

Abstract
Introduction
The plate counting method widely used at present to discern viable from non-viable Brucella in the host or cell is time-consuming and laborious. Therefore, it is necessary to establish a rapid, simple method for detecting and counting viable Brucella organisms.
Material and Methods
Using propidium monoazide (PMA) to inhibit amplification of DNA from dead Brucella, a novel, rapid PMA-quantitative PCR (PMA-qPCR) detection method for counting viable Brucella was established. The standard recombinant plasmid with the target BCSP31 gene fragment inserted was constructed for drawing a standard curve. The reaction conditions were optimised, and the sensitivity, specificity, and repeatability were analysed.
Results
The optimal exposure time and working concentration of PMA were 10 min and 15 μg/mL, respectively. The correlation coefficient (R2) of the standard curve was 0.999. The sensitivity of the method was 103 CFU/mL, moreover, its specificity and repeatability also met the requirements. The concentration of B. suis measured by the PMA-qPCR did not differ significantly from that measured by the plate counting method, and the concentrations of viable bacteria in infected cells determined by the two methods were of the same order of magnitude.
Conclusion
In this study, a rapid and simple PMA-qPCR counting method for viable Brucella was established, which will facilitate related research.
Keywords: Brucella, viable bacteria, BCSP31gene, propidium monoazide, quantitative PCR
Introduction
Brucellosis is one of the zoonotic infections caused by the intracellular Brucella bacteria infecting humans and a variety of livestock and wildlife (11, 12). Diseased animals always show related clinical symptoms like night sweats, joint pain, abortion in pregnant females, and testicular inflammation in males. Brucellosis leads to huge economic losses and serious public health problems, and has been classified as a class B infectious disease in China (28, 29). The drawbacks of the conventional brucellosis testing methods such as bacteriology and serology are their heavy time demand and their production of false positives to some extent. Molecular biology technology has been widely used in the detection of pathogenic bacteria due to its rapidity and accuracy. With the development of the research on pathogenic mechanism of Brucella, it is increasingly important to investigate the equilibrium between the status and quantity of intracellular Brucella and the host or cell functions. In particular, the presence or absence of viable Brucella in the body or cells has become one of the most important indicators for assessing the virulence of Brucella (15) and the existence of brucellosis or effectiveness of vaccination against it (1). At present, a plate counting method is used for finding the number of viable Brucella, but it has some drawbacks like a heavy time demand, labour-intensiveness, and a personal subjectivity factor (16). Additionally, in vivo or intracellular bacteria which are viable but not culturable (VBNC) need an unconventional counting plate for bacterial culture. Although the traditional plate counting method is considered to be an effective method for counting viable bacteria, it cannot completely reflect the actual number of viable cells as
VBNC bacteria cannot be identified in this way (27, 30). Therefore, it is necessary to establish a rapid, accurate, and simple method for determining the viable number of Brucella for the demands of studying the intracellular survival mechanism of Brucella and evaluating the protective capability of attenuated brucellosis vaccines.
It was demonstrated in previous reports that the BCSP31 gene of the 31 kDa Brucella cell surface protein, an important outer membrane structural protein, is present and conserved in the genome of all Brucella species (2, 17). Many researchers used the BCSP31 gene as an important target molecule for detection and identification of Brucella (3). In this study, we detected viable Brucella by targeting its BCSP31 gene and did so by combining qPCR with propidium monoazide (PMA). PMA is a DNA-binding dye which can enter the small groove of DNA molecules. It can be photolysed by bright visible light to produce a nitrene, which is able to form a covalent cross-link with the PMA-bound DNA to inhibit its PCR amplification (18). Usually, dead bacteria do not have a complete membrane structure; thereby, PMA can only penetrate dead bacterial cell membranes to inhibit DNA amplification. As a result, viable bacteria can be specifically detected using PMA-PCR (4). Some reports show that excessive PMA has a suppressive effect on the DNA amplification and detection of viable cells, but excessive PMA can be inactivated by exposure to bright light, guaranteeing viable bacterial DNA amplification. Therefore, it is necessary to optimise the working concentration and exposure time of PMA (19, 24).
Since the advent of real-time quantitative PCR (qPCR) technology, it has been widely used in the detection of pathogenic bacteria due to its time and labour resource economy and high accuracy. By analysing the sensitivity, specificity, and repeatability of a novel, rapid, and simple PMA-qPCR method for detecting and counting viable Brucella, a new laboratory tool was provided. The PMA-qPCR method will facilitate rapid viable intracellular bacteria counting in the Brucella-related study fields.
Material and Methods
Bacteria, cells, and chemicals. Brucella ovis (BNCC 340736), attenuated B. suis strain S2 (CVCC 22), a model Escherichia coli strain (CMCC 44817), Salmonella typhimurium (CGMCC 1.1194), Yersinia enterocolitica (CMCC 52208), Vibrio parahaemolyticus (CGMCC 1.1616), and murine macrophage cell line RAW264.7 were available from the Institute of Zoonosis, Jilin University (Changchun, China). The extracted genomic DNAs from B. abortus strains 2308 and A19 and B. melitensis strains M5 and 16M were provided by the Institute of Veterinary Medicine (Changchun, China). B. suis S2 strain was selected as the experimental strain, and others were control strains. A bacterial genomic DNA extraction kit, TIANgel MIdi purification kit, and plasmid extraction purification kit were purchased from TIANGEN BIOTECH Co. (Beijing, China). Propidium bromide (PMA) was sourced from Biotium (Fremont, CA, USA). Fast Start Universal SYBR Green Master (Rox) was obtained from Roche (Basel, Switzerland). Brucella broth was ordered from Qingdao Rishui Biotechnology Co. (Qingdao, China). Triton X-100 was procured from Shanghai Shenggong Technology Co. (Shanghai, China).
Primer design. A pair of specific primers for an amplified fragment of 200 bp in length was designed using Premier Primer 5 software and synthesised by Comate Biosciences Co. (Changchun, China). They were based on the sequence of Brucella outer membrane protein gene BCSP31 in the NCBI GenBank with accession number M204404.1 and comprised the upstream sequence 5′-AAACATCAAATCGGTCGC-3′ and the downstream sequence 5′-CCGCCCACAAAG AAATAG-3′.
Extraction of genomic DNA from bacteria. The Brucella strain S2 was inoculated into 5 mL trypticase soy broth (TSB) medium, and the other control bacteria were inoculated into 5 mL Luria-Bertani (LB) broth medium. After shaking at 37°C for 12 h, the genome DNA was extracted as a template for the qPCR using the bacterial genomic DNA extraction kit according to the manufacturer’s instructions, and its concentration was determined by an Epoch microplate reader (BioTek, Winooski, VT, USA). The sample was stored at −20°C.
Preparation of recombinant plasmid standards and establishment of standard curve. The target fragment was amplified by PCR using the extracted genome DNA as a template. The PCR product was analysed by 1% agarose gel electrophoresis and purified by the TIANgel MIdi Purification kit. After the target DNA fragment was ligated into the pMD-18T vector (TaKaRa Bio, Dalian, China) (13, 26); the identified positive bacteria with the standard recombinant plasmid were sequenced. The concentration of plasmids was determined, and the corresponding copy number of the standard plasmids was calculated according to the following formula (1):
As the BCSP31 gene is a single copy gene in Brucella genome (23), the copy concentration of the standard plasmids is equal to the concentration of Brucella.
The annealing temperature and primer concentration were optimised respectively using 50– 60°C and 0.25–0.75 μmol/L. The standard plasmids were diluted with double distilled water (ddH2O) in a tenfold series, and the qPCR reaction mixture was composed of 10 μL of Fast Start Universal SYBR Green Master (Rox) (Roche, Mannheim, Germany), 2 μL of standard plasmid template, 1 μL of upstream and downstream primers separately, and 6 μL of ddH2O in a total volume of 20 μL. The optimal reaction conditions for qPCR were judged by performing said qPCR with pre-denaturation at 94°C for 2 min and 40 cycles of denaturation at 94°C for 45 s, annealing at 55°C for 30 s, and extension at 72°C for 32 s. PCR was repeated with each sample three times. The logarithm of the copy number concentration was plotted as the abscissa (X axis), the cycle threshold (CT) was plotted as the ordinate (Y axis), and the amplification efficiency was calculated according to the formula:
where E is the amplification efficiency, and k the slope of the standard curve (2).
Optimisation of PMA treatment. The extent of PMA binding DNA is associated with the exposure time and the concentration of PMA, which need to be optimised (7, 19). The optimal condition of completely inhibiting the DNA amplification of dead bacteria by PMA was determined by qPCR described as above. Firstly, heat-killed bacteria of B. suis S2 strain were treated with PMA to a final concentration of 15 μg/mL, and a part of the same bacteria were left untreated with PMA as a control. The optimal exposure time was determined by exposing the samples to a 650 W halogen lamp (OSRAM, Munich, Germany) for different times, following which the qPCR reactions were carried out. And then, the heat-killed bacteria of B. suis S2 strain with added PMA to different final concentrations were exposed for the optimal exposure time to determine the optimal concentration of PMA completely inhibiting the DNA amplification of dead bacteria by the qPCR. Additionally, the viable bacteria of B. suis S2 strain had PMA added to different final concentrations, and the optimal PMA concentration unaffecting the amplification of live bacterial genomic DNA was determined by qPCR following the optimal exposure time as described above.
Sensitivity analysis of PMA-qPCR detection method. The optimal PMA-treatment conditions were applied to the known concentration of B. suis S2 strain, and then the bacterial genomic DNAs were extracted. After that, the extracted DNAs diluted in a tenfold series were used as the template for detection by the qPCR described above. The sensitivity of the PMA-qPCR method was analysed in comparison with the PMA-treatment-free conventional qPCR described above and the conventional PCR described as follows. The reaction mixture was composed of 10 μL of Premix Taq (Ex Taq Version 2.0) (TaKaRa Bio), 2 μL of the extracted genomic DNA template, 1 μL of upstream and downstream primers separately, and 6 μL of ddH2O in a total volume of 20 μL, and the reaction conditions were: pre-denaturation at 94°C for 2 min; 40 cycles of denaturation at 94°C for 45 s, annealing at 55°C for 35 s, and extension at 72°C for 40 s; and final extension at 72°C for 3 min.
Specificity analysis of the PMA-qPCR detection method. The extracted genomic DNAs from B. abortus (strains 2308 and A19), B. melitensis (strains M5 and 16M), B. ovis, E. coli, S. typhimurium, Y. enterocolitica, and V. parahaemolyticus were used to analyse the specificity of the PMA-qPCR method. RNase-free water was used as a negative control, and the conventional PCR experiments for analysing the method specificity were also conducted.
Repeatability analysis of the PMA-qPCR detection method. The DNA was extracted from the known concentration of B. suis S2 strain treated with the optimal concentration of PMA, and diluted in a series of 105, 106, and 107 as templates for qPCR. The repeatability of the PMA-qPCR method was determined, and the coefficient of variation (CV) was calculated from inter- and intra-assay PMA-qPCR data.
Counting viable bacteria stained with the different ratios of dead bacteria by PMA-qPCR. Viable B. suis at a known concentration was mixed with dead bacteria in different proportions. DNA extracted from 1 mL of each viable bacterial proportion treated with PMA was used for the PMA-qPCR detection, and the same from each viable bacterial proportion without PMA was used as a control.
Comparison of PMA-qPCR and plate counting methods for viable bacteria. The concentration of viable B. suis S2 strain cultured in TSB (named pure bacterial liquid) was measured by the PMA-qPCR and plate counting methods, which were compared by using them to quantitatively compare and analyse the numbers of viable B. suis in infected mouse macrophage RAW 264.7 cells. Each well of a 12-well cell culture plate was inoculated with 2 × 105 of mouse macrophage RAW 264.7 cells in DMEM with 10% foetal bovine serum (FBS) containing 100 units/mL of penicillin and 100 μg/mL of streptomycin, and the DMEM with penicillin-streptomycin was changed for new penicillin-streptomycin-free DMEM (10% FBS) at 1 h before infection. Then, RAW264.7 cells were infected with B. suis S2 strain and incubated for 1 h under the conditions of 37°C and 5% CO2. After that, the cells were washed twice with DMEM, and the infected macrophages were collected for intracellular viable bacteria counting by the PMA-qPCR and standard plate count methods (25). Cells in a 12-well cell-culture plate were infected with 100 μL of different concentrations of B. suis S2 strain in two groups, A and B (group A with 1.28 × 1012 CFU/mL and group B with 0.64 × 1012 CFU/mL). A total of six replicate wells were set up in each group (three were analysed by the PMA-qPCR assay and three by the plate count method). The PMA-qPCR detection was carried out according to the optimal PMA treatment conditions, and the concentration of the viable bacteria was calculated according to the standard curve of the relationship between the CT values and bacterial concentrations.
Statistical analysis. The obtained data were statistically analysed through Student’s t-test or one-way analysis of variance (ANOVA) using SPSS 19.0 software (IBM SPSS, Armonk, NY, USA).
Results
Preparation of plasmid standards and standard curve. The 200 bp of PCR product amplified from genomic DNA of B. suis was consistent with the expected results (Fig. 1). The sequencing results also showed that the recombinant plasmid pMD-18T-BCSP31 was constructed successfully. The concentration of the extracted recombinant plasmid was 398.408 ng/μL, and the calculated copy number was approximately 6.28 × 1010 copies/μL according to the formula (1). The optimum annealing temperature was determined as 55°C by the gradient PCR. Based on 6.28 × 100–6.28 × 108 copies/μL of the gradient copy concentration of standard plasmids diluted in a tenfold series, the standard curve was drawn according to the results of the qPCR detecting amplification (Fig. 2) with the regression equation y = -3.492x + 36.303, where the y represents the CT value and x the logarithm of the gene copy concentration. The correlation coefficient calculated from the regression equation is R2 = 0.999, the linear relationship between the CT value and the logarithm of the copy concentration fully met the requirement, and the limit of detection was 6.28 × 100 copies/μL. According to the formula (2), the amplification efficiency was 93.36%.
Fig. 1.
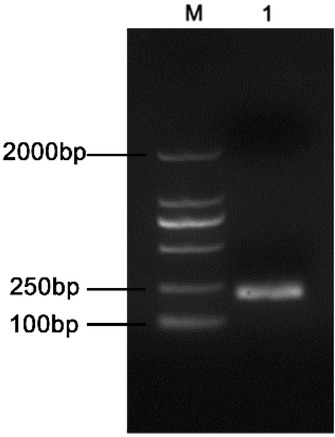
The target BCSP31 fragment of PCR amplification from Brucella suis S2 strain. M – DL2000 DNA Marker (TaKaRa Bio); lane 1 – DNA fragment of PCR amplification
Fig. 2.
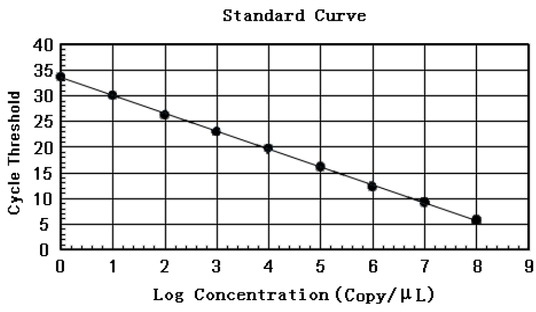
The standard curve for calculating Brucella concentration according to CT of qPCR based on the template of recombinant standard plasmid pMD-18T-BCSP31
Optimal PMA conditions. The exposure time and concentration of PMA were optimised (Fig. 3). As shown in Fig. 3A, the CT value increased continuously as the exposure time increased. There were no significant differences in CT values at each exposure time point equal to or longer than 10 min (P > 0.05). The concentration of PMA fully inhibiting the DNA amplification of the dead bacteria was ≥15 μg/mL, as shown in Fig. 3B. While the concentrations of PMA unaffecting the amplification of viable bacteria DNAs were shown in Fig. 3C, there was no significant difference in the CT value of each PMA concentration (P > 0.05) ranging from 0 to 15 μg/mL, and the CT value increased significantly as the PMA concentration increased continuously over 20 μg/mL. It was indicated that higher concentrations of PMA affected the detection of viable bacteria. Therefore, it was determined that the optimal conditions of the exposure time and working concentration of PMA were respectively 10 min and 15 μg/mL.
Fig. 3.
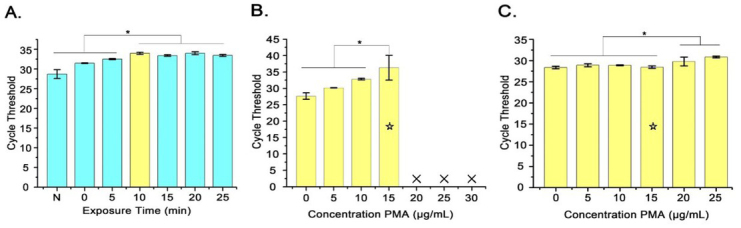
Optimisation of PMA treatment conditions. A – Effect of different exposure time on qPCR amplification; B – Different concentrations of PMA treating dead bacteria; C – Different concentrations of PMA treating viable bacteria; * – P < 0.05; Yellow columns – optimal PMA treatment time of 10 min; Yellow columns with ☆ – optimal PMA treatment concentration of 15 μg/mL; × – not detected
Sensitivity. Comparing different bacterial concentrations ranging from 103 CFU/mL to 108 CFU/mL measured by the PMA-qPCR and PMA-free conventional qPCR (Fig. 4A), their CT values were similar. Nevertheless, there was a slight discrepancy between the CT values of the two methods. The standard curve for the PMA-qPCR showed a good linear relationship (R2 = 0.986 and 0.991) with a sensitivity of 103 CFU/mL of bacteria (Fig. 4A) and consistency with the sensitivity of the standard curve constructed by plasmid standards (Fig. 2), while the detection sensitivity of the conventional PCR (Fig. 4B) was 105 CFU/mL. It was confirmed that the sensitivity of the PMA-qPCR for detection of B. suis S2 strain was 100 times higher than that of the conventional PCR, and that viable Brucella could be better quantified by the PMA-qPCR than by the qPCR.
Fig. 4.
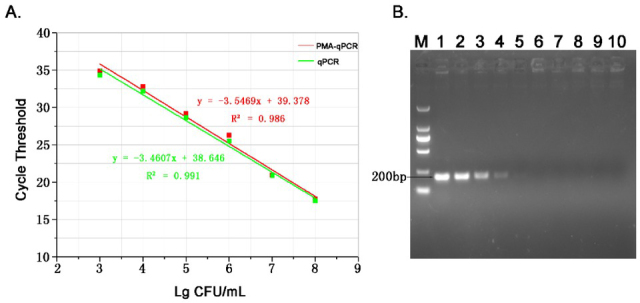
The sensitivity of detecting B. suis S2 strain. A – sensitivity by the PMA-qPCR; B – sensitivity by the conventional PCR, comprising M – DL2000 DNA Marker; lanes 1–10 – 108 – 10-1 CFU/mL
Specificity. Different bacterial genomic DNAs and RNase-free water were used as templates for amplification to analyse the specificity of the PMA-qPCR method. As seen in Fig. 5, genomic DNAs of different Brucella species including B. suis, B. abortus, B. melitensis, and B. ovis could be amplified, while amplification results of bacteria other than Brucella and water were negative. Meanwhile, there were no primer dimers or non-specific amplification, with the melting curve showing a single peak (Fig. 5B) at 87.87 – 88.36°C, which was the Tm value of the qPCR amplification product. It was indicated that the specificities of the primers and the method met the detection requirement. The common PCR also confirmed that the PMA-qPCR detection method was specific (Figs 5C and 5D).
Fig. 5.
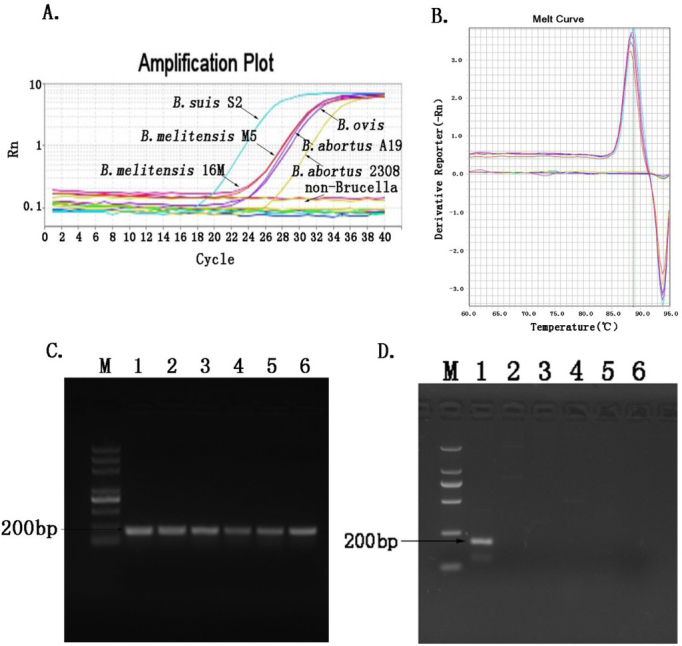
Analysis of the specificity of PMA-qPCR. A – amplification plot; B – melting curves of the qPCR amplification product; C – agarose gel electrophoresis of different Brucella species detected by the normal PCR, comprising M – DL5000 DNA Marker; lane 1 – B. suis S2; lane 2 – B. abortus 2308; lane 3 – B. abortus A19; lane 4 – B. melitensis M5; lane 5 – B. melitensis 16M; and lane 6 – B. ovis; D – agarose gel electrophoresis of species other than Brucella amplified by the conventional PCR, comprising M – DL2000 DNA Marker; lane 1 – B. suis S2; lane 2 – E. coli; lane 3 – S. typhimurium; lane 4 – Y. enterocolitica; lane 5 – V. parahaemolyticus; and lane 6 – RNase-free water
Repeatability. Three dilutions (105, 106, and 107) of bacterial DNAs were selected for intra- and inter-assay experiments to determine the repeatability and coefficient of variation (CV) of the PMA-qPCR method. As Table 1 shows, the stability and repeatability of the PMA-qPCR method met the detection requirements, borne out by the CVs ranging from 0.63 % to 2.04 % in the experimental results.
Table 1.
Coefficient of variation for intra- and inter-batch experiments
| DNA dilution times | Intra-assay | Inter-assay | ||||
|---|---|---|---|---|---|---|
| Average value of CT | SD | CV | Average value of CT | SD | CV | |
| 107 | 17.55277 | 0.13205 | 0.75 | 17.95203 | 0.26862 | 1.50 |
| 106 | 21.9137 | 0.26965 | 1.23 | 22.35123 | 0.45614 | 2.04 |
| 105 | 23.80697 | 0.15085 | 0.63 | 23.82857 | 0.43607 | 1.83 |
SD – standard deviation; CV – coefficient of variation
Detection of different ratios of viable bacteria to dead bacteria by PMA-qPCR. Viable or dead bacteria of known concentrations were mixed in different proportions for PMA-qPCR detection, and samples without PMA treatment served as controls. It can be observed from Fig. 6 that the amplification of the dead bacterial DNAs was completely inhibited by PMA, as the CT value could not be detected when the viable bacterial proportion in the bacterial mixture was zero. The CT value gradually decreased as the proportion of viable bacteria increased, but still was higher than the CT value in the PMA-untreated groups. When the content of viable bacteria was 100%, there was no significant difference of CT value between the PMA treatment and PMA-untreated groups detected by PMA-qPCR (P > 0.05). It indicated that PMA-qPCR could accurately detect the viable bacteria regardless of the presence or lack of dead bacteria.
Fig. 6.
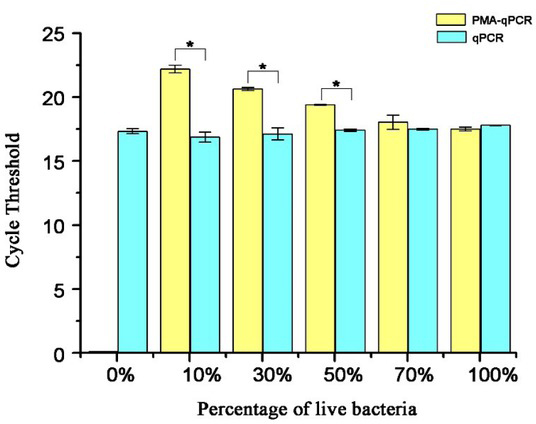
Analysis of different ratios of viable to dead B. suis S2 strain using the PMA-qPCR method and the qPCR method. * – P < 0.05
Comparison of PMA-qPCR and plate counting methods for detection of viable bacteria. As Fig. 7 shows, the concentration of B. suis S2 strain cultured in TSB was measured by the PMA-qPCR and plate counting methods without significant differences (P = 0.07). Subsequently, the number of viable bacteria determined by the two methods were significantly different with P = 0.048 in group A and P = 0.042 in group B among S2-infected cells. However, the numbers of viable cells assayed by the two methods were the in same order of magnitude and showed a consistent trend.
Fig. 7.
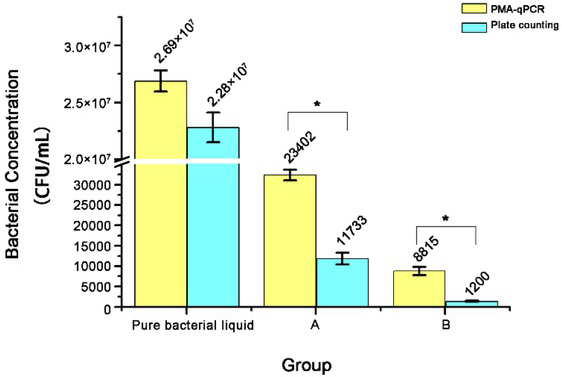
Determination of the amount of the viable B. suis S2 strain by PMA-qPCR and plate counting methods. Pure bacterial liquid – B. suis cultured in TSB; A – B. suis harbouring in mouse macrophage RAW 264.7 cells infected with 1.28 × 1012 CFU/mL of B. suis S2 strain; B – B. suis harbouring in mouse macrophage RAW 264.7 cells infected with 0.64 × 1012 CFU/mL of B. suis S2 strain; * P < 0.05
Discussion
At present, bacteriology, serology, and molecular biology methods are widely used for detection of Brucella spp. and diagnosis of brucellosis. The “gold standard” for the detection of Brucella, bacteriological testing, has the disadvantage of being a time-consuming and dangerous operation. Serological tests mainly include the Rose Bengal test (RBT), milk ring test (MRT), serum agglutination test (SAT), and complement fixation test (CFT). These methods are sensitive and specific, but with shortcomings such as giving false positive results (6, 22). PCR and its derivative technologies are the main detection methods of molecular biology. Traditional PCR can only calculate the starting amount according to the amount of product, and system errors occur easily during these operations, which can only achieve the goal of semi-quantitation. Real-time quantitative PCR can monitor the fluorescence signal of each round of DNA amplification to achieve absolute or relative quantification, and due to its higher sensitivity and accuracy, the real-time PCR method is currently widely used in quantitative detection (5). The practice of detecting Brucella long-used in research on the pathogenic mechanism of Brucella, especially for counting viable Brucella in cell cultures, the plate counting method, is typically time-consuming, laborious, and experience-dependent. Therefore, a rapid, simple, and easily standardised detection method of counting viable Brucella is urgently needed. Outer membrane protein 25 (omp25), 16S rRNA, IS711, and BCSP31 are the main target genes used for detection of Brucella. Since Baily et al. (2) firstly selected BCSP31 as a target to detect Brucella spp. in 1992, a single copy of BCSP31 gene has been used for Brucella nucleic acid detection, due to its specificity, sensitivity, stability, and its sequence being highly conserved. This was confirmed by many reports (8, 21). In this study, we selected BCSP31 gene as a detection target molecule.
From the moment Nogva et al. (20) used azide dye and PCR together to detect viable bacteria for the first time, this viable bacterial detection technology has been widely used (9, 10). However, there have been no reports on counting the number of viable Brucella by penetrating damaged cell membranes with PMA to inhibit DNA amplification of dead Brucella. Brucella is an intracellular parasite and as such, when viable Brucella still keep their complete cell membrane structure after being processed by cell lysate, thus PMA cannot bind to the DNA of viable Brucella under the protection of a complete bacterial plasma membrane, while PCR amplification of dead Brucella DNA can be inhibited by PMA binding to it. After the interaction of PMA with DNA from dead bacteria, the free PMA in the system was inactivated through exposure. Then, the viable bacterial DNA was extracted and amplified for detection of viable bacteria. Thus, the interference of the dead bacterial DNA could be eliminated. However, it is necessary to optimise the concentration of PMA, in order to ensure that there is adequate PMA to inhibit dead Brucella DNA amplification, but not so much that it impacts the amplification of viable Brucella DNA. In addition, the exposure can inactivate the free PMA and promote the irreversible covalent cross-linking of DNA and PMA to better inhibit the amplification of the dead Brucella DNA.
The detection sensitivities of PMA-qPCR and PMA-free conventional qPCR are shown by their CT values, which were different but similar. This may result from certain amounts of dead bacteria existing in the sample (Fig. 4A). In addition, quantitative detection of intracellular viable Brucella was performed on Brucella-infected cell samples to investigate the applicability of the established PMA-qPCR method. B. suis S2 strain cultured in TSB was counted at similar bacterial concentrations using the PMA-qPCR and plate counting methods. However, the intracellular viable Brucella concentrations measured by the PMA-qPCR method and the plate counting method in infected cells showed statistically significant difference (P < 0.05), albeit they were of the same order of magnitude. Fig. 7 presents how the bacterial concentration measured by the plate count method was less than that of the PMA-qPCR method. One of the reasons for this may be that personal subjective factors may cause experimental errors in the plate counting method. Also, the cell lysate may have a certain inhibitory effect on the growth of viable bacteria, so it is necessary to culture the bacteria on the plate for more than 48 h to form colonies. Some viable Brucella in infected cells may enter the VBNC state, which can thwart culturing Brucella in vitro on a plate or require longer incubation time to form visible colonies (14). In addition, a higher value measured in PMA-qPCR than in the plate counting method may result from incomplete inhibition by PMA of dead Brucella DNAs due to incomplete exposure of intracellular dead Brucella to the PMA, and from over-reliance on the standard curve of absolute qPCR leading to experimental errors.
Based on the conserved BCSP31 gene of Brucella as the detection target, a novel, rapid, and simple method for counting viable Brucella was established using PMA combined with qPCR technology. The sensitivity of the detection method was 103 CFU/mL with good repeatability and specificity. The time necessary for counting the viable Brucella was greatly shortened, and quantitative and qualitative detection of viable Brucella was completed synchronously. This study made a worthwhile exploration of a new method for rapid detection of viable Brucella counts which will benefit Brucella-related research.
Footnotes
Conflict of Interest
Conflict of Interests Statement: The authors declare that there is no conflict of interests regarding the publication of this article.
Financial Disclosure Statement: This work was supported by grants from the National Key R&D Program of China (nos. 2018YFD0500900 and 2018YFD0500903), the Science and Technology Development Project of Jilin Province, China, and the Fundamental Research Funds for the Central Universities.
Animal Rights Statement: None required.
References
- 1.Arenas-Gamboa A.M., Ficht T.A., Kahl-McDonagh M.M., Gomez G., Rice-Ficht A.C.. The Brucella abortus S19 DeltavjbR live vaccine candidate is safer than S19 and confers protection against wild-type challenge in BALB/c mice when delivered in a sustained-release vehicle. Infect Immun. 2009;77:877–884. doi: 10.1128/IAI.01017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baily G.G., Krahn J.B., Drasar B.S., Stoker N.G.. Detection of Brucella melitensis and Brucella abortus by DNA amplification. J Trop Med Hyg. 1992;95:271–275. [PubMed] [Google Scholar]
- 3.Bounaadja L., Albert D., Chenais B., Henault S., Zygmunt M.S., Poliak S., Garin-Bastuji B.. Real-time PCR for identification of Brucella spp.: a comparative study of IS711, bcsp31 and per target genes. Vet Microbiol. 2009;137:156–164. doi: 10.1016/j.vetmic.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 4.Chang C.W., Lin M.H.. Optimization of PMA-qPCR for Staphylococcus aureus and determination of viable bacteria in indoor air. Indoor Air. 2018;28:64–72. doi: 10.1111/ina.12404. [DOI] [PubMed] [Google Scholar]
- 5.Chen J., Zhao Z., Chen Y., Zhang J., Cao W.. Development and application of a SYBR green real-time PCR for detection of the emerging avian leukosis virus subgroup K. Poult Sci. 2018;97:2568–2574. doi: 10.3382/ps/pey086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dos Santos L.S., Sa J.C., Dos Santos Ribeiro D.L., Chaves N.P., Da Silva Mol J.P., Santos R.L., Da Paixao T.A., De Carvalho Neta A.V.. Detection of Brucella spp. infection through serological, microbiological, and molecular methods applied to buffaloes in Maranhao State, Brazil. Trop Anim Health Prod. 2017;49:675–679. doi: 10.1007/s11250-017-1238-3. [DOI] [PubMed] [Google Scholar]
- 7.Fittipaldi M., Nocker A., Codony F.. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J Microbiol Methods. 2012;91:276–289. doi: 10.1016/j.mimet.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Imaoka K., Kimura M., Suzuki M., Kamiyama T., Yamada A.. Simultaneous detection of the genus Brucella by combinatorial PCR. Jpn J Infect Dis. 2007;60:137–139. [PubMed] [Google Scholar]
- 9.Kibbee R.J., Ormeci B.. Development of a sensitive and false-positive free PMA-qPCR viability assay to quantify VBNC Escherichia coli and evaluate disinfection performance in wastewater effluent. J Microbiol Methods. 2017;132:139–147. doi: 10.1016/j.mimet.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Liang N., Dong J., Luo L., Li Y.. Detection of viable Salmonella in lettuce by propidium monoazide real-time PCR. J Food Sci. 2011;76:M234–M237. doi: 10.1111/j.1750-3841.2011.02123.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu N.N., Liu Z.S., Hu P., Zhang Y., Lu S.Y., Li Y.S., Yang Y.J., Zhang D.S., Zhou Y., Ren H.L.. Full-Length cDNA Cloning, Molecular Characterization and Differential Expression Analysis of Lysophospholipase I from Ovis aries. Int J Mol Sci. 2016;17:1206. doi: 10.3390/ijms17081206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N.N., Liu Z.S., Lu S.Y., Hu P., Li Y.S., Feng X.L., Zhang S.Y., Wang N., Meng Q.F., Yang Y.J.. Full-length cDNA cloning, molecular characterization and differential expression analysis of peroxiredoxin 6 from Ovis aries. Vet Immunol Immunopathol. 2015;164:208–219. doi: 10.1016/j.vetimm.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Liu N.N., Liu Z.S., Lu S.Y., Hu P., Zhang Y., Fu B.Q., Li Y.S., Zhou Y., Zhang Y., Ren H.L.. Isolation and characterisation of peroxiredoxin 6 promoter from sheep Ovis aries. J Vet Res. 2016;60:315–321. [Google Scholar]
- 14.Liu Y., Wang C., Tyrrell G., Li X-F.. Production of Shiga-like toxins in viable but nonculturable Escherichia coli O157:H7. Water Res. 2010;44:711–718. doi: 10.1016/j.watres.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 15.Martirosyan A., Moreno E., Gorvel J.P.. An evolutionary strategy for a stealthy intracellular Brucella pathogen. Immunol Rev. 2015;240:211–234. doi: 10.1111/j.1600-065X.2010.00982.x. [DOI] [PubMed] [Google Scholar]
- 16.Masters C.I., Shallcross J.A., Mackey B.M.. Effect of stress treatments on the detection of Listeria monocytogenes and enterotoxigenic Escherichia coli by the polymerase chain reaction. J Appl Microbiol. 2010;77:73–79. doi: 10.1111/j.1365-2672.1994.tb03047.x. [DOI] [PubMed] [Google Scholar]
- 17.Mayfield J.E., Bricker B.J., Godfrey H., Crosby R.M., Knight D.J., Halling S.M., Balinsky D., Tabatabai L.B.. The cloning, expression, and nucleotide sequence of a gene coding for an immunogenic Brucella abortus protein. Gene. 1988;63:1–9. doi: 10.1016/0378-1119(88)90540-9. [DOI] [PubMed] [Google Scholar]
- 18.Nocker A., Camper A.K.. Novel approaches toward preferential detection of viable cells using nucleic acid amplification techniques. FEMS Microbiol Lett. 2009;291:137–142. doi: 10.1111/j.1574-6968.2008.01429.x. [DOI] [PubMed] [Google Scholar]
- 19.Nocker A., Richter-Heitmann T., Montijn R., Schuren F., Kort R.. Discrimination between live and dead cellsin bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int Microbiol. 2010;13:59–65. doi: 10.2436/20.1501.01.111. [DOI] [PubMed] [Google Scholar]
- 20.Nogva H.K., Drømtorp S.M., Nissen H., Rudi K.. Ethidium monoazide for DNA-based differentiation of viable and dead bacteria by 5′-nuclease PCR. BioTechniques. 2003;34:804–813. doi: 10.2144/03344rr02. [DOI] [PubMed] [Google Scholar]
- 21.Queipo-Ortuno M.I., Colmenero J.D., Reguera J.M., Garcia-Ordonez M.A., Pachon M.E., Gonzalez M., Morata P.. Rapid diagnosis of human brucellosis by SYBR Green I-based real-time PCR assay and melting curve analysis in serum samples. Clin Microbiol Infect. 2005;11:713–718. doi: 10.1111/j.1469-0691.2005.01202.x. [DOI] [PubMed] [Google Scholar]
- 22.Sabrina R., Mossadak H.T., Bakir M., Asma M., Khaoula B.. Detection of Brucella spp. in milk from seronegative cows by real-time polymerase chain reaction in the region of Batna, Algeria. Vet World. 2018;11:363–367. doi: 10.14202/vetworld.2018.363-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanjuan-Jimenez R., Colmenero J.D., Bermudez P., Alonso A., Morata P.. Amplicon DNA melting analysis for the simultaneous detection of Brucella spp. and Mycobacterium tuberculosis complex. Potential use in rapid differential diagnosis between extrapulmonary tuberculosis and focal complications of brucellosis. PLoS One. 2013;8:e58353. doi: 10.1371/journal.pone.0058353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soejima T., Iida K., Qin T., Taniai H., Seki M., Yoshida S.. Method to detect only live bacteria during PCR Amplification. J Clin Microbiol. 2008;46:2305–2313. doi: 10.1128/JCM.02171-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L.L., Chen X.F., Hu P., Lu S.Y., Fu B.Q., Li Y.S., Zhai F.F., Ju D.D., Zhang S.J., Shui Y.M., Chang J., Ma X.L., Su B., Zhou Y., Liu Z.S., Ren H.L.. Host Prdx6 contributing to the intracellular survival of Brucella suis S2 strain. BMC Vet Res. 2019;15:304. doi: 10.1186/s12917-019-2049-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L.L., Lu S.Y., Hu P., Fu B.Q., Li Y.S., Zhai F.F., Ju D.D., Zhang S.J., Su B., Zhou Y., Liu Z.S., Ren H.L.. Construction and activity analyses of single functional mouse peroxiredoxin 6 (Prdx6) J Vet Res. 2019;63:99–105. doi: 10.2478/jvetres-2019-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan M., Xu L., Jiang H., Zhou Z., Zhou S., Zhang L.. PMA-LAMP for rapid detection of Escherichia coli and shiga toxins from viable but non-culturable state. Microb Pathog. 2017;105:245–250. doi: 10.1016/j.micpath.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y.J., Liu Z.S., Lu S.Y., Hu P., Li C., Ahmad W., Li Y.S., Xu Y.M., Tang F., Zhou Y., Ren H.L.. Cloning and differential expression analyses of Cdc42 from sheep. J Vet Res. 2018;62:113–119. doi: 10.1515/jvetres-2018-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y.J., Liu Z.S., Lu S.Y., Li C., Hu P., Li Y.S., Liu N.N., Tang F., Xu Y.M., Zhang J.H.. Molecular cloning, expression and characterization of programmed cell death 10 from sheep Ovis aries. Gene. 2015;558:65–74. doi: 10.1016/j.gene.2014.12.040. [DOI] [PubMed] [Google Scholar]
- 30.Zhong Q., Tian J., Wang B., Wang L.. PMA based real-time fluorescent LAMP for detection of Vibrio parahaemolyticus in viable but nonculturable state. Food Control. 2016;63:230–238. [Google Scholar]