechanisms. J. Mol. Biol. 2016;428:3831–3849. doi: 10.1016/j.jmb.2016.06.020. [DOI] [PubMed] [Google Scholar]
- Györffy B., Lanczky A., Eklund A.C., Denkert C., Budczies J., Li Q., Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Hagan C.R., Daniel A.R., Dressing G.E., Lange C.A. Role of phosphorylation in progesterone receptor signaling and specificity. Mol. Cell. Endocrinol. 2012;357:43–49. doi: 10.1016/j.mce.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helzer K.T., Hooper C., Miyamoto S., Alarid E.T. Ubiquitylation of nuclear receptors: new linkages and therapeutic implications. J. Mol. Endocrinol. 2015;54:R151–R167. doi: 10.1530/JME-14-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill K.K., Roemer S.C., Jones D.N.M., Churchill M.E.A., Edwards D.P. A progesterone receptor co-activator (JDP2) mediates activity through interaction with residues in the carboxyl-terminal extension of the DNA binding domain. J. Biol. Chem. 2009;284:24415–24424. doi: 10.1074/jbc.M109.003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen B.M., Horwitz K.B. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol. Cell. Endocrinol. 2012;357:18–29. doi: 10.1016/j.mce.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner P., Krust A., Turcotte B., Stropp U., Tora L., Gronemeyer H., Chambon P. Two distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J. 1990;9:1603–1614. doi: 10.1002/j.1460-2075.1990.tb08280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougioumtzi A., Tsaparas P., Magklara A. Deep sequencing reveals new aspects of progesterone receptor signaling in breast cancer cells. PLoS One. 2014;9:e98404. doi: 10.1371/journal.pone.0098404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson T.P., Lange C.A. Tracking progesterone receptor-mediated actions in breast cancer. Pharmacol. Ther. 2014;142:114–125. doi: 10.1016/j.pharmthera.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S.S., Chen D., Lee Y.H., Stallcup M.R. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J. Biol. Chem. 2001;276:1089–1098. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- Kovacevic Z., Menezes S.V., Sahni S., Kalinowski D.S., Bae D.-H., Lane D.J.R., Richardson D.R. The metastasis suppressor, N-MYC downstream-regulated gene-1 (NDRG1), down-regulates the ErbB family of receptors to inhibit downstream oncogenic signaling pathways. J. Biol. Chem. 2016;291:1029–1052. doi: 10.1074/jbc.M115.689653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C.A., Shen T., Horwitz K.B. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc. Natl. Acad. Sci. U S A. 2000;97:1032–1037. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Romancer M., Treilleux I., Leconte N., Robin-Lespinasse Y., Sentis S., Bouchekioua-Bouzaghou K., Goddard S., Gobert-Gosse S., Corbo L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol. Cell. 2008;31:212–221. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Liu L.-M., Sun W.-Z., Fan X.-Z., Xu Y.-L., Cheng M.-B., Zhang Y. Methylation of C/EBPα by PRMT1 inhibits its tumor-suppressive function in breast cancer. Cancer Res. 2019;79:2865–2877. doi: 10.1158/0008-5472.CAN-18-3211. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf D.J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H., Russell I.A., Stark R., Rueda O.M., Hickey T.E., Tarulli G.A., Serandour A.A., Serandour A.A.A., Birrell S.N., Bruna A. Progesterone receptor modulates ERα action in breast cancer. Nature. 2015;523:313–317. doi: 10.1038/nature14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaig C., Potter L., Abramczyk O., Murray J.T. Phosphorylation of NDRG1 is temporally and spatially controlled during the cell cycle. Biochem. Biophys. Res. Commun. 2011;411:227–234. doi: 10.1016/j.bbrc.2011.06.092. [DOI] [PubMed] [Google Scholar]
- Menezes S.V., Sahni S., Kovacevic Z., Richardson D.R. Interplay of the iron-regulated metastasis suppressor NDRG1 with epidermal growth factor receptor (EGFR) and oncogenic signaling. J. Biol. Chem. 2017;292:12772–12782. doi: 10.1074/jbc.R117.776393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métivier R., Penot G., Hübner M.R., Reid G., Brand H., Kos M., Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Migliaccio A., Piccolo D., Castoria G., Di Domenico M., Bilancio A., Lombardi M., Gong W., Beato M., Auricchio F. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 1998;17:2008–2018. doi: 10.1093/emboj/17.7.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morettin A., Baldwin R.M., Côté J. Arginine methyltransferases as novel therapeutic targets for breast cancer. Mutagenesis. 2015;30:177–189. doi: 10.1093/mutage/geu039. [DOI] [PubMed] [Google Scholar]
- Mulac-Jericevic B., Lydon J.P., DeMayo F.J., Conneely O.M. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc. Natl. Acad. Sci. U S A. 2003;100:9744–9749. doi: 10.1073/pnas.1732707100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulard C., Treilleux I., Lavergne E., Bouchekioua-Bouzaghou K., Goddard-Léon S., Chabaud S., Trédan O., Corbo L., Le Romancer M. Activation of rapid oestrogen signalling in aggressive human breast cancers. EMBO Mol. Med. 2012;4:1200–1213. doi: 10.1002/emmm.201201615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulard C., Rambaud J., Le Romancer M., Corbo L. Proximity ligation assay to detect and localize the interactions of ERα with PI3-K and Src in breast cancer cells and tumor samples. Methods Mol. Biol. 2014;1204:135–143. doi: 10.1007/978-1-4939-1346-6_12. [DOI] [PubMed] [Google Scholar]
- Poulard C., Corbo L., Le Romancer M. Protein arginine methylation/demethylation and cancer. Oncotarget. 2016;7:67532–67550. doi: 10.18632/oncotarget.11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulard C., Jacquemetton J., Pham T.H., Le Romancer M. Using proximity ligation assay to detect protein arginine methylation. Methods. 2020;175:66–71. doi: 10.1016/j.ymeth.2019.09.007. [DOI] [PubMed] [Google Scholar]
- Read L.D., Snider C.E., Miller J.S., Greene G.L., Katzenellenbogen B.S. Ligand-modulated regulation of progesterone receptor messenger ribonucleic acid and protein in human breast cancer cell lines. Mol. Endocrinol. 1988;2:263–271. doi: 10.1210/mend-2-3-263. [DOI] [PubMed] [Google Scholar]
- Robin-Lespinasse Y., Sentis S., Kolytcheff C., Rostan M.-C., Corbo L., Le Romancer M. hCAF1, a new regulator of PRMT1-dependent arginine methylation. J. Cell. Sci. 2007;120:638–647. doi: 10.1242/jcs.03357. [DOI] [PubMed] [Google Scholar]
- Shen T., Horwitz K.B., Lange C.A. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol. Cell. Biol. 2001;21:6122–6131. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S.E., Mellor P., Ward A.K., Kendall S., McDonald M., Vizeacoumar F.S., Vizeacoumar F.J., Napper S., Anderson D.H. Molecular characterization of breast cancer cell lines through multiple omic approaches. Breast Cancer Res. 2017;19:65. doi: 10.1186/s13058-017-0855-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.-J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.-G. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Stallcup M.R., Chen D., Koh S.S., Ma H., Lee Y.H., Li H., Schurter B.T., Aswad D.W. Co-operation between protein-acetylating and protein-methylating co-activators in transcriptional activation. Biochem. Soc. Trans. 2000;28:415–418. [PubMed] [Google Scholar]
- Vicent G.P., Nacht A.S., Zaurin R., Font-Mateu J., Soronellas D., Le Dily F., Reyes D., Beato M. Unliganded progesterone receptor-mediated targeting of an RNA-containing repressive complex silences a subset of hormone-inducible genes. Genes Dev. 2013;27:1179–1197. doi: 10.1101/gad.215293.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignon F., Bardon S., Chalbos D., Rochefort H. Antiestrogenic effect of R5020, a synthetic progestin in human breast cancer cells in culture. J. Clin. Endocrinol. Metab. 1983;56:1124–1130. doi: 10.1210/jcem-56-6-1124. [DOI] [PubMed] [Google Scholar]
- Wang H., Huang Z.Q., Xia L., Feng Q., Erdjument-Bromage H., Strahl B.D., Briggs S.D., Allis C.D., Wong J., Tempst P. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- Yang Y., Bedford M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Zhang L., Tran N.-T., Su H., Wang R., Lu Y., Tang H., Aoyagi S., Guo A., Khodadadi-Jamayran A., Zhou D. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife. 2015;4:e07938. doi: 10.7554/eLife.07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been submitted to Gene Expression Omnibus (GEO) and are available with the GSE134194 submission number.
Original membranes for all the immunoblots presented in the paper have been deposited to Mendeley Data: [https://doi.org/10.17632/rgctdkjyzx.2].