

Abstract
A new era of science and technology has emerged in pharmaceutical research with focus on developing novel drug delivery systems for oral administration. Conventional dosage forms like tablets and capsules are associated with a low bioavailability, frequent application, side effects and hence patient noncompliance. By developing novel strategies for drug delivery, researchers embraced an alternative to traditional drug delivery systems. Out of those, fast dissolving drug delivery systems are very eminent among pediatrics and geriatrics. Orally disintegrating films are superior over fast dissolving tablets as the latter are assigned with the risk of suffocation. Due to their ability of bypassing the dissolution and the first pass effect after oral administration, self-emulsifying formulations have also become increasingly popular in improving oral bioavailability of hydrophobic drugs. Osmotic devices enable a controlled drug delivery independent upon gastrointestinal conditions using osmosis as driving force. The advances in nanotechnology and the variety of possible materials and formulation factors enable a targeted delivery and triggered release. Vesicular systems can be easily modified as required and provide a controlled and sustained drug delivery to a specific site.
This work provides an insight of the novel approaches in drug delivery covering the critical comparison between traditional and novel “advanced-designed” systems.
Keywords: Fast dissolving, Nanoparticulate, Osmotic, Vesicular, Selfemulsifying
Graphical abstract
1. Introduction
“To liberate the drug at the right time in a right amount of concentration at a specified target site” is the major objective of a drug delivery system (Vijaya Shanti and Mrudula, 2011). The requirements for a successful drug delivery are usually determined by the physicochemical characteristics of the therapeutic agent and bio-barriers like the skin and membrane of body organs. Depending on size, chemical composition, hydrophilicity and ability to bind specific receptor, drug properties may vary greatly even when used to treat the same symptoms. Many drugs suffer from an insufficient bioavailability due to insolubility in physiological fluids and low permeability of different body organs. Hence, the therapeutic performance is not merely dependent on the activity of the applied drug, but also, on the bioavailability at the target side according to evidence (Mbah et al., 2014).
In the past decades, the treatment of serious diseases or chronic illnesses has mainly consisted of rapid acting and simple compound that are administered conventionally in form of as tablets, pills, capsules, cremes, liquids, aerosols, suppositories, injectables or ointments (Vijaya Shanti and Mrudula, 2011; Khan and Irchhaiya, 2016). These conventional drug delivery systems represent the classical method for delivery of drugs orally. These common dosage forms are often accompanied by systemic adverse effects that are primarily attributable to their unspecified bio-distribution and missing controllability of the drug release characteristics (Liu et al., 2016). Furthermore, conventional drug delivery systems have been found to have severe constraints including non-controlled release, higher doses and a frequent application (Liu et al., 2016; Bhagwat and Vaidhya, 2013). Another major challenge in the formulation of drugs is the improvement of bioavailability (MH a et al., 2013).
To overcome the limitations of conventional drug delivery systems, pharmaceutical companies focused on the development and design of novel drug delivery systems. The need for high performance, flexibility and controlled release systems are provoked by the compelling advancements in patient compliance, clinical efficacy, prolonged product life through a controlled drug release and economic aspects like reduced frequency and expenses of administration. For this reason, novel drug delivery systems might be among the fastest expanding segments in the drug industry (Vijaya Shanti and Mrudula, 2011).
Novel drug delivery systems are engineered according to a rational design to enhance the delivery and the performance of existing drugs with respect to traditional systems. Novel drug delivery systems in comparison to traditional ones combine advanced techniques and new dosage forms in order to target, control and modulate the delivery of drugs. By the evolution of a drug from a conventional to a novel drug delivery system the performance regarding efficacy, safety and patient compliance can be remarkably improved (Vijaya Shanti and Mrudula, 2011). There are two prerequisites that novel drug carriers aim to fulfill: the delivery of the drug to the specific target site at a pace and extent geared by the demands of the body and the monitoring of the active unit directly during the treatment. In contrast, the term “drug delivery system” is limited to only those systems that involves the delivery of drug to a target site for a specific period. The main rationale for the advancement of novel drug delivery systems is to enable a sustained and controlled drug delivery, to maintain efficient drug level and simultaneously reduce adverse effects (Jain et al., 2014; Namdeo et al., 2014; Akhtar, 2014).
Amid the different novel drug delivery systems, fast dissolving drug delivery systems have acquired remarkable importance regarding oral route of administration. Initially developed as alternative to tablets, capsules and syrups for pediatric and geriatric patients with the fear of suffocation, fast dissolving drug delivery systems have the major benefit of a quick disintegration or dissolution in the salvia without the need of additional liquid (Heer et al., 2013). Amid the various approaches to improve oral bioavailability of hydrophobic drugs, self-emulsifying drug delivery systems (SEDDSs) also possess significant potential. After oral administration, dispersion in gastrointestinal fluid is formed and produces micro-emulsified or nano-emulsified drug that easily gets absorbed via lymphatic pathways and hence bypasses the first past metabolism in the liver (Mahapatra et al., 2014). Traditional oral formulations have almost no control over drug release and the effective concentration at target site, which may lead to fluctuations in plasma concentration. By using osmotic pressure as driving force, osmotic devices allow a controlled drug delivery independent upon gastrointestinal conditions (Sowjanya et al., 2017; Singh et al., 2016). Part of this emerging interest in novel drug delivery systems has also been stimulated by the advances in nanotechnology and the variety of nanoscale platforms. Due to their size in the nanoscale, nanoplatforms can selectively accumulate and specifically bind to the target site with a controlled release behavior (Crommelin and Florence, 2013). Drugs can also be successfully targeted and released in a controlled or sustained manner trough encapsulation in vesicular structures. A large number of vesicular systems has been developed, whereby every newly vesicular system is advantageous over the former one (Namdeo et al., 2014).
This work provides an insight of the novel approaches in drug delivery covering fast-dissolving, self-emulsifying, osmotic, nanoparticulate and vesicular drug delivery systems.
2. Fast dissolving drug delivery systems
2.1. Notable characteristics
The oral route is the most favored route for drug delivery for medical practitioners and manufacturers due to cost effectiveness, ease of administration and hence the highest level of patient compliance. Tablets and capsules are the most popular oral solid dosage forms. Although these have numerous benefits like precise dosing, painlessness and self-medication compared to other administration routes, they remain problematic. While tablets and capsules are hard to swallow especially for geriatric, pediatric and dysphagic patients with fear of suffocation, the major challenge for syrups and other liquid orals is accurate dosing. Furthermore, special cases such as kinetosis, allergic shocks or simply the unavailability of water show the need of a novel oral drug delivery system. In 1970s fast dissolving drug delivery systems were developed mainly to solve swallowing problems. About 25 years later the first fast dissolving tablet (FDT) containing the antihistamine loratadine called Zydis ODT was approved by the FDA (Bhattarai and Gupta, 2016).
FDTs are also called porous tablets, fast melting/disintegrating tablets or orodispersible tablets in subject-related literature. Without the requirement of additional liquid and mastication in the administration process, the dissolution or disintegration takes place within one minute after being moistened by the salvia. By immediate absorption of the released drug and hence a direct entry to the systemic circulation the first pass metabolism is avoided. This way a better alternative to conventional oral dosage forms, particularly for patients suffering nausea and vomiting as well as bedridden patients, is provided. Anyways FDTs are still associated with the fear of choking. Several surrounding conditions like expensive packaging, poor formulations causing unpleasant tastes, friability and difficult handling during manufacturing and transportation led to the development of fast dissolving oral films (FDOFs) (Heer et al., 2013; Bala et al., 2013; Irfan et al., 2016).
Research focused on the concept of transdermal patches to develop a better dosage form. In the first place, FDOFs also known as oral strips or oral wafers, were introduced to the market as breath strips for oral care and are nowadays also available as over-the-counter drugs and prescription-free medication as displayed in Table 1.
Table 1.
Examples for already marketed fast dissolving oral films covering the supplied drug, the indication and the dose strength. This table was revised from Heer et al. (2013).
| Trade name | Drug carried | Application | Dose level |
|---|---|---|---|
| Benadril | Diphenhydramine HCl | Antihistaminic oral strips | 12.5 mg |
| Donepezil | Donepezil HCl | Alzheimer's disease | 5/10 mg |
| Sudafed PE | Phenylephrine HCl | Decongestant oral strips | 10 mg |
| Theraflu | Dextromethorphan HBr | Prolonged cough | 12.5 mg |
Since the design has evolved according to the principle of FDTs, water or chewing aren't required for intake. Oral strips allow administration via the buccal, sublingual or intragastric route causing a local action or a systemic delivery (Heer et al., 2013; Bhattarai and Gupta, 2016). Fast dissolving films can be classified in flash release, mucoadhesive melt-away wafers and mucoadhesive, sustained release wafers, which differ e.g. in thickness, structure and application (Bala et al., 2013). Conventional methods for preparation are solvent casting and semi-solid casting, hot melt and solid dispersion extrusion or rolling (Irfan et al., 2016).
Oral strips consist of a group of flat elegant films, which can be compared to postage stamps in relation to their object properties such as shape and thickness. The size ranges from 1 to 20 cm2 depending on the incorporated drug and dose level. A single dose up to 30 mg is possible (Bala et al., 2013). An ideal film should have the following qualities: flexibility, elasticity, softness together with good physicochemical abilities (Karki et al., 2016). While the water-soluble polymer is used to integrate drug in the form of a matrix, there are many other excipients affecting the characteristics of the strips as listed in Table 2 (Irfan et al., 2016).
Table 2.
Standard ingredients for the formulation of fast dissolving oral films including the concentration, purpose of use and examples (Bhattarai and Gupta, 2016; Bala et al., 2013; Irfan et al., 2016; Karki et al., 2016).
| Ingredients | Concentration | Examples | Purpose of use |
|---|---|---|---|
| Drug | 5–30% | Antiallergic, antidepressants, antiemetic | Active pharmaceutical agent |
| Hydrophilic polymer | 40–50% | Pectin, pullulan and polyvinyl alcohol | As film forming agents |
| Plasticizer | 0–20% | Citrate derivatives, glycerol, PEG | To increase the elasticity and to reduce fragility |
| Sweetener | 2–6% | Glucose, saccharin, stevioside | For a sweet taste and to enhance palatability |
| Salvia stimulating agent | 3–6% | Ascorbic acid, citric acid, lactic acid | To stimulate the salvia production |
| Surfactant | q.s. | Poloxamer 407, sodium lauryl sulfate, tween | For a rapid dissolution and hence to release the API |
| Flavors, colors, fillers | q.s. | Peppermint oil, FD&C colors, natural colors | To better the aesthetic character |
API: Active pharmaceutical components, FD&C: Food, drugs and cosmetics (certified color additives for the use in food, drugs and cosmetics), PEG: Polyethylene glycol.
Efficacy at low doses, a pleasant taste and a sufficient stability in both water and salvia and an adequate permeability are ideal properties of the active pharmaceutical ingredient (API). Hydrophilic polymers are used to form films. The molecular weight is directly related to the dissolution rate i.e. an increase in weight leads to a reduction of the quantity to be disintegrated. The mechanical features, a quick dissolution upon contact with a wet surface along with a good mouth feel are affected by the choice of the hydrophilic polymer. Similar to APIs, there are certain criteria hydrophilic polymers must meet: besides being non-toxic, low-priced and well-tolerated, good spreadability and wetting properties are assumed (Bhattarai and Gupta, 2016).
Along with the ease of administration FDOFs have many advantages compared to traditional oral dosage forms such as a higher dissolution rate due to a larger surface area and a quick disintegration leading to an enhanced bioavailability especially for lipophilic, insoluble drugs. By avoiding the first pass effect due to a direct entry to the blood stream the bioavailability is improved. Additionally, there is no need of water for oral administration, an unpleasant taste of the drug can be overcome and the risk of suffocation is eliminated. This drug carrier enables an enhanced stability as well as dosing accuracy and is easy to manufacture, transport and package. FDOFs still suffers from a few limitations. In comparison to fast FDTs it is only possible to integrate low doses. Furthermore, there are some technical issues and the main target is to achieve dose uniformity (Heer et al., 2013; Bhattarai and Gupta, 2016; Bala et al., 2013).
2.2. Recent advances and clinical aspects
Today FDOFs are the state-of-the-art in rapid dissolving drug delivery systems and are becoming increasingly important lately. Amitriptyline hydrochloride, which is administered to treat severe depression has a poor bioavailability of 30–60% due to a significant first pass metabolism. Salman et al. presented a study to enhance the bioavailability and patient compliance and accordingly optimize the therapeutic effect of amitriptyline hydrochloride by developing oral films. Ten formulations were produced, made of various kinds of polymers, plasticizers and surfactants using the solvent casting method. After visual inspection, the thickness, drug content uniformity, folding endurance and tensile strength were evaluated as well as the surface pH was calculated to prevent oromucosal irritation. Additionally, in vitro/
in vivo disintegration tests and an in vitro dissolution study were conducted. The formulation containing 22.67% w/w maltodextrin and HPMC 15cp each showed the best results concerning an in vitro/in vivo disintegration time of 16.8/13.2 s, 80% drug release within 1.1 min and 89.77% of the drug dissolved after two minutes along with satisfying mechanical properties. As proof of concept a cross-over study using rabbits was designed to compare the pharmacokinetic data of the optimized formulation with a commercially available solution (Amitriptyline Hydrochloride). The bioavailability study showed a rise of the peak blood concentration (0.927 μg/mL) in a short time (2 h) which suggests a fast absorption. In conclusion fast dissolving films of amitriptyline HCl are appropriate to treat depression if rapid onset of action and increased patient compliance is desired (Salman et al., 2014).
Numerous other studies were performed to evaluate fast dissolving films as novel drug carrier for multiple drugs demonstrating their importance as novel drug delivery systems, which are shown in Table 3.
Table 3.
Studies performed to maintain the ideal formulation of fast dissolving oral films for the delivery of various drugs followed by its evaluation and comparison to a current product on the market:
| Drug | Study performed | Proof of concept and study design | Results obtained compared to reference formulation | References |
|---|---|---|---|---|
| Aprepitant |
In vitro characterization In vitro disintegration time, wetting time and drug release |
In vivo comparative pharmacokinetic animal study | Optimized formulation containing 40–45% pullulan and 15–20% PEG 400 showed a shorter disintegration time (20 s), a greater dissolution rate (88.87%) and alike pharmacokinetic values |
Bala and Sharma, 2018 |
| Two period, two sequence, cross-over | ||||
| Sumatriptan succinate |
In vitro characterization In vitro disintegration time and dissolution study |
In vivo comparative pharmacokinetic studies in healthy human volunteers | Optimized formulation containing 60% PVA 20000 and 24% PEG 4400 showed a higher peak concentration (10.78 ng/mL), shorter disintegration time (0.25 h) and an increased AUC |
Tayel et al., 2016 |
| Randomized, two treatment, two period, cross-over | ||||
| Terbutaline sulphate |
In vitro characterization In vitro/in vivo disintegration time and dissolution study |
Bioavailability studies in healthy human volunteers | Optimized formulation containing HPMC-Na alginate-maltodextrin, PEG and water showed a quick disintegration time (25 s), a higher plasma concentration of 12.525 μg/mL and a greater AUC |
Sayed et al., 2013 |
| Randomized, single dose, cross-over |
AUC: Area under the curve, HPMC: Hydroxypropyl methylcellulose, PEG: Polyethylene glycol, PVA: Polyvinyl alcohol.
The joint purpose of these studies was to accomplish direct access to the systemic circulation in order to avoid the first pass effect indicating higher levels of bioavailability. The solvent casting was used for the preparation of oral films in each study. The characterization parameter mainly involved the physicochemical properties as well as the surface pH and moisture content (Bala and Sharma, 2018; Tayel et al., 2016; Sayed et al., 2013).
Furthermore, orally soluble films were investigated in some clinical trials to compare their bioavailability with a marketed product. Ondansetron (OND) is used to prevent patients from vomiting and nausea after chemotherapy, radiotherapy or surgery. To overcome swallowing issues of dysphagic patients and consumers suffering from dry mouth a novel oral dosage form of OND was formulated. A study to compare the bioequivalence between the oral soluble film (OSF) 8 mg (Zuplenz) and the orally disintegrating tablet (ODT) 8 mg (Zofran) was performed in form of three individual open-label, randomized, cross-over studies. In each, healthy adult subjects (men and woman) were treated with a single dose of OND OSF 8 mg and OND ODT 8 mg each. The drug was administered under fasted conditions (study 1 with 48 subjects), under fed conditions (study 2 with 48 subjects) and fasted with or without water (study 3 with 18 subjects) followed by a 7-day (study 1 and 2) respectively 3-day washout period (study 3). Blood samples were collected 1 h before and 24 h after treatment to receive pharmacokinetic data. The results revealed a maximum plasma concentration within the 80–125% range, similar clinical efficacy and safety profile as well as a corresponding bioequivalence between OND OSF 8 mg and OND ODT 8 mg. To conclude OND OSF 8 mg are an appropriate alternative to the conventional tablets (Dadey, 2015).
3. Osmotic drug delivery systems
3.1. Unique properties
Conventional drug carriers often lack in control regarding the drug release and the effective concentration at site of action. This may engender unanticipated, variable plasma concentrations (Sowjanya et al., 2017; Patra et al., 2013). Although research has shown that frequent dosing leads to a low patient compliance, standard drug therapy in terms of dosage level and frequency is designed to provide plasma concentration within the therapeutic range (Patel and Parikh, 2017). Apart from that, some drug substances suffer from a poor oral bioavailability due to solubility and permeability difficulties (Sowjanya et al., 2017). Thus, remarkable attention was paid to the development of a new drug delivery system that provides a controlled release of the API over an prolonged period of time and is not affected by gastrointestinal conditions (Sowjanya et al., 2017; Patel and Parikh, 2017).
The design of controlled drug delivery systems facilitates an ongoing release of the bioactive component at a predestined rate over a defined, extended time with forestalled and replicable kinetics (Syed et al., 2015; Ratnaparkhi et al., 2013). While typical controlled dosage forms like matrix systems or reservoir systems are reliant on pH-value, motility of the GIT and the presence of food, osmotic drug delivery systems (ODDSs) are irrespective of physiological conditions (Patra et al., 2013).
Among the several pharmaceutical attempts to develop a long-acting pharmaceutical form for a single administration per day, osmotic devices are the most dependable ones. Osmotic pressure acts as driving force to release the API in a monitored manner (Syed et al., 2015). Both oral and parenteral administration are possible, whereby a distinction is made between gastrointestinal therapeutic systems, respectively oral osmotic pumps and implantable pumps (Sharma et al., 2018).
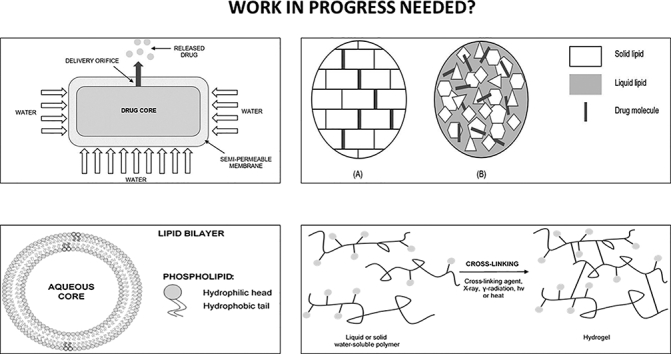
Osmosis can be conventionally described as the net motion of water across a semi-permeable membrane created by the disparity in osmotic pressure across this membrane. The selectivity of the membrane allows only the passing of water, but declines the entrance to most solute molecules and ions (Patel and Parikh, 2017). The release of bioactive agents from osmotic devices is regulated by the osmotic pressure impelled through the penetration of liquid from external surroundings. Moreover, the extent of drug release is directly proportional to the osmotic pressure in the core (Patra et al., 2013). Solubility, osmotic pressure, dimension of the delivery orifice and membrane properties mainly affect the drug release from ODDSs. An important factor is to attain a steady osmotic pressure gradient between the inner and external compartment by maintaining saturation of the osmotic agent in the compartment (Patra et al., 2013). In 1955 the first drug delivery technology based on the concept of osmotic pressure was invented by Rose and Nelson, approximately 75 years after the osmosis principle was discovered (Sowjanya et al., 2017; Patel and Parikh, 2017). The simplest form of osmotic pump, the elementary osmotic pump, was developed by Theeuws in the 70s. The concept comprises a core enclosed by a semi-permeable membrane with one or several delivery pores as seen in Fig. 1.
Fig. 1.
Structure of elementary osmotic pump consisting of drug core, semi-permeable membrane and delivery orifice. Illustration of drug liberation after imbibing water. This figure was adapted from Kumar et al. (2018) and Swojanya et al. (2017) (Sowjanya et al., 2017; Bala and Sharma, 2018).
Hence, many new ODDSs, as for example the push-pull osmotic pump and oral osmotic release systems have been investigated to address the restrictions of the elementary osmotic pump (Yang et al., 2016; Missaghi et al., 2014; Ranjan et al., 2014).
There are three main components in osmotic systems: the drug, the osmotic agent and the semi-permeable membrane. The ideal selection of API should exhibit a brief biological life.
(3–6 h), a high potency and is suitable for long-term treatment. That means especially drugs to treat chronic diseases are good candidates for ODDSs. Further, the API should show a water-solubility within 50–300 mg/L for an optimized drug release. Osmotic agents, or respectively osmogents, are either from organic or inorganic nature, as for example sodium chloride, sodium sulphate and methylcellulose. Semi-permeable membranes have already been used in pharmaceutical industry before. An adequate wet strength and modulus, an ample water permeability and biocompatibility are some of the ideal characteristics for the formulation of ODDSs (Patel and Parikh, 2017; Sharma et al., 2018).
Generally, ODDSs can be classified in single chamber osmotic systems, multi chambered osmotic pumps and specific type of osmotic systems as shown in Table 4 (Syed et al., 2015).
Table 4.
General classification of osmotic drug delivery systems and further subdivision (Sowjanya et al., 2017; Syed et al., 2015).
| Type of osmotic pump | Subtype |
|---|---|
| Single chamber | Elementary osmotic pump |
| Multi-chambered | Push-pull osmotic pumps |
| Sandwiched osmotic pump | |
| Osmotic pump with non-expanding second chamber | |
| Specific type | Controlled porosity osmotic pump |
| Monolithic osmotic pumps tablet | |
| Colon targeted oral osmotic system | |
| Asymmetrical membrane osmotic tablet | |
| Liquid oral osmotic system | |
| Effervescent osmotic pump tablet | |
| Multiparticulate delayed release system | |
| Self-emulsified osmotic tablet | |
| Telescopic capsule for delayed release |
Besides the previously mentioned advantages such as independency on physiological conditions in the GIT, this delivery system benefits from a zero-order release after a primarily retardation, an entirely foreseeable, programmable drug release rate and the possibility of a delayed or pulsed drug delivery. Further, this highly understood and characterized delivery system reduces adverse effects, enhances bioavailability and is suitable for long-term treatment. However, there are still some restrictions that must be considered including high expenses, poorly manufactured films leading to dose dumping, size of delivery orifice, the impact of food intake, no possibility for retrieval therapy and a quick development of tolerance (Sowjanya et al., 2017; Patel and Parikh, 2017; Sharma et al., 2018).
3.2. New approaches to research
A current trend in the design of novel drug delivery systems is the usage of strategies based on two steps. The first phase is aimed to improve the solubility of the API by e.g. micronization, while the second step enables the control of drug liberation by using osmotic systems (Liu et al., 2014a).
In a recent study, Li et al. designed and evaluated a new osmotic pump capsule comprising pH-modulated solid dispersion for the controlled release of flurbiprofen (FP). Flurbiprofen, a non-steroidal anti-inflammatory drug, is among the 40% of drugs with a poor water-solubility and thus limited clinical application. The purpose of this study was to enhance the solubility and oral bioavailability of FP and at the same time to minimize fluctuations in plasma concentration. The preparation of the pH-modulated solid dispersion was conducted by using solvent evaporation method. Then, the osmotic pump capsule was amassed by a semi-permeable capsule shell of cellulose acetate produced by perfusion approach and filled with tableted solid dispersion, penetration enhancer and suspending agents. To optimize the formulation and to assess the formulation aspects a two factor, five level central composite design was used. Various methods were utilized for physical characterization of the optimized formulation including spectrophotometer, differential scanning calorimetry, power X-ray diffraction, Fourier-transform infrared spectroscopy and microscope observation. In addition, the in vitro dissolution in consonance with USP paddle method, the morphologies employing scanning electron microscopy and in vitro drug release were studied. The final formulation contained FP as bioactive compound, Kollidion® 12 PF as hydrophilic device and Na2CO3 as alkalizer in a ratio of 1:4.5:0.02 with a zero-order release profile and a completed drug delivery. The outcome of differential scanning calorimetry and power X-ray diffraction displayed a transition from crystalline structure into amorphous form of the drug. To prove the concept in vivo, a randomized, two period cross-over study was implemented in beagle dogs to determine the pharmacokinetic parameters compared to marketed tablets and a washout period of two weeks. In comparison to commercially available tablets, the osmotic pump capsule showed an increased relative bioavailability of 133.99% and a reduced peak plasma concentration of 59.26 μg/mL. To sum up, the consolidation of solid dispersion and osmotic pump enabled a controlled delivery, reduced administration frequency, enhanced bioavailability and reduced mean peak plasma concentration of the insoluble drug (Li et al., 2015).
Similar to FP, the application of carvedilol is limited by a poor water-solubility and a broad first pass effect in the liver leading to a poor oral bioavailability. Although oral bioavailability can be improved through formulating nanosuspensions, an initial release of drug and extensive fluctuations in plasma concentration are still occurring. To address these drawbacks, Liu et al. prepared and evaluated novel osmotic pump capsules for controlled delivery of carvedilol nanosuspension. Therefore, carvedilol-loaded nanosuspension was produced using freeze-drying method. Next, the capsules assembled by semi-permeable capsule shells of cellulose acetate were filled with carvedilol-loaded nanosuspension drying powder, penetration amplifiers and suspension promoters. In order to predetermine the optimal constitution, central composite design and response surface methodology were employed. The physicochemical characterization of nanosuspension involving particle size, distribution of size, zeta potential and morphology was performed by laser diffraction method and transmission electron microscope (TEM) analysis. Furthermore, the in vitro dissolution was examined in conformity with USP paddle method. The preliminary investigations predicted formulation contained 200 mg Plasdone S-630, 94 mg mannitol and 2,34 g PEG 400 in the solution for coating. The constitution of the coating solution as well as the temperature for lyophilization had a great impact on the homogeneity, elasticity and shell color. Additionally, a bias of about 1% was revealed indicating an appropriate correlation between predicted and actual values. The findings of physicochemical analysis revealed a globular shape in TEM pictures, an average size of 252.19 nm and negative zeta potential. As proof of concept a randomized, two period cross-over study was conducted to obtain pharmacokinetic profiles of eight healthy beagle dogs with a-week long washout period. The in vivo results exhibited a bettered relative bioavailability of 203.5% and a lower peak plasma concentration of 706.59 ± 187.71 ng/mL compared to commercially available tablets. In conclusion, the combined strategy of nanosuspension and osmotic pump is promising in increasing oral bioavailability, reducing administration frequency and attenuating maximum plasma concentration (Liu et al., 2014b).
4. Nanoparticulate drug delivery systems
4.1. Properties of nanodevices
Nanoparticulate drug vehicles are solid, colloidal systems with a high surface-to-volume ratio due to their small size (1–1000 nm) and properties and morphology determined by the design. Since nanomaterials are either composed of lipids and polymers (synthetic or natural) or inorganic metals, the division can take place in inorganic and organic nanodevices as seen in Figure 2 (Thakor and Gambhir, 2013; Martinelli et al., 2019; Rizvi and Saleh, 2018).
Fig. 2.

Classification of nanomedicines based on the materials used for synthetization. This figure was modified from Martinelli et al. (2019) (Martinelli et al., 2019).
Nanoparticulate drug delivery systems are usually composed of two fundamental constituents: the nanoparticle itself and the carried therapeutic agent. The drug is either covalently attached to the surface or alternatively, entrapped and encapsulated by the nanoparticle in order to be protected from demotion and denaturing (Thakor and Gambhir, 2013). The optimum particle size is about 100 nm small, so that instantaneous clearance by the lymphatic system is averted, the blood brain barrier is penetrated and an adequate amount of drug is delivered due to a large surface area. More recently, polymer coating with water-soluble polymers such as polyethylene glycol (PEG) or polysorbate 80 was invented to prolong circulation in the blood stream (Thakor and Gambhir, 2013; Rizvi and Saleh, 2018). Common approaches for the synthesis of nanoparticles are the top-down method and the bottom-up method (Khan et al., n.d.).
Besides the capability of integrating lipophilic and hydrophilic drugs, nanoparticles have a great stability, high drug loading capacity and numerous possible administration routes due to an adjustable size, shape and surface properties. However, there are safety concerns in regard to the application such as a slow dissolution rate and a poor degradation in the human body (Thakor and Gambhir, 2013; Martinelli et al., 2019). Momentous effort has been made in the design of novel nanodevices for the delivery of API especially in the field of nano-oncology as shown in Table 5 (Kakkar et al., 2017).
Table 5.
Examples for already approved nanodevices for different anticancer drugs. This table was modified from Martinelli et al. (2019) (Martinelli et al., 2019).
| Trade name | Material description | Indications | Year of approval |
|---|---|---|---|
| Abraxane | Albumin-bound paclitaxel | Metastatic breast cancer | 1995 |
| Doxil | Liposomal doxorubicin | HIV-related sarcoma, metastatic breast and ovarian cancer | 2005 |
| Oncaspar | Polymeric PEG-L-asparaginase | Acute lymphoblastic tumor | 1994 |
| Onivyde | Liposomal irinotecan | Pancreatic cancer | 2015 |
| Mepact | Liposomal mifamurtide | Osteosarcoma | 2009 |
| Myocet | PEGylated liposomal doxorubicin | Lymphoma, leukemia, carcinoma and sarcoma |
2000 |
| Nanotherm | Iron oxide | Glioblastoma | 2010 |
HIV: Human immunodeficiency virus, PEG: Polyethylene glycol.
4.2. Targeted delivery and triggered release
Nanocarriers can be designed to enhance the efficacy and at the same time to minimize adverse effects by delivering the API to a certain target-site. In anticancer therapy, for example, nanoparticles can take advantage of the enhanced permeability and retention effect of tumor cells due to their small size and leave the systemic circulation in order to get into the extravascular space to amass in tumor tissues. But multiple limitations are associated with passive targeting like a poor drug diffusion and controllability, which led to the development of active targeting. Active targeting is based on the molecular recognition via antigen-antibody or ligand-receptor interactions and is achieved with the help of surface modification through attaching different ligands such as peptides, antibodies or oligosaccharides. This assumes that the targeted molecule must be overexpressed on the target site, i.e. imperceptible in healthy cells. Furthermore, nanocarriers can be devised to respond to a certain stimulus and therefore release the drug locally by altering their structure. Triggered release profits from a very specific, controlled drug delivery enabling protection of healthy tissues from perturbation. Such triggers include oxidative stress, pH value, ultrasounds and temperature (Thakor and Gambhir, 2013; Martinelli et al., 2019; Patra et al., 2018).
4.3. Types of nanoscale drug delivery systems
4.3.1. Polymeric nanoparticles
While polymeric nanoparticle is a generic term for different types of polymer nanoparticles, nanospheres and nanocapsules are mainly described (Thakor and Gambhir, 2013; Patra et al., 2018). Basically, nanospheres are spherical, solid particles with a size ranging from 10 to 200 nm, based on a matrix system and a homogeneous structure throughout (Mamo, 2015; Khalil et al., 2017). In comparison nanocapsules are vesicular systems consisting of a rather oily than aqueous liquid core surrounded by a polymer membrane or coating (Thakor and Gambhir, 2013; Frank et al., 2015). In the inner core the drug is encapsulated either in dispersed or dissolved form, in the polymeric membrane entrapped and amid the pseudo-phase distributed. The wall forming polymer is mostly made of a biodegradable material of natural or synthetic origin. Frequently employed polymers are gelatin, chitosan, polylactide and polylactide-co-glycolic (Frank et al., 2015; Lai et al., 2014). While nanoprecipitation is the most popular approach for the production of nanocapsules, a total of six methods is reported: polymer-coating, interfacial deposition of preformed polymer, solvent displacement, emulsion coacervation, double emulsification and emulsion-diffusion.
Since nanocapsules as drug vehicles have been investigated in various studies for different routes of administration indicating their diversity, several other benefits can be achieved by the entrapment of the API. On the on hand the chemical stability along with photoprotection are provided due to the polymer in the nanocapsules interface. Further, there is an enhanced interaction with tissues and cells since the therapeutic agent is usually taken up while being entrapped within the nanocapsules. By using nanocapsules as drug delivery system, the bioavailability and efficacy is improved and at the same side effects are reduced. While a large number of research has been published, only a few products are currently available in the market (Frank et al., 2015; Yurgel et al., 2013). The clinical use of conventional formulations of anticancer drugs is associated with severe limitations leading to an extensive research in the field of nanocarriers.
In this area, Gonzalo et al. investigated the potential of polyamino acid nanocapsules for nano-oncological therapy to improve the toxicity and efficacy ratio of plitidepsin. These biodegradable nanocapsules were produced using an adapted solvent displacement technique whereby the polyglutamic acid (PGA) was electrostatically applied onto the oily core. The physicochemical characterization was performed using photo correlation spectroscopy, laser doppler anemometry and TEM. The findings revealed a mean particle size of 200 nm, a negative zeta potential and an encapsulation efficacy over 90%. Furthermore, the nanocapsules could be lyophilized and showed an improved long-term stability during storage. As proof of concept the pharmacokinetic and toxicity profile of PGA and PGA-PEG nanocapsules was compared to that of the control formulation. The result obtained from healthy mice after i.v. administration of a single dose revealed extended blood circulation and significant reduction of toxicity. Overall, the findings of the study highlighted the ability of polyamino acid nanocapsules as drug delivery systems for anticancer drugs (Gonzalo et al., 2013).
4.3.2. Solid lipid-based nanoparticles
Solid lipid-based nanoparticles were designed with the intention to accomplish a substitute drug delivery system to polymeric nanoparticles, liposomes and emulsions. In fact, there are two key types differing in the constitution of the solid particle matrix as seen in Fig. 3: solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) (Yoon et al., 2013; Naseri et al., 2015; KH et al., 2013; Ganesan and Narayanasamy, 2017).
Fig. 3.

Illustrative representation of (A) Solid lipid nanoparticles and (B) Nanostructured lipid carriers. This figure was adapted from Yoon et al. (2013) (Yoon et al., 2013).
While SLNs are referred to the first generation of solid lipid-based nanoparticles, NLCs belong to a new era in developing solid lipid-based nanoparticles and each can be further divided in three subtypes as seen in Table 6 (Yoon et al., 2013; Ganesan and Narayanasamy, 2017).
Table 6.
Classification of solid lipid nanoparticles and nanostructured lipid carriers based on the distinct character of the matrix. This table was modified from Ganesan and Narayanasamya (2017) (Ganesan and Narayanasamy, 2017).
| Solid lipid nanoparticles |
Nanostructured lipid carriers |
||
|---|---|---|---|
| Type | Nature of matrix | Type | Nature of matrix |
| I | Homogenous matrix model | Imperfect | Imperfectly structured solid matrix |
| II | Drug enriched shell model | Amorphous | Structureless solid amorphous matrix |
| III | Drug enriched core model | Multiple | Multiple oils in fat in water |
SLNs have a spherical morphology with a size in the nanoscale and remain in solid state at human body or room temperature compared to physiological lipids. While hydrophilic drugs are separated externally from the lipid matrix as a result of being thermodynamically immiscible, lipophilic drugs disperse easily in the lipid matrix (Yoon et al., 2013). Primarily, these lipids deployed to form the matrix involve triglycerides, complex glyceride mixtures, fatty acids and waxes. Moreover, a significant impact is exerted by the choice of the lipid component on the particle size, long-term stability during depository as well as on the drug loading capacity and the drug release profile (Naseri et al., 2015; KH et al., 2013; Ganesan and Narayanasamy, 2017). Additional components added to stabilize and to prolong systemic circulation the matrix are surfactants and polymers (Yoon et al., 2013). Most popular approaches for the manufacturing of SLNs are micro emulsification and high pressure homogenization at low and high temperatures (KH et al., 2013).
Initially developed to combine the advantages and to conquer the difficulties of several drug carriers, SLNs offer many reasons to be considered as promising drug delivery system. By replacing the liquid with a solid lipid not only a controlled release of the bioactive agent is enabled, but also, the chemical degradation is reduced due to the decrease of mobility in the solid matrix. Additionally, the biocompatibility and biodegradability of the employed lipids lead to a reduced acute and chronic toxicity and an improved bioavailability of the incorporated drug. From the fabrication perspective, SLNs are advantageous due to a cost-effective synthesis through high pressure homogenization e.g. and the possibility of large-scale production. Despite these benefits, a limited payload due to a tight lipid crystal structure and drug expulsion during storage caused by polymorphic transition must be considered.
To overcome said complications the next generation of lipid nanocarriers was developed: nanostructured lipid carriers. The matrix of NLCs comprises a mixture of solid and fluid lipids but remain in a solid condition at room and human body temperature. Firstly, variations in the structure of the solid and liquid lipid lead to an imperfect crystal structure allowing an increased drug loading capacity. Secondly, the presence of liquid drug release is inhibited. But at the same time no significant reduction of the cytotoxicity is reported (Naseri et al., 2015; Jaiswal et al., 2016; García-Pinel et al., 2019; Poovi and Damodharan, 2018).
Recently, Cirri et al. investigated NLCs as inventive oral formulation of hydrochlorothiazide for pediatric use to enhance therapeutic efficiency (Cirri et al., 2018). In a former study, hydrochlorothiazide-loaded low-dosage liquid SLNs already improved therapeutic efficacy and prolonged drug release (Cirri et al., 2017). The object of this study was to appraise the actual benefits compared to SLNs. Hereto, the performance of several synthetic and natural lipids was studied. In order to determine their influence on the properties of NLCs two methods were deployed for the preparation: namely homogenization-ultrasonication and microemulsion. Besides the physicochemical characterization using dynamic light scattering, the drug entrapment efficacy and drug loading capacity, the long-term stability upon storage as well as in vitro drug release were examined. The optimized formulation was prepared by microemulsion and contained Precirol®ATO5 as solid lipid along with tween 80 and tween 20 in a ratio of 1:4 as surfactants. The results presented a mean particle size of 327.6 ± 3.7 nm, a polydispersity index of 0.38 ± 0.03, negative zeta potential and long-term stability over 3 months during storage. As in vivo proof of concept a pharmacological study was performed in rats to determine the diuretic activity after single treatment including a control group. To conclude, nanostructured lipid carriers > solid lipid nanoparticles in regard to a higher drug entrapment efficacy, exhibit a prolonged drug release of 6 h and better diuretic acitivity (Cirri et al., 2018).
4.3.3. Gels
Hydrogels consist of a three-dimensional network with porous characteristics made of cross-linked, hydrophilic polymers from natural or synthetic sources, imbibing large amounts of water and therefore have high levels of flexibility (Ullah et al., 2015; Caló and Khutoryanskiy, 2015). The resemblance to living tissue in the swollen state permits high biocompatibility and makes them suitable for numerous applications. The term “smart” or respectively “stimuli-responsive” refers to hydrogels that can respond to changes in the environment by altering their volume (Lee et al., 2013; Vashist et al., 2014). The use of glucose sensors such as lectin in order to control swelling – or respectively deswelling - allows a self-regulated release of insulin, for example (Lee et al., 2013). Polymers obtained from natural or synthetic sources, can either be chemically or physically cross-linked as demonstrated in Fig. 4.
Fig. 4.

Preparation of hydrogels made of liquid or solid water-soluble polymers via cross-linking. This figure was adapted from Caló et Khutoryanskiy (2015) (Caló and Khutoryanskiy, 2015).
Recently, physical hydrogel has gained more attention, as often toxic cross-linking agents are not required for the production. This process is attained through chain aggregation, crystallization, hydrophobic association, and hydrogen bonding for instance and is usually reversible as a result of conformational adjustments (Ullah et al., 2015; Caló and Khutoryanskiy, 2015). Among the different kinds of classification for hydrogels, a frequent one takes place based on their preparation methods as follows: homopolymer, copolymer, semi-interpenetrating and interpenetrating network (Ullah et al., 2015; Das, 2013).
Nanogels, also called the next era of hydrogels, have similar structure and characteristics to hydrogels, apart from their size in the nanoscale. The classification is either based on the type of cross-linking of the three-dimensional network or on the behavior towards an explicit stimulus (Neamtu et al., 2017; Yadav et al., 2017). Most noteworthy are pH or temperature sensitive nanogels exhibiting ideal drug loading and drug release properties due to their swelling and shrinking property (Neamtu et al., 2017). For example, by employing polymers with deionizable functional groups in the synthesis pH-responsive nanogels are prepared (Yadav et al., 2017).
In addition to previously mentioned advantages, due to their small particle size, nanogels provide the following possibilities in relation to other drug delivery systems including nanocarriers. Nanogels are inert in the aqueous milieu as well as in the blood and hence enable prevention of an immunogenic response. The drug delivery via nanogels improves biocompatibility and biodegradability, whereas the latter avoids toxicity and side effects caused by aggregation of nanomaterials. Due to functional groups on the polymeric network and the ease of drug incorporation, a greater level of drug payload is possible. However, even if the production is an affordable process, the removal of the surfactant and solvent in the end can be expensive (Yadav et al., 2017; Jain et al., 2019).
Lately, Schütz et al. examined the favorable effect of surface modification of positively charged chitosan-loaded nanoparticulate complexes with polyanions to build negatively charged particles in contrast to chitosan hydrogels. To explore the advantages of nanogels, El-Feky et al. prepared and assessed silver sulfadiazine-loaded chitosan nanogels with sodium alginate (ALG) coating. Due to its antibacterial effect silver sulfadiazine (SSD) is widely used to treat burn wound infections. Conventional creme formulations are associated with the lack of controllability of drug release and painful removal due to poor biodegradability. To define process criteria and to optimize respective process settings a two-level design of experiment was chosen. By deploying 32 factorial design nine disparate batches of nanogel formulations were prepared with the percentage of ALG and SSD as independent variables. The characterization of SSD-loaded nanogels included particle size, zeta potential, drug entrapment efficacy and in vitro drug release. All nanogels showed an initial blast followed by a gradual and sustainable drug release. The optimized formulation predicted by JMP® software contained 0.4% ALG and 0.414% SSD with an average size of 960 ± 98 nm, negative zeta potential and encapsulation efficacy of 62.65 ± 2.98%. The in vivo study was performed to determine the therapeutic efficacy in the treatment of infected burn wounds. Herein, the animals were divided into three groups and respectively treated with no medication, freeze-dried nanogel powder and commercially available creme. The findings revealed a higher therapeutic efficacy compared to marketed formulation (El-Feky et al., 2017).
5. Vesicular drug delivery systems
5.1. Distinctive features
Highly ordered units of one or more concentrical lipid bilayers formed when amphiphilic building blocks are in contact with water are called vesicular systems. Frequently used materials for the preparation are cholesterol, phospholipids and non-ionic surfactants. Additionally, there is a varied assortment of amphiphilic components. The efficacy is heavily affected by the form, size, construction, lamellarity and encapsulation capacity (Shilakari et al., 2013). Vesicular drug delivery systems (VDDSs) are favorable over conventional dosage forms due to the fact that both lipophilic and hydrophilic drugs can be entrapped in the bilayer, respectively in the aqueous core. Furthermore, the positives include an improved bioavailability, especially of hardly dissolvable drugs, a retarded metabolization, a prolonged systemic circulation and a reduced toxicity (Jain et al., 2014; Namdeo et al., 2014). Among these benefits, VDDDs still have to deal with several drawbacks concerning the drug loading capacity and amount of drug leaked during production, conservation and in vivo transportation (Namdeo et al., 2014).
Today VDDSs are also known as “rebirth systems”, as every newly developed system brings advantages in comparison to the existing systems (Namdeo et al., 2014; Kamboj et al., 2013). The design offers numerous opportunities through delivery a defined amount of drug to a specific target-site and navigation of the effective unit at the site of infection. In this manner multiple administration routes, drug targeting as well as a sustained or controlled release of drugs are supplied (Namdeo et al., 2014; Kamboj et al., 2013; Pattni et al., 2015). A possible categorization of VDDSs can be done from liposomes according to the composition as demonstrated in Fig. 5.
Fig. 5.

Classification of vesicular drug delivery systems starting from liposomes according to the main components into lipid-based and non-lipid-based analogues (Kamboj et al., 2013).
5.2. Liposomes
Liposomes are self-assembling, globular blisters composed of an aqueous core surrounded by one or several concentric lipid bilayers ranging from 20 nm up to a few micrometers. Since the main component are phospholipids there is a high inclination of forming membranes in aqueous environments as illustrated in Fig. 6.
Fig. 6.
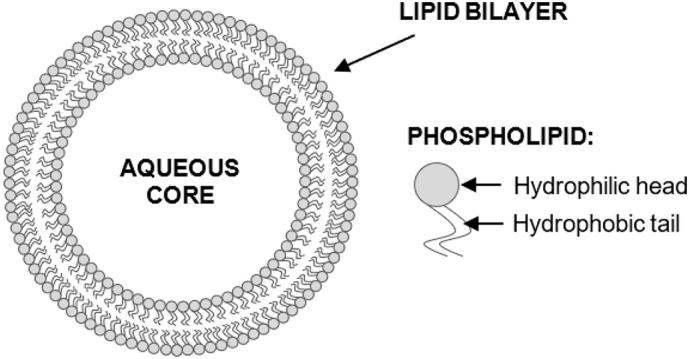
Schematic illustration of liposomes. Structure of phospholipids with hydrophilic head and hydrophobic tail (Bozzuto and Molinari, 2015).
The amphiphilic character of the lipids making them particularly suitable as drug carriers for drugs and cells (Bozzuto and Molinari, 2015; Goyal and Liposomes, 2014). Among the most widely used preparation approaches are the bulk method and the film method (Patil and Jadhav, 2014). A frequently-used categorization of liposomes is done based on the structural design or on the composition, as shown in Table 7 (Pattni et al., 2015).
Table 7.
Classification of liposomes based on size, lamellarity and composition. This table was adapted from Pattni et al. (2015).
| Lamellarity and size |
Composition |
|
|---|---|---|
| Small unilammellar vesicles | 20–100 nm | Conventional liposomes |
| Large unilammellar vesicles | >100 nm | Long-circulating liposomes |
| Giant unilammelar vesicles | >1000 nm | Cationic liposomes |
| Oligolamellar vesicles | 100–1000 nm | Stimuli-responsive liposomes (pH, temperature, magnetic field) |
| Multilamellar vesicles | >500 nm | Immunoliposomes |
Three decades after being first documented by Bingham in the 1960s the first liposomal formulation containing the anticancer drug doxorubicin was launched in the market under the name Doxil (Fan and Zhang, 2013; Estanqueiro et al., 2015). To conquer limitations associated with the first generation of liposomes, research focused on the lipid arrangement, vesicle size and surface charge to implement a new era of liposomes. The attachment of cholesterol to the lipid bilayer of liposomes improves stability and reduces permeability due to a tighter package of phospholipids. Pharmacological restrictions such as a swift clearance from circulation are evaded by surface modification via PEGylation. Although this way an enhanced stability and prolonged circulation time are accomplished, the efficacy of PEGylated liposomes is confined by the absence of specificity. By additional adjustments through target-specific ligands or antibodies PEGylated liposomes can be actively addressed. For a triggered release of the therapeutic agents, stimuli-responsive components can be further installed (Pattni et al., 2015; Bozzuto and Molinari, 2015).
While liposomes have been studied for several decades, a growing interest is noted in the last two decades (Pattni et al., 2015). Conventional treatment of glaucoma consists of timolol maleate (TM)-loaded eye drops to reduce the ocular pressure. A downside of this treatment though is a low bioavailability, the need for frequent application and an accordingly patient compliance.
Yu et al. studied the potential of liposomes dispersed in ion-sensitive in-situ-gels for the ophthalmic delivery of TM to optimize its bioavailability and histocompatibility. For preparation of the liposomal formulation the pH-gradient method was combined with reversed evaporation. In the next step, the TM-loaded liposomes were dispersed into deacylated gellan gum gels. Besides the physicochemical characterization, the drug loading efficacy was examined. Additionally, in vitro release studies and in vitro permeability of the cornea isolated from rabbits using the Franz-cell-type were investigated. Furthermore, in vivo fluorescence imaging as well as eye-irritation studies for single and multiple doses were performed in rabbits. The optimized formulation containing TM 0.25% (w/w), cholesterol 0.75% (w/w), soy phosphatidylcholine 2.0% (w/w) and deacetylated gellan gum 4.0% (w/w) showed a uniform and spherical shape in TEM pictures. Moreover, a vesicle size of 136 nm, an encapsulation efficacy of 47% and no eye-irritation were shown. As proof of concept an in vivo pharmacodynamic study in comparison to traditional eyedrops was carried out in rabbits before and after water-induced hypertension to measure the intraocular pressure. The results from 30 to 180 min after water loading revealed a significant reduction of interocular pressure with a minimum of
13.61 ± 0.95 mmHg at 2 h and longer duration of effect than observed with TM eyedrops. This is probably the outcome of a longer retention time and a 1.93 times greater permeability demonstrating a great cornea penetration (Yu et al., 2015).
Another study was aimed to develop a novel drug carrier for targeted delivery of the anticancer drug paclitaxel to overcome adverse effects and restrictions with passive targeting. Herein, Ravar et al. produced paclitaxel-loaded hyaluronic acid-coated liposomes by thin film method. The physicochemical characterization included the encapsulation efficacy and the drug release profile. In addition, uptake studies, flow cytometry analysis and an in vitro cytotoxicity assay were conducted under respective use of T47D and 4 T1 cells. The liposomal formulation had a spherical shape in TEM pictures, a small vesicle size of 106.6 ± 3.2 nm, a satisfactory encapsulation efficacy of 92.1 ± 1.7% and released 95% of paclitaxel in buffer within 40 h. The results of the confocal laser scanning microscopy and the flow cytometry analysis revealed a higher cellular internalization in respect of free coumarin and an improved cell uptake led to a greater cytotoxic activity compared to free paclitaxel. As proof of concept the antitumor efficacy and biodistribution were investigated in 4 T1 tumor bearing mice to compare the liposomal formulation with a commercially available solution (Intaxel®). The in vivo animal studies showed an increase of the tumor accumulation and also acceptable antitumor activity by the use of active targeting with the aid of hyaluronic acid coated liposomes (Ravar et al., 2016).
However, current approaches in the era of liposomal drug delivery are promising but still suffer from several impediments especially in the transfer to large-scale production and to clinical usage (Pattni et al., 2015; Bozzuto and Molinari, 2015).
5.3. Non-lipid-based analogue
5.3.1. Niosomes
Niosomes are considered as alternative to liposomes regarding the similarity in terms of structure and physical features but slightly differ in composition. Due to a high susceptibility and cost intensity of lipids included in the first vesicular drug carriers, niosomes are formulated by using non-ionic surfactants. The decisive difference is a better chemical and physical stability as well as lower expenses (Khan and Irchhaiya, 2016). This system is characterized by a bilayer structure, which is devised through self-assembling of non-ionic surfactants. Similar to liposomes, niosomes can be categorized in small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicels (Ag Seleci et al., 2016). Furthermore, cholesterol can be added to the formation to provide rigidness to the bilayer and hence restrain its drug leakage (Akhtar, 2014). The attachment of charged groups to the bilayer enhances the stability of niosomes by improving the surface charge density and therefore providing prevention from aggregation (Ag Seleci et al., 2016). The properties of niosomes are strongly dependent on the method of preparation (Khan and Irchhaiya, 2016). Common approaches for production are e.g. thin film hydration, reverse phase evaporation and microfluidization (Ag Seleci et al., 2016).
Niosomes offer the following merits including a wide range of biocompatible, biodegradable and immunogenic surfactants, various routes of administration namely oral, parenteral, ocular and topical routes with an enhanced bioavailability as well as osmotic activity and stability over traditional liposomal formulations and other DDSs.
Niosomes have already conquered the cosmetic industry and are now being explored to determine the potential for further commercial applications (Khan and Irchhaiya, 2016; Akhtar, 2014). Recently, Asthana et al. studied the capability of niosomes for controlled delivery of clarithromycin. The results showed that a sustained and extended drug delivery along with an enhanced bioavailability were provided by the niosomal formulation (Asthana et al., 2016). Another study presented by Fathalla et al. aimed to formulate and evaluate aceclofenac-loaded niosomal gels for sustained delivery. Further, the impact of the extent of non-ionic surfactant, cholesterol and concentration of aceclofenac on the encapsulation efficacy was investigated. Aceclofenac is an anti-inflammatory drug used for the treatment of osteoarthritis and rheumatoid arthritis, but the application is restricted by a short biological lifetime and a low therapeutic index. The reverse phase evaporation method was used for the preparation. The characterization of the formulations was performed using different types of techniques including TEM, optical microscope, differential scanning calorimetry and Fourier-transform infrared spectroscopy. Elected aceclofenac-loaded niosomal formulations were incorporated in several gel bases like HPMC, PEG 600 and ALG and investigated for in vitro drug release and in vitro skin permeation. The findings showed a spherical shape with a certain inner aqueous core of niosomes and an increased drug encapsulation efficiency. In comparison to free drug loaded gel formulations, the niosomal gels provided a sustained drug release up to 6 h and a larger skin permeation. The in vivo comparative efficacy study in carrageenan-induced rats using paw edema test revealed an enhanced anti-inflammatory activity along with an prolonged release of aceclofenac suggesting that niosomal gel formulations are appropriate drug carriers (Fathalla, 2015).
5.4. Lipid-based analogues
5.4.1. Transfersomes
The concept of transfersomes was first invented in 1990s and describes an utmost malleable vesicle with an elastic nature that enables penetration through pores minor than its own size (Shilakari et al., 2013). Conveyance of therapeutic agents through skin is considered as an advanced and fortunate route for drug delivery since the skin is the largest human organ in terms of surface with
2.5–3 m2 (Sachan et al., 2013; Sarmah, 2013). Conventional liposomes and niosomes are not capable of deep penetration and therefore larger portions remain in in the upper skin layers due to the lack of flexibility (Garg et al., 2016). The mechanism of action of transfersomes is based on the osmotic gradient across the many skin layers (Garg et al., 2016).
Apart from phospholipids, edge activators such as tween 80 or span 60 are the main constituents in the formulation of transfersomes. This single chain surfactants effect the destabilization of the lipid bilayers leading to an increase in its malleability making them particularly suitable for skin penetration (Sarmah, 2013; Garg et al., 2016). Common techniques for the preparation of transfersomes are the thin film hydration method and the modified hand shaking, or respectively lipid film hydration method (Sachan et al., 2013).
Transfersomes are considered advantageous in topical and systemic drug delivery for the following distinctive features. On the one hand, transfersomes offer a great encapsulation efficacy up to 90% of drugs with a low or high molecular weight and a large variety in solubility. Moreover, the API is protected from biodegradation and a laggard, incrementally drug release is enabled due to depot function. Regarding production, an easy expansion to large-scale is possible. Despite these benefits, transfersomes still suffer from some shortcomings such as tendency of oxidative degradation, a range in purity of phospholipids from natural origin and an expensive production (Sachan et al., 2013; Sarmah, 2013).
The oral route is unfavorable for the application of asenapine maeleate (AM) for antipsychotic treatment of bipolar disorder and schizophrenic due to an extensive hepatic metabolism. A current study presented by Shreya et al. examined the potential of nano-transfersomes for the delivery of AM via transdermal route. The aim of the study was to improve bioavailability through bettering skin permeation by a combination of chemical and transfersomal attempts. The transfersomes were produced by thin film hydration technique. Vesicle size, zeta potential, incorporation efficacy, polydispersity index, as well as surface morphology were included in physicochemical characterization. An in vitro skin permeation study of AM-loaded transfersomes was conducted and different kinds of chemical enhancers were used to increase transdermal transportation. The optimized transfersomal formulation was composed of AM, soy phosphatidylcholine and sodium deoxycholate in a weight ratio of 5:75:10. Further, a spherical shape with an average vesicle size of 126.0 nm and a drug entrapment efficiency of 54.96% were revealed. The cumulative extent of AM penetrated within 24 h was 160 μ, 132.9 μg and 309.3 μg indicating a synergetic effect of chemical enhancer (ethanol 20% (v/v)) and transfersomes. As proof of concept the pharmacokinetic profiles obtained from rats after transdermal administration were compared to oral route. A 1.16 times bigger plasma concentration was revealed in comparison to the reference substance as well as a significant drop in bioavailability. Finally, the combination of permeation enhancer and transfersomes enabled an increase of transdermal permeation and therefore bioavailability of AM (Shreya et al., 2016).
5.4.2. Ethosomes
Similar to transfersomes, ethosomes can improve the penetration through the stratum corneum barrier due to a quick permeation and greater transdermal flow (Parashar et al., 2013). The second generation of novel vesicular drug carriers are represented by these spherical, lipid blisters mainly composed of phospholipids, ethanol and water. The high alcohol content of up to 45% is the main distinguishing feature from liposomes enabling a decrease in size and elasticity when same method of preparation is used. In order to reach deeper tissues and cause a systemic action the penetration of the natural skin barrier and the magnitude of transdermal permeation are influenced. Further adjuvants added to the ethosomal formulation are cholesterol to improve stability or gel markers for increased residence time (Mbah et al., 2014; Garg et al., 2016; Abd El-Alim et al., 2019). In general, the approaches for preparation of ethosomes could be assorted in solvent evaporation and mechanical dispersion (Abd El-Alim et al., 2019).
Besides the aptitude for transdermal and dermal drug delivery, the most noteworthy advantages of ethosomes include a great patient convenience due to a semi-solid dosage, a wide range of applicability and the possibility for instant commercialization. Further, ethosomes enable a passive, noninvasive drug delivery including larger therapeutic agents and are an easy attempt compared to phonophoresis or iontophoresis. However, the major challenge is to achieve stability of ethosomes especially during storage due to oxidation sensitivity of the lipid component (Parashar et al., 2013; Abd El-Alim et al., 2019).
Recently, Jain et al. prepared and evaluated ethosomal hydrogels for transdermal delivery of diclofenac to improve its anti-inflammatory activity and simultaneously to comprehend the correlation of formulation parameters with physicochemical features and permeation flux. Various approaches to better transdermal drug uptake through different permeation enhancers and drug carriers are problematic due to painful and valuable treatment and lasting skin harm. The rotary evaporation method was used for manufacturing of diclofenac-loaded ethosomes and liposomal control formulations. The findings of physicochemical characterization and in vitro skin permeation study demonstrated the influence exerted by the interplay of variable components - especially size and flexibility - plus controllability through its manipulation. A concentration of 22.9% ethanol and a ratio of soy phosphatidylcholine to cholesterol of 88.4:11.6 resulted in an optimal composition with a size of 144 ± 5 nm, an elasticity of 2.48 ± 0.75 and an encapsulation efficacy of 71 ± 4%. The optimized formulation showed a significant increase of the in vitro permeation as well as a strengthened anti-inflammatory effect in vivo animal studies compared to the control formulations. In conclusion, diclofenac-loaded ethosomal hydrogels can improve the therapeutic efficacy compared to liposomes and plain hydrogel (Jain et al., 2015).
6. Self-emulsifying drug delivery systems
6.1. Remarkable qualities
Amidst the different administration routes, the oral is the most popular one due to a high patent convenience and consequentially superior compliance. For absorption from the GIT, the dissolution of the API in gastrointestinal fluid is necessary (Mu et al., 2013). Almost 40% of recently developed drugs are affected by a poor water-solubility leading to an insufficient oral bioavailability, large intra- and inter-subject variety as well as missing dose proportionality (Pathak et al., 2013). While various technologies were exploited to address these shortcomings, lipid-based drug delivery systems have gained considerable attention lately.
The term lipid-based formulation covers a wide range from basic lipid solutions to well-advanced SEDDSs. In fact, the encapsulation of the API into inert lipid devices can improve its oral bioavailability (Mu et al., 2013; Kalepu et al., 2013; Rahman et al., 2013). The selection of lipid excipient has not only an impact on the solubility of the API in the formulation, but also affects the drug solubilizing in the GIT during digestion of lipid in addition to the absorption and bioavailability of the drug (Mu et al., 2013). To interpret the in vivo behavior of the lipid preparation with regard to specific physicochemical aspects of the drug, the “lipid formulation classification model” (LCFS) was introduced in 2000 and updated in 2006 by Pouton. For this purpose, lipid formulations were divided into four types in accordance to their constitution and conceivable effects regarding dissolution and digestion (Mahapatra et al., 2014; Kalepu et al., 2013).
Among the various approaches to augment oral bioavailability of poorly water-soluble drugs, SEDDSs appears promising (Balakumar et al., 2013a). After oral administration, dispersion in gastrointestinal fluid is formed and produces micro-emulsified or nano-emulsified drug that effortlessly gets absorbed via lymphatic system avoiding the first pass effect in the liver. The agitation needed for self-emulsification is supplied by the motility of digestion in the stomach and intestines (Kalepu et al., 2013).
Isotropic compounds of API, lipid and surfactants, mostly with one or several co-dissolvers or co-emulsifying agents are referred to as SEDDS. These self-emulsifying oil formulations can promptly build fine oil-in-water emulsions when introduced into aqueous media upon gentle agitation. SEDDS is a general term for delivery systems that usually builds emulsions with a dot size ranging from a couple nanometers to some micrometers. While self-micro-emulsifying drug delivery system (SMEDDS) describes transparent microemulsions with oils droplet size of 100 to 250 nm, self-nano-emulsifying drug delivery system (SNEDDS) is a more recent concept with a spherical size of below 100 nm (MH a et al., 2013; Mahapatra et al., 2014). There are some differences between SEDDS, SMEDDS and SNEDDS that need to be understood. These differences are summarized in Table 8.
Table 8.
Main discrepancies between self-emulsifying, self-micro-emulsifying and self-nanoemulsifying drug delivery systems. This table was adapted from Doaknia et Joshi. (2015) (Dokania and Joshi, 2015).
| Character | SEDDS | SMEDDS | SNEDDS |
|---|---|---|---|
| Dimension | >300 nm | <250 nm | <100 nm |
| Occurrence | Murky | Visually clear | Visually clear |
| HLB level of surfactant | <12 | >12 | >12 |
| Classification by LCFS | Type II | Type IIIB | Type IIIB |
| Amount of oil | 40–80% | >20% | >20% |
| Amount of surfactant | 30–40% | 40–80% | 40–80% |
HLB: Hydrophile-lipophile balance, LCFS: Lipid classification formulation system, SEDDS: Self-emulsifying system, SMEDDS: Self-micro-emulsifying system, SNEDDS: Self-nano-emulsifying system.
In accordance with LCFS, SEDDSs are isotropic mixtures of oil or mixtures of oil and surfactant (type II), but are further modified to SMEDDS and SNEDDS classified as type IIIa and IIIb, which additionally contain one or several co-surfactants or hydrophilic co-solvents (Chatterjee et al., 2016). While the prime mover for the formation of microemulsions is the extremely low interfacial tense achieved by at least two emulsifiers, an energy input is required for nano-emulsion formation provided by either chemical potential or mechanical equipment. As stated by Reiss, the process of self-emulsification appears when the change in entropy is bigger than the energy necessary to improve the surface are of the dispersion. Additionally, released energy of an ordinary emulsion formation is a direct function of the energy needed to build a novel surface between the oil and water phase (MH a et al., 2013; Balakumar et al., 2013a).
SEDDS are a possible representative, alternatively, to traditional oral formulations of lipophilic components. SEDDS can be regarded as isotropic solutions of oil, surfactant, co-surfactant, and drug forming oil–water (o/w) emulsions when being exposed to fluids and motility of the GI tract. The oil is one of the most essential excipients used in the formulation of SEDDS, not merely because considerable quantities of lipophilic drugs are solubilized, or self-emulsification facilitated, but also and primarily the amount of lipophilic drug transported through the intestinal lymphatic system can be increased. Triglyceride oils with a long or medium chain length and variable levels of saturation can be used for the formulation. To form and sustain an emulsion status in the GIT a surfactant concentration of 30–60% (w/w) is required. The most frequently recommended surface-active agents are non-ionic ones with a moderately high hydrophilic-lipophilic equilibrium like tween 80. Furthermore, co-solvents such as PEG or ethanol are added to allow the dissolution of larger amounts of hydrophilic surfactants or drugs in the lipid base. In order to prevent either drugs or unsaturated fatty acid chains from oxidation, lipophilic antioxidants like α-tocopherol or β-carotene could be involved in formulations (Mahapatra et al., 2014).
By forming fine oil-in-water emulsions or microemulsions when in contact with gastrointestinal fluid upon gentle agitation, self-emulsifying oil formulations are offering an enhancement in rate and degree of absorption and enable more easily reproducible plasma concentrations. In general, the oral bioavailability of hydrophobic drugs can be improved (Mahapatra et al., 2014; Rahman et al., 2013).
In addition, the more recent SNEDDS are advantageous because sensitive drugs are preserved, drug payload is improved and the storage is simplified due to thermodynamic stability. This novel delivery system selectively aligns the drug to a specific absorption window in the GIT, embellishes oral bioavailability enabling lower drug doses and minimizes irritation caused by prolonged contact between bulk-drugs and intestinal wall. Furthermore, the size in nanoscale leads to a larger surface to partition the drug in between oil and water compared to oily solutions. Several drawbacks must be considered including the lack of suitable in vitro models for the evaluation of formulations and the several formulations factors affecting self-emulsification (MH a et al., 2013; Kazi et al., 2019).
6.2. Current trends in research
Talinolol (TAL), a poorly water-soluble, long-acting beta-blocker, suffers from a fluctuating bioavailability most likely due to precipitation in the GIT, inchoate and irregular absorption as well as P-glycoprotein imparted efflux transport in the gut. The object of a recent study was to evolve and examine the performance of self-nano emulsifying formulations of TAL. Herein, Kazi et al. produced several formulations using different kinds of natural and semisynthetic oils, lipophilic and hydrophilic surfactants and water-soluble co-solvents. The characterization involving droplet size, PDI and zeta potential were performed using laser light diffraction analysis method. To attain the maximum drug loading equilibrium solubility of TAL was conducted in anhydrous and diluted SNEDDS. Further, in vitro dissolution studies, ex vivo permeation experiments and in vitro red blood cell toxicity test were carried out to compare the developed formulations with the plain drug and a commercially available product. The findings from characterization and solubility studies revealed a higher stability with smaller droplet sizes and a greater TAL solubility. In comparison to reference substances, a significant increase of TAL release of more than 97% within 2 h was achieved by SNEDDS in the dissolution studies. The selected composition appeared to be the most stable formulation with droplet size of 35.99 nm, low PDI of 0.18 upon watery dilution as well as a negative zeta potential. As proof of concept, an in vivo comparative bioavailability study was completed in rats. A 4-fold increase of gut permeability and 1,58-fold improved oral bioavailability of TAL compared to plain drug were shown in ex vivo permeability tests and in vivo pharmacokinetic study. To summarize, TAL-laden SNEDDSs enabled a better drug payload, drug dissolution, intestinal permeation and oral bioavailability along with a decreased/no human red blood cell toxcitiy (Kazi et al., 2019).
In another study, Balakumar et al. formulated and evaluated rosuvastatin calcium (ROC)- loaded SNEDDS with the aim of enhancing its solubility and oral bioavailability. The therapeutic performance of ROC, a lipid reducing agent, is limited by a poor oral bioavailability of 20% due to poor water-solubility and extensive hepatic metabolism. Different kinds of oils involving essential oils were investigated for their self-emulsification property with surfactants and co-surfactants. To optimize the system ternary phase diagram was created based on solubility analysis of ROC. The produced formulations were characterized for their self-emulsification time, robustness against dilution, droplet size, particle size, zeta potential and in vitro drug release using various techniques including laser diffraction analysis and phase contrast microscope. The mixture consisting of 30% cinnamon oil, 60% Labrasol, 10% Capmul MCM C8 had the best self-emulsification property of ROC, a driblet size of below 200 nm resulting in improved drug solubility and an appropriate zeta potential of −29.5 ± 0.63 mV for stability. In comparison to the marketed product the self-emulsifying formulation showed a clear incline in dissolution as shown by in vitro drug release tests. To proof the results obtained from in vitro studies, an in vivo comparative pharmacokinetic study was conducted in rats and the findings were determined by HPLC method. The recorded findings revealed a 2.45 times higher bioavailability and an increased peak plasma concentration of 7.83 ± 2.61 μg/mL than ROC in suspension. Finally, the optimized self-emulsified formulation enabled enhanced oral bioavailability compared to marketed product probably due to collective mechanism of nanosized dispersion with greater surface area (Balakumar et al., 2013b). Further advances in the era of SNEDDS and SMEDDS are summarized in Table 9.
Table 9.
In vitro/ In vivo studies conducted to obtain an optimized self-emulsifying formulation for an improved oral bioavailability of various drugs.
| Formulation | Study performed | Proof of concept and study design | Results obtained compared to reference formulation | References |
|---|---|---|---|---|
| Darunavir-loaded solid SNEDDS | Liquid state characterization Solid state characterization |
In vivo comparative pharmacokinetic studies in rats | Optimized formulation (L-SNEDDS) containing 16.6% Capmul MCM C8, 41.7% tween 80 and 41.7% Transcutol-P showed a 3-times higher dissolution rate and augmented oral bioavailability | Inugala et al., 2015 |
| Ziyuglycoside I- loaded SMEDDS |
Physicochemical characterization In vitro drug release |
In vivo activity study in mice In vivo comparative pharmacokinetic study in rats |
Optimized formulation containing Obleique CC497, tween 20, and Transcutol HP in the ratio of 0.25:0.45:0.30 showed a 6.94-greater absolute bioavailability of 21.94 ± 4.67% |
Xiong et al., 2019 |
Capmul MCM: Mono-diglyceride of medium chain fatty acids, SMEDDS: Self-micro-emulsifying drug delivery systems, SNEDDS: Self-nano-emulsifying drug delivery systems.
7. Conclusion
The development of novel drug delivery systems plays a leading role in pharmaceutical sector nowadays. Conventional dosage forms such as tablets, capsules and emulsions are limited by several drawbacks. To address these shortcomings of traditional drug delivery systems research mainly focused on improving bioavailability and patient compliance while reducing toxicity and side effects. A variety of drug delivery technologies has been developed and evaluated, and numerous strategies for a controlled and targeted delivery have been explored. The performance of the drug in terms of bioavailability, safety and efficacy can be positively changed by alteration of an already existing drug into a novel delivery technology.
Amid the administration routes the oral one is the most popular one with the highest levels of patient compliance, but oral bioavailability is often limited because of a poor water-solubility, low permeability or high first pass metabolism. By developing fast-dissolving formulations comprising FDTs and later FDOFs, researchers embraced an alternative to traditional oral dosage forms. The distinctive features such as the ease of administration, a rapid disintegration and a quick action within 60 s making it a promising delivery especially for geriatric, pediatric and dysphasic patients who find it hard to swallow capsules or tablets. Since almost 40% of newly discovered drugs are encountered with a poor water-solubility causing a poor bioavailability and missing dose proportionality, the use of self-emulsifying formulations has become increasing popular for preclinical studies. The design of SEDDS, SMEDDS and SNEDDS enable bypassing the dissolution step after oral administration and the first pass effect indicating higher levels of bioavailability. Based on the utilization of osmosis as driving force, osmotic drug devices have come a long way since its origin about 25 years ago. By modulating the formulation factors including dissolubility and osmotic pressure of the gist, membrane characteristics and the orifice size, ODDSs can deliver multiple API in a controlled manner and irrespective of physiological conditions. Nanotechnology is also one exciting sector in drug delivery that has experienced growth at imposing rate lately. Ranging from organic to inorganic nanoparticles, a large number of materials and formulation aspects are possible and allow great versatility, controllable size and shape, the functionalization to a targeted delivery and triggered release as well as the loading of various active agents. Drugs can also be successfully addressed to a specific target site by using lipoidal vesicular systems such as liposomes, transfersomes and ethosomes or non-lipoidal vesicular systems like niosomes. In this manner toxic or undesirable effects to other sites can be prevented. This system has been investigated over the years in drug deliverer, particularly for transdermal drug delivery, due to its flexibility to be modified for different desired purposes. Finally, VDDSs enable an improved bioavailability, a prolonged systemic circulation and enhanced patient compliance.
In conclusion, the evolution of drug delivery systems has come a long way and will proceed to grow at an extraordinary rate. Plentiful therapeutic as well as commercial merits are provided by the incorporation of drug molecules in novel drug delivery systems. It is evident that new pathways have been paved and doors opened in the delivery of already existing as well as new drugs. But some aspects including translation to clinical use, costly production and limited drug payload must be addressed in more extensive manner. However, there is still room for improvement and all newly developed drug delivery systems will need to be thoroughly characterized and investigated before being approved to be used in humans.
8. Future perspectives and critical point of view
Today's drug delivery technologies enable the embodiment of the drug into novel delivery devices and hence facilitate various therapeutic and commercial benefits. Ranging from fast dissolving to nanoparticulate drug delivery systems, a variety of novel delivery systems have been developed and evaluated and numerous strategies for a controlled drug delivery to a specific target-site have been researched. However, there are several challenges remaining.
One of the permanent features of drug delivery technologies, is the important part that polymers play in navigating the drug liberation as well as in manufacturing drug carriers. Progress, especially in the field of nanotechnology, is limited by the availability of suitable biocompatible polymers. An ongoing interest in new polymer synthesis has occurred due to the demand for polymers with aimed physical and biological features. For this reason, a wide array of biodegradable polymers form natural or synthetic origin has been studied for their ability of an extended drug liberation and targeted drug delivery. So far, only a small number of them are found to be biocompatible. From the manufacturing perspective, conventional methods have the merit of easy scale-up but are likely to lose conciseness in monitoring over particle characteristics. Although top-down techniques would allow to regulate size and shape, they are only applicable to a few drug delivery systems.
Several other approaches have been made including in the treatment of diabetes mellitus to address the limitations with the administration of insulin. By using the glucose modulation, the insulin delivery rates can be regulated enabling a self-regulated drug delivery. The major challenge is to develop a delivery system that exhibits the natural pattern of insulin release in vivo.
Critically concluded, the need for new materials for their quality of being biodegradable, biocompatible and low toxicity will be met in future and in combination with novel fabrication techniques will provide significant advantages in drug delivery. Tomorrow's drugs will definitely be more challenging in regard to drug delivery and the pharmaceutic science will have a really difficult task ahead.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abd El-Alim S.H., Kassem A.A., Basha M., Salama A. Comparative study of liposomes, ethosomes and transfersomes as carriers for enhancing the transdermal delivery of diflunisal: in vitro and in vivo evaluation. Int. J. Pharm. 2019;563:293–303. doi: 10.1016/j.ijpharm.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Ag Seleci D., Seleci M., Walter J.G., Stahl F., Scheper T. Niosomes as Nanoparticular Drug Carriers: Fundamentals and recent applications. J. Nanomater. 2016;2016:1–13. doi: 10.1155/2016/7372306. [DOI] [Google Scholar]
- Akhtar N. Vesicles: a recently developed Novel carrier for Enhanced Topical Drug delivery. Curr Drug Deliv. 2014;11(1):87–97. doi: 10.2174/15672018113106660064. [DOI] [PubMed] [Google Scholar]
- Asthana G.S., Sharma P.K., Asthana A. In Vitro and in Vivo Evaluation of Niosomal Formulation for Controlled delivery of Clarithromycin. Scientifica (Cairo). 2016;2016 doi: 10.1155/2016/6492953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bala R., Sharma S. Formulation optimization and evaluation of fast dissolving film of aprepitant by using design of experiment. Bull Fac Pharmacy, Cairo Univ. 2018;56(2):159–168. doi: 10.1016/j.bfopcu.2018.04.002. [DOI] [Google Scholar]
- Bala R., Pawar P., Khanna S., Arora S. Orally dissolving strips: a new approach to oral drug delivery system. Int J Pharm Investig. 2013;3(2):67–76. doi: 10.4103/2230-973X.114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakumar K., Vijaya Raghavan C., Tamil Selvan N., Habibur Rahman S.M. Self emulsifying drug delivery system: Optimization and its prototype for various compositions of oils, surfactants and co-surfactants. J. Pharm. Res. 2013;6(5):510–514. doi: 10.1016/j.jopr.2013.04.031. [DOI] [Google Scholar]
- Balakumar K., Raghavan C.V., selvan N.T., prasad R.H., Abdu S. Self nanoemulsifying drug delivery system (SNEDDS) of Rosuvastatin calcium: Design, formulation, bioavailability and pharmacokinetic evaluation. Colloids Surf. B: Biointerfaces. 2013;112:337–343. doi: 10.1016/j.colsurfb.2013.08.025. [DOI] [PubMed] [Google Scholar]
- Bhagwat R., Vaidhya I. Novel Drug Delivery Systems: An Overview. Int. J. Pharm. Sci. Res. 2013;4(3):970–982. [Google Scholar]
- Bhattarai M., Gupta A.K. Fast Dissolving Oral Films: a Novel Trend to Oral Drug delivery System. Sunsari Tech Coll J. 2016;2(1):58–68. doi: 10.3126/stcj.v2i1.14802. [DOI] [Google Scholar]
- Bozzuto G., Molinari A. Liposomes as nanomedical devices. Int. J. Nanomedicine. 2015;10:975–999. doi: 10.2147/IJN.S68861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caló E., Khutoryanskiy V.V. Biomedical applications of hydrogels: a review of patents and commercial products. Eur. Polym. J. 2015;65:252–267. doi: 10.1016/j.eurpolymj.2014.11.024. [DOI] [Google Scholar]
- Chatterjee B., Hamed Almurisi S., Ahmed Mahdi Dukhan A., Mandal U.K., Sengupta P. Controversies with self-emulsifying drug delivery system from pharmacokinetic point of view. Drug Deliv. 2016;23(9):3639–3652. doi: 10.1080/10717544.2016.1214990. [DOI] [PubMed] [Google Scholar]
- Cirri M., Mennini N., Maestrelli F., Mura P., Ghelardini C., di Cesare Mannelli L. Development and in vivo evaluation of an innovative “Hydrochlorothiazide-in Cyclodextrins-in Solid Lipid Nanoparticles” formulation with sustained release and enhanced oral bioavailability for potential hypertension treatment in pediatrics. Int. J. Pharm. 2017;521(1–2):73–83. doi: 10.1016/j.ijpharm.2017.02.022. [DOI] [PubMed] [Google Scholar]
- Cirri M., Maestrini L., Maestrelli F. Design, characterization and in vivo evaluation of nanostructured lipid carriers (NLC) as a new drug delivery system for hydrochlorothiazide oral administration in pediatric therapy. Drug Deliv. 2018;25(1):1910–1921. doi: 10.1080/10717544.2018.1529209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crommelin D.J.A., Florence A.T. Towards more effective advanced drug delivery systems. Int. J. Pharm. 2013;454(1):496–511. doi: 10.1016/j.ijpharm.2013.02.020. [DOI] [PubMed] [Google Scholar]
- Dadey E. Bioequivalence of Ondansetron Oral Soluble Film 8 mg(ZUPLENZ) and Ondansetron Orally Disintegrating Tablets 8 mg (ZOFRAN) in healthy adults. Am. J. Ther. 2015;22(2):90–97. doi: 10.1097/MJT.0000000000000174. [DOI] [PubMed] [Google Scholar]
- Das N. Preparation methods and properties of hydrogel: a review. Int J Pharm Pharm Sci. 2013;5(3):112–117. [Google Scholar]
- Dokania S., Joshi A.K. Self-microemulsifying drug delivery system (SMEDDS)-challenges and road ahead. Drug Deliv. 2015;22(6):675–690. doi: 10.3109/10717544.2014.896058. [DOI] [PubMed] [Google Scholar]
- El-Feky G.S., El-Banna S.T., El-Bahy G.S., Abdelrazek E.M., Kamal M. Alginate coated chitosan nanogel for the controlled topical delivery of silver sulfadiazine. Carbohydr. Polym. 2017;177(August):194–202. doi: 10.1016/j.carbpol.2017.08.104. [DOI] [PubMed] [Google Scholar]
- Estanqueiro M., Amaral M.H., Conceição J., Sousa Lobo J.M. Nanotechnological carriers for cancer chemotherapy: the state of the art. Colloids Surf. B: Biointerfaces. 2015;126(2015):631–648. doi: 10.1016/j.colsurfb.2014.12.041. [DOI] [PubMed] [Google Scholar]
- Fan Y., Zhang Q. Development of liposomal formulations: from concept to clinical investigations. Asian J Pharm Sci. 2013;8(2):81–87. doi: 10.1016/j.ajps.2013.07.010. [DOI] [Google Scholar]
- Fathalla D. In-vitro and In-vivo Evaluation of Niosomal Gel Containing Aceclofenac for Sustained Drug delivery. Int. J. Pharm. Sci. Res. 2015;1:1):1–11. doi: 10.15344/2394-1502/2014/105. [DOI] [Google Scholar]
- Frank L.A., Contri R.V., Beck R.C.R., Pohlmann A.R., Guterres S.S. Improving drug biological effects by encapsulation into polymeric nanocapsules. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology. 2015;7(5):623–639. doi: 10.1002/wnan.1334. [DOI] [PubMed] [Google Scholar]
- Ganesan P., Narayanasamy D. Lipid nanoparticles: different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain. Chem. Pharm. 2017;6(May):37–56. doi: 10.1016/j.scp.2017.07.002. [DOI] [Google Scholar]
- García-Pinel B., Porras-Alcalá C., Ortega-Rodríguez A. Lipid-based Nanoparticles: Application and recent advances in Cancer Treatment. Nanomaterials. 2019;9(4):638. doi: 10.3390/nano9040638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg V., Singh H., Bimbrawh S. Ethosomes and Transfersomes: Principles, Perspectives and Practices. Curr Drug Deliv. 2016;14(5):613–633. doi: 10.2174/1567201813666160520114436. [DOI] [PubMed] [Google Scholar]
- Gonzalo T., Lollo G., Garcia-Fuentes M. A new potential nano-oncological therapy based on polyamino acid nanocapsules. J. Control. Release. 2013;169(1–2):10–16. doi: 10.1016/j.jconrel.2013.03.037. [DOI] [PubMed] [Google Scholar]
- Goyal T.G., Liposomes A.K. Targeted and Controlled delivery System. Drug Deliv Lett. 2014;1(4):62–71. [Google Scholar]
- Heer D., Aggarwal G., Kumar S.L.H. Recent trends of fast dissolving drug delivery system - an overview of formulation technology. Pharmacophore. 2013;4:1):1–9. [Google Scholar]
- Inugala S., Eedara B.B., Sunkavalli S. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) of darunavir for improved dissolution and oral bioavailability: in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2015;74:1–10. doi: 10.1016/j.ejps.2015.03.024. [DOI] [PubMed] [Google Scholar]
- Irfan M., Rabel S., Bukhtar Q., Qadir M.I., Jabeen F., Khan A. Orally disintegrating films: a modern expansion in drug delivery system. Saudi Pharm J. 2016;24(5):537–546. doi: 10.1016/j.jsps.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Jain V., Mahajan S.C. Lipid based Vesicular Drug delivery Systems. Adv. Pharm. 2014;2014:1–12. doi: 10.1155/2014/574673. [DOI] [Google Scholar]
- Jain S., Patel N., Madan P., Lin S. Quality by design approach for formulation, evaluation and statistical optimization of diclofenac-loaded ethosomes via transdermal route. Pharm. Dev. Technol. 2015;20(4):473–489. doi: 10.3109/10837450.2014.882939. [DOI] [PubMed] [Google Scholar]
- Jain S., Ancheria R.K., Shrivastava S., Soni S.L., Sharma M. An Overview of Nanogel –Novel Drug Delivery System. Asian J Pharm Res Dev. 2019;7(2):47–55. doi: 10.22270/ajprd.v7i2.482. [DOI] [Google Scholar]
- Jaiswal P., Gidwani B., Vyas A. Nanostructured lipid carriers and their current application in targeted drug delivery. Artif Cells, Nanomedicine Biotechnol. 2016;44(1):27–40. doi: 10.3109/21691401.2014.909822. [DOI] [PubMed] [Google Scholar]
- Kakkar A., Traverso G., Farokhzad O.C., Weissleder R., Langer R. Evolution of macromolecular complexity in drug delivery systems. Nat Rev Chem. 2017;1:1–18. doi: 10.1038/s41570-017-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalepu S., Manthina M., Padavala V. Oral lipid-based drug delivery systems – an overview. Acta Pharm. Sin. B. 2013;3(6):361–372. doi: 10.1016/J.APSB.2013.10.001. [DOI] [Google Scholar]
- Kamboj S., Saini V., Magon N., Bala S., Jhawat V. Vesicular drug delivery systems: a novel approach for drug targeting. Int J Drug Deliv. 2013;5(2):121–130. [Google Scholar]
- Karki S., Kim H., Na S.J., Shin D., Jo K., Lee J. Thin films as an emerging platform for drug delivery. Asian J Pharm Sci. 2016;11(5):559–574. doi: 10.1016/j.ajps.2016.05.004. [DOI] [Google Scholar]
- Kazi M., Al-Swairi M., Ahmad A. Evaluation of Self-Nanoemulsifying Drug delivery Systems (SNEDDS) for Poorly Water-Soluble Talinolol: Preparation, in vitro and in vivo Assessment. Front. Pharmacol. 2019;10:1–13. doi: 10.3389/fphar.2019.00459. (May) [DOI] [PMC free article] [PubMed] [Google Scholar]
- KH R., SA S., SN D. Solid lipid nanoparticles- a review. Int J Appl Pharm. 2013;5(2):8–18. doi: 10.9790/3013-26103444. [DOI] [Google Scholar]
- Khalil I.R., Burns A.T.H., Radecka I. Bacterial-derived polymer poly-γ-glutamic acid (γ-PGA)-based micro/nanoparticles as a delivery system for antimicrobials and other biomedical applications. Int. J. Mol. Sci. 2017;18(2):313. doi: 10.3390/ijms18020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R., Irchhaiya R. Niosomes: a potential tool for novel drug delivery. J Pharm Investig. 2016;46(3):195–204. doi: 10.1007/s40005-016-0249-9. [DOI] [Google Scholar]
- Khan I., Saeed K., Khan I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2017;12:908–931. [Google Scholar]
- Lai P., Daear W., Löbenberg R., Prenner E.J. Overview of the preparation of organic polymeric nanoparticles for drug delivery based on gelatine, chitosan, poly(d,l-lactide-co-glycolic acid) and polyalkylcyanoacrylate. Colloids Surf. B: Biointerfaces. 2014;118:154–163. doi: 10.1016/j.colsurfb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- Lee S.C., Kwon I.K., Park K. Hydrogels for delivery of bioactive agents: a historical perspective. Adv. Drug Deliv. Rev. 2013;65(1):17–20. doi: 10.1016/j.addr.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wang X., Wang Y. A novel osmotic pump-based controlled delivery system consisting of pH-modulated solid dispersion for poorly soluble drug flurbiprofen: in vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2015;41(12):2089–2099. doi: 10.3109/03639045.2015.1078348. [DOI] [PubMed] [Google Scholar]
- Liu X., Wang S., Chai L. A two-step strategy to design high bioavailable controlled-release nimodipine tablets: the push-pull osmotic pump in combination with the micronization/solid dispersion techniques. Int. J. Pharm. 2014;461(1–2):529–539. doi: 10.1016/j.ijpharm.2013.12.023. [DOI] [PubMed] [Google Scholar]
- Liu D., Yu S., Zhu Z. Controlled delivery of carvedilol nanosuspension from osmotic pump capsule: in vitro and in vivo evaluation. Int. J. Pharm. 2014;475(1):496–503. doi: 10.1016/j.ijpharm.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Liu D., Yang F., Xiong F., Gu N. The smart drug delivery system and its clinical potential. Theranostics. 2016;6(9):1306–1323. doi: 10.7150/thno.14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra A.K., Murthy P.N., Swadeep B., Swain R.P. Self-emulsifying drug delivery systems (SEDDS): an update from formulation development to therapeutic strategies. Int J PharmTech Res. 2014;6(2):546–568. [Google Scholar]
- Mamo B. Literature review on Biodegradable Nanospheres for Oral and Targeted Drug delivery. Asian J Biomed Pharm Sci. 2015;5(51):01–12. doi: 10.15272/ajbps.v5i51.761. [DOI] [Google Scholar]
- Martinelli C., Pucci C., Ciofani G. Nanostructured carriers as innovative tools for cancer diagnosis and therapy. APL Bioeng. 2019;3(1) doi: 10.1063/1.5079943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbah C.C., Builders P.F., Attama A.A. Nanovesicular carriers as alternative drug delivery systems: ethosomes in focus. Expert Opin Drug Deliv. 2014;11(1):45–59. doi: 10.1517/17425247.2013.860130. [DOI] [PubMed] [Google Scholar]
- MH a Makadia, Bhatt M.A.Y., Parmar R.B., Paun M.J.S., Tank H.M. Self-nano Emulsifying Drug delivery System (SNEDDS): Future Aspects. Asian JPharm Res. 2013;3(1):21–27. [Google Scholar]
- Missaghi S., Patel P., Farrell T.P., Huatan H., Rajabi-Siahboomi A.R. Investigation of critical Core Formulation and Process Parameters for Osmotic Pump Oral Drug delivery. AAPS PharmSciTech. 2014;15(1):149–160. doi: 10.1208/s12249-013-0040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu H., Holm R., Mul̈lertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013;453(1):215–224. doi: 10.1016/j.ijpharm.2013.03.054. [DOI] [PubMed] [Google Scholar]
- Namdeo G.S., Nagesh H.A., Ajit S.K. Recent advances in Vesicular Drug delivery System. Res J Pharm Dos Forms Technol. 2014;6(2):110–120. [Google Scholar]
- Naseri N., Valizadeh H., Zakeri-Milani P. Solid lipid nanoparticles and nanostructured lipid carriers: Structure preparation and application. Adv Pharm Bull. 2015;5(3):305–313. doi: 10.15171/apb.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neamtu I., Rusu A.G., Diaconu A., Nita L.E., Chiriac A.P. Basic concepts and recent advances in nanogels as carriers for medical applications. Drug Deliv. 2017;24(1):539–557. doi: 10.1080/10717544.2016.1276232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parashar T., Sachan R., Singh V., Singh G., Tyagi S. International Journal of Research and Development in Pharmacy and Life Sciences. Vol. 2. 2013. Patel C. Ethosomes : A Recent Vesicle Of Transdermal Drug Delivery System; pp. 285–292. [Google Scholar]
- Patel H.J., Parikh V.P. An overview of Osmotic Drug delivery System: an update review. Int J Bioassays. 2017;6(7):5426. doi: 10.21746/ijbio.2017.07.001. [DOI] [Google Scholar]
- Pathak C.V., Gujarathi N.A., Rane B.R., Pawar S.P. A review on self microemulsifying drug delivery system. Pharma Sci Monit. 2013;4(1):3628–3648. [Google Scholar]
- Patil Y.P., Jadhav S. Novel methods for liposome preparation. Chem. Phys. Lipids. 2014;177:8–18. doi: 10.1016/j.chemphyslip.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Patra C.N., Swain S., Sruti J. Osmotic Drug delivery Systems: Basics and Design Approaches. Recent Pat Drug Deliv Formul. 2013;7(2):150–161. doi: 10.2174/1872211311307020007. [DOI] [PubMed] [Google Scholar]
- Patra J.K., Das G., Fraceto L.F. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnol. 2018;16(1):1–33. doi: 10.1186/s12951-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattni B.S., Chupin V.V., Torchilin V.P. New Developments in Liposomal Drug delivery. Chem. Rev. 2015;115(19):10938–10966. doi: 10.1021/acs.chemrev.5b00046. [DOI] [PubMed] [Google Scholar]
- Poovi G., Damodharan N. Lipid nanoparticles: a challenging approach for oral delivery of BCS Class-II drugs. Futur J Pharm Sci. 2018;4(2):191–205. doi: 10.1016/J.FJPS.2018.04.001. [DOI] [Google Scholar]
- Rahman M.A., Hussain A., Hussain M.S., Mirza M.A., Iqbal Z. Role of excipients in successful development of self-emulsifying/ microemulsifying drug delivery system (SEDDS/SMEDDS) Drug Dev. Ind. Pharm. 2013;39(1):1–19. doi: 10.3109/03639045.2012.660949. [DOI] [PubMed] [Google Scholar]
- Ranjan O.P., Nayak U.Y., Reddy M.S., Dengale S.J., Musmade P.B., Udupa N. Osmotically controlled pulsatile release capsule of montelukast sodium for chronotherapy: Statistical optimization, in vitro and in vivo evaluation. Drug Deliv. 2014;21(7):509–518. doi: 10.3109/10717544.2013.853209. [DOI] [PubMed] [Google Scholar]
- Ratnaparkhi P., Jyoti G.P., Mukesh Sustained Release Oral Drug delivery System -an Overview. Int J Pharma Res Rev IJPRR. 2013;2(23):11–21. [Google Scholar]
- Ravar F., Saadat E., Gholami M. Hyaluronic acid-coated liposomes for targeted delivery of paclitaxel, in-vitro characterization and in-vivo evaluation. J. Control. Release. 2016;229:10–22. doi: 10.1016/j.jconrel.2016.03.012. [DOI] [PubMed] [Google Scholar]
- Rizvi S.A.A., Saleh A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm J. 2018;26(1):64–70. doi: 10.1016/j.jsps.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachan R., Parashar T., Singh V., Singh G., Tyagi S., Patel C. Drug carrier transfersomes: a novel tool for transdermal drug delivery system. Int J Res Dev Pharm LIFE Sci. 2013;2(2):309–316. www.ijrdpl.com (Accessed May 22, 2019) [Google Scholar]
- Salman Z.D., Maraie N.K., Alabbassi M.G., Ghareeb M.M. In Vitro/in Vivo Evaluation and Bioavailability Study of Amitriptyline Hydrochloride from the Optimized Oral Fast Dissolving Films. UK J Pharm Biosci. 2014;2(6):32. doi: 10.20510/ukjpb/2/i6/91171. [DOI] [Google Scholar]
- Sarmah Prasurjya Jyoti. Bhupen Kalita AKS. Transfersomes based transdermal drug delivery: an overview. Int J Adv Pharm Res. 2013;4(12):2555–2563. [Google Scholar]
- Sayed S., Ibrahim H.K., Mohamed M.I., El-Milligi M.F. Fast-dissolving sublingual films of terbutaline sulfate: Formulation and in vitro/in vivo evaluation. Mol. Pharm. 2013;10(8):2942–2947. doi: 10.1021/mp4000713. [DOI] [PubMed] [Google Scholar]
- Sharma A., Kumar D., Painuly N. Formulation and Evaluation of Sublingual Tablet of Losartan Potassium. Asian J Pharm Res Dev. 2018;6(4):101–109. doi: 10.22270/ajprd.v6i4.383. [DOI] [Google Scholar]
- Shilakari G., Singh D., Asthana A. Novel vesicular carriers for topical drug delivery and their application’s. Int J Pharm Sci Rev Res. 2013;21(1):77–86. [Google Scholar]
- Shreya A.B., Managuli R.S., Menon J. Nano-transfersomal formulations for transdermal delivery of asenapine maleate: in vitro and in vivo performance evaluations. J Liposome Res. 2016;26(3):221–232. doi: 10.3109/08982104.2015.1098659. [DOI] [PubMed] [Google Scholar]
- Singh K., Walia M.K., Agarwal G., Harikumar S.L. Osmotic pump drug delivery system: a noval approach. J Drug Deliv Ther. 2016;3(5):156–162. doi: 10.22270/jddt.v3i5.636. [DOI] [Google Scholar]
- Sowjanya M., Venkata Prasada Rao Ch., P Srinivasa Babu P.K. Osmotic drug delivery systems: a review. Pharma Times. 2017;2017(3):1–9. [Google Scholar]
- Syed Shoaeb Mohammad, Farooqui Z., Mohammed M., Dureshahwar K., Farooqui M. Osmotic Drug delivery System: an Overview. Int J Pharm Res Allied Sci. 2015;4(3):10–20. [Google Scholar]
- Tayel S.A., El Nabarawi M.A., Amin M.M., Abou Ghaly M.H. Sumatriptan succinate sublingual fast dissolving thin films: Formulation and in vitro/in vivo evaluation. Pharm. Dev. Technol. 2016;21(3):328–337. doi: 10.3109/10837450.2014.1003655. [DOI] [PubMed] [Google Scholar]
- Thakor A.S., Gambhir S.S. Nanooncology: the future of cancer diagnosis and therapy. CA Cancer J. Clin. 2013;63(6):395–418. doi: 10.3322/caac.21199. [DOI] [PubMed] [Google Scholar]
- Ullah F., Othman M.B.H., Javed F., Ahmad Z., Akil H.M. Classification, processing and application of hydrogels: a review. Mater. Sci. Eng. C. 2015;57:414–433. doi: 10.1016/j.msec.2015.07.053. [DOI] [PubMed] [Google Scholar]
- Vashist A., Vashist A., Gupta Y.K., Ahmad S. Recent advances in hydrogel based drug delivery systems for the human body. J. Mater. Chem. B. 2014;2(2):147–166. doi: 10.1039/c3tb21016b. [DOI] [PubMed] [Google Scholar]
- Vijaya Shanti B., Mrudula T.P.K.V. An Imperative note on Novel Drug delivery Systems. J Nanomed Nanotechnol. 2011;2(7) doi: 10.4172/2157-7439.1000125. [DOI] [Google Scholar]
- Xiong Y., Zou Y., Chen L., Xu Y., Wang S. Development and in Vivo Evaluation of Ziyuglycoside I–Loaded Self-Microemulsifying Formulation for activity of increasing Leukocyte. AAPS PharmSciTech. 2019;20(3) doi: 10.1208/s12249-019-1313-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav H.K.S., Anwar N., Halabi A., Alsalloum G.A. Nanogels as Novel Drug delivery Systems - a Review Properties of Nanogels Keywords : Introduction Advantages of Nanogels. Insight Pharma Res. 2017;1(5):1–8. http://www.imedpub.com/articles/nanogels-as-novel-drug-delivery-systems--a-review.php?aid=18950 (Accessed May 20, 2019) [Google Scholar]
- Yang Y., Zhao Z., Wang Y. A novel asymmetric membrane osmotic pump capsule with in situ formed delivery orifices for controlled release of gliclazide solid dispersion system. Int. J. Pharm. 2016;506(1–2):340–350. doi: 10.1016/j.ijpharm.2016.04.061. [DOI] [PubMed] [Google Scholar]
- Yoon G., Park J.W., Yoon I.S. Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs): recent advances in drug delivery. J Pharm Investig. 2013;43(5):353–362. doi: 10.1007/s40005-013-0087-y. [DOI] [Google Scholar]
- Yu S., Wang Q.M., Wang X. Liposome incorporated ion sensitive in situ gels for opthalmic delivery of timolol maleate. Int. J. Pharm. 2015;480(1–2):128–136. doi: 10.1016/j.ijpharm.2015.01.032. [DOI] [PubMed] [Google Scholar]
- Yurgel V., Collares T., Seixas F. Developments in the use of nanocapsules in oncology. Braz. J. Med. Biol. Res. 2013;46(6):486–501. doi: 10.1590/1414-431X20132643. [DOI] [PMC free article] [PubMed] [Google Scholar]